
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConformational landscape of the epidermal growth factor receptor kinase reveals a mutant specific allosteric pocket†
Srinivasaraghavan
Kannan
 *a,
Gireedhar
Venkatachalam
b,
Hong Hwa
Lim
bc,
Uttam
Surana
bcd and
Chandra
Verma
*aef
*a,
Gireedhar
Venkatachalam
b,
Hong Hwa
Lim
bc,
Uttam
Surana
bcd and
Chandra
Verma
*aef
aBioinformatics Institute (BII), A*STAR, 30 Biopolis Street, 07-01 Matrix, Singapore 138671. E-mail: raghavk@bii.a-star.edu.sg; chandra@bii.a-star.edu.sg; Fax: +65 6478 9048; Tel: +65 6478 8353 Tel: +65 6478 8273
bInstitute of Molecular and Cell Biology (IMCB), A*STAR, 61 Biopolis Drive, 06-01 Proteos, Singapore 138673
cBioprocessing Technology Institute, A*STAR, 20 Biopolis Way, Singapore 138668, Singapore
dDepartment of Pharmacology, National University of Singapore, 16 Medical Drive, Singapore 117660, Singapore
eSchool of Biological Sciences, Nanyang Technological University, 60 Nanyang Drive, Singapore 637551, Singapore
fDepartment of Biological Sciences, National University of Singapore, 14 Science Drive 4, Singapore 117543, Singapore
First published on 16th May 2018
Abstract
Activating mutations within the epidermal growth factor receptor (EGFR) kinase domain give rise to several cancers including Non-Small Cell Lung Cancer (NSCLC). Small molecule inhibitors targeted at these mutants have proven to be clinically successful drugs. These molecules are ATP competitive and rapidly result in the emergence of resistance. Recently Jia et al. [Nature, 2016, 534, 129–132] reported a small molecule inhibitor (called EAI045) that binds at an allosteric pocket, does not compete with ATP and displays high potency and selectivity towards certain activating mutants (L858R, T790M, L858R/T790M) of EGFR, with IC50 values ranging from 3 nM to 49 nM. We present here a study combining extensive molecular dynamics simulations with binding assays to provide a structural basis underlying the mechanism of binding of this molecule. It appears that in mutants, conformational destabilization of the short helix (that carries Leu858 in the wildtype), is key to the exposure of the allosteric pocket which otherwise is occluded by a set of sidechains including L858. We extend this hypothesis to show that a similar mechanism would enable the molecule to inhibit EGFRL861Q which is another oncogenic mutant and validate this with binding experiments. The screening of the human structural kinome revealed at least 12 other oncogenic kinases which carry at least one activating mutant in this disorder-prone region and hence would be amenable to allosteric inhibition by molecules such as EAI045. Our study characterizes a druggable allosteric pocket which appears to be specific to certain oncogenic mutants of the EGFR and holds therapeutic potential.
Introduction
Diverse activating mutations within the epidermal growth factor receptor (EGFR) kinase domain give rise to several cancers including Non-Small Cell Lung Cancer (NSCLC) which accounts for ∼80% of all lung cancers; L858R and exon 19 deletions are the most common of these mutations.1 Small molecule tyrosine kinase inhibitors (TKIs) targeting tumours harbouring these activating mutations have demonstrated success in treating a subset of patients with NSCLC. TKIs such as gefitinib and erlotinib inhibit the tyrosine kinase activity of EGFR by competing with ATP for the ATP-binding site, located in the kinase domain, and are used for the treatment of advanced NSCLC.1 However, most NSCLC patients who initially respond to TKIs acquire resistance through secondary mutations. One of the most common of these mutations occurs at the Thr790 position which is located deep in the ATP site and is found to be mutated to Met.2–4 The longer side chain of Met partly blocks the drug from optimal interactions and simultaneously results in an increase in the relative affinity for ATP,2 thus rendering the ATP-competitive inhibitors ineffective. The Thr790Met (T790M) mutation is hence referred to as the gatekeeper mutation. The realization that a free thiol group in a Cys residue (Cys797) at the mouth of the ATP binding pocket could be targeted by a class of inhibitors that could covalently bind to Cys797 through its sidechain sulphur resulted in the development of “irreversible” inhibitors that demonstrated the ability to effectively inhibit the EGFRT790M mutants. Success in clinical trials led to their approval for the treatment of NSCLC positive patients harbouring the T790M mutations.5–7 However, the rapid emergence of a Cys to Ser mutation (C797S) rendered these molecules also ineffective.8 This resulted in a depressing scenario where none of the available EGFR TKIs were effective against the EGFRT790M/C797S mutant. The emergence of resistance against inhibitors that target the active sites is not surprising because enzymes (such as kinases) are under evolutionary pressures to bind to the substrate (ATP in the case of kinases) and hence will mutate towards achieving this function, albeit at the cost of some stability. Hence, there is an urgent need to find alternative mechanisms for inhibiting the activity of such enzymes.Recently, Jia et al.9 reported a breakthrough in the identification of a potent small molecule inhibitor (called EAI045) of the L858R/T790M mutant of EGFR, with an IC50 of 3 nM and a ∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 fold selectivity over EGFRWT. The inhibitor did not compete with ATP, and subsequent crystallographic analysis showed that a close analogue of EAI045 (EAI001) bound to an allosteric site that was distinct from the ATP binding site. Although the compound was not effective in blocking EGFR driven cell proliferation, combination with the anti-EGFR antibody cetuximab resulted in great efficacy in mouse models of lung cancer driven by the mutant EGFRL858R/T790M/C797S. The observation that this pocket in EGFR partially overlaps with a pocket in MEK1, which has successfully been targeted by allosteric inhibitors (currently in clinical trials),10 raises hopes for similar allosteric inhibitors of other kinases. What is intriguing is that Jia et al.9 could not crystallize EAI001 (analogue of EAI045) in a complex with the mutant EGFRL858R/T790M, even though the compound binds to this mutant with sub-nM potency. They did successfully co-crystallize it with the T790M/V948R mutant (PDB id: 5D41); the V948R mutation was designed to prevent the dimerization of the kinase.11 Given that EAI045 is an analogue of EAI001, we assume that EAI045 also binds to the same pocket. Kinases are known to adopt two distinct conformations that represent their active and inactive forms and are detailed in the ESI (Fig. S1†). The inactive state of the EGFR kinase is characterized by a short α-helix (residues G857–G863) in the activation loop (A-loop) sandwiched between a glycine rich P-loop and helix αC, with the latter adopting an ‘αC-Out’ conformation (see the ESI†). As a result, the sidechains of three residues (L858, L861, and L862) project into a cavity that is created by the outward movement of αC (Fig. 1A and S2†); the side chain of Leu858, the most highly conserved residue in the region, is the most buried into the cavity, preventing the αC moving into the αC-in conformation (characteristic of the active state of the kinase). The cavity itself is very hydrophobic (made up of residues V726, K745, I759, E762, A763, M766, C775, L777, L788, T790, D855, F856, G857, L858, L861 and L862, Fig. S2†). The crystal structure of EAI001 complexed to the T790M/V948R mutant EGFR (PDB: 5D41) shows EAI001 bound at an allosteric site that is created by the outward displacement of αC. This conformation characterizes the inactive state, even though the T790M mutation is known to stabilize the kinase in the active or the αC-in conformation. With the A-loop coordinates missing, presumably due to increased mobility, what is not clear is whether the kinase is in an inactive state (due to the outward movement of the αC helix) or in an active state (stabilized by the activating T790M mutation) or perhaps it adopts an intermediate conformation. An examination of the PDB revealed several crystal structures of EGFR (WT, L858R, T790M and L858R/T790M) in active and inactive forms. Most crystallographic structures lack well-defined density for the A-loop and none of the structures showed an accessible allosteric pocket.
