
Low intensity focused ultrasound (LIFU) triggered drug release from cetuximab-conjugated phase-changeable nanoparticles for precision theranostics against anaplastic thyroid carcinoma†
Yang
Wang
a,
Guoqing
Sui
a,
Dengke
Teng
a,
Qimeihui
Wang
a,
Jia
Qu
a,
Lingyu
Zhu
a,
Haitao
Ran
 b,
Zhigang
Wang
b,
Chunxiang
Jin
a and
Hui
Wang
*a
b,
Zhigang
Wang
b,
Chunxiang
Jin
a and
Hui
Wang
*a
aDepartment of Ultrasound, China-Japan Union Hospital of Jilin University, Changchun, Jilin Province 130033, P. R. China. E-mail: wanghuicjuh@sohu.com
bInstitute of Ultrasound imaging of Chongqing Medical University, Chongqing 400010, P. R. China
First published on 25th October 2018
Abstract
Currently, multifunctional nanotechnology is strongly expected to improve the prospect of treatment and diagnosis. In this study, we synthesized epidermal growth factor (EGFR)-targeted phase-changeable polymer nanoparticles (C-HPNs) loaded with two drugs (C225 and 10-HCPT) for specific tumor targeting, synergistic chemotherapy, and ultrasound imaging under low-intensity focused ultrasound (LIFU). Cetuximab (C225), an EGFR-targeted monoclonal antibody, was conjugated on the surface of the nanoparticles, leading to a significantly high binding affinity to EGFR-overexpressing anaplastic thyroid C643 cells both in vitro and in vivo. As expected, an increase of more than 3- to 4-fold in the release rates of 10-HCPT was observed after LIFU irradiation in vitro, demonstrating that LIFU could enhance the release of drugs from the nanoparticles. Combined treatment with C-HPNs and LIFU showed excellent inhibition of cell proliferation in vitro, as well as a remarkable therapeutic effect in vivo. Moreover, the combined treatment simultaneously enhanced ultrasound imaging by LIFU-induced acoustic droplet vaporization (ADV). In conclusion, C225-modified and phase-changeable nanoparticles combined with LIFU exhibited great promise for concurrent targeted ultrasound molecular imaging and effective synergistic antitumor therapy.
1. Introduction
Anaplastic thyroid carcinoma (ATC) is relatively uncommon but one of the most malignant carcinomas, characterized by rapid proliferation, invasion of the neck structures, and distant metastasis.1–3 The grave prognosis of ATC is due to the rapid progress of the tumor before diagnosis.4 However, current treatment relies on multimodal combinations of surgery with chemotherapy, and external beam radiotherapy has failed to improve survival,5 resulting in a median survival of 3–5 months and a 1-year survival rate of less than 20%.6–8 Therefore, there are compelling reasons to develop a novel theranostic strategy to realize early diagnosis and effective therapy against ATC.Recently, triggerable drug-loaded nanocarriers combined with various internal or external stimuli, such as pH, temperature, ultrasound, laser, and microwave radiation, to facilitate controlled release have been widely investigated for personalized treatment and hold great potential to provide an improved antitumor therapy effect and reduced systemic toxicity.4,9–11 As one of the potential external triggers, low-intensity focused ultrasound (LIFU), which is noninvasive and exhibits considerable tissue-penetrating ability, has been intensively studied for tumor therapy and imaging diagnosis.12–16 In particular, LIFU in which low-intensity ultrasound (US) energy could focus on a specific site can control drug release accurately, thus significantly increasing chemotherapeutic efficacy, preventing damage to the surrounding tissues and minimizing adverse side effects.16–18 However, LIFU-triggered drug release from nanocarriers and further tumor therapy are still not satisfactory, which is mainly attributed to the relatively low accumulation efficiency of drug-loaded nanocarriers at tumor sites.19 On this ground, a large number of targeting ligands have been widely investigated to promote the aggregation of nanocarriers in tumor areas without severe side effects.16,20–24
