
Direct synthesis of acrylate monomers in heterogeneous continuous flow processes†
Jatuporn
Salaklang
a,
Veronique
Maes
a,
Matthias
Conradi
b,
Rudy
Dams
b and
Tanja
Junkers
 *ac
*ac
aInstitute for Materials Research, Polymer Reaction Design Group, Universiteit Hasselt, Agoralaan, Gebouw D, BE-3590 Diepenbeek, Belgium. E-mail: tanja.junkers@uhasselt.be
bMaterials Resource Division, 3M Belgium BVBA, Haven 1005, Canadastraat 11, BE-2070 Zwijndrecht, Belgium. E-mail: rdams1@mmm.com
cIMEC associated lab division IMOMEC, Wetenschapspark 1, 3590 Diepenbeek, Belgium
First published on 4th December 2017
Abstract
An efficient, safe and convenient laboratory scale continuous flow process for synthesis of (meth)acrylate monomers is described. (Meth)acryloyl chloride is reacted with n-octanol and other alcohols in the presence of triethyl amine in a tubular reactor, resulting in excellent conversions of alcohols to their corresponding esters within 0.5 to 5 minutes of reaction times. The formation of an unwanted side-compound, 3-chloropropionyl ester, is significantly minimized to less than 1% and the formed slurry can be processed without difficulty using an ultrasonication-assisted flow strategy. The synthesis is carried out over a period of 4 h without clogging or pressure build-up, with an operation throughput of 78.6 g h−1 of n-octyl acrylate monomer on laboratory scale, with 95% isolated yield. This procedure provides an efficient alternative synthetic route of functional (meth)acrylate esters, minimizes the side product and facilitates the handling of the formed slurry in the process. Synthesis of various (meth)acrylates is demonstrated, including biobased terpenyl acrylates. This development will play an important role for an on-demand production of (meth)acrylate monomers in fast, reliable and energy-efficient process.
Introduction
Acrylates, methacrylates and their polymers are manufactured on multimillion-ton per year levels due to their increasing global demand and broad utility in consumer products, healthcare, pharmaceuticals and generally the chemical industry.1,2 The process to obtain these functional materials by reaction of an alcohol with (meth)acryloyl chloride is a common procedure. Yet, the reaction remains a significant challenge due to the high reactivity of the acid chloride and the exothermicity of the reaction, which makes the reaction difficult to control.3,4Continuous flow processes are currently used in large volume production of petrochemical and bulk chemicals. Still, companies with a broad product portfolio, especially in the pharmaceutical, fine chemical or specialty materials sector, very often choose more flexible, but less energy and resources efficient batch production over continuous flow processes. During the last decade, the interest in flow technology is also on this scale growing with the increasing availability of new and more versatile flow reactor technology that provide more sustainable and inherently green process approaches.5–7 Also on laboratory scale, continuous flow technology gains more and more interest as reliable and efficient synthesis processes.
The production of the functional acrylates is often carried out via reaction of (meth)acryloyl chloride and alcohols. In a conventional batch procedure, the highly reactive acid chloride is added dropwise in stoichiometric amounts to the alcohol in presence of a non-nucleophilic hydrogen halide acceptor such as pyridine, triethyl amine or 4-N,N dimethylaminopyridine. The reaction temperature must typically be kept low (around 0–5 °C) and slow addition of the acid chloride is required in order to avoid overheating within the reactor, resulting in extended reaction times and lowered product yields due to side product formation.8 Exothermic reactions benefit especially from continuous flow application, as heat is dissipated more efficiently, providing safer synthesis conditions and more stable operation.
The formation of esters in the presence of base in conventional batch procedures is well known.9–12 Use of continuous flow (micro) reactor technology for highly exothermic reactions e.g. the synthesis of acryloyl chloride,13 the condensation of acid halides and alcohols in the presence of base as well as many other known esterification were already reported.14–17 Furthermore, a fully homogenous base-free continuous flow procedure for the condensation of acid chlorides and alcohols was investigated and developed by Van Waes et al.18 This approach is for continuous flow application highly interesting as the HCl salt adducts are typically insoluble and lead to heterogeneous reaction conditions, rendering pumping of reaction mixtures through tubular reactors a challenge. Although this particular study reported the production of the corresponding esters with high isolated yield, the reaction was performed only with certain acid chlorides (benzoyl chloride, butyryl chloride and 4-bromobenzoyl chloride), where the presence of an HCl-acceptor in situ was not required. The presence of HCl gas dissolved in the reaction medium is largely inconsequential for synthesis of saturated esters. For acrylates or methacrylates though, addition of HCl to the double bond can occur, leading to significant side product formation. For the formation of functional (meth)acrylates by reacting alcohols with (meth)acryloyl chloride, presence of an acid acceptor is hence required, as will be demonstrated below. Heterogeneous conditions can in consequence not be avoided, which leads to a major challenge – handling of the slurry-like products and salt formation during the flow process. Via conventional approaches, such type of reaction is considered almost impossible to be carried out in a continuous microflow reactor due to the risk of blockages inside the reaction tube. To overcome this hurdle, and therefore developing a flow process for (meth)acrylate synthesis is of high interest.
