
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsight into wild-type and T1372E TET2-mediated 5hmC oxidation using ab initio QM/MM calculations†
Hedieh
Torabifard
 a and
G. Andrés
Cisneros
*b
a and
G. Andrés
Cisneros
*b
aDepartment of Chemistry, Wayne State University, Detroit, MI 48202, USA
bDepartment of Chemistry, University of North Texas, Denton, TX 76203, USA. E-mail: andres@unt.edu
First published on 11th September 2018
Abstract
Ten-eleven translocation 2 (TET2) is an Fe/α-ketoglutarate (α-KG) dependent enzyme that dealkylates 5-methylcytosine (5mC). The reaction mechanism involves a series of three sequential oxidations that convert 5mC to 5-hydroxy-methylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC). Our previous biochemical and computational studies uncovered an active site scaffold that is required for wild-type (WT) stepwise oxidation (Nat. Chem. Bio., 13, 181). We showed that the mutation of a single residue, T1372 to some amino acids, such as Glu, can impact the iterative oxidation steps and stop the oxidation of 5hmC to 5fC/caC. However, the source of the stalling at the first oxidation step by some mutant TET proteins still remains unclear. Here, we studied the catalytic mechanism of oxidation of 5hmC to 5fC by WT and T1372E TET2 using an ab initio quantum mechanical/molecular mechanical (QM/MM) approach. Our results suggest that the rate limiting step for WT TET2 involves a hydrogen atom abstraction from the hydroxyl group of 5hmC by the ferryl moiety in the WT. By contrast, our calculations for the T1372E mutant indicate that the rate limiting step for this variant corresponds to a second proton abstraction and the calculated barrier is almost twice as large as for WT TET2. Our results suggest that the large barrier for the 5hmC to 5fC oxidation in this mutant is due (at least in part) to the unfavorable orientation of the substrate in the active site. Combined electron localization function (ELF) and non-covalent interaction (NCI) analyses provide a qualitative description of the evolution of the electronic structure of the active site along the reaction path. Energy decomposition analysis (EDA) has been performed on the WT to investigate the impact of each MM residue on catalytic activity.
Introduction
Cytosine methylation is an important epigenetic modification, which is directly involved in the modulation of transcriptional activity and other genome functions.1–5 Ten-eleven translocation (TET) enzymes that possess enzymatic activity toward 5-methylcytosine (5mC) have essential roles in regulating gene expression and maintaining cellular identity.6–8 TET enzymes include TET1–3 and the AID/APOBEC subfamily. TET2 catalyzes three iterative oxidation steps and converts 5mC into 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC).9–16 The TET enzymes belong to the Fe(II) and α-ketoglutarate (α-KG) dependent dioxygenase superfamily of enzymes17 and its overall catalytic mechanism has been largely inferred from related proteins such as TauD,18,19E. coli AlkB20 and its two human homologues, hABH2 and hABH3.21To initiate the oxidation, an O2 molecule needs to displace a water molecule from the primary coordination sphere of Fe. According to the conserved reaction mechanism of Fe(II)/α-KG dependent dioxygenases, the first step after the oxygen diffusion is the attack of the Fe-bound dioxygen on the carbonyl carbon (C2) of α-KG and the formation of an Fe(II)–OO–R (peroxy bridge). This process occurs concomitantly with decarboxylation of the α-KG and formation of succinate. TET enzymes use this general mechanism for three stepwise oxidation reactions to oxidize 5mC into 5hmC, 5fC, and 5caC. Thymine DNA glycosylase (TDG) can excise the last two oxidation products (fC and caC)22 and eventually results in the removal of DNA methylation markers via the base excision repair (BER) pathway.23
Most of the studies have so far focused on the oxidation of 5mC to 5hmC.24,25 Some studies suggest that 5hmC is stable in mammalian genome DNA, and it does not act as an intermediate for the next oxidation steps.16,25,26 They proposed that the hydrogen abstraction mechanism is more efficient on 5mC than 5hmC and 5fC.16,25–27 However, these studies do not explain the existence of 5fC and 5caC, and the fact that these are the substrates for the base excision by TDG.9,28–30
Recently, we reported combined experimental and computational studies on WT TET2 and a series of TET2 mutants. Our results revealed that the active site is shaped to enable higher-order oxidation states of the substrate.31 Our previous research showed that the scaffold established by T1372 and Y1902 in the active site is required for WT stepwise oxidation. T1372 forms a direct hydrogen bond with Y1902 and orients Y1902 to promote non-bonded interaction with 5hmC. This non-bonded interaction aligns 5hmC in the active site properly to go through the oxidation steps. We showed that the perturbation of this scaffold results in a “hmC-stalling” phenotype for several mutants, i.e., the mutants can only perform one round of oxidation, stalling at 5hmC.
For instance, the scaffold by T1372–Y1902 is eliminated by mutation of T1372 to glutamate, which results in the formation of a new hydrogen bond between E1372 and 5hmC. This hydrogen bond stabilizes 5hmC and changes its orientation with respect to Y1902, resulting in the misalignment of 5hmC in the active site. We proposed that the loss of the scaffold and formation of a direct hydrogen bond between the mutation site and 5hmC results in the unique stalling phenotype of this mutant. This study provides a qualitative image of the stepwise oxidation path in TET2. However, it raises new questions such as whether oxidation of 5hmC to 5fC is kinetically favorable. To gain further insight into the oxidation of 5hmC to 5fC, we have employed ab initio quantum mechanical/molecular mechanical (QM/MM) simulations on WT TET2 and T1372E mutant as an example of “hmC-stalling” mutants. The remainder of the paper is organized as follows: Section 2 presents the details for the methods employed for the QM/MM calculations and further analyses. This is followed by results and discussion in Section 3. Subsequently, the concluding remarks are presented in Section 4.