Currently, several reports have demonstrated that epidermal growth factor receptor (EGFR) overexpression is common in ATC25,26 and is strongly related to tumor progression, migration, and invasion.27,28 EGFR-based immunotherapy with antibodies or small molecules can significantly enhance the therapeutic effect against ATC.26,29–32 Cetuximab (also known as Erbitux, C225) is a human-murine chimeric EGFR-targeted monoclonal antibody that has a high affinity for the extracellular domain of human EGFR and inhibits its epidermal growth factor signals in cells by blocking the normal function of the receptor.33,34 Cetuximab has been approved by the Food and Drug Administration for the therapy of EGFR-expressing malignant tumors, such as head and neck squamous cell carcinoma and colorectal cancer, in both preclinical studies and clinical trials.35–37 Thus, C225 could be an appropriate target moiety for the nanocarrier system to enhance the therapeutic effect of ATC. Interestingly, some studies have shown that the combination of C225 with camptothecin analogs such as 10-hydroxycamptothecin (HCPT) exhibits a significantly synergetic antitumor effect for a broad spectrum of cancers.38–40 Thus, 10-HCPT combined with C225 may lead to an effective treatment for ATC. However, due to the slow vascular diffusion of C225 and the hydrophobicity of 10-HCPT, the drug permeability into the tumor and the drug dosage within the tumor area were intrinsically limited, which tremendously weakened their antitumor efficacies.41–43 Fortunately, all of these issues may be improved by integrating 10-HCPT and C225 into a single nanocarrier to achieve the combination chemotherapy of C225 and 10-HCPT while simultaneously affording nanocarrier targeting ability.
In addition, medical imaging is essential for the early diagnosis and the monitoring of tumor progression.44,45 Several reports have suggested that LIFU has the potential to realize simultaneous ultrasound imaging and targeted drug delivery,14–18 fulfilling the current need for early diagnosis and therapy against ATC. However, conventional ultrasound agents, such as microbubbles, show excellent US contrast imaging ability but are not suitable for drug delivery to realize the tumor theranostic strategy due to the instability and large sizes of the microbubbles.46 To circumvent this dilemma, phase-changeable nanomedicines that could be triggered by LIFU have been intensively studied.47 The phase-changeable nanoparticles exhibited a US-triggered drug release profile under LIFU exposure, providing significant advantages for companion tumor-targeting ultrasound imaging and ultrasound-triggered drug delivery in tumor theranostics.48 This novel strategy affords the potential to improve cancer therapy, significantly addressing the current need for theranostics against ATC.49–52
The overriding aim of this study was to fabricate a C225-modified nanocarrier to precisely theranostically inhibit ATC that could accumulate in the tumor tissues via the high tumor-homing properties of C225 in addition to the EPR effect. The payload of 10-HCPT could be released and it exhibited synergistic chemotherapy with C225 triggered by LIFU, which could significantly maximize the therapeutic effectiveness, enhance ultrasound imaging and reduce the side effects of chemotherapeutic treatment. As shown in the scheme in Fig. 1, to fulfill these requirements, we used a biodegradable polymer (lactic acid-co-glycolic acid) (PLGA) as the shell structure of the nanocarrier due to its excellent biodegradability and biocompatibility.53–56 Then, we synthesized phase-changeable NPs with liquid perfluoropentane (PFP, with a boiling-point of 29 °C).47 Meanwhile, 10-HCPT was loaded into the NPs, at the same time as the conjugation of C225 on the surface of the PNs to afford C225-conjugated 10-HCPT-loaded phase-transformation PLGA nanoparticles (C-HPNs). As far as we know, this study is the first report of a LIFU-mediated C225-modified nanosystem that integrates tumor-targeted US imaging and US-triggered drug delivery against ATC. It will introduce a novel theranostic strategy for ATC.