In here, we describe a versatile, scalable laboratory process platform for on-demand (meth)acrylate monomer synthesis starting from (meth)acryloyl acid chloride and the corresponding alcohols. Amongst numerous methods for handling slurry products, ultrasonication is reported as a method of choice to prevent reactor fouling and was successfully employed for prevention of clogging.19–23 Synthesis of a selection of (meth)acrylates is presented, including biobased monomers made from terpineols.
Results and discussion
To develop said flow process, different reaction conditions i.e. reagent concentrations, reaction temperature, residence time, solvents and flow setups were evaluated in order to identify the most suitable process conditions. Preliminary information on reaction parameters was first obtained by screening reaction conditions using a chip-based microflow reactor.Screening of reaction conditions in a microreactor
A Labtrix start Flex® microreactor (internal volume 10 or 19.5 μl) was used for screening of the reaction conditions for acrylic ester formation. The flow parameters were chosen as previously reported by Van Waes et al.18 Acryloyl chloride (1) and n-octanol (2, R = C8H17) were condensed in solvent in the absence of a base at 195 °C. The screening was carried out at different reaction times. The results show that a full conversion of the acid chloride was obtained after 5 minutes (Table 1). However, the HCl adduct 4 (octyl 3-chloropropionate) was observed as main compound in the crude product mixture. By shortening the reaction time, the amounts of adduct compound could be reduced, but not eliminated. HCl addition to the double bond is at high temperatures clearly a kinetically competitive reaction to the esterification.Next, the influence of temperature was investigated; prior to reaction, n-octanol was first diluted in a dry THF due to its high viscosity at low T. At 10 °C, the starting material was not consumed completely in the reaction, leading to a low product conversion within the tested reaction time window. Slightly higher temperature were thus required. Esterification proceeds still at a considerable rate at 25 °C, while HCl addition is significantly slowed down. A conversion from acid chloride to acrylate of 92% could be reached at 1 min residence time. Yet, complete avoidance of side product 4 formation could not be reached. Extension of residence times only lead to a further conversion of product 3 into the unwanted compound 4. The process hence yields reasonable yields, yet requires tedious product isolation via distillation, which must be avoided for a truly efficient process. As the data for 10 °C demonstrate, lowering of temperature and accepting longer residence times does not solve the problem of side product formation, as the extended reaction times also lead to HCl addition (Scheme 1).
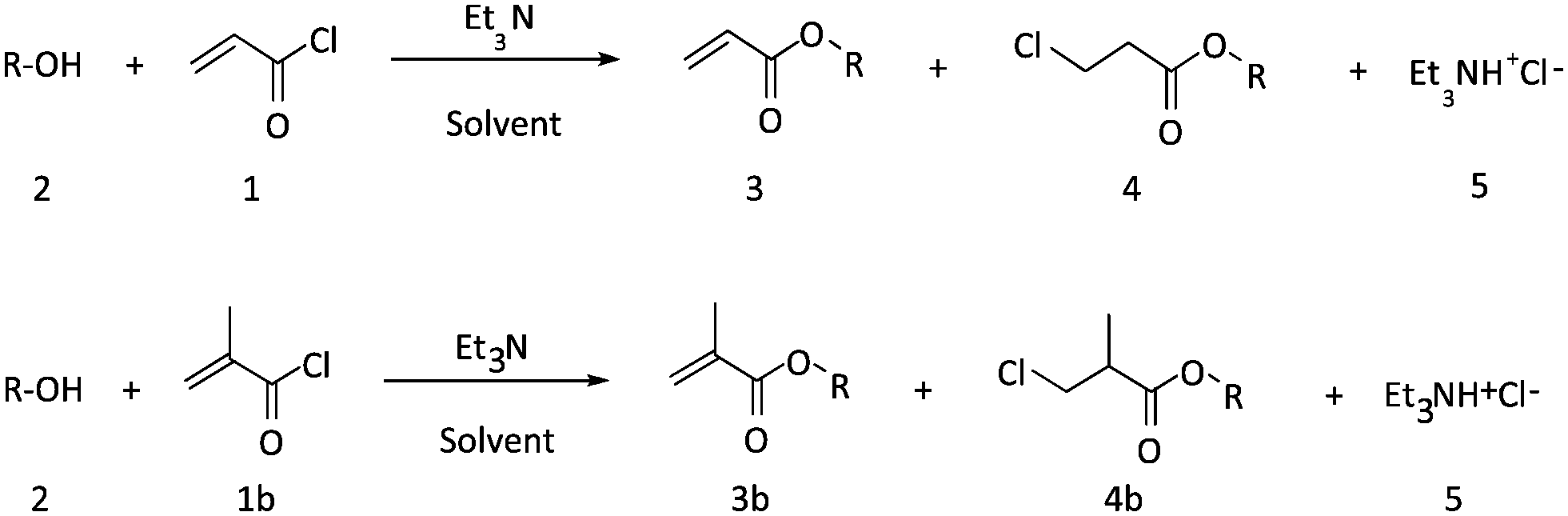 | ||
| Scheme 1 General synthesis of acrylates 3 and methacrylates 3b. | ||
Slurry processing in a flow tubular reactor