Methods
Two systems corresponding to TET2 with 5hmC and 5fC were generated and equilibrated based on the TET2 crystal structure with 5mC (PDBID: 4NM6 (ref. 27)) as previously reported.31 Several stable structures were extracted from the MD simulation to perform QM/MM optimizations on the reactant state (with 5hmC). The water molecule coordinated to the iron atom (trans to H1881) was replaced by an oxyl moiety. The α-KG molecule was also replaced by a succinate molecule since the present work is focused on the mechanism after the formation of the ferryl moiety and succinate. The mechanism studied in this paper is presented in Scheme 1. The snapshot with the lowest energy was selected for further QM/MM calculations. All the QM/MM calculations were performed with LICHEM,32 which is able to interface Gaussian09 (ref. 33) and TINKER34 to perform additive QM/MM with electrostatic embedding.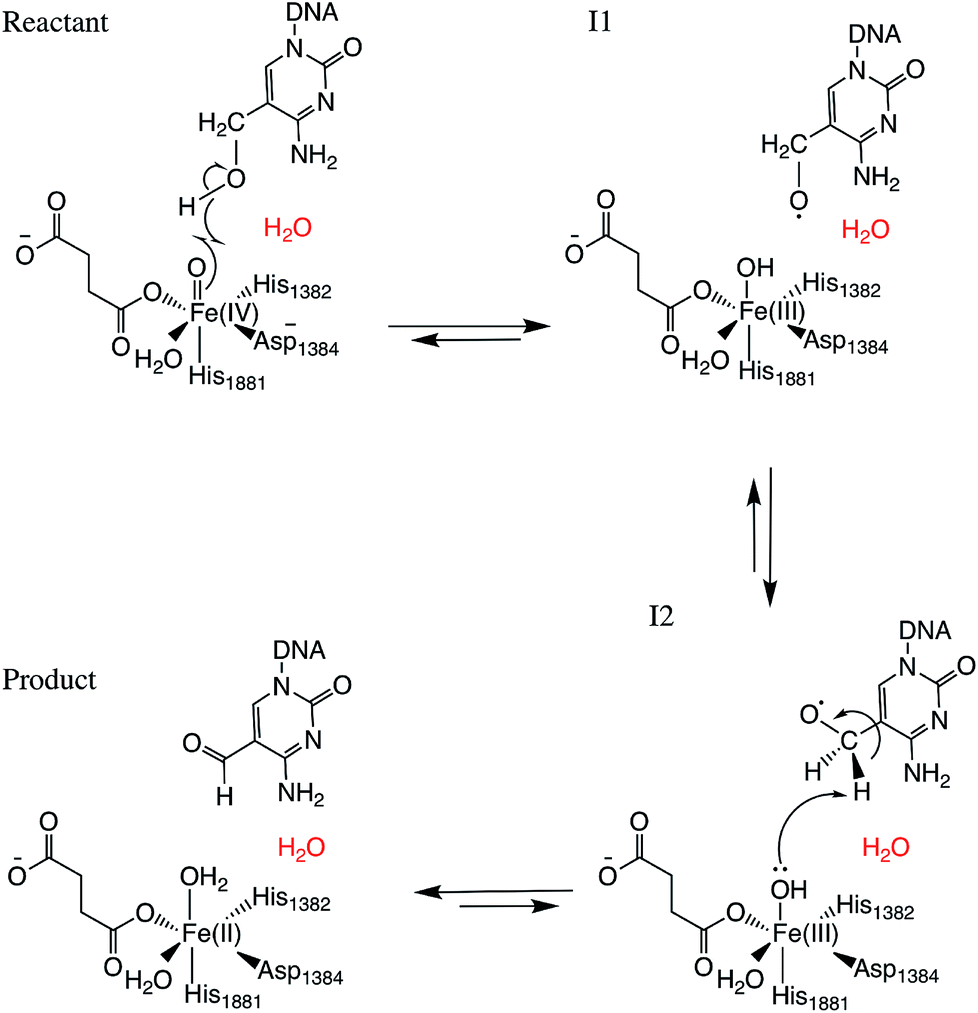 | ||
| Scheme 1 Overall mechanism for 5hmC to 5fC oxidation in TET2. The “2nd-shell” water molecule is shown in red. | ||
For each model, the QM part contains the Fe atom, the side chain of the residues coordinated to the iron (H1382, D1384 and H1881), the oxyl moiety, the succinate molecule, the water molecule (coordinated to the iron at an equatorial position) and substrate base (5hmC or 5fC). As the residues T1372 and Y1902 were found to establish a scaffold which is essential for iterative oxidation, they are also included in the QM regions. Fang et al. showed that the existence of a water molecule which is located near Fe(IV) = O is important for the mechanism of AlkB, another Fe/α-KG dependent enzyme.35 We observed from our MD simulations, one water molecule from the solvent diffuses into the active site and occupies the vacancy between D1384 and 5hmC for almost the entire simulation time. Therefore, we included that water molecule in our QM region. This water will be named as the “2nd-shell” water molecule in the subsequent discussion to distinguish it from the coordinated water at the equatorial position.
The QM region was modeled at the ωB97XD/6-311G(d,p) level of theory.35–37 The recent studies on the hydrogen abstraction mechanism by cytochrome P450 and AlkB have shown that this level of theory provides an accurate description of the systems under study.38–40 The remainder of the system (MM region) was treated with the AMEBR99SB force field.41 In LICHEM, the geometry optimizations are performed by adding the MM point-charges to the effective QM Hamiltonian,42,43 where the QM and MM atoms are optimized separately.32 To have a smooth connection at the QM/MM interface, the boundary atoms were modeled by the pseudobond approach.44Fig. 1 shows the QM and pseudobond atoms in the reactant state and the “2nd-shell” water molecule is circled in red. All calculations were carried out in the quintet electronic state since previous spectroscopic45 and computational8,35,46 studies found that mononuclear iron enzymes were in the high-spin (S = 2) electronic ground state configurations and quintet Fe(IV)-oxo species was the most reactive toward C–H bond activation.
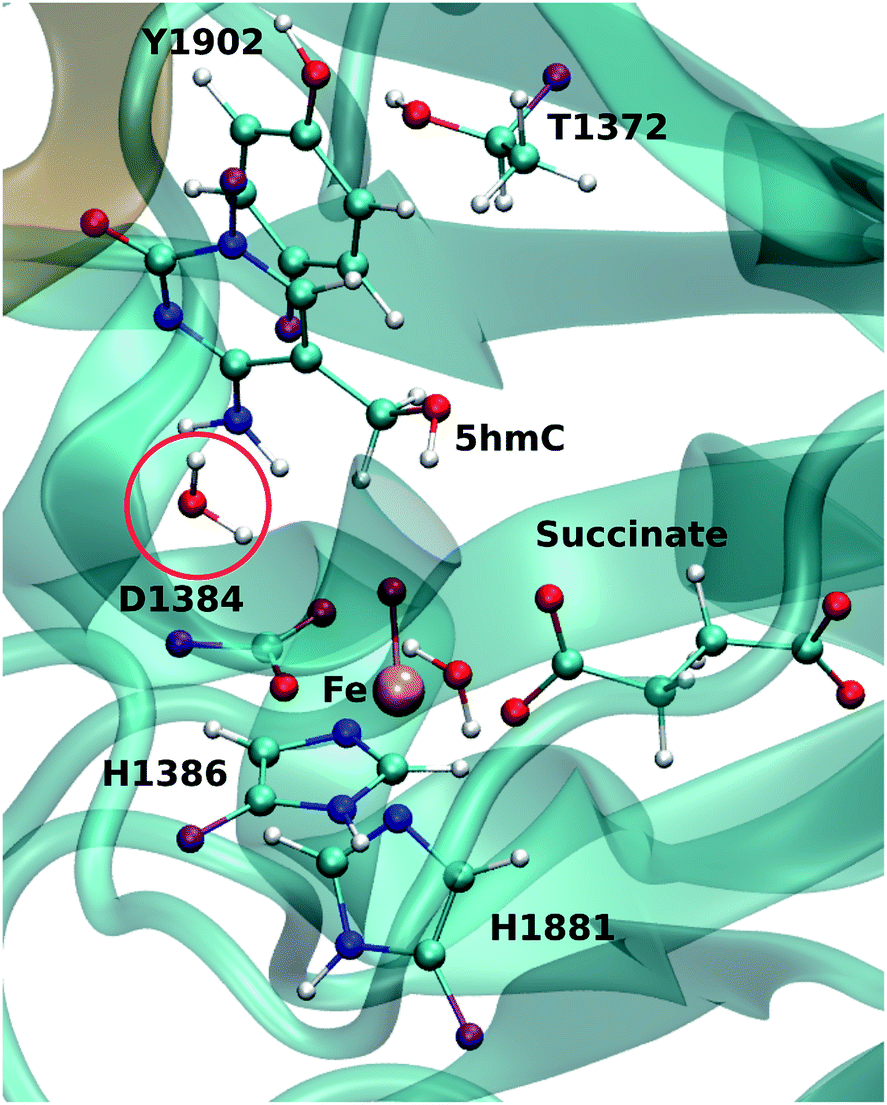 | ||
| Fig. 1 QM/MM active site for wild-type TET2. The protein (DNA) in MM region is shown in cyan (brown). The purple atoms are the pseudobond atoms. The “2nd-shell” water molecule is shown by a red circle. | ||
After the reactants and products were fully optimized, the Nudged Elastic Band (NEB) method47–49 as implemented in LICHEM was used for path optimizations between critical structures. Frequency calculations were performed for all critical point structures, and it was found that reactants and intermediates have no imaginary frequencies. The transition states in the NEB method are approximation to the true TS since the NEB version employed does not provide explicit TS optimization. Therefore, the frequency calculations have not been performed for the TS structures. In the case of the WT, only one chain with 17 beads connecting the reactant to the product was employed. For the T1372E mutant, each elementary step was optimized separately resulting in a string of 35 beads. The larger string for the mutant was necessary due to the significantly different orientation of the substrate (see below).