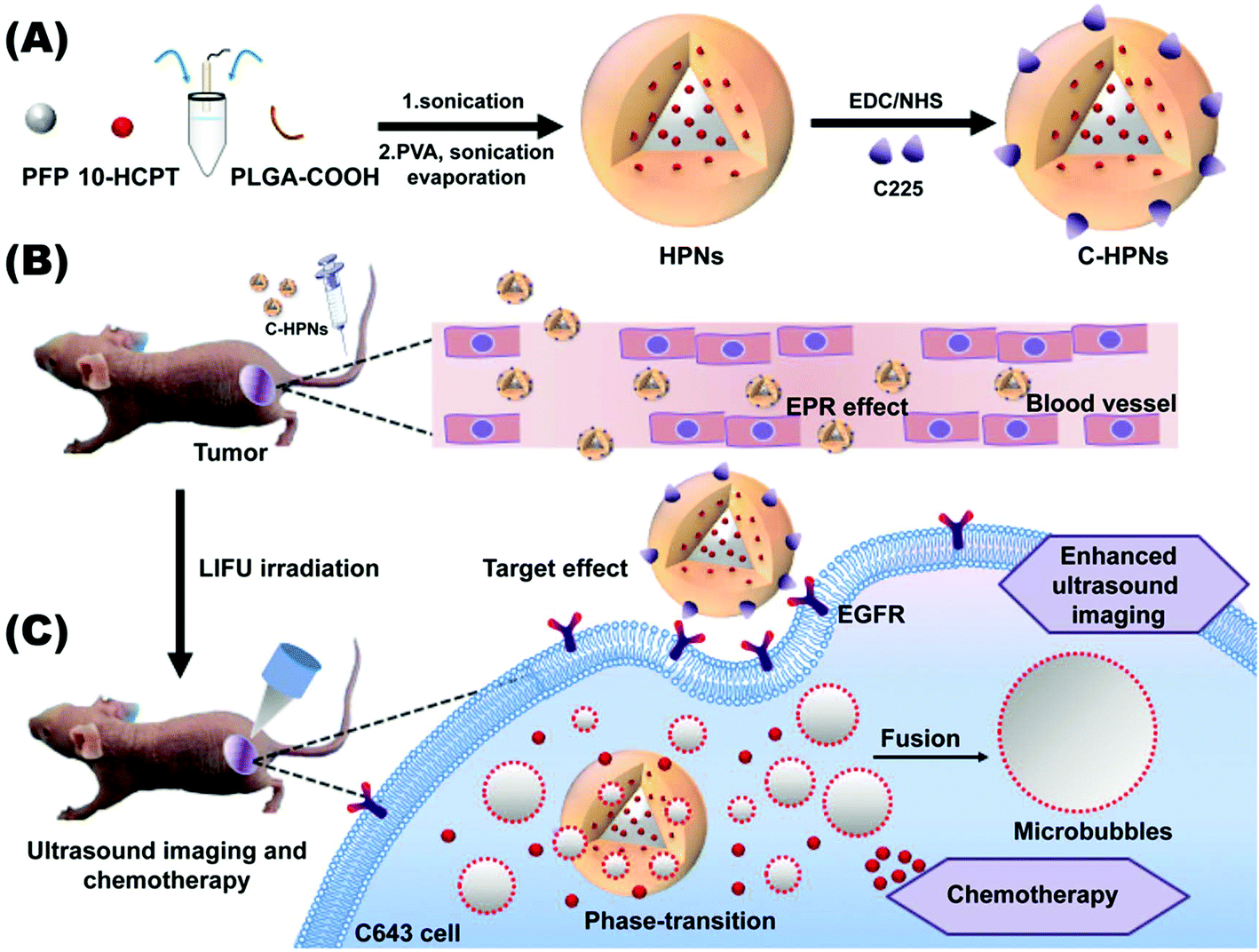 | ||
| Fig. 1 Schematic illustration of the multifunctional C225-modified theranostic system. (A) Synthetic process for C-HPNs via a double emulsion method and EDC/NHS chemistry. (B) With the nanoscale size, C-HPNs can pass through the tumor endothelial cell gap. (C) C-HPNs can attach to EGFR overexpressing cells via the high targeting property of C225. With LIFU assistance at the tumor site, the liquid PFP core in the nanoparticles vaporizes and transforms into microbubbles, enhancing tumor ultrasound molecular imaging for tumor diagnosis. The intracellular “explosion effect” is induced by ADV and UTMD, and causes the release of 10-HCPT into tumor for effective chemotherapy. | ||
2. Experimental section
2.1 Materials
PLGA–COOH (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, [MW] = 12.3 kDa) was purchased from Jinan Daigang Biomaterial Co. (Shandong, China). 10-HCPT (purity = 99.9%) was purchased from Lanbei Technology (Chengdu, China). Perfluoropentane (PFP, boiling point of 29 °C), polyvinyl alcohol (PVA, [MW] = 26 kDa), 2-(N-morpholino)ethanesulfonic acid (MES monohydrate), N-(3-dimethyl-aminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), and fluorescent dyes (1,1′-dioctadecyl-3,3,3′,3′ tetramethylindocarbocyanine perchlorate (DiI), 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindo-tricarbocyanine iodide (DiR) and 4′,6-diamidino-2-phenylindole (DAPI)) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Cetuximab (C225, Erbitux) was purchased from Merck KGaA Co. (Frankfurter, Germany). N-Hydroxysuccinimide (NHS) was purchased from Shanghai Medpep Co. (Shanghai, China). Tween 80 was purchased from BBI Life Sciences Co. (Shanghai, China). Goat anti-human IgG Fc (FITC) was purchased from Abcam Co. (Shanghai, China). RPMI-1640 medium (1640) and fetal bovine serum (FBS) were purchased from Gibco Co. (Carlsbad, CA, USA). Cell Counting Kit-8 (CCK-8) was obtained from Dojindo Molecular Technology (Shanghai, China). Trichloromethane, methanol, isopropanol, and dimethyl sulfoxide (DMSO) were purchased from Chongqing Chuandong Chemicals (Chongqing, China). All other reagents used in this work were of analytical grade and were used as received.
:50, [MW] = 12.3 kDa) was purchased from Jinan Daigang Biomaterial Co. (Shandong, China). 