The above described pre-tests demonstrate clearly that a different strategy must be used when side product formation is to be avoided. Removal of HCl gas from a continuous flow reactor is difficult – if not impossible by conventional techniques. Hence, further research focused on slurry processing and addition of a hydrogen halide acceptor to capture the formed HCl. In a conventional batch procedure, triethylamine (Et3N) or diisopropylethylamine (DIPEA) are often added to the reaction mixture.24 Another method is to purge an inert gas through the reaction mixture and neutralizing it, a strategy that is only feasible for batch operation.A number of studies on continuous flow processes of suspensions and particulates are reported in literature.25–28 One way of transportation of suspensions in these flow processes is to employ either ultrasonication or pulsed agitation. Prior the actual synthesis in a flow reactor setup, acrylates at different concentrations were prepared via a batch procedure29 in two different solvents (0.05 M to 1 M in CH2Cl2 and THF). In order to observe the transport behaviour of slurries in a flow setup, the acrylate slurry of an individual batch reaction was then pumped through a tubular reactor (0.8 mm ID) from a peristaltic pump. The results showed that slurries in CH2Cl2 with a concentration up to 0.5 mol L−1 could flow through a tubular reactor without clogging the reactor. Clogging was observed only when the product concentration was increased to 1 mol L−1. The coiled reactor was merged in the water bath and ultrasonication was then applied. In combination with ultrasonication, no blockage of the reactor was observed (Table 2).
| Solvent | Concentration (mol L−1) | Applied sonication | Remark |
|---|---|---|---|
| CH2Cl2 | 0.05 | — | No blockage of reactor |
| 0.1 | — | No blockage of reactor | |
| 0.5 | — | No blockage of reactor | |
| 1 | — | Blockage of reactor | |
| 1 | ✓ | Blockage of reactor | |
| THF | 1 | ✓ | No blockage of reactor |
Flow synthesis of octyl acrylate in an ultrasonicated flow reactor
Octyl acrylate preparation was carried out in a PFA coiled reactor (0.75 mm ID, internal volume 2.2 mL), submerged in a sonication bath. Fig. 1 depicts the reactor setup. A stream of acryloyl chloride solution (A) in dry THF or CH2Cl2, was mixed with a stream of n-octanol solution (B) and triethyl amine in dry THF or CH2Cl2, through a T-piece by means of two HPLC pumps. The feed concentration of 1 mol L−1 in THF was chosen as a starting test condition. The concentration was then increased in order to determine the maximum amount which allows the free transport of slurries (see Table 3). The reactor could be operated without reactor blockage using an ultrasonication assisted flow setup. In this setup, the coiled reactor is submerged in a conventional laboratory ultrasonication bath at 37 Hz. Ultrasonication hereby helps to avoid reactor fouling and supports slurry agitation. On the other side, it also aids in breaking down salt particles. The particle size of salt adducts was determined to be around 800 nm via dynamic light scattering.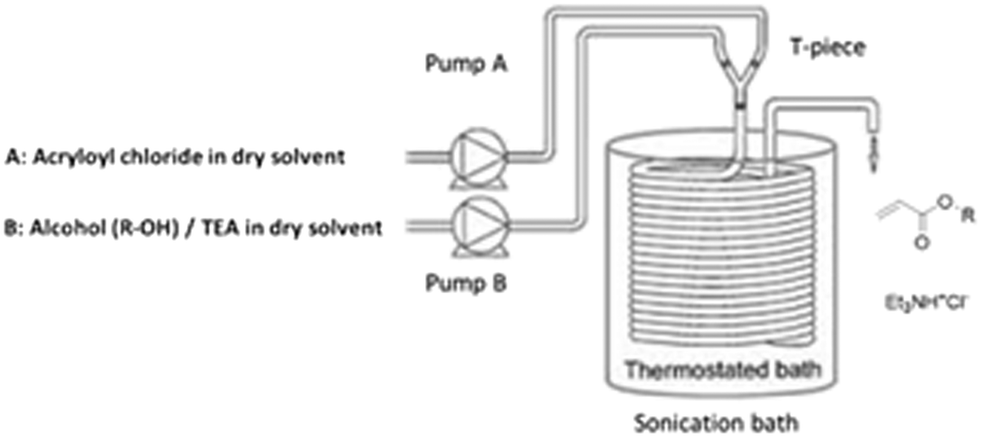 | ||
| Fig. 1 Continuous flow set-up for the continuous synthesis of acrylates. | ||
| Solvent/[M]a | Residence time (min) | 3 (R = C8H17) | 4 (R = C8H17) | Yield after extractione (%) | |
|---|---|---|---|---|---|
| a Reactions were performed in a 2.2 mL PFA coiled reactor (0.75 mm ID) in a sonication bath (37 Hz; 30%). b Reaction temperature = 40 °C. c The power of the sonication bath had to be increased in order to avoid blockage of the reactor. d Reaction temperature in CH2Cl2 = 25–30 °C. e Samples were collected on 10% NaCl (aq) and extracted with a suitable solvent (diethyl ether or CH2Cl2). | |||||