000 fold selectivity over EGFRWT. The inhibitor did not compete with ATP, and subsequent crystallographic analysis showed that a close analogue of EAI045 (EAI001) bound to an allosteric site that was distinct from the ATP binding site. Although the compound was not effective in blocking EGFR driven cell proliferation, combination with the anti-EGFR antibody cetuximab resulted in great efficacy in mouse models of lung cancer driven by the mutant EGFRL858R/T790M/C797S. The observation that this pocket in EGFR partially overlaps with a pocket in MEK1, which has successfully been targeted by allosteric inhibitors (currently in clinical trials),10 raises hopes for similar allosteric inhibitors of other kinases. What is intriguing is that Jia et al.9 could not crystallize EAI001 (analogue of EAI045) in a complex with the mutant EGFRL858R/T790M, even though the compound binds to this mutant with sub-nM potency. They did successfully co-crystallize it with the T790M/V948R mutant (PDB id: 5D41); the V948R mutation was designed to prevent the dimerization of the kinase.11 Given that EAI045 is an analogue of EAI001, we assume that EAI045 also binds to the same pocket. Kinases are known to adopt two distinct conformations that represent their active and inactive forms and are detailed in the ESI (Fig. S1†). The inactive state of the EGFR kinase is characterized by a short α-helix (residues G857–G863) in the activation loop (A-loop) sandwiched between a glycine rich P-loop and helix αC, with the latter adopting an ‘αC-Out’ conformation (see the ESI†). As a result, the sidechains of three residues (L858, L861, and L862) project into a cavity that is created by the outward movement of αC (Fig. 1A and S2†); the side chain of Leu858, the most highly conserved residue in the region, is the most buried into the cavity, preventing the αC moving into the αC-in conformation (characteristic of the active state of the kinase). The cavity itself is very hydrophobic (made up of residues V726, K745, I759, E762, A763, M766, C775, L777, L788, T790, D855, F856, G857, L858, L861 and L862, Fig. S2†). The crystal structure of EAI001 complexed to the T790M/V948R mutant EGFR (PDB: 5D41) shows EAI001 bound at an allosteric site that is created by the outward displacement of αC. This conformation characterizes the inactive state, even though the T790M mutation is known to stabilize the kinase in the active or the αC-in conformation. With the A-loop coordinates missing, presumably due to increased mobility, what is not clear is whether the kinase is in an inactive state (due to the outward movement of the αC helix) or in an active state (stabilized by the activating T790M mutation) or perhaps it adopts an intermediate conformation. An examination of the PDB revealed several crystal structures of EGFR (WT, L858R, T790M and L858R/T790M) in active and inactive forms. Most crystallographic structures lack well-defined density for the A-loop and none of the structures showed an accessible allosteric pocket.
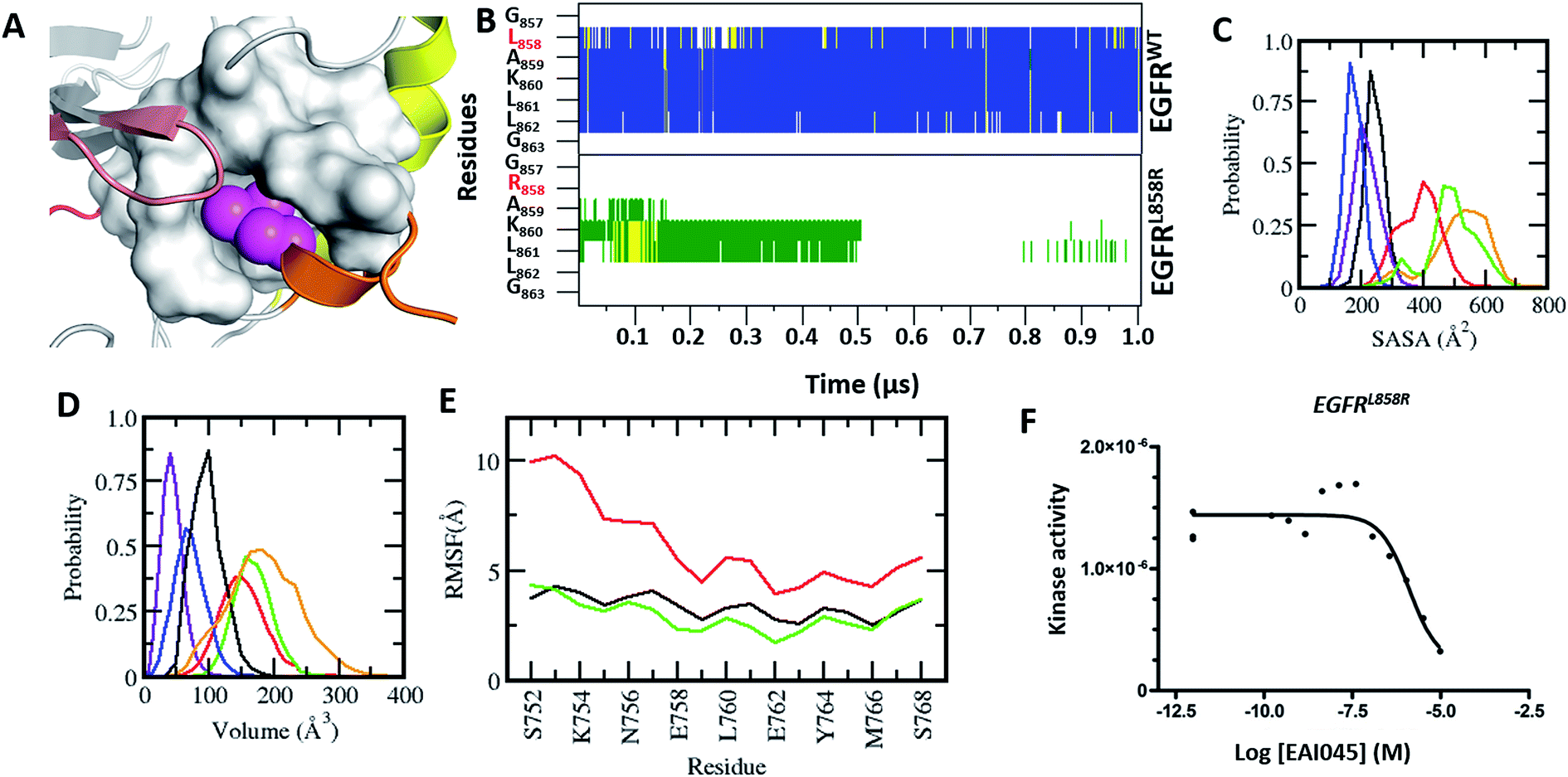 | ||
| Fig. 1 Characterization of the druggable allosteric pocket in EGFRL858R: (A) structure of the EGFRWT kinase in its inactive form with the αC-helix (yellow), short α-helix (orange), P-loop (brown), hinge (red) and residues that form the hydrophobic allosteric pocket shown in surface (grey) with Leu858 (magenta spheres) buried. (B) Secondary structure evolution (blue: α-helix, yellow: β-strand, grey: 310-helix, green: turn, and white: coil) of the short α helix (y-axis) during the MD simulations (x-axis) of EGFRWT (top) and EGFRL858R (bottom). Probability distributions of the (C) accessible surface area (ASA) of the short α-helix (D) volume of the allosteric pocket calculated from conformations sampled during the MD simulations of EGFRWT (black), EGFRL858R (red), EGFRL858R–EAI045 (green), EGFRdel19 (purple), EGFRT790M (blue) and EGFRL858R/T790M (orange). (E) Root mean squared fluctuations (RMSF) of conformations of the αC-helix sampled during the MD simulations of EGFRWT (black), EGFRL858R (red) and EGFRL858R–EAI045 (green). (F) Binding affinity (Kd) of EAI045 with EGFRL858R measured experimentally using KINOMEscan™ at DiscoverX. | ||
So then how does EAI045 bind to the kinase? To answer this question, we present here a study combining extensive molecular dynamics simulations with experiments that validate our findings. Our study suggests a mechanism that couples the amino acid substitutions with changes in the conformational landscape of EGFR and its mutants, resulting in mutant specific binding of EAI045.
Results
Experiments carried out by Jia et al.9 demonstrated that EAI045 binds specifically to EGFR carrying activating mutations. However, in the active state, the αC-in conformation renders the EAI045 pocket inaccessible (Fig. S3†). None of the active forms of EGFR in the PDB have this pocket accessible. We carried out MD simulations of the active forms of EGFRWT, EGFRT790M, EGFRL858R, and EGFRT790M-L858R to see if the pocket can be induced through fluctuations but found the ‘αC-In’ conformation to be stable and the EAI045 pocket remained occluded (Fig. S3†).The crystal structures of the inactive forms of EGFRWT or mutants such as T790M (in one instance this activating mutant was found to have crystallized in the inactive state) that adopt the inactive state upon complexation with inhibitors all show the allosteric pocket to be occluded by the insertion of the sidechain of L858. Once again, our attempts to induce the pocket to get exposed through MD simulations failed; the overall fold of EGFRWT remained stable (rmsd ∼ 3.5 Å, Fig. S4A†) and the short α-helix (residues G857–G863) in the A-loop remained well preserved (Fig. 1B) with low amplitude fluctuations (Fig. S4B†) and buried between the P-loop and αC (with an ASA of ∼200 Å3, Fig. 1C). The side chain of Leu858 remained stably buried in the hydrophobic allosteric pocket with an ASA of ∼20 Å3 and occupied the pocket where EAI045 is known to bind in EGFRT790M/V948R.9 The occlusion of this allosteric pocket in both active and inactive conformations of WT EGFRWT likely explains the lack of inhibition of EGFRWT by EAI045 in the study by Jia et al.9 We further confirmed this by carrying out binding studies and showed that the compound does not bind to EGFRWT, even at concentrations as high as 10 μM.