Energy decomposition analysis was used for the WT system to calculate the non-bonded inter-molecular interaction energies along the path for the critical points using energy decomposition analysis (EDA). This is accomplished by considering the changes in Coulomb and van der Waals interaction energies between individual residues and the QM subsystem when the system goes from reactants to the other critical points (intermediates and TSs): ΔE = 〈Enon-bondedi/QM〉MM,CP − 〈Enon-bondedi/QM〉MM,reactant, where i represents an individual residue in the MM subsystem, Enon-bondedi/QM represents the Coulomb or van der Waals interaction energy between residue i and the QM subsystem, CP stands for critical point (TS1, I1, TS2, I2, TS3 or product), and 〈⋯〉MM,CP and 〈⋯〉MM,reactant represent the averages over the ensembles obtained for the MM subsystem sampled with the QM subsystem corresponding to the respective point for an ensemble of structures of 500 ps (reactant or CP). This analysis provides a qualitative assessment of the role of individual residues in the reaction steps, all EDA calculations were carried out with an in-house FORTRAN90 program.50–52 This analysis has been previously employed for QM/MM and MD simulations to study a number of protein systems.35,53–57
The electron localization function (ELF) analysis58–60 is a topological analysis that can measure the electron localization in molecular systems. It was initially proposed on the basis of the Hartree–Fock (HF) approach58 and then was extended for DFT.61 The details on the topological analysis of ELF and its calculation methods in enzyme systems have been discussed previously.35,40,57 In summary, the critical points of ELF (attractors) are located on atoms, bonds, and lone pairs, while the molecular space can be divided into regions, named basins. Basins contain all points whose ELF gradient field converges toward the same attractor. The different basins do not overlap, and they can be denoted as C() for core electrons and V() for valence electrons. Core basins belong to a single nucleus. Valence basins are defined as belonging to either a single atom (e.g. lone pair) or two (or three) atoms (e.g. bonds). All ELF calculations for WT TET2 were performed with the TopMod package62 with a 2003 au grid, a step size of 0.1 au, and all wave functions are truncated, including only the QM atoms. The isovalue for the visualization is 0.5.
Non-covalent interaction (NCI) analysis is based on the analysis of the relation between the electronic density and the reduced density gradient (RDG) in regions of low electron density.63,64 The results obtained from the analysis consist of surfaces between the interacting molecules. These surfaces are assigned specific colors to denote the strength and characteristic of the interactions: green surfaces denote weak interactions (for example, van der Waals (VdW)), blue surfaces strong attractive interactions (for example, hydrogen bonds), and red surfaces strong repulsive interactions. All NCI calculations were performed with the NCI-Plot program63,64 using the same truncated wave functions as for the ELF calculations.
Results and discussion
Optimization of the reactant structures
Fourteen and six stable structures from the MD simulation of WT and T1372E mutant, respectively, were extracted to perform QM/MM optimizations on the reactant state (5hmC). Fig. S1† shows the relative energies of QM/MM optimized snapshots for WT and T1372E mutant. The lowest energy snapshots were selected for further calculations (snapshot number 2 in WT and number 3 in T1372E mutant). The lowest energy structures for the TET2/5fC WT system were used to model the product state for each selected snapshot. We also optimized the product state corresponding to snapshot number 1 in the T1372E mutant due to its relatively low energy and different optimized geometry (Fig. S1†). The optimized geometries of the active site of the TET2-5hmC/fC complex in the WT and the T1372E mutant along with NCI results are presented in Fig. 2–4.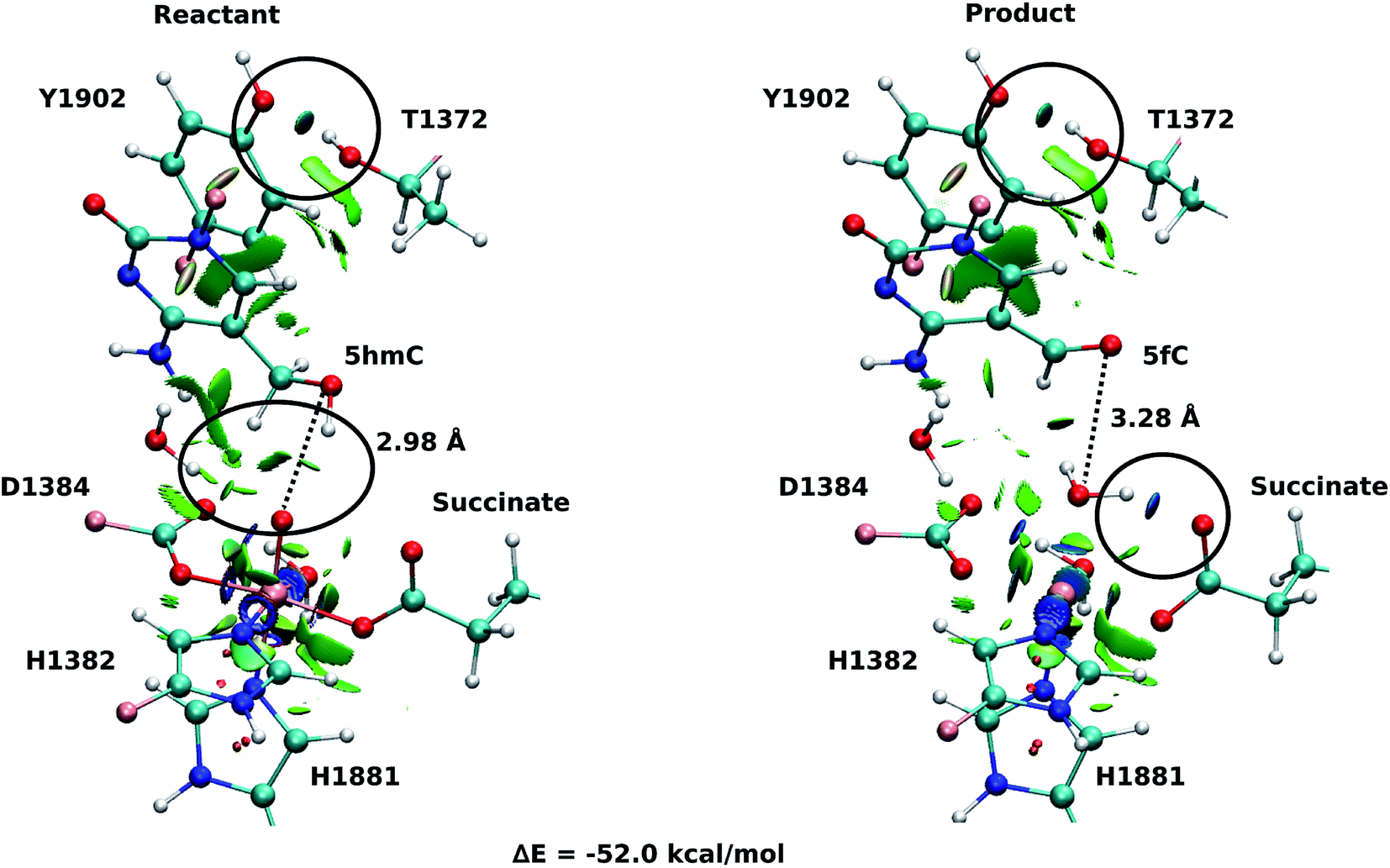 | ||
| Fig. 2 NCI plot for optimized geometries of the active site of wild-type TET2 with 5hmC/fC substrate. The hydrogen bonds are shown in black circle. The dashed line shows the distance between the oxygen atom of 5hmC/fC and the oxyl moiety. The MM subsystem is omitted for clarity. | ||
In the WT reactant state (Fig. 2) the oxyl moiety forms a hydrogen bond with the “2nd-shell” water molecule (similar to AlkB35), and another hydrogen bond with the hydroxyl group of 5hmC, simultaneously. These hydrogen bonds align the ferryl moiety and 5hmC in the active site for further oxidation. In the product, the succinate forms a hydrogen bond with the newly formed water molecule (at the axial position) to stabilize the product. The calculated reaction energy for the oxidation of 5hmC to 5fC in the WT is −52.0 kcal mol−1. The spin densities on the iron and the oxyl moiety in the reactant state are 3.25 and 0.54, respectively. This result suggests that there are 3 alpha electrons in the d orbitals of the iron, and an alpha electron in the p orbital of the oxyl moiety. Therefore, the electron configuration for the reactant is ISFe(III)–OF (Table 1 and Fig. S2†), which is consistent with the electron configuration of the iron-oxyl moiety in the AlkB reactant.35
| Mulliken spin density | Reactant | TS1 | I1 |
|---|---|---|---|
| Fe(III) | 3.25 | 4.13 | 4.35 |
| Oxyl O | 0.54 | 0.01 | 0.25 |
| Hydroxyl O | 0.00 | −0.43 | −0.86 |
| Hydroxyl H | 0.00 | 0.04 | 0.01 |
| Electron configuration | ISFe(III)–OF | HSFe(III)–OAF | HSFe(III)–OAF |
For snapshot number 3 of the T1372E mutant, the oxygen atom of the “2nd-shell” water molecule is oriented toward the oxyl moiety (Fig. 3). This is in contrast with the orientation of this water molecule in the WT (Fig. 2). Consequently, the spin densities on the iron and the oxyl moiety change and indicate the existence of 4 alpha electrons on the iron and a beta electron on the oxyl moiety. This spin densities correspond to an HSFe(III)–OAF electron configuration (Table S2, Fig. S2†). The corresponding product is highly stabilized due to a hydrogen bond formed between the “2nd-shell” water molecule and the newly formed water molecule. In addition, the new water molecule forms a hydrogen bond with formyl group of 5fC. These new interactions provide a large stabilization of the product with a resulting reaction energy of −62.8 kcal mol−1 for the T1372E mutant.