| THF/[1 M]b | 1c | 81 | 0.5 | — | |
| 5 | 88 | 0.4 | 56 | ||
| 7 | 90 | 0.4 | — | ||
| THF/[2 M] | 1c | 94 | 0 | — | |
| 5 | 94 | 1 | — | ||
| 10 | 94 | 0.5 | — | ||
| THF/[3 M] | 5c | 93 | 2 | 80 | |
| 10 | 98 | 1 | — | ||
| THF/bulk | 5 | Reactor blocked | — | ||
| Diisopropyl ether/[3 M] | 5 | 99 | Reactor blocked | 1 | — |
| 10 | |||||
| CH2Cl2d/[3 M] | 5 | Reactor blocked | — | ||
| CH2Cl2/[2 M] | 5 | Reactor blocked | — | ||
| CH2Cl2/[1.5 M] | 5 | 97 | 0.5 | 70 | |
| CH2Cl2/[1 M] | 5 | 98 | — | 38 | |
Adding of triethylamine as HCl capturing agent resulted as expected in reduction of the formation of HCl-adduct (4) to less than 1% in the crude mixture. Furthermore, a feed concentration of up to 3 mol L−1 in THF was successfully applied using otherwise the same reactor setup and reaction parameters. During this process, the system pressure increased to 9 bar at the highest feed concentration, which is well within limits for most lab-based flow reactors. Clogging was only observed at higher feed concentrations or when ultrasonication was shut down. The reaction could be operated for 4 h without interruptions without a change in product composition at room temperature with a residence time of 5 minutes. The process yielded n-octyl acrylate up to 80% isolated yield after work-up. The reaction in another solvent i.e. CH2Cl2 was also tested. The synthesis of n-octyl acrylate in CH2Cl2 was performed using the reaction parameters as used in the reaction with THF. The process with feed concentrations larger than 1.5 mol L−1 was not able to proceed due to clogging of the tubing. At this maximum feed concentration, the reaction provides, however, an excellent conversion of 98% n-octyl acrylate. The experiment was repeated a few times in order to determine the isolated yield and to test the robustness of the system. The extraction of the product during the work-up was, however, not straightforward due to difficult phase separation between the organic and aqueous phase, resulting also here only in an isolated yield of around 70%.
Since the above described reactor was of relatively small scale, a first increase in reactor volume was investigated. Further, a liquid–liquid separator (Zaiput Flow Technologies, 1 μm pore-size, hydrophobic PTFE membrane) was coupled at the outlet of the reactor. Before entering the separator unit, a 10% NaCl aqueous solution was mixed into the product stream to dissolve the salt contents. This aqueous phase was then separated off, allowing for a faster work up of the crude mixture. This separation unit has been shown to minimize the required work-up in several organic processes. It helps to break up emulsions, while improving the separation efficiency and increasing the isolated yield of the product. By increasing the internal diameter of the tubular reactor from 0.75 mm to 2.00 mm (to 5 mL reactor volume), the reaction could proceed without clogging during the process when ultrasonication was further used. The optimum working concentration on the extended scaled process was identified at 1.5 mol L−1 in THF. As a result, the tubular reactor setup with a 2.00 mm internal diameter produced 1.313 g min−1 (78.6 g h−1) of n-octyl acrylate and yielded up to 95% of isolated product.