10-HCPT (purity = 99.9%) was purchased from Lanbei Technology (Chengdu, China). Perfluoropentane (PFP, boiling point of 29 °C), polyvinyl alcohol (PVA, [MW] = 26 kDa), 2-(N-morpholino)ethanesulfonic acid (MES monohydrate), N-(3-dimethyl-aminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), and fluorescent dyes (1,1′-dioctadecyl-3,3,3′,3′ tetramethylindocarbocyanine perchlorate (DiI), 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindo-tricarbocyanine iodide (DiR) and 4′,6-diamidino-2-phenylindole (DAPI)) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Cetuximab (C225, Erbitux) was purchased from Merck KGaA Co. (Frankfurter, Germany). N-Hydroxysuccinimide (NHS) was purchased from Shanghai Medpep Co. (Shanghai, China). Tween 80 was purchased from BBI Life Sciences Co. (Shanghai, China). Goat anti-human IgG Fc (FITC) was purchased from Abcam Co. (Shanghai, China). RPMI-1640 medium (1640) and fetal bovine serum (FBS) were purchased from Gibco Co. (Carlsbad, CA, USA). Cell Counting Kit-8 (CCK-8) was obtained from Dojindo Molecular Technology (Shanghai, China). Trichloromethane, methanol, isopropanol, and dimethyl sulfoxide (DMSO) were purchased from Chongqing Chuandong Chemicals (Chongqing, China). All other reagents used in this work were of analytical grade and were used as received.
2.2 Synthesis of 10-HCPT- and PFP-loaded polymer nanoparticles
10-HCPT-loaded PLGA nanoparticles encapsulating PFP (HPNs) were fabricated using a water/oil/water double emulsion process. For this purpose, 2 mg of 10-HCPT was dissolved in a mixture of 2 mL of trichloromethane and 1 mL of methanol, and 50 mg of PLGA–COOH was added to the solution when 10-HCPT was completely dissolved. The obtained organic solution was then emulsified using an ultrasonic probe (VCX-130; SONICS & MATERIALS Inc.; USA) with 25% intensity for 3 min (5 s on and 5 s off) after PFP (200 μL) was gradually added into the solution, and the resulting emulsion was poured into 5 mL of 4% PVA and sonicated at 20% intensity for 5 min (5 s on and 5 s off) to form a double emulsion. Finally, the emulsion was mixed with 10 mL of isopropanol (2%, v/v) and constantly stirred using a magnetic stirrer (HJ-1, Ronghua, China) for 3–4 h. Subsequently, the obtained solution was centrifuged (centrifuge, 5804R; Eppendorf, Hamburg, Germany) at 10000 rpm for 8 min and washed three times using PBS to remove the unloaded drugs. Finally, HPNs were collected, sealed, and stored at 4 °C for future use. All steps were carried out in an ice bath to maintain a low temperature due to the relatively low boiling point of PFP (29 °C). Pure PNs were fabricated according to the same procedures described above without the addition of 10-HCPT and were used as control groups. In addition, to prepare the 10-HCPT solution, approximately 2 mg of 10-HCPT was dissolved in 200 μL of DMSO, and 100 μL of Tween 80 was added to the mixture to promote complete dissolution. A further dilution was carried out using 0.9% NaCl solution.