 | ||
| Fig. 3 NCI plot for optimized geometries of the active site of the snapshot number 3 in the T1372E mutant with 5hmC/fC substrate. The hydrogen bonds are shown in black circle. The dashed line shows the distance between the oxygen atom of 5hmC/fC and the oxyl moiety. The MM subsystem is omitted for clarity. | ||
As explained above, the product corresponding to snapshot number 1 of the T1372E mutant was also optimized to investigate whether there are other possible paths for the oxidation of this mutant. In this alternative orientation, the hydrogen atom of the “2nd-shell” water molecule in this snapshot is pointed toward the oxyl moiety (similar to the orientation observed in the WT), therefore, it can not form a hydrogen bond with the newly formed water molecule to stabilize the product (Fig. 2 and 4). The spin densities for the iron-oxyl moiety confirm the electron configuration of this system is similar to the WT (ISFe(III)–OF) (Table S2†). These results support the important role of the “2nd-shell” water molecule in the oxidation mechanism. Although the electron configuration of the Fe-oxyl moiety is similar to the WT, the reaction energy for this snapshot is highly endothermic (31.3 kcal mol−1). Thus, this alternative structure for the T1372E mutant was not considered for subsequent reaction path calculations.
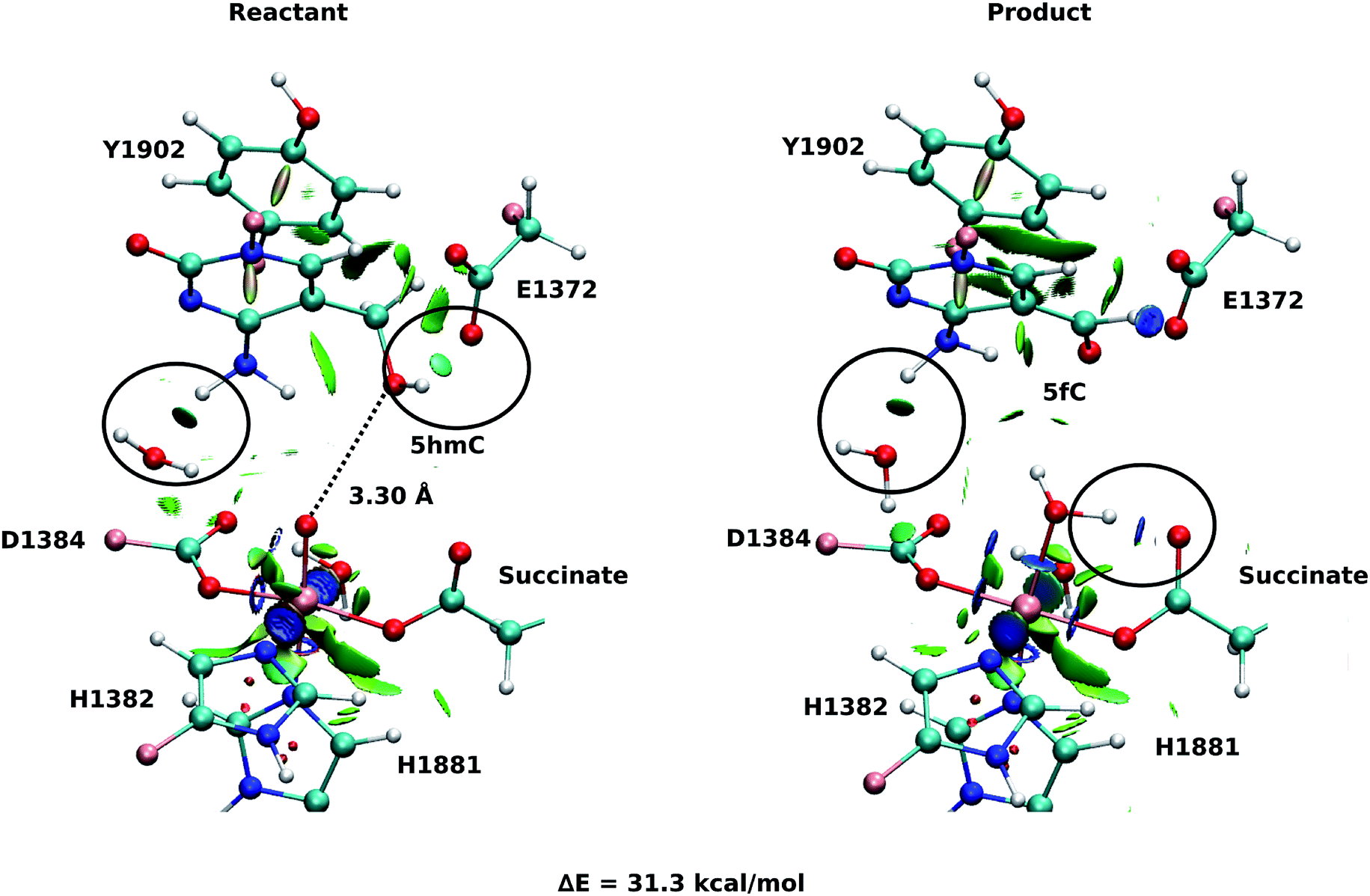 | ||
| Fig. 4 NCI plot for optimized geometries of the active site of the snapshot number 1 in the T1372E mutant with 5hmC/fC substrate. The hydrogen bonds are shown in black circle. The dashed line shows the distance between the oxygen atom of 5hmC/fC and the oxyl moiety. The MM subsystem is omitted for clarity. | ||
Our QM/MM optimized structures for both TET2-5hmC/fC complexes in the WT show that T1372 forms a hydrogen bond with Y1902 (Fig. 2). In T1372E, this hydrogen bond is eliminated by mutating T1372 to glutamate. Instead, the mutated residue (E1372) forms a hydrogen bond directly with 5hmC in the active site (Fig. 3). This new hydrogen bond stabilizes 5hmC and disrupts the non-bonded interaction between 5hmC and Y1902 (more details are provided in our previous study31). Therefore, the 5hmC orientation toward the ferryl moiety changes and the distance between the hydroxyl group and the oxyl moiety increases in comparison with the WT (Fig. 2 and 3). The hydrogen bond formed between the amino group of 5hmC and water molecules in the reactant and the product of the T1372E mutant confirms the misalignment of 5hmC in the active site (Fig. 3). These observations help explain the large energy barrier calculated for the oxidation of 5hmC to 5fC in this mutant compared with the WT-catalyzed reaction barrier as detailed below.