Broadening the scope of the synthesis towards other acrylate monomers
The process concept was further evaluated towards the synthesis of different acrylates (see Table 5). A variety of alcohols were applied to the reaction, resulting in the acrylate product 3 of the corresponding alcohol 2-methyl-1-butanol, stearyl alcohol, β-citronellol, α-terpineol and 4-hydroxybenzaldehyde (PHBA).The acrylate derived of 2-methyl 1-butanol was obtained by applying similar reaction condition as described for the synthesis of n-octyl acrylate. A conversion of 90% was obtained. The isolated yield of 2-methyl-1-butyl acrylate was, however, only between 20–40%. The relative low yield of the product was due to the solubility of the 2-methyl-1-butyl acrylate in the aqueous layer. Due to low solubility of stearyl alcohol CH2Cl2, the synthesis of stearyl acrylate was optimized in THF (at 0.75 mol L−1), resulting in 75% of product conversion. Bio-based acrylate monomers were successfully synthesized using β-citronellol, α-terpineol and 4-hydroxybenzaldehyde (PHBA). High product conversions (90 to 100% based 1H NMR data) were obtained within a residence time of 1–5 minute with isolated yields up to 75% (Table 4).
| Solvent/[M]a | R–OH 2 | Reaction time (min) | 3 (%) | Yield after extractions (%) |
|---|---|---|---|---|
| a Reactions were performed in a 2.2 mL PFA coiled reactor (0.75 mm ID) in a sonication bath. b T = 40 °C. c T = 25–30 °C. | ||||
| THF/[0.75 M]b | Stearyl alcohol | 10 | 75 | 65 |
| THF/[0.75 M]b | 2-Methyl-1-butanol | 5 | 90 | 20–40 |
| CH2Cl2/[0.5 M]c | β-Citronellol | 1 | 94 | 75 |
| 5 | 93 | — | ||
| 10 | 87 | — | ||
| THF/[0.5 M]b | β-Citronellol | 0.5 | 90 | 35–50 |
| 1 | 85 | |||
| CH2Cl2/[1.5 M]c | β-Citronellol | 1 | 89 | |
| 5 | 90 | 50–70 | ||
| CH2Cl2/[1.0 M]c | α-Terpineol | 1 | 98 | 55 |
| 5 | 100 | 65 | ||
| THF/[1.0 M]b | α-Terpineol | 1 | 100 | 38–50 |
| CH2Cl2/[2.0 M]c | α-Terpineol | 1 | 90 | 70 |
| CH2Cl2/[1.0 M]c | PHBA | 1 | 95 | 68 |
Synthesis of octyl methacrylate
In the next step, the developed flow procedure was extended for the synthesis of n-octyl methacrylate 3b. Methacrylates differ only slightly in structure from their corresponding acrylates, yet the additional methyl group can have significant effect on product mixture viscosities. In fact, application of the same reaction conditions led to blocking of the reactor. Lowering the feed concentrations, increasing the length (total volume) of the tubular reactor or increasing the flow rates did not result in better processing of the slurry.Therefore, the reaction was tested using a coiled PFA reactor with a larger internal diameter of 1.00 mm. Two different reactor volumes and reaction times were tested. (Table 5) The result showed that shortening the residence times (i.e. increasing the flow rates) did not improve in better processing of the slurry. However, slurry processing was greatly improved when the reactor volume was increased from 2.36 mL to 3.93 ml at 1 min (or lower) residence time, hence combining higher flow rates with steady residence times. This effect could be potentially due to higher shear forces playing a role in the larger reactor (where a higher flow rate is then applied to keep residence times constant). The synthesis of n-octyl methacrylate was then consequently further performed in a 3.93 ml PFA reactor (1.00 mm ID). To achieve 90% conversion the reaction was carried out at a residence time of 10 min.