So how does EAI045 inhibit the activating mutants EGFRL858R, EGFRT790M and EGFRL858R/T790M given that the active conformation does not contain the allosteric pocket? To investigate this, we first generated structural models of these mutants in their inactive states (by replacing Leu858 with Arg858 in the EGFRWT structure and in the crystal structure of the inactive form of EGFRT790M from its co-crystal structure with dacomitinib, pdb: 4I24) and found that the longer Arg858 sidechain in the L858R containing mutants occludes the allosteric pocket; however this model does not incorporate conformational effects resulting from the mutation. Given this discrepancy with the experimental observation which shows unambiguously that EAI045 does inhibit the mutants L858R, T790M and L858R/T790M (and does so with sub-nanomolar potency9), it was clear that these mutants must undergo some structural change (relative to the model we constructed) that enables the binding of EAI045. The T790M mutation is thought to stabilize the active form of the kinase by stabilizing the hydrophobic spine,12,13 while the L858R mutation is known to destabilize the inactive form and stabilize the kinase in an intermediate form.14–20 We therefore subjected our models of the T790M, L858R and L858R/T790M mutants in their inactive states to multiple μs MD simulations. The simulations very quickly show that the L858R substitution, either as a single point mutation or in combination with T790M, is not tolerated as modelled based on the wild type conformation, i.e. with the Arg858 sidechain buried in the allosteric pocket. The destabilization of the short α-helix (Fig. 1B) occurs, and it undergoes rapid unfolding, with its solvent exposure increasing (from an ASA of ∼200 Å3 in the wildtype to ∼400 Å3 in the mutant; Fig. 1C). The helix was not seen to refold over the 1 μs simulation nor in extensions to 4 μs. As a result, Arg858 becomes more solvent exposed, with its ASA reaching values as high as 130 Å3, interacting transiently with several residues from the activation loop. The unfolding of the short α helix is coupled with high fluctuations and destabilization of the αC-helix (Fig. 1E). Increased flexibility and destabilization of the αC-helix have also been reported in earlier computational studies.14–20 This clearly points to the structural requirement of a helical motif in this region (the short helix) to stabilize the kinase in its inactive state. Apart from the increased flexibility observed for the A-loop, the αC-helix and the P-loop (Fig. S4B and D†), the rest of the kinase domain of these mutants of EGFR remains stable (rmsd ∼ 4 Å, Fig. S4A†). We next constructed the models of the complexes of EAI045 with EGFRL858R and with EGFRL861Q and subjected them to MD simulations (as outlined in the Methods). In these simulations, the short α helix remained unfolded and the αC-out conformation was stabilized with dampened fluctuations similar to those seen in the inactive EGFRWT simulations.
In contrast to simulations of the systems containing L858R, EGFRT790M in its inactive form remained stable with no major structural perturbations and with the short α-helix retained in its helical conformation (Fig. S5†). This suggests that at least the Arg mutation at the position of Leu858 induces the allosteric pocket to open. So, this raises the question as to how does the T790M mutation bind EAI045? Given that the V948R mutation, which abrogates dimer formation, had to be introduced into T790M to crystallize the complex with EAI045, we speculate that the conformational changes are most likely independent of dimer formation.
We next calculate the volume of the allosteric pocket (defined by residues that are within 6 Å of the EAI045 inhibitor binding site in the crystal structure 5D41.pdb) from the simulated trajectories (uncomplexed states of EGFRWT, EGFRL858R, EGFRT790M, EGFRL858R/T790M and the EGFRL858R–EAI045 complex). The distribution of pocket volumes (Fig. 1D) calculated over the entire trajectory revealed that the pocket in EGFRL858R and EGFRL858R/T790M samples large volumes (ranging from 70 to 300 Å3, averaging 190 Å3 and 150 Å3 respectively). The broad range of the pocket volumes reflects that the pocket is quite dynamic and large enough to accommodate molecules as large as EAI045 (the volume of the pocket in the complex simulation is 170 Å3 and is clearly well sampled in the apo states of the mutants, Fig. 1C). This clearly suggests that the allosteric pocket already exists in the apo form of the L858R and L858R/T790M mutants; the EGFRT790M mutant samples only smaller pockets (∼90 Å3). However, once again we face a contradiction: the experimental data of Jia et al.9 show that EAI045 inhibits the activity of EGFRT790M with an IC50 of 49 nM. The short α helix remains stable, similar to what we see in the inactive EGFRWT simulations, where the pocket volume sampled is also very small, ranging from ∼10 Å3 to 100 Å3, averaging at ∼45 Å3, and therefore unable to accommodate EAI045. In a control simulation where we modelled the missing A-loop in the crystal structure 5D41.pdb (T790M + V948R with AMP-NP and EAI001), removed the AMP-NP and EAI001, and simulated the apo form, we see that the N-terminal region of the A-loop (containing L858) quickly adopts a helical conformation (Fig. S6†), and the L858 sidechain inserts into the allosteric pocket and remains stable with the allosteric pocket too small again to accommodate EAI001 (Fig. S6†). Our binding assays showed that EAI045 is a weak binder of EGFRT790M with a Kd of ∼5 μM (Fig. S5†), supporting our atomic model, and this contrasts with the IC50 value of 49 nM reported by Jia et al.9; we do not understand the reason for this difference.
We next probe the energetics of the formation of the allosteric pocket by constructing the associated free energy surfaces (FES). For constructing the FES we used three parameters: (1) backbone dihedrals (phi and psi angles or the Ramachandran map) of residues that form the α-helix; (2) pocket volume (as described above); (3) rmsd of the short α-helix with respect to its starting folded structure. From the FES of phi and psi distributions (Fig. 2A), a single minimum was observed in the case of EGFRWT (Fig. 2A) and this was localized to the α helical region in the Ramachandran map. In contrast, for EGFRL858R we see a broad distribution of phi and psi angles, representing multiple minima, populating largely the non-helical regions in the Ramachandran map (Fig. 2B). These minima are separated by low energy barriers, suggesting that the residues (Gly857, Leu858/Arg858, Ala859, Lys860, Leu861, Leu862, and Gly863) interconvert easily on the simulation timescales. Surprisingly the minima (corresponding to the α-helix) observed in the EGFRWT have mostly disappeared in EGFRL858R, further evidence that the region assumes disordered states.