NEB path calculations for the oxidation of 5hmC to 5fC
Based on the optimized reactant and product structures, the potential energy surface of the whole reaction pathway of the TET2 mediated oxidation on 5hmC in the WT and the T1372E mutant were obtained and compared. The Fe(IV)-oxo moiety in the reactant state in our study is generated after O–O heterolysis. The high valence Fe(IV)-oxo species is highly reactive and can, therefore, activate the O–H bond of the substrate and mediate the hydrogen abstraction reaction.The hydrogen abstraction step is known to be the rate-limiting step in the catalysis of the Fe(II)/α-KG dependent enzyme, AlkB.35Fig. 5 shows the NEB path for the 5hmC to 5fC oxidation in the WT TET2. The calculated reaction path shows that 5hmC converts to 5fC via three steps. The first step is the rate-limiting step with a 20.1 kcal mol−1 barrier. The experimental barrier energy for the rate-limiting step of 1-methyladenine (1meA) oxidation mediated by AlkB is 19.8 kcal mol−1.65 In addition, a comparison of the calculated barrier and the reaction energies in the WT TET2, AlkB and ABH2 is presented in Table S3.† In the second step, the substrate shifts in the active site to reduce the distance between the second proton to be abstracted and the ferryl moiety, to properly orient the substrate for the next step (Fig. 5). The proton transfer from the (–CH2–) group of 5hmC occurs via a third transition state to form 5fC (Fig. 5). An animation of the reaction mechanism is provided for the NEB-calculated path for 5hmC/fC oxidation in the WT TET2 in ESI (Movie S1†).
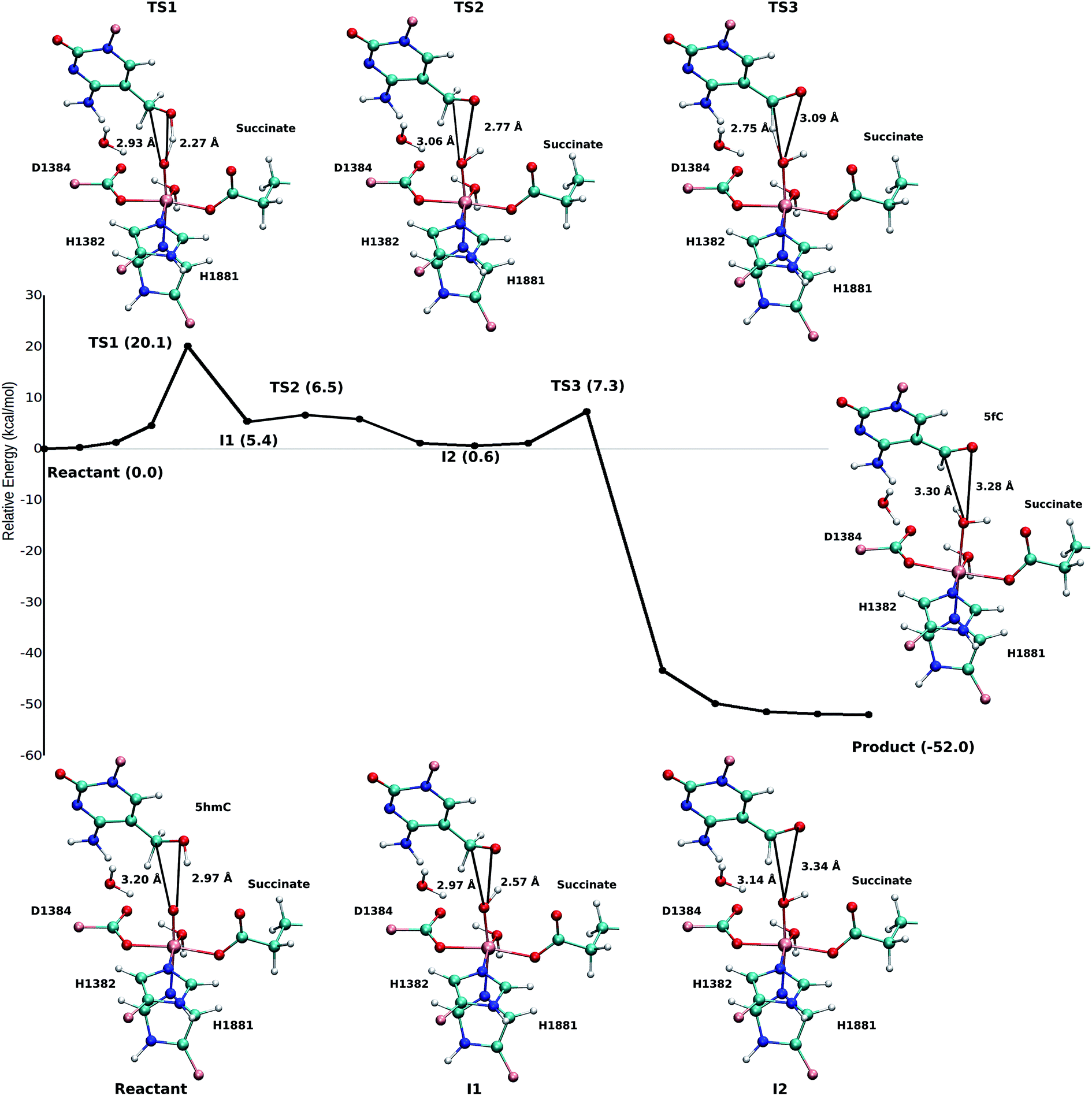 | ||
| Fig. 5 NEB path and the optimized geometries of critical structures for the wild-type. The distance between the carbon and hydrogen atoms of the DNA substrate and the oxyl moiety is shown in solid black line. | ||
The calculated path for the T1372E mutant (Fig. 6) indicates that the oxidation of 5hmC to 5fC occurs via four steps. The corresponding optimized critical structures are presented in Fig. S3.† Initially, the hydroxyl group of 5hmC is oriented toward E1372 to form a hydrogen bond in the reactant. The first step involves the breakage of this hydrogen bond, followed by rotation of the OH group toward the oxyl moiety to initiate the oxidation via TS1 with a barrier of 13.0 kcal mol−1. Once the hydrogen bond between 5hmC and E1372 breaks, the hydrogen abstraction occurs via second transition state with 14.1 kcal mol−1 barrier. Since the second hydrogen atom is not in a suitable position to be transferred to the Fe-hydroxyl moiety, the oxymethyl group of the substrate undergoes a rotation with a 25.4 kcal mol−1 barrier. After this rotation, the proton transfer occurs via a fourth transition state with an associated barrier of 39.6 kcal mol−1 barrier corresponding to the rate-limiting step. ESI Movie S2† shows the animation corresponding to the path for 5hmC/fC oxidation by T1372E TET2. This result suggests that the oxidation of 5hmC to 5fC by T1372E is kinetically unfavorable compared with WT TET2. Since the rate-limiting step for the calculated potential energy path of T1372E is significantly larger (almost 2 times as large) than WT, we will concentrate only on WT TET2 for the subsequent analyses presented below.