| [M] | R–OH 2 | Reaction time (min) | 3b (%) | Yield after extractions (%) |
|---|---|---|---|---|
| Reactions in CH2Cl2 were performed using a 3.93 mL PFA coiled reactor (1.00 mm ID) in a sonication bath ata room temperature;b T = 40 °C. | ||||
| [1.5 M]a | β-Citronellol | 1 | 89 | 75 |
| 5 | 95 | 65 | ||
| 10 | 96 | 80 | ||
| [1.5 M]b | β-Citronellol | 1 | 90 | — |
| 5 | 88 | — | ||
| [2.0 M]a | α-Terpineol | 1 | 97 | 70 |
| [1.0 M]a | PHBA | 1 | 94 | 55 |
Variation of alcohols for the synthesis of various methacrylate monomers
The developed flow methodology was also applied to obtain other methacrylates accordingly. As an example, the reaction between β-citronellol and methacryloyl chloride was carried out at room temperature within 1–5 min of residence time, yielding 89% to 96% of the corresponding methacrylate at a maximum concentration of 1.5 mol L−1 (Table 6).| Solvent/[M] | R–OH 2 | Reaction time (min) | 3b (%) | |
|---|---|---|---|---|
| a Reactions were performed in a 2.2 mL PFA coiled reactor (0.75 mm ID) in a sonication bath. b Reactions were performed in a 2.36 mL PFA coiled reactor (1.00 mm ID) in a sonication bath. c Reactions were performed in a 3.93 mL PFA coiled reactor (1.00 mm ID). | ||||
| CH2Cl2/[1.5 M]a | n-Octanol | 5 | Blocked reactor | |
| CH2Cl2/[1.5 M]b | n-Octanol | 1, 3, 5 | Blocked reactor | |
| 10 | 75 | |||
| CH2Cl2/[0.5 M]b | n-Octanol | 1 | Blocked reactor | |
| 3 | 88 | |||
| 10 | 90 | |||
| CH2Cl2/[0.5 M]c | n-Octanol | 1 | 33 | |
| 3 | 88 | |||
| 10 | 93 | |||
Conclusions
A convenient lab-scale process for the synthesis of acrylates and methacrylates using an ultrasonication-assisted continuous flow procedure is demonstrated. The process works well for a broad variety of (meth)acrylate products with high yield and high product conversion, within a short residence time at room temperature. Addition of a base to remove free acid from the reaction solution and hence to avoid HCl addition to the (meth)acrylate double bond is required, leading to a slurry process. Yet, by applying a conventional laboratory ultrasonicator, good processing of the slurry in small diameter tubular reactors could be achieved, and allows for high yield synthesis of the acrylates with ease. The reactor is operated under stable conditions, and a first (laboratory scale) upscale by increasing tubing diameters and overall internal volume leads to significant production capacity of the residual (meth)acrylates. This enables an on-demand synthesis of specialized monomers, which is often a tedious step in polymer development. This is specifically demonstrated on the synthesis of biobased monomers based on terpineols, which hold great promise for future polymer developments. Further, with the growing interest in continuous flow polymerization in microstructured reactors, it is a logical consequence to also develop facile monomer synthesis strategies that can later be combined.Experimental section
Materials
Acryloyl chloride (Merck, stabilized with phenothiazine), methacryloyl chloride (Acros Organics, stabilized with 200 ppm MEHQ), and 1-octanol (Acros Organics, 99%) were all used as received. The other alcohols i.e. 2-methyl-1-butanol, 1-octadecanol, (bio-based) β-citronellol, α-terpineol and 4-hydroxybenzaldehyde were purchased from TCI Europe N.V. Dry dichloromethane and THF were obtained from a MB-SPS-800 solvent drying system.Microreactor set-up
Screening of reaction conditions was performed using a Labtrix® Start R2.2 Flex system (Chemtrix BV, The Netherlands) fitted with a glass micro reactor chip (3223, reactor volume = 10 μl; 3227, reactor volume = 19.5 μl) containing a SOR-2 static micromixer. Reagent solutions were introduced into a chip reactor by means of two syringe pumps (Chemyx) via two gas-tight syringes (SGE). The operation flow rates were set between 0.1 and 25 μl min−1. The temperature of the reactor was controlled by a thermoelectric cooler temperature controller MTTC1410 (Melcor Thermal Solutions). The system was maintained at 20 bar using a preset ultralow dead-volume (6 μl) back pressure regulator, which was placed via a T-piece in between the tubing from the outlet of the reactor to the collecting vial.Continuous tubular reactor set-up
The continuous tubular reactor set-up consists of two HPLC pumps (Knauer): an Azura P2.1S pump with ceramic 10 mL pump head for handling the corrosive acid chloride (pump A) and a BlueShadow 20P pump with a pressure sensor and 10 mL pump head for delivering the alcohol (pump B). The solutions of acid chloride (A) and alcohol (B) were stored in brown laboratory bottles with a GL-45 screw cap with an argon inlet. The outlets of both pumps were connected through a T-piece (0.75 mm hole) or a Y-piece (1.00 mm hole) to the inlet of the PFA tubular reactor (Advanced Polymer Technology GmbH; 0.75 mm ID or 1.00 mm ID, OD 1/16′′). For ultrasonic assisted process, the coiled tubular reactor was submerged in a thermostatic ultrasonic bath (Elma®) at 37 Hz. The delivery of the solutions was carried out by means of the two HPLC pumps, while the reagents was kept in the containers under Ar during the continuous operation. The product was quenched and collected with 10% (w/v) NaCl. The product was then analysed by 1H-NMR.General procedure for screening of reaction conditions for synthesis of n-octyl acrylate (3) via the microreactor set-up