 | ||
| Fig. 2 Characterization of the energy landscape of the allosteric pocket in EGFRL858R: 2D free energy landscape as a function of (A, B) backbone dihedrals of residues that form the short α helix at the N-terminus of the A-loop and (C, D) root mean squared deviation (RMSD) of the short α-helix and the volume of the allosteric pocket. All the conformations sampled during the MD simulations of EGFRWT (A, C) and EGFRL858R (B, D) are included in the 2D free energy calculations. RMSD was calculated against the starting folded structure. Cartoon representation of MD snapshots of EGFRWT and EGFRL858R structures are shown with the αC-helix (yellow), short α-helix (orange), Leu858/Arg858 in sticks (top) and residues that form the hydrophobic allosteric pocket in spheres (bottom). | ||
Next, we compare the relationship between the volume of the pocket and the structural deviation from the starting state (helical state). The FES (Fig. 2C) is characterized by a single minimum for EGFRWT which is small both in volume and in the deviations from helicity. In contrast, EGFRL858R shows a broader distribution both in rmsd and in pocket volume and shows the sampling of larger volumes and structural deviations from the helical state. The absence of energy minima with RMSD < 2.5 Å suggests that the region undergoes rapid destabilization/unfolding, accompanied by an increase in the pocket volume; the rmsd of the region reaches ∼4.5 Å while the pocket volume reaches ∼250 Å3 (Fig. 2D). Closer scrutiny shows that there are two minima in EGFRL858R where the volume is high at ∼150 Å3 but the rmsd is either 3.3 Å or 4.8 Å suggesting that the pocket opens to its maximal value even when the helix is only partially unfolded. The magnitude of the maximal pocket volume is similar to the volume of the pocket that stabilizes EAI045 in the simulations of the EGFRL858R–EAI045 complex (Fig. 1D). Together this suggests that the allosteric pocket exists in the apo state of the EGFRL858R mutant and the unfolding of the α-helix makes the pocket accessible to the inhibitor. To confirm this, we constructed a model of the complex of EAI045 with EGFRL858R and separately with EGFRL858R in their inactive states and show that the mutations result in the shift of the conformations of the kinase away from the inactive to the intermediate state where the allosteric pocket is exposed and EAI045 fits snugly into this pocket, stabilized by van der Waals packing and h-bond interactions with the side chains of catalytic Lys745 and of Thr854 (Fig. 3). We next carried out binding assays of EAI045 against EGFRL858R and found that it does bind with a Kd of 1.2 μM (Fig. 1F), thus supporting our atomic model. Surprisingly, this value contrasts with the IC50 value of 3 nM reported by Jia et al.9 pointing to the complexity of comparing results from different assays (binding (Kd) vs. enzymatic (IC50) for example).
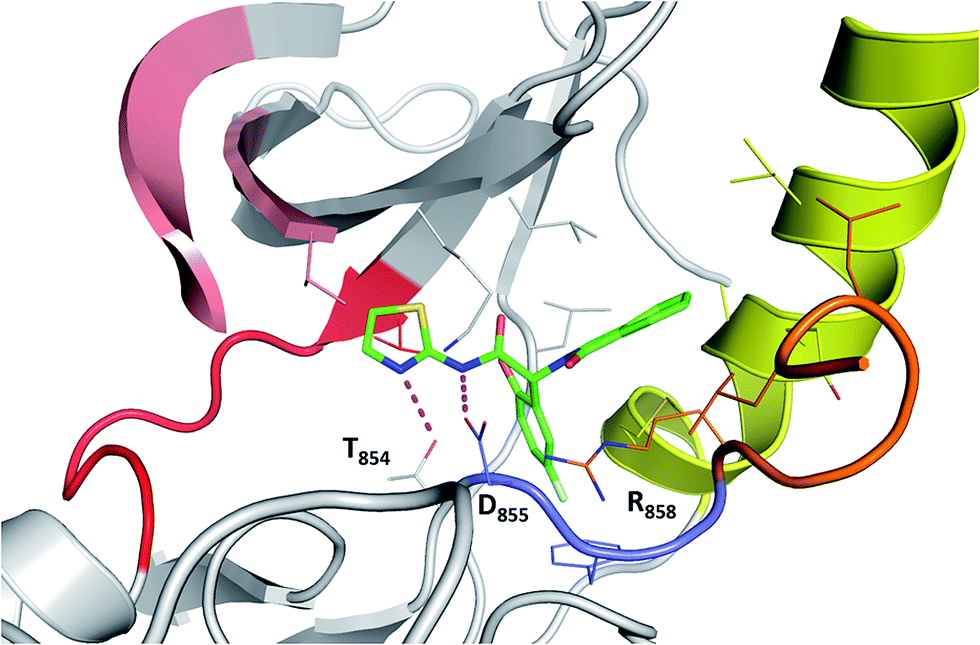 | ||
| Fig. 3 Binding mode of EAI045 to EGFRL858R: snapshot from the MD simulation of the complex between EGFRL858R and EAI045 (green carbon, stick representation). All the interacting residues are shown as thin sticks and all the interactions are highlighted as dotted lines (magenta). Colouring of the kinase structure is the same as in Fig. 1 with the DFG motif highlighted in blue colour. | ||
Allosteric pocket in exon 19 deletion
We next asked whether the other major activating mutation found in NSCLC, the exon 19 deletion (found in 40% of NSCLC patients), also results in the creation of this druggable allosteric pocket. This mutation is also known to destabilize the inactive state in favour of the active state.20 We model the EGFR19del mutant by removing 5 amino acids (746ELREA750) from the β3-αC-helix loop that connects the αC-helix and β-strand (β3), from the EGFRWT in its inactive state. During the simulations of this mutant in its apo state, the volume of the allosteric pocket is much smaller as compared to that of the EGFRL858R/T790M and also smaller than that of the EGFRWT (Fig. 1D). Due to the deletion of five amino acids in the EGFR19del the αC-helix is pulled towards the ATP binding site, resulting in constraining the allosteric site to a small volume. Based on these results we hypothesized that EAI045 will not bind to this mutant (this was also speculated by Jia et al.9) and we further confirmed our hypothesis by carrying out binding measurements which showed the lack of binding even at concentrations as high as 10 μM.Allosteric pocket in L861Q
L861Q is the second most common mutation in exon 21 and accounts for ∼2% of the NSCLC patients.21 Like L858R, L861Q also displays enhanced kinase activity.21 However, in contrast to L858R, which has increased sensitivity to type-I inhibitors (gefitinib, erlotinib and afatinib), partially because it has a reduced affinity for ATP, the L861Q mutant does not show increased drug sensitivity towards the inhibitors as it retains high affinity for ATP.21 Hence, there is an urgent need for inhibitors against this mutant. Leu861 is also located in the small α-helix that stabilizes the kinase in its auto-inhibited state, with the side chain of Leu partially buried into the same hydrophobic cavity as Leu858 (Fig. 4A). We wondered whether the substitution at Leu861 by Gln could result in the creation of the allosteric pocket, as was also speculated by Jia et al.9 As there are no experimental structures of EGFRL861Q, we generated a model by substituting the Leu at the 861 position with Gln in the structure of EGFRWT and subjected it to MD simulations for 1 μs. The substitution of Leu861 with Gln in the inactive form of the kinase is not tolerated, and the short α-helix unfolds rapidly during the simulations (Fig. 4B). As a result, both the αC-helix in its ‘αC-out’ conformation and the A-loop undergo increased fluctuations and destabilize the inactive form of the mutant kinase (Fig. 4D). Together, these changes result in the