 | ||
| Fig. 6 NEB path for the T1372E mutant. The corresponding optimized geometries of critical structures are presented in Fig. S3.† | ||
Spin states of critical structures in the path
According to the proposed reaction mechanism, the ferryl moiety bears two positive charges. Ideally, the charge of the iron should be 4+, and the charge of the oxygen should be 2−. However, an electron can transfer between the Fe and O atoms, and both Fe(IV)–O2− and Fe(III)–O− could be possible. In O− the electron can be spin-down or spin-up, therefore there are two substrates for the quintet and triplet surfaces, respectively, corresponding to antiferro- or ferromagnetic coupling to the iron. Fang et al.35 studied spin density of the ferryl moiety in AlkB extensively. They showed that the minimum energy path for AlkB oxidation involves an inter-system crossing (ISC) between the reactant and the first transition state from the anti-ferromagnetically coupled Fe(III)–O− quintet to the ferromagnetically coupled Fe(III)–O− quintet in WT AlkB.Table 1 shows the Mulliken spin densities and electron configuration for the first step of the WT path. The Mulliken spin density on the iron-oxyl moiety in the reactant correspond to the intermediate spin iron coupled ferromagnetically to the oxygen (consistent with the reactant in AlkB). The spin density on the oxygen atom of the hydroxyl group in the first intermediate (I1) is −0.86, indicating the existence of a beta electron in its p orbital. This suggests that an alpha electron from the oxygen atom of the hydroxyl group transfers to the p orbital of the oxyl group to form an O–H bond. However, the electron in the p orbital of the oxyl moiety is spin-up. Therefore, this transfer is spin-forbidden and it is not conducive to form the O–H bond, unless the electron configuration of the iron-oxyl moiety changes. The spin densities for the first transition state (TS1) indicate that iron is in a high spin +3 state, anti-ferromagnetically coupled to the oxygen. Therefore, the electron in the p orbital of the oxyl moiety is spin-down and is now available to form the O–H bond with the alpha electron of the oxygen atom of the hydroxyl group (5hmC). The change in electron configuration from the reactant to the first transition state suggests the existence of an ISC between these two surfaces. These results are in agreement with the results obtained for AlkB. Table S4† shows the comparison between spin densities of the WT in AlkB and ABH2 with TET2. The difference in the spin densities of oxygen in hmC and carbon in 1meA is due to the fact that hydrogen is abstracted from an electronegative atom in TET2. Table S5† shows the Mulliken spin densities for all the critical points of the WT path.
As discussed above, the electron configurations of two selected snapshots of T1372E mutant are different due to their different optimized geometry and the orientation of the water molecule in active site. The electron configuration in snapshot number 1 with endothermic reaction energy is ISFe(III)–OF. The orientation of water molecule in this snapshot, and consequently, its electron configuration is similar to those in the WT. However, the oxidation mechanism for this snapshot is thermodynamically unfavorable due to its unstable product. In the snapshot with highly stable product (snapshot number 3), the electron configuration is HSFe(III)–OAF.
Electron localization function and non-covalent interaction analysis on critical structures
ELF and NCI together can be used to investigate the localization of electrons and the interactions between molecules.40 ELF can indicate the positions of the electrons in the system and reveal strong interactions, e.g. atomic centers, lone pairs, and covalent bonds. NCI provides information on weaker, non-covalent interactions such as hydrogen bonding and van der Waals forces. The combination of these two analyses provides a qualitative view of the evolution of the electron structure without the need for complicated orbital analyses.The basin populations for the bonds involved in the first hydrogen transfer (V(O(hmC), H1), V(O(oxo), H1), V(C(hmC), H2), V(O(oxo), H2) and V(O(hmC), C(hmC))) for the wild-type-catalyzed oxidation are shown in Table 2. The population of the O(hmC)–H1 basin is observed to decrease as the system transits from the reactant to I1, while the population in O(oxo)–H1 increases. This suggests that the H atom is transferred from the hydroxyl group of 5hmC to the ferryl moiety, as the O(oxo)–H1 bond is formed. At the TS1, the basin is shared by O(oxo), H1 and O(hmC) to form a three-center two-electron bond between these atoms. This confirms that the hydrogen atom is abstracted in this step. ELF basins of the iron-oxyl moiety in reactant, TS1 and I1 are presented in Table S6.†
| V(O(hmC),H1) | V(O(oxo),H1) | V(C(hmC),H2) | V(O(oxo),H2) | V(O(hmC),C(hmC)) | |
|---|---|---|---|---|---|
| Reactant | 1.35 | 0 | 2.17 | 0 | 0.96 |
| TS1 | 0.5 (V(O(hmC),H1,O(oxo)) | 2.16 | 0 | 0.87 | |
| I1 | 0 | 1.18 | 2.21 | 0 | 0.86 |
| TS2 | 0 | 1.22 | 2.19 | 0 | 0.90 |
| I2 | 0 | 1.24 | 2.17 | 0 | 0.92 |
| TS3 | 0 | 1.26 | 2.26 | 0 | 0.95 |
| Product | 0 | 1.34 | 0 | 1.35 | 0.97–1.14 |
By contrast with the first hydrogen abstraction, the second hydrogen is transferred as a proton. At TS3, there is no shared basin between C(hmC), H2 and O(oxo). This suggests that there is no electron that transfers with the hydrogen atom. In addition, the comparison of basin populations on C(hmC)–H2 and O(oxo)–H2 in I2 and the product confirms that a proton is transferred from 5hmC to Fe-hydroxyl moiety. There are two basin populations on O(hmC)–C(hmC) in the product, suggesting the formation of double bond for 5fC. This process is also presented in Fig. 7 and Movie S3.†
 | ||
| Fig. 7 ELF and NCI surfaces for the active site of (a) reactant; (b) TS1; (c) TS3; (d) product in the wild-type TET2-5hmC/fC complex. The isovalue for ELF and NCI is 0.5. For the NCI surfaces, the color scale is chosen so that blue indicates relatively strong attraction, green indicates relatively weak interaction, and red indicates relatively strong repulsion in the region of non-covalent interactions. The MM region is not shown for clarity. | ||
The NCI analysis shows 6 blue surfaces around the iron atom, indicating its octahedral structure (Fig. 7). In addition, there is an NCI surface between the oxo ligand and the first hydrogen being transferred. This surface becomes bluer, meaning the attraction increases as the reaction process toward TS1 (Movie S3†). Subsequently, this surface disappears to form a shared ELF basins between O(hmC), H and O(oxo) atoms. As the reaction proceeds forward, the basin between H and O(oxo) forms. There is also a similar NCI surface between oxo moiety and the second hydrogen being transferred at the beginning of the reaction. This surface vanishes when the first hydrogen is transferred and forms again before transferring the second hydrogen. Then, it disappears again while second hydrogen is transferring. The second hydrogen is transferred without sharing an ELF surface between three atoms, suggesting a proton transfer. When water molecule forms, one blue surface appears between the water and the iron, showing the water is coordinated to the iron. In addition, an ELF surface forms on the oxygen atom of the water molecule as oxygen lone pair. Beside, new water forms hydrogen bond with succinate while its interaction with the “2nd-shell” water molecule decreases.