2.3 C225 conjugation
Conjugation of C225 to the 10-HCPT-loaded nanoparticles was performed using carbodiimide chemistry. Briefly, the prepared HPNs were dissolved in 5 mL of MES buffer solution (0.1 M, pH 5.5) together with a mixture of 3 mg of EDC and 10 mg of NHS, and then incubated vigorously for a period of 1 h on a gentle shaker. The resulting solution was centrifuged and washed three times with PBS to remove unreacted EDC and NHS. Then, the sediment was redissolved in 5 mL of MES buffer solution (0.1 M, pH 8.0). Next, excess C225 was dropped into the above solution and stirred on a gentle shaker for another 2 h. After the reaction was completed, C-HPNs were obtained by centrifugation, washed thrice with PBS again to remove unconjugated C225 and preserved at 4 °C before use. All the aforementioned procedures were carried out in an ice bath. PNs with C225 conjugation (C-PNs) were also prepared using the same procedures.2.4 Characterization of C-HPNs
The morphology and particle distribution of C-HPNs were observed by optical microscopy (CKX41; Olympus, Tokyo, Japan) and confocal laser scanning microscopy (CLSM) (Nikon A1, Tokyo, Japan). The mean particle size, polydispersity index (PDI) and zeta potential of various forms of NPs were determined by using a dynamic light scattering analyzer (DLS) (Malvern Instruments, Malvern, UK). The morphological characterization of the NPs was performed using transmission electron microscopy (TEM) (H-7500; Hitachi, Tokyo, Japan). To better illuminate the stability of the polymer nanoparticles, the mean particle size of the nanoparticles was determined by DLS tested within 7 days. The absorbance spectra of NPs were recorded by using a UV-VIS spectrophotometer (UV2550, Shimadzu, Japan). Additionally, by increasing the temperature from 29 °C to 55 °C with a hot plate, the temperature-stimuli phase changeable nanoparticles in the hot plate could be monitored under an optical microscope. Moreover, thermotropic phase transition experiments on a saline solution and C225-10-HCPT-NPs without PFP were conducted with the same parameters.2.5 Evaluation of binding between C225 and HPNs
To detect the binding between C225 and HPNs, DiI-labeled C-HPNs and HPNs were incubated with FITC-labeled goat anti-human IgG (10 μL) for a period of 2 h in an ice bath on a gentle shaker. CLSM was used to observe the conjugation. Flow cytometry analysis (FCM; FACSVantage, BD, USA) was applied to further analyze the binding efficiency. Eventually, the bicinchoninic acid assay (BCA) was applied.572.6 Evaluation of encapsulation efficiency and drug-loading efficiency
To measure the encapsulation efficiency (EE) and loading efficiency (LE) of 10-HCPT, C-HPNs were completely dissolved in DMSO. High performance liquid chromatography (HPLC, LC-2010AHt; Shimadzu, Kyoto, Japan) was utilized to depict the standard concentration curve and assay the concentration of 10-HCPT. The drug EE and LE were calculated using the following equations:58–61| LE = W1/W2 × 100% |
| EE = W1/W3 × 100% |
2.7 In vitro 10-HCPT release
The dialysis bag diffusion technique was used to evaluate the release profile. An acoustic frequency of 1.0 MHz, an acoustic intensity of 1.2 W cm−2, a 50% duty cycle, pulse wave mode, a power of 6 W and a duration of 3 min were applied as LIFU parameters for sonication. Briefly, a C225-10-HCPT-PFP-NP resuspension was immersed in a dialysis bag ([MW] cut-off = 7000 Da) and then immediately treated with LIFU irradiation. The distal-sealed dialysis bag was then completely immersed in 200 mL of PBS containing 1.5% Tween 80 with stirring at 100 rpm in a thermostatic rotary shaker (37 °C). At predetermined time intervals, 2 mL of the release medium was collected to analyze the concentration of 10-HCPT, and an equal amount of fresh release medium was added into the container to maintain the sink condition. The concentration of 10-HCPT was analyzed by HPLC at 360 nm. C-HPNs at the same concentration and volume were used as the contrast group, and the procedures were performed according to the above steps but without LIFU irradiation.2.8 In vitro ADV and ultrasound imaging