emergence of the cryptic allosteric pocket that now assumes volumes similar to those observed in the L858R mutation (Fig. 4C). We modelled the complex of EAI045 with L861Q and observed that EAI045 is likely to have a higher affinity for L861Q than for L858R because the shorter Gln861 side chain forms a unique hydrogen bond with EAI045 (Fig. 4E); in L858R, the longer side chain of Arg858 is stabilized by Glu758 of the αC-helix and is thus not free to engage EAI045. Binding energy calculations (Fig. S7†) using a protocol (outlined in the Methods) show that EAI045 binds more favourably to the L861Q mutant (by ∼9 kcal mol−1) compared to the L858R mutant; the unique hydrogen bond that EAI045 forms with the L861Q mutant is reflected in the higher electrostatic contribution (Fig. S7†). We next carried out binding assays of EAI045 with the L861Q mutant and found that as predicted from our computational studies the compound indeed binds to the mutant, albeit with a Kd of 300 nM (Fig. 4F), and its affinity for L861Q is much stronger compared to its affinity for L858R (Kd of 1.2 μM).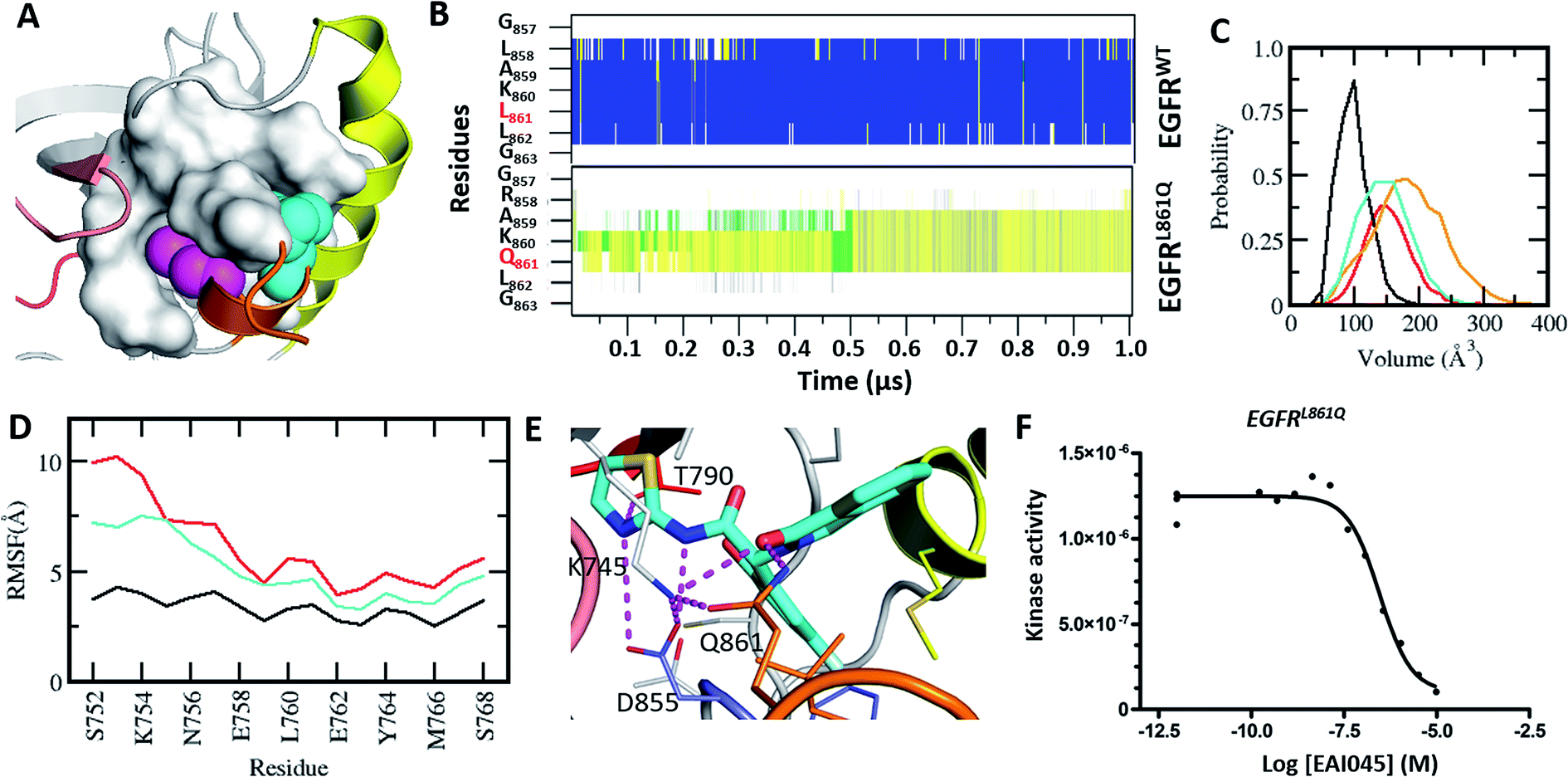 | ||
| Fig. 4 Characterization of the druggable allosteric pocket in EGFRL861Q: (A) structure of the EGFRWT kinase in its inactive form with the αC-helix (yellow), short α-helix (orange) and the hydrophobic allosteric pocket shown in surface with buried residues Leu858 (shown as magenta spheres) and Leu861 (shown as cyan spheres); (B) secondary structure evolution (blue: α-helix, yellow: β-strand, gray: 310-helix, green: turn, and white: coil) of the short α helix (y-axis) during the MD simulations of EGFRWT and EGFRL861Q. (C) Volume of the allosteric pocket calculated from the conformations sampled during the MD simulations of EGFRWT (black), EGFRL858R (red), EGFRL861Q (cyan), and EGFRL858R/T790M (orange). (D) RMSF of the αC-helix during the MD simulations of EGFRWT (black), EGFRL858R (red) and EGFRL861Q (cyan). (E) Snapshot from the MD simulation of the complex between EGFRL861Q and EAI045 (cyan carbon, stick representation). All the interacting residues are highlighted as thin sticks and all the interactions are highlighted as dotted lines (magenta). Colouring of the kinase structure is the same as in Fig. 1. (F) Binding affinity (Kd) of EAI045 for EGFRL861Q measured experimentally using KINOMEscan™ at DiscoverX. | ||
SNPs at the allosteric site
We next asked whether any Single Nucleotide Polymorphisms (SNPs) have been reported at the allosteric site and if so would they interfere with the ability of EAI045 to inhibit the mutants. We found 7 SNPs (D855G, F856Y, G857E, I759V, L777Q, L788F, and M766F) that are located in the vicinity of the allosteric pocket22 [Fig. 5]. Simple modelling of the SNPs by mutating the sidechains suggests that the associated size changes will likely impact the binding of EAI045. We next carried out MD simulations (250 ns in triplicates for each SNP) of individual SNPs in EGFRWT and in EGFRL858R (we used the inactive conformations for both because the inactive state is the only state relevant for the emergence of the pocket as we have shown earlier in this study). In the EGFRWT, the allosteric pocket remains inaccessible and appears to be agnostic to the nature of the SNP, with the pocket sampling volumes ranging from ∼50 to 100 Å3 (Fig. 5A). Indeed, since most SNPs involve small to large amino acid changes, the volume of the potential pocket is further occluded compared to the WT. | ||
| Fig. 5 Characterization of the allosteric pocket in the presence of SNPs in EGFR volume of the allosteric pocket in (A) EGFRWT (black) and (B) EGFRL858R (red) with the various SNPs modelled (dashed lines). (C) Model of EAI045 (green carbon, sticks) bound to EGFRL858R. Hydrogen bond (magenta) interactions are shown as dotted lines. All the allosteric pocket residues are highlighted as sticks. Various SNPs (D855G, F856Y, G857E, I759V, L777Q, L788F, and M766F) located within the allosteric pocket are shown as magenta sticks. | ||
In the L858R mutant, we modelled the SNPs onto a conformation selected from the simulation of the L858R mutant described earlier in this study; in this conformation, the short α-helix was already unfolded and the allosteric pocket was present. However, we saw a rapid collapse of the allosteric pocket during the MD simulations of all 7 SNPs; the volumes of the allosteric pockets sampled were 50–100 Å3 as compared to the volume of the pocket of ∼150 Å3 in the case of EGFRL858R (Fig. 5B). This is not surprising since all SNPs (except the D855G) examined here involve a small to large sidechain substitution. Additionally these larger sidechains are all hydrophobic and occupy the pocket and stabilize it, or rather result in occluding the pocket from binding EAI045 (Fig. 5C). The D855G mutation results in the removal of a charged sidechain which was engaged in a salt bridge with Lys745. Upon its removal, the lysine sidechain now occupies the pocket with its charged head group stabilized by a combination of large fluctuations and new hydrogen bonds with the carbonyl backbones of Phe856 and the sidechain oxygen of Thr845. Therefore, we speculate that despite the short α-helix stabilized in an unfolded state in the L858R mutant, making it accessible to EAI045, such compounds may not be effective if the patient carries any of the SNPs studied here or indeed develops resistance against the EAI045 type of compounds due to the emergence of such mutations. Indeed patients carrying the L777F/M SNP in EGFR (or L785F in Her2) are known to be resistant to the clinical EGFR/Her2 inhibitors lapatinib or neratinib, part of which occupies this allosteric pocket.23,24 If this is experimentally validated for molecules such as EAI045, the approach presented here of combining computational models with some binding assays could provide a very powerful filter to select patients for maximal benefit as part of ongoing efforts in precision medicine.