Energy decomposition analysis
To gain a qualitative understanding of the effects of each residue in the MM environment on the QM subsystem, an energy decomposition analysis (EDA) at the residue level has been performed for the WT system. This analysis is based on the subtraction of the average intermolecular interaction energy (ΔE) between each residue in the MM region and the QM subsystem at all critical structures.55,56,66–69Fig. 8a–c show the EDA results for TS1, TS2 and TS3 with respect to the reactant. Fig. S4† shows the energy difference for the rest of the critical structures with respect to the reactant. The sign of this energy difference may be used to qualitatively termine the catalytic role of the residues. A positive(negative) ΔΔE indicates that this residue raises(lowers) the reaction barrier. In this study we consider the residues that have a |ΔΔE| ≥ 2 kcal mol−1 (Fig. 8 and Table 3). Table S7† shows all the residues with |ΔΔE| ≥ 1 kcal mol−1. | ||
| Fig. 8 Difference of total, Coulomb and vdW energies of (a) TS1, (b) TS2, (c) TS3 and reactant. A positive(negative) ΔΔE indicates that this residue raises(lowers) the reaction barrier. | ||
| Residue | ΔΔE | ΔΔE | ΔΔE |
|---|---|---|---|
| (TS1-reactant) | (TS2-reactant) | (TS3-reactant) | |
| K1142 | — | −2.6 | — |
| R1253 | 2.3 | — | — |
| T1259 | — | 3.3 | — |
| R1261 | −9.7 | −15.9 | −4.0 |
| R1262 | — | −5.2 | — |
| E1279 | — | — | 4.4 |
| K1299 | −6.2 | — | — |
| K1321 | 2.5 | — | 2.1 |
| R1383 | — | −2.1 | — |
| H1386 | 3.4 | 3.1 | 4.6 |
| N1387 | 3.1 | 2.9 | 3.2 |
| N1403 | — | — | 3.2 |
| K1409 | 2.4 | — | 2.3 |
| E1874 | −2.3 | — | — |
| R1896 | — | — | 3.8 |
| E1923 | — | −2.2 | −2.2 |
| K1924 | — | 2.5 | — |
The EDA based on the optimized structures of the TS1 and the reactant reveals eight residues have a |ΔΔE| ≥ 2 kcal mol−1. These residues include R1261, R1253, K1299, K1321, H1386, N1387, K1409, E1874. The EDA on the TS2 and TS3 indicate the following residues are involved in catalysis: K1142, T1259, R1261, R1262, E1279, K1321, R1381, H1386, N1387, N1403, K1409, R1896, E1923 and E1924. Residues R1261, H1386 and N1387 are common in all three elementary steps. The position of these residues in the protein are shown in Fig. 9 (see Table 3 for ΔΔE values). The sequence alignment of TET1–3 (Fig. S5†) shows that these residues are (at least partially) conserved in several TET homologues.
 | ||
| Fig. 9 Positions of the residues obtained from ΔΔE of TS1/2/3 relative to the QM region. The residues obtained from TS1/2/3 are shown in blue/brown/black. The purple residues are in common for all EDA. G* is the one of the linkers. The QM atoms are shown in ball and stick. | ||
Hu et al. investigated the impact of a series of TET2 mutants on 5mC oxidation, and showed that the K1299E/S1303N double mutant significantly decreases TET2 enzymatic activity,27 while R1262A has a minor affect on TET2 activity. Our results are consistent with these experimental findings given the effect of K1299 on the stabilization of TS1 and the (smaller) stabilizing effect of R1262 on the TS2. Another result from Hu et al. indicates that R1261G reduces TET2 activity, our calculation suggests that R1261 has a large stabilizing effect on all TSs. This high impact of R1261 on the catalytic activity is because of its interaction with α-KG (or NOG in the crystal structure27). Other possible interesting targets for mutagenesis include H1386 and N1387, which show destabilizing effects on all three transition states.
Conclusion
The WT and T1372E TET2 mediated oxidation of 5hmC has been investigated by means of QM/MM calculations. Based on the results of our previous molecular dynamics (MD) study on TET2, we performed reaction path calculations on the oxidation of 5hmC to 5fC to understand the source of substrate preference in the WT and a ‘5hmC-stalling’ mutant (T1372E). The calculated energy barrier of the rate-limiting step in the WT for the oxidation of 5hmC to 5fC is in good agreement with other Fe/α-KG dependent enzymes. ELF/NCI results show that the first hydrogen transfers as a hydrogen atom, while the second transfer involves a proton. The results also compared with available experimental and computational values for AlkB and ABH2. The comparison of the optimized structure of the reactant, product, and their corresponding electron configurations between the WT and the T1372E mutant reveals that the orientation of the water molecule in the active site is essential for iterative oxidation. Two possible oxidation paths have been investigated for the T1372E mutant, which show that one path is kinetically unfavorable, and the other one is not viable thermodynamically. Thus, our calculations suggest that the reason for the experimentally observed stalling phenotype in the T1372E TET2 variant is due to the formation of a hydrogen bond between the substrate and the mutation site (absent in WT), which results in an improper orientation of the substrate leading to a much larger energy barrier. EDA analysis provides insights on over 18 residues that significantly impact the catalytic step including three residues that have been experimentally shown to impact catalysis. Our calculations provide new targets for mutagenesis studies of human TET2.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Institutes of Health, grant numbers R01GM108583 and R01GM118501 to GAC. Computing time from Wayne State's C&IT is gratefully acknowledged.References
- R. Jaenisch and A. Bird, Nat. Genet., 2003, 33, 245–254 CrossRef PubMed
.
- S. Simonsson and J. Gurdon, Nat. Cell Biol., 2004, 6, 984–990 CrossRef PubMed
- X.-J. He, T. Chen and J.-K. Zhu, Cell Res., 2011, 21, 442–465 CrossRef PubMed
- A. P. Bird, Nature, 1986, 321, 209–213 CrossRef PubMed
- E. Li, C. Beard and R. Jaenisch, Nature, 1993, 366, 362–365 CrossRef PubMed
- S. C. Wu and Y. Zhang, Nat. Rev. Mol. Cell Biol., 2010, 11, 607–620 CrossRef PubMed
- C. Dahl, K. Grønbæk and P. Guldberg, Clin. Chim. Acta, 2011, 412, 831–836 CrossRef PubMed
- M. Tahiliani, K. P. Koh, Y. Shen, W. A. Pastor, H. Bandukwala, Y. Brudno, S. Agarwal, L. M. Iyer, D. R. Liu and L. Aravind, Science, 2009, 324, 930–935 CrossRef PubMed
- Y.-F. He, B.-Z. Li, Z. Li, P. Liu, Y. Wang, Q. Tang, J. Ding, Y. Jia, Z. Chen and L. Li, Science, 2011, 333, 1303–1307 CrossRef PubMed
- F. Mohr, K. Döhner, C. Buske and V. P. Rawat, Exp. Hematol., 2011, 39, 272–281 CrossRef PubMed