The screening was performed using a Labtrix® Start R2.2 Flex and the experiment was set up as described earlier. Before performing the reaction, the micro reactor was primed with the solvent which was used for the reaction. Syringe A was filled with 12.3 mol L−1 acryloyl chloride under nitrogen atmosphere, whereas syringe B was filled with a mixture of n-octanol in dry THF (3.16 mol L−1). Prior to the screening, the system was primed with solution B for 20 min. The reactions were then carried out at 10 °C, 25 °C and 195 °C. The samples of an individual reaction condition were collected after passing the reagents through the reactor (3× of the residence time). Samples of 5–10 μl were collected and analysed by 1H-NMR.Procedure for the synthesis of n-octyl acrylate (3) and other acrylate monomers using the continuous tubular flow reactor
Before starting the reaction, the system was primed with dry dichloromethane. The two HPLC pumps were connected to a coiled PFA reactor using a T-piece or Y-piece (IDEX Health & Science Technologies). Two solutions of acid chloride and alcohol were prepared and purged for 5 min with argon. Solution A: 1.5 mol L−1 of acryloyl chloride (6.1 mL, 0.075 mol) in dry dichloromethane (43.9 mL) and a solution B; a mixture of 1.5 mol L−1 of 1-octanol (12 mL, 0.075 mol) and triethyl amine (10.5 mL, 0.075 mol) in dry dichloromethane (27.5 mL). In order to avoid a (pre) reaction between acryloyl chloride and triethylamine before reacting with an alcohol, the base was premixed with the solvent containing alcohol. Both solutions were pumped via HPLC pumps into the PTFE T- or Y-mixers to a coiled tubular reactor (reactor volume = 2.2 mL; 0.75 mm ID). The frequency of the sonication waves was set at 37 Hz (power of the sonication bath was between 30–60%), with a controlled temperature of 25 °C. The residence time, in general, was carried out at 1 min, 5 min and 10 min in the reactor at the flow rates of 0.22 mL min−1 (for the residence time of 5 min) for each pump. The product was extracted, dried and further analysed via1H-NMR. The reaction mixture was collected in a 10 w% NaCl aqueous solution. n-Octyl acrylate was obtained as a slightly yellow oil in 70–95% yield after drying.Procedure for the synthesis of n-octyl methacrylate (3b) using a continuous tubular flow reactor
The reactor set-up was primed with dry CH2Cl2. The synthesis of various methacrylate products was prepared from the solutions of methacryloyl chloride and a mixture of alcohol with triethylamine solution using the similar setup for acrylates synthesis, except for the tubular reactor. For the synthesis of methacrylate, a coiled PFA tubular reactor of 3.93 mL (1.00 mm ID) was used during the process. The reaction temperature and residence time were controlled during the investigation in order to obtain the monomers at the most optimum reaction condition. For the synthesis of n-octyl methacrylate, a solution of 0.62 mol L−1 methacryloyl chloride (3.0 mL, 0.031 mol) in dry CH2Cl2 (43 mL) and a mixture of 0.5 mol L−1 of 1-octanol (4.0 mL, 0.025 mol) and triethyl amine (3.5 mL, 0.025 mol) in dry CH2Cl2 (42.5 mL) were prepared and purged under Ar for 5 min. Both solutions were pumped via HPLC pumps through a PTFE, Y-mixer into a PFA coiled reactor. The frequency of the sonication bath was set at 37 Hz, with 40% power. The water temperature of the sonication bath was kept between 25–30 °C. The reaction time was at 10 min (at operating flow rates of 0.196 mL min−1 for each pump). The product after processing was then collected on 10% NaCl (aq). The organic and aqueous layer was separated and the organic layer then washed with water. After drying the organic phase product over Na2SO4 and filtration, the organic solvent was removed under reduced pressure. Methacrylate compounds were obtained as a slightly yellow oil after drying.Analysis
1H-NMR spectra are recorded in CDCl3 (Oxford Instruments Ltd. Using a Varian probe) and chemical shifts (δ, in ppm) are determined relative to the residual solvent signals (7.26 ppm for CDCl3).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors are grateful for funding of the research via the Flanders Innovation Hub for Sustainable Chemistry (FISCH), now Catalisti Belgium.References
- K. Nagai, Appl. Catal., A, 2001, 221(1–2), 367–377 CrossRef CAS
.