Conservation of the allosteric pocket across the human kinome
Does this allosteric pocket exist in other kinases? The ∼518 kinases from the human kinome share sequence homology ranging from very low (10%) to very high (90%); however they all adopt a similar fold. Most of the residues that form this allosteric pocket are part of the αC-helix and the A-loop, both of which are conserved structural and functional features across the kinome (Fig. 6). Using a global sequence alignment of the human kinome25 we first extracted the residues from all the kinases that align with the residues forming the allosteric pocket in the EGFR kinase. While the catalytic Lys and Glu and the DFG motif are mostly conserved, there are variations in the other positions (Fig. 6A); residues in the αC-helix and the A-loop are least conserved. However, 15 out of the 23 amino acids that make up the allosteric pocket are conserved in polarity in more than 75% of the sequences including residues in the A-loop (Fig. 6A). Indeed, the hydrophobic nature of the allosteric pocket across the human kinome is conserved with a hydrophobicity score26 averaging at ∼12.0 (the analogous score for EGFR in 12.75; Fig. 6B). Given the high conservation of the allosteric pocket we next investigated the accessibility of the pocket in the human kinome. However, crystal structures are only available for a third of all kinases in the human kinome. In addition, there is further paucity of data since the densities for the activation loop are missing in most of the crystal structures; this is not surprising given the flexibility of this region. So we decided to generate simplified models of the allosteric pocket in all the kinases based on the crystal structure of the inactive form of the EGFR kinase. The allosteric pockets were generated by simply replacing/substituting each residue comprising the allosteric pocket in the structure of the inactive form of the EGFR kinase with a homologous residue from the target kinase. The modelled structure was then energy minimized to remove steric clashes and the volume of the allosteric pocket was calculated from the minimized structure. We find that most of the kinases in the human kinome have a very small allosteric pocket with an average volume of ∼20 Å3 as compared to the EGFR (volume of ∼ 40 Å3; Fig. 6C); 15% of the kinome has pockets with volumes greater than 40 Å3. This may partly explain the observation by Jia et al.9 in their screens that EAI045 showed no inhibitory activity against a panel of 250 WT kinases.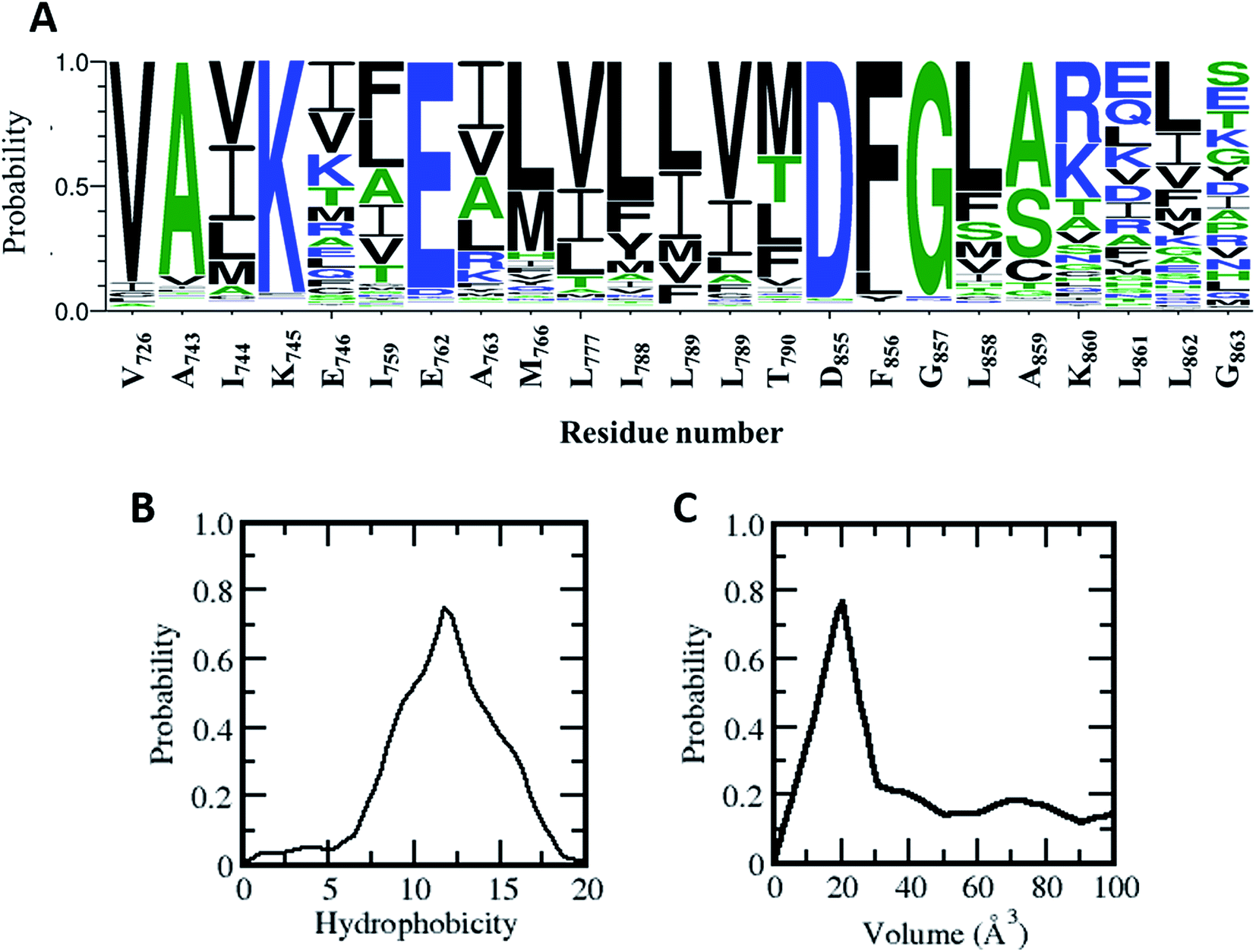 | ||
| Fig. 6 Conservation of hydrophobic allosteric pocket residues across the human kinome: (A) sequence logos made using the WebLogo server (http://weblogo.berkeley.edu) show the conservation and variation of hydrophobic allosteric pocket residues as found in multiple sequence alignments of the catalytic domains of the kinases from the human kinome (residue number corresponds to the EGFRWT kinase). (B) Distribution of hydrophobicity of the allosteric pocket across the human kinome (hydrophobicity of EGFRWT corresponds to 12.75). (C) Distribution of allosteric pocket volume across the human kinome. The allosteric pocket in each kinase was modelled using the 3D coordinates of the allosteric pocket in the EGFRWT as the template, and the pocket volume was calculated for the minimized modelled structures. | ||
Of the residues that form the allosteric pocket, it is clear that positions 858 and 861 are important in its genesis. EAI045 displays high selectivity towards the L858R and L861Q mutations in EGFR. We observed earlier that the leucines bury into the hydrophobic pocket and occlude it. However, upon mutation to Arg or Gln, the pocket is rendered accessible. Leu at the 858 position is highly conserved across the human kinome (present in ∼50% of kinases) while ∼80% have a hydrophobic residue at this position (Fig. S8†). Seven kinases contain a charged residue (Asp in five kinases, Arg in one kinase and Glu in one kinase) at this position but they lack the DFG motif and hence are unlikely to be catalytically active. In contrast, Leu at the 861 position is conserved in only 25% of the kinases, but ∼50% have a hydrophobic amino acid at this position and ∼30% have a charged residue. When the positions 858 and 861 have non-hydrophobic residues such as Arg or Gln, it is clear that they result in destabilizing the inactive state (which was stabilized by the short αC-helix).
Therefore, we examined other oncogenic kinases that may have similar mutations in the short α-helix which is part of the A-loop. A recent study by Dixit et al.27 has shown that the distribution of oncogenic mutations across the kinase domain is biased towards specific functional regions, with a high frequency in the functionally important A-loop. We found, from the COSMIC database,22 48 substitutions in this region in 12 different kinases that are known to be highly oncogenic including ABL, ALK, BRAF, BTK, EGFR, ErbB2, FLT3, KIT, KDR, MET, PDGFRA, and RET (Fig. 7). The mapping of these oncogenic mutations onto the structure of the catalytic domain of EGFR revealed that all these mutations are localized at the structurally conserved short α-helix (residues 857–864 in EGFR), making it a significant hotspot. We constructed models of these 12 kinases and found that the allosteric pocket is well conserved (Fig. S9†). We hypothesize that all these oncogenic mutant kinases will have similar exposed allosteric sites, albeit of differing sizes, and therefore could potentially be targeted by allosteric inhibitors.