- K. P. Koh, A. Yabuuchi, S. Rao, Y. Huang, K. Cunniff, J. Nardone, A. Laiho, M. Tahiliani, C. A. Sommer and G. Mostoslavsky, Cell Stem Cell, 2011, 8, 200–213 CrossRef PubMed
- N. Bhutani, J. J. Brady, M. Damian, A. Sacco, S. Y. Corbel and H. M. Blau, Nature, 2010, 463, 1042–1047 CrossRef PubMed
- S. Kriaucionis and N. Heintz, Science, 2009, 324, 929–930 CrossRef PubMed
- S. Ito, A. C. D'Alessio, O. V. Taranova, K. Hong, L. C. Sowers and Y. Zhang, Nature, 2010, 466, 1129–1133 CrossRef PubMed
- T. Pfaffeneder, B. Hackner, M. Truß, M. Münzel, M. Müller, C. A. Deiml, C. Hagemeier and T. Carell, Angew. Chem., 2011, 123, 7146–7150 CrossRef
- S. Ito, L. Shen, Q. Dai, S. C. Wu, L. B. Collins, J. A. Swenberg, C. He and Y. Zhang, Science, 2011, 333, 1300–1303 CrossRef PubMed
- R. P. Hausinger, Crit. Rev. Biochem. Mol. Biol., 2004, 39, 21–68 CrossRef PubMed
- P. K. Grzyska, E. H. Appelman, R. P. Hausinger and D. A. Proshlyakov, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3982–3987 CrossRef PubMed
- E. Eichhorn, J. R. van der Ploeg, M. A. Kertesz and T. Leisinger, J. Biol. Chem., 1997, 272, 23031–23036 CrossRef PubMed
- S. C. Trewick, T. F. Henshaw, R. P. Hausinger, T. Lindahl and B. Sedgwick, Nature, 2002, 419, 174–178 CrossRef PubMed
- D.-H. Lee, S.-G. Jin, S. Cai, Y. Chen, G. P. Pfeifer and T. R. O'Connor, J. Biol. Chem., 2005, 280, 39448–39459 CrossRef PubMed
- Z. Liutkevičiūtė, G. Lukinavičius, V. Masevičius, D. Daujotytė and S. Klimašauskas, Nat. Chem. Biol., 2009, 5, 400–402 CrossRef PubMed
-
C. Walsh and G. Xu, DNA Methylation: Basic Mechanisms, Springer, 2006, pp. 283–315 Search PubMed
- H. Yardimci and Y. Zhang, Nat. Struct. Mol. Biol., 2015, 22, 656–661 CrossRef PubMed
- L. Hu, J. Lu, J. Cheng, Q. Rao, Z. Li, H. Hou, Z. Lou, L. Zhang, W. Li and W. Gong, Nature, 2015, 527, 118–122 CrossRef PubMed
- M. Bachman, S. Uribe-Lewis, X. Yang, M. Williams, A. Murrell and S. Balasubramanian, Nat. Chem., 2014, 6, 1049–1055 CrossRef PubMed
- L. Hu, Z. Li, J. Cheng, Q. Rao, W. Gong, M. Liu, Y. G. Shi, J. Zhu, P. Wang and Y. Xu, Cell, 2013, 155, 1545–1555 CrossRef PubMed
- A. Maiti and A. C. Drohat, J. Biol. Chem., 2011, 286, 35334–35338 CrossRef PubMed
- A. R. Weber, C. Krawczyk, A. B. Robertson, A. Kuśnierczyk, C. B. Vågbø, D. Schuermann, A. Klungland and P. Schär, Nat. Commun., 2016, 7, 10806–10819 CrossRef PubMed
- J. Lu, L. Hu, J. Cheng, D. Fang, C. Wang, K. Yu, H. Jiang, Q. Cui, Y. Xu and C. Luo, Phys. Chem. Chem. Phys., 2016, 18, 4728–4738 RSC
- M. Y. Liu, H. Torabifard, D. J. Crawford, J. E. DeNizio, X.-J. Cao, B. A. Garcia, G. A. Cisneros and R. M. Kohli, Nat. Chem. Biol., 2017, 181–187 CrossRef PubMed
- E. G. Kratz, A. R. Walker, L. Lagardère, F. Lipparini, J.-P. Piquemal and G. A. Cisneros, J. Comput. Chem., 2016, 1019–1029 CrossRef PubMed
-
M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, Gaussian 09, revision D. 01, 2009 Search PubMed
-
J. Ponder, TINKER-Software Tools for Molecular Design, 2008 Search PubMed
- D. Fang, R. L. Lord and G. A. Cisneros, J. Phys. Chem. B, 2013, 117, 6410–6420 CrossRef PubMed
- J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC
- R. Lonsdale, J. N. Harvey and A. J. Mulholland, J. Phys. Chem. Lett., 2010, 1, 3232–3237 CrossRef
- R. Lonsdale, J. N. Harvey and A. J. Mulholland, J. Chem. Theory Comput., 2012, 8, 4637–4645 CrossRef PubMed
- D. Fang, R. Chaudret, J.-P. Piquemal and G. A. Cisneros, J. Chem. Theory Comput., 2013, 9, 2156–2160 CrossRef PubMed
- D. A. Case, T. E. Cheatham, T. Darden, H. Gohlke, R. Luo, K. M. Merz, A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef PubMed
- H. Liu, Z. Lu, G. A. Cisneros and W. Yang, J. Chem. Phys., 2004, 121, 697–706 CrossRef PubMed
- Y. Zhang, H. Liu and W. Yang, J. Chem. Phys., 2000, 112, 3483–3492 CrossRef
- J. M. Parks, H. Hu, A. J. Cohen and W. Yang, J. Chem. Phys., 2008, 129, 10B613 CrossRef PubMed
- J. C. Price, E. W. Barr, B. Tirupati, J. M. Bollinger and C. Krebs, Biochemistry, 2003, 42, 7497–7508 CrossRef PubMed
- S. Ye and F. Neese, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1228–1233 CrossRef PubMed
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef
- H. Jónsson, G. Mills and K. W. Jacobsen, Nudged elastic bond method for finding minimum energy paths of transitions, Classical and Quantum Dynamics in Condensed Phase Simulations, World Scientific Press, Singapore, 1998, pp. 385–404 Search PubMed.
- S. E. Graham, F. Syeda and G. A. Cisneros, Biochemistry, 2012, 51, 2569–2578 CrossRef PubMed
- A. A. Elias and G. A. Cisneros, Adv. Protein Chem. Struct. Biol., 2014, 96, 39–75 Search PubMed
- S. W. Dewage and G. A. Cisneros, J. Phys. Chem. B, 2015, 119, 3669–3677 CrossRef PubMed
- Q. Cui and M. Karplus, Adv. Protein Chem., 2003, 66, 315–372 CrossRef PubMed
- S. Martí, J. Andrés, V. Moliner, E. Silla, I. Tuñón and J. Bertrán, Chem.–Eur. J., 2003, 9, 984–991 CrossRef PubMed
- H. M. Senn, D. O'Hagan and W. Thiel, J. Am. Chem. Soc., 2005, 127, 13643–13655 CrossRef PubMed
- G. A. Cisneros, L. Perera, R. M. Schaaper, L. C. Pedersen, R. E. London, L. G. Pedersen and T. A. Darden, J. Am. Chem. Soc., 2009, 131, 1550–1556 CrossRef PubMed
- D. Fang and G. A. Cisneros, J. Chem. Theory Comput., 2014, 10, 5136–5148 CrossRef PubMed
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef
- J.-P. Piquemal, J. Pilmé, O. Parisel, H. Gérard, I. Fourré, J. Berges, C. Gourlaouen, A. De La Lande, M.-C. Van Severen and B. Silvi, Int. J. Quantum Chem., 2008, 108, 1951–1969 CrossRef
- B. Silvi and A. Savin, Nature, 1994, 371, 683–686 CrossRef
- A. Savin, O. Jepsen, J. Flad, O. K. Andersen, H. Preuss and H. G. von Schnering, Angew. Chem., Int. Ed. Engl., 1992, 31, 187–188 CrossRef
- S. Noury, X. Krokidis, F. Fuster and B. Silvi, Comput. Chem., 1999, 23, 597–604 CrossRef
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef PubMed
- P. Koivisto, T. Duncan, T. Lindahl and B. Sedgwick, J. Biol. Chem., 2003, 278, 44348–44354 CrossRef PubMed
- G. A. Cisneros, H. Liu, Y. Zhang and W. Yang, J. Am. Chem. Soc., 2003, 125, 10384–10393 CrossRef PubMed
- G. A. Cisneros, M. Wang, P. Silinski, M. C. Fitzgerald and W. Yang, Biochemistry, 2004, 43, 6885–6892 CrossRef PubMed
- G. Li and Q. Cui, J. Am. Chem. Soc., 2003, 125, 15028–15038 CrossRef PubMed
- G. A. Cisneros, L. Perera, M. García-Doíaz, K. Bebenek, T. A. Kunkel and L. G. Pedersen, DNA Repair, 2008, 7, 1824–1834 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02961j |
| This journal is © The Royal Society of Chemistry 2018 |