-
(a) H. Sugiyama, U. Fischer, K. Hungerbühler and M. Hirao, AIChE J., 2008, 54(4), 1037–1053 CrossRef CAS
- S. B. Evans, J. E. Mulvaney and H. K. Hall, J. Polym. Sci., Part A: Polym. Chem., 1990, 28(5), 1073–1078 CrossRef CAS
- S. G. Newman and K. F. Jensen, Green Chem., 2013, 15, 1456–1472 RSC
-
(a) C. Wiles and P. Watts, Green Chem., 2012, 14, 38–54 RSC
- E. García-Verdugo, B. Altava, M. I. Burguete, P. Lozanob and S. V. Luisa, Green Chem., 2015, 17, 2693–2713 RSC
- C. Wiles and P. Watts, Green Chem., 2014, 16, 55–62 RSC
- J. Warneke, M. Plaumann, Z. Wang, E. Boehler, D. Kemken, S. Kelm, D. Leibfritz and V. A. Azov, Tetrahedron Lett., 2015, 56, 1124–1127 CrossRef CAS
- K. Ishihara, H. Kurihara and H. Yamamoto, J. Org. Chem., 1993, 58(15), 3791–3793 CrossRef CAS
- A. R. Hajipour, M. Kargosha and A. E. Ruoho, Synth. Commun., 2009, 39(6), 1084–1091 CrossRef CAS
- T. Sano, K. Ohaschi and T. Oreyama, Synthesis, 1999, 7, 1141–1144 CrossRef
- J. K. Twibanire and T. Bruce Grindley, Org. Lett., 2011, 13(12), 2988–2991 CrossRef CAS PubMed
- M. Movsisyan, T. Heugebaert, R. Dams and C. Stevens, ChemSusChem, 2016, 9(15), 1945–1952 CrossRef CAS PubMed
- C. Wiles, P. Watts, S. Haswell and E. Pombp-Villar, Tetrahedron, 2003, 59, 10173–10179 CrossRef CAS
- O. Naef, M. Roch and T. Chappuis, Chimia, 2010, 61(12), 889–891 CrossRef
- G. Pipus, J. Chem. Eng., 2000, 76, 239 CrossRef CAS
- E. Sinkovec, A. Pohar and M. Krajnc, Microfluid. Nanofluid., 2013, 14, 489–498 CrossRef CAS
- F. Van Waes, J. Drabowicz, A. Cukalovic and C. V. Stevens, Green Chem., 2012, 14, 2776–2779 RSC
- D. L. Browne, B. J. Deadman, R. Ashe, I. R. Baxendale and S. V. Ley, Org. Process Res. Dev., 2011, 15, 693–697 CrossRef CAS
- J. Sedelmeier, S. V. Ley, I. R. Baxendale and M. Baumann, Org. Lett., 2010, 12, 3618–3621 CrossRef CAS PubMed
- T. Horie, M. Sumino, T. Tanaka, Y. Matsushita, T. Ichimura and J. Yoshida, Org. Process Res. Dev., 2010, 14, 405–410 CrossRef CAS
- R. L. Hartman, Org. Process Res. Dev., 2012, 16, 870–887 CrossRef CAS
- N. P. Tu, J. E. Hochlowski and S. W. Djuric, Mol. Diversity, 2012, 16(1), 53–58 CrossRef CAS PubMed
- S. B. Evans, J. E. Mulvaney and H. K. Hall, J. Polym. Sci., Part A: Polym. Chem., 1990, 28(5), 1073–1078 CrossRef CAS
- B. J. Deadman, D. L. Browne, I. R. Baxendale and S. V. Ley, Chem. Eng. Technol., 2015, 38(2), 259–264 CrossRef CAS
- H. Mohapatra, M. Kleiman and A. P. Esser-Kahn, Nat. Chem., 2017, 9, 135–139 CrossRef CAS
- N. Hengl, Y. Jin, F. Pignon, S. Baup, R. Mollard, N. Gondrexon, A. Magnin, L. Michot and E. Paineau, Ultrason. Sonochem., 2014, 21, 1018–1025 CrossRef CAS PubMed
- J. Jordensa, A. Honingsa, J. Degrèvea, L. Braekenb and T. Van Gervena, Ultrason. Sonochem., 2013, 20(6), 1345–1352 CrossRef PubMed
- T. Terashima, T. Sugita, K. Fukae and M. Sawamoto, Macromolecules, 2014, 47, 589–600 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7re00120g |
| This journal is © The Royal Society of Chemistry 2018 |