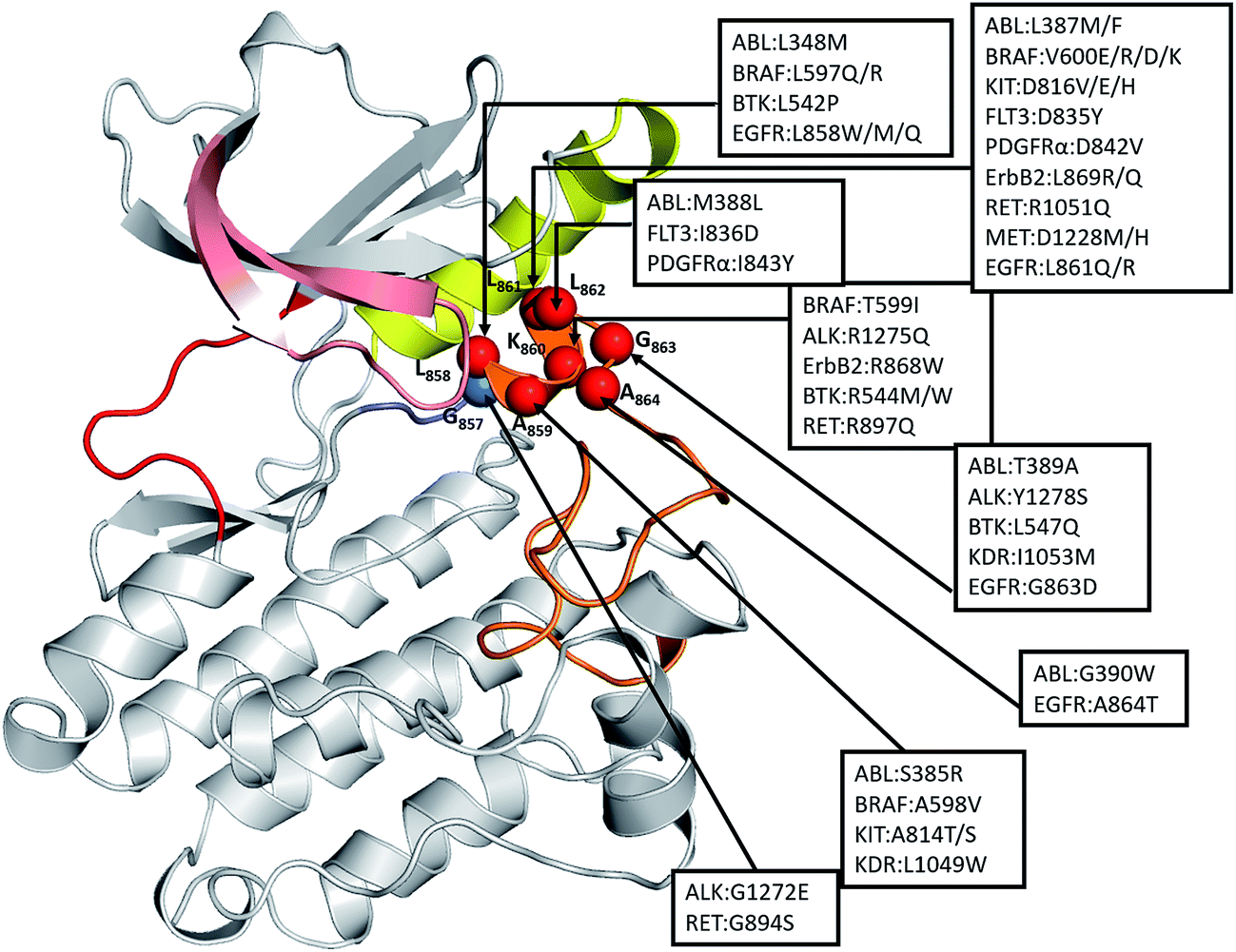 | ||
| Fig. 7 Structural mapping of oncogenic mutations in the short α-helix across the human kinome: the spatial distribution of highly oncogenic mutations in the catalytic core of kinases (ABL1, BRAF, KIT, FLT3, PDGFRα, ALK, ErbB2, BTK, MET, KDR, and EGFR). The kinase mutations with known high oncogenic potential were mapped onto the kinase catalytic domain of EGFRWT in its inactive form. Structurally conserved hotspots of kinase cancer mutations are annotated (number corresponds to the EGFRWT kinase) with spheres and their locations is indicated by arrows. | ||
Discussion
Kinases have traditionally been viewed as existing in an active or an inactive state and have been exploited successfully resulting in several clinically successful inhibitors.28 The inactive conformation undergoes large conformational changes, often upon phosphorylation of residues in the A-loop, to adopt a catalytically competent active form.29,30 Several mutations within the catalytic domain of kinases, particularly characteristic of cancers, have been found to stabilize the active state.29,30 The molecular details of the transition between active and inactive states could provide opportunities to drug intermediate conformations, but remain elusive. Advanced sampling methods such as metadynamics and Markov state models have been used to model these pathways for proteins such as Src, the EGFR kinases.15,31–33 Sutto L. et al.15 investigated the effects of oncogenic mutations (L858R and T790M) on the conformational free-energy landscape of the EGFR kinase domain through extensive MD simulations, parallel tempering and metadynamics. They proposed that while the EGFRWT mostly exists in the inactive conformation, all the oncogenic mutants destabilize the inactive conformation in favour of the active conformation. In an unbiased, all-atom MD simulation for ∼25 μs, the D. E. Shaw group31 observed spontaneous transition from the active to the so-called “Src-like inactive” conformation of the EGFRWT kinase funnelled across two different pathways; they confirmed their findings by carrying out hydrogen–deuterium (H/D) exchange measurements. Combining targeted MD, unbiased MD and Bayesian clustering, Li et al.32 also proposed two distinct pathways between the active and inactive forms of the EGFR kinase domain. Using Markov state models to explore the conformational landscape of the c-src tyrosine kinase, Shukla D. et al.33 identified key structural intermediates along the transition path between the active and inactive states and identified potentially druggable allosteric sites in the intermediates.Recently, Jia et al.9 identified a small molecule EAI045 which displayed high specificity towards the oncogenic L858R, T790M and L858R/T790M mutants of EGFR. Upon co-crystallization of the molecule EAI001 with EGFRT790M/V948R they found that the compound binds at a novel allosteric site which is located between the αC-helix and the A-loop. The activity of the inhibitor against the activating mutations seems paradoxical since the pocket cannot be formed in the active state of EGFR; indeed this pocket is not accessible even in the inactive state of EGFR, at least not in the crystal structures available in the PDB. We show, using a combination of modelling and extensive MD simulations, that mutations such as L858R, L858R-T790M and L861Q result in structural perturbations that destabilise the inactive state of the EGFR and in the process create a transient intermediate state where the allosteric pocket is fully formed and accessible to binding by EAI045; we confirm our findings with binding assays. It appears that the specificity of this molecule arises as a result of the intrinsic flexibility of the mutant kinases. The mutations are located in the short α helix of the activation loop, resulting in destabilization around this region and the subsequent expansion and exposure of the allosteric pocket. Although an allosteric pocket exists in the inactive state of EGFRWT, the highly stable α-helix in the N-terminal region of the activation loop restricts the allosteric pocket to be stabilized by hydrophobic interactions to much smaller sizes, thus making it inaccessible to EAI045.
All kinases undergo conformational changes during the transition from the inactive to the active form; the short α helix unfolds and adopts an extended conformation and the αC-helix is displaced. This transition is induced by phosphorylation and/or the binding of a partner protein at the N-lobe which pushes the αC-helix towards its αC-in conformation. However, the allosteric pocket does exist in the unphosphorylated monomeric form, as reported by Jia et al.,9 for the T790M/V948R mutant of the EGFR. This mutant is in its activated state as a result of the T790M mutation and cannot dimerize due to the V948R mutation;11 binding assays also suggest that the allosteric pocket exists in EGFRT790M and in EGFRL858R. We now show through structural modelling, validated by binding assays, that the presence of Arg at the 858 position results in structural perturbations that result in the opening of the allosteric pocket to which EAI045 binds. EGFRL858R is known to destabilize the inactive state and it is highly likely that this allosteric pocket emerges transiently during the transition from the inactive to the active state.
We extended the structural modelling to investigate L861Q, another oncogenic mutation, which is located in the same α-helix as L858R and found that its inactive state is destabilized in a manner similar to that seen in L858R, resulting in an exposed allosteric site that can bind EAI045. We next confirmed this by carrying out binding assays. We speculate that the unfolding of the helix and increased flexibility of the αC-helix observed here characterize intermediates between the inactive and active states. Such destabilization of α helices has also been reported in earlier experimental and computational studies employing advanced sampling methodologies.14–21,31,32 We further extend the structural modelling to show that this allosteric pocket is absent in the exon 19 deletion, another activating mutation that is commonly found in cancers and known to destabilize the inactive form of EGFR. The deletion of the five amino acids in this mutant imposes structural restraints that prevent the αC from adopting the ‘αC-Out’ conformation that is needed to create the accessible allosteric pocket.
It appears that the allosteric site is specific to EGFR carrying activating mutations such as L858R or L861Q, i.e. mutations known to destabilise the hydrophobic cluster that stabilises the inactive form of the kinase; the mechanism underlying the observed binding to the EGFRT790M remains unclear. Jia et al.9 showed that EAI045 binds only when the dimers have been disabled, for example in the presence of antibodies such as cetuximab. While this work was carried out using cell lines that carried only the T790M/L858R mutations, it would be interesting to explore this in cell lines carrying the wild type or indeed other mutations. The observation that the pocket is targeted by these molecules in only certain mutants that destabilize the inactive state opens a new window of opportunity to stratify patient populations in precision medicine programs.
Finally we scanned the structural kinome and identified 12 other oncogenic kinases (clinically reported) which have activating mutations in this short α-helical region. We speculate that these kinases will have exposed allosteric sites, albeit of differing sizes, and could potentially be targeted by molecules such as EAI045.
Methods
Computational methods
Atomistic molecular dynamics (MD) simulations are used to investigate the structural dynamics modulating the mutant specific binding of EAI045 to EGFR. The structure for the kinase domain (KD) was taken from the protein structure database or modelled using standard methods.34 We model the KD of EGFRWT, EGFRL858R, EGFRT790M, EGFRL858R/T790M, EGFRL861Q and EGFR19del in active and inactive forms. The crystal structures of apo EGFRWT are available in both active and inactive forms. The apo forms of EGFRL858R, EGFRT790M, and EGFRL858R/T790M in the active forms were derived from the corresponding liganded crystal structures. The inactive forms of these 3 mutants were generated using the apo form of the inactive EGFRWT structure. The structures of EGFR19del and EGFRL861Q were similarly modelled using the apo EGFRWT structures. We constructed the models of the complexes of EAI045 with EGFRL858R and with EGFRL861Q. The modelled structures were then subjected to atomistic MD simulations using standard protocols.20 All the structures and templates used in this study are listed in Table S1† and the methodology used to construct the models and carry out the MD simulations and analyses, including the binding energy calculations, is summarized in the ESI.†Experimental methods
In vitro binding assay was performed using KINOMEscan™ at DiscoverX (details of the methods are outlined in the ESI†).Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank the National Super Computing Center (NSCC) for computing facilities and A*STAR, Singapore for support.References
- D. S. Tan, et al., The International Association for the Study of Lung Cancer consensus statement on optimizing management of EGFR mutation-positive non-small cell lung cancer: status in 2016, J. Thorac. Oncol., 2016, 11, 946–963 CrossRef PubMed.
- C. H. Yun, et al., The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2070–2075 CrossRef PubMed.
- C. H. Yun, et al., Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity, Cancer Cell, 2007, 11, 217–227 CrossRef PubMed.
- K. D. Carey, et al., Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor erlotinib, Cancer Res., 2006, 66, 8163–8171 CrossRef PubMed.
- D. A. Cross, et al., AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer, Cancer Discovery, 2014, 4, 1046–1061 CrossRef PubMed.
- D. Singh, B. K. Attri, R. K. Gill and J. Bariwal, Review on EGFR inhibitors: critical updates, Mini-Rev. Med. Chem., 2016, 16, 1134–1166 CrossRef PubMed.
- A. O. Walter, et al., Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC, Cancer Discovery, 2013, 3, 1404–1415 CrossRef PubMed.
- K. S. Thress, et al., Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M, Nat. Med., 2015, 21, 560–562 CrossRef PubMed.
- Y. Jia, et al., Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors, Nature, 2016, 534, 129–132 CrossRef PubMed.
- Y. Zhao and A. A. Adjei, The clinical development of MEK inhibitors, Nat. Rev. Clin. Oncol., 2014, 11, 385–400 CrossRef PubMed.
- X. Zhang, J. Gureasko, K. Shen, P. A. Cole and J. Kuriyan, An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor, Cell, 2006, 125, 1137–1149 CrossRef PubMed.
- A. P. Kornev, N. M. Haste, S. S. Taylor and L. F. Eyck, Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17783–17788 CrossRef PubMed.
- M. Azam, M. A. Seeliger, N. S. Gray, J. Kuriyan and G. Q. Daley, Activation of Tyrosine Kinases by Mutation of the Gatekeeper Threonine, Nat. Struct. Mol. Biol., 2008, 15, 101109–111810 Search PubMed.
- Y. Shan, et al., Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization, Cell, 2012, 149, 860–870 CrossRef PubMed.
- L. Sutto and F. L. Gervasio, Effects of oncogenic mutations on the conformational free-energy landscape of EGFR kinase, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10616–10621 CrossRef PubMed.
- C. J. Tsai and R. Nussinov, The free energy landscape in translational science: how can somatic mutations result in constitutive oncogenic activation?, Phys. Chem. Chem. Phys., 2014, 16, 6332–6341 RSC.
- R. Nussinov and C. J. Tsai, Allostery in disease and in drug discovery, Cell, 2013, 153(2), 293–305 CrossRef PubMed.
- R. Nussinov and C. J. Tsai, Unraveling structural mechanisms of allosteric drug action, Trends Pharmacol. Sci., 2014, 35, 256–264 CrossRef PubMed.
- S. Wan, D. W. Wright and P. V. Coveney, Mechanism of drug efficacy within the EGF receptor revealed by microsecond molecular dynamics simulation, Mol. Cancer Ther., 2012, 11, 2394–2400 CrossRef PubMed.
- S. Kannan, et al., Hydration effects on the efficacy of the epidermal growth factor receptor kinase inhibitor afatinib, Sci. Rep., 2017, 7, 1540 CrossRef PubMed.
- R. K. Kancha, et al., The Epidermal Growth Factor Receptor-L861Q Mutation Increases Kinase Activity without Leading to Enhanced Sensitivity toward Epidermal Growth Factor Receptor Kinase Inhibitors, J. Thorac. Oncol., 2011, 6, 387–392 CrossRef PubMed.
- S. A. Forbes, et al., COSMIC: exploring the world's knowledge of somatic mutations in human cancer, Nucleic Acids Res., 2015, 43, D805–D811 CrossRef PubMed.
- E. Vizienyte, R. Ward and A. Garner, Comparison of the EGFR resistance mutation profiles generated by EGFR-targeted tyrosine kinase inhibitors and the impact of drug combinations, Biochem. J., 2008, 415, 197–206 CrossRef PubMed.
- Z. Sun, et al., Analysis of different HER-2 mutations in breast cancer progression and drug resistance, J. Cell. Mol. Med., 2015, 19, 2691–2701 CrossRef PubMed.
- G. Manning, D. B. Whyte, R. Martinez, T. Hunter and S. Sudarsanam, The Protein Kinase Complement of the Human Genome, Science, 2002, 298, 1912–1934 CrossRef PubMed.
- H. B. Bull and K. Breese, Surface tension of amino acid solutions: a hydrophobicity scale of the amino acid residues, Arch. Biochem. Biophys., 1974, 161, 665–670 CrossRef PubMed.
- A. Dixit and G. M. Verkhivker, The Energy Landscape Analysis of Cancer Mutations in Protein Kinases, PLoS One, 2011, 6(10), e26071 Search PubMed.
- D. Singh, B. K. Attri, R. K. Gill and J. Bariwal, Review on EGFR inhibitors: critical updates, Mini-Rev. Med. Chem., 2016, 16, 1134–1166 CrossRef PubMed.
- L. N. Johnson, M. E. Noble and D. Owen, Active and inactive protein kinases: structural basis for regulation, Cell, 1996, 85, 149–158 CrossRef PubMed.
- M. Huse and J. Kuriyan, The conformational plasticity of protein kinases, Cell, 2002, 109, 275–282 CrossRef PubMed.
- Y. Shan, et al., Transitions to catalytically inactive conformations in EGFR kinase, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7270–7275 CrossRef PubMed.
- Y. Li, et al., Conformational Transition Pathways of Epidermal Growth Factor Receptor Kinase Domain from Multiple Molecular Dynamics Simulations and Bayesian Clustering, J. Chem. Theory Comput., 2014, 10, 3503–3511 CrossRef PubMed.
- D. Shukla, et al., Activation pathway of Src kinase reveals intermediate states as targets for drug design, Nat. Commun., 2014, 5, 3397 CrossRef PubMed.
- A. Sali and T. L. Blundell, Comparative protein modelling by satisfaction of spatial restraints, J. Mol. Biol., 1993, 234, 779–815 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01262h |
| This journal is © The Royal Society of Chemistry 2018 |