According to the relevant research in our laboratory,15 we investigated LIFU-stimulated phase changeable C-HPNs at a power of 4–7 W and a duration of 1–4 min, and the parameters were as described above. MyLab 90 (Esaote, Italy) with an LA523 linear probe (5–12 MHz) and an agar gel phantom (4% agar w/v in distilled water) was used to evaluate the ADV and ultrasound imaging of C-HPNs in vitro. The B-mode and contrast-enhanced ultrasound (CEUS)-mode images were captured before and after LIFU irradiation, and the pre-images were acquired as controls. After 1 mL of C-HPNs (1.25 mg mL−1) was added in the gel phantom, the LIFU transducer was placed on the center of the phantom. The mean grayscale values within the ROI in the B mode and CEUS mode were analyzed using the DFY software (Chongqing Key Laboratory of Ultrasound Molecular Imaging, Chongqing, China). Moreover, ultrasound imaging of the saline solution and C225-10-HCPT-NPs without PFP was performed with the same LIFU parameters and duration.Furthermore, optical microscopy was used to observe the liquid collected from the gel mold immediately after LIFU irradiation. To determine whether the generation of microbubbles is due to mechanical oscillation or temperature increase with LIFU exposure, 1 mL of C-HPNs (1.25 mg mL−1) was placed in a 96-well plate, and then irradiated with LIFU at a power of 4–7 W and a duration of 1–5 min. PBS was used as the control group. An infrared (IR) thermal imaging system (Fluke Ti32, Fluke Corporation, USA) was applied to determine the temperature during LIFU irradiation.
2.9 Cell culture and nude mice
A human ATC line (C643) that overexpresses the EGFR was obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China).25,26,29 The cells were cultured in RPMI-1640 medium containing 10% FBS and 1% penicillin–streptomycin at 37 °C in humidified air containing 5% CO2. Female BALB/C mice and nude mice (weighing approximately 20 g, 4 weeks) were purchased from and bred at the Laboratory Animal Centre of Chongqing Medical University (Chongqing, China). All animals in our experiments were obtained from the Laboratory Animal Center in Chongqing Medical University and maintained under the guidelines approved by the Animal Ethics Committee of Chongqing Medical University (Chongqing, China). In addition, all the animal experimental operations were in strict accordance with the policy of the Institutional Animal Care and Use Committee (IACUC) of Chongqing Medical University, and the IACUC has approved this study.To establish an anaplastic thyroid tumor model in nude mice, C643 cells in the logarithmic growth phase were collected, washed twice with serum-free RPMI-1640 medium and injected subcutaneously to the left flank of every mouse (4 × 106 cells in 200 μL of serum-free RPMI-1640 medium per mouse). The length and width of the tumors were measured by using a Vernier caliper, and the tumor volume was calculated using the following equation: volume = (length × width2)/2.62
2.10 In vitro cell experiments
2.11 Biosafety evaluation of C-HPNs
Healthy BALB/C mice (weighing approximately 20 g, 4 weeks) were intravenously injected with C-HPNs (200 μL, 5 mg mL−1), and mice administered with saline solution were used as controls. The mice were sacrificed on the 1st, 7th, and 15th day (five mice per group) after injection, and the blood samples were collected for biochemical and hematological examination. The major organs were collected and stained with H&E (hematoxylin and eosin) for histological analysis.2.12 In vivo fluorescence imaging in xenograft tumors
C643 tumor-bearing mice were administered with a consistent dose of DiR-labeled HPNs and C-HPNs (2.5 mg mL−1, 200 μL). All mice were completely narcotized with 1% pentobarbital, and fluorescence images were obtained before the injection and 3 h, 6 h, and 24 h post-injection. The changes in the fluorescence intensity within the tumor regions were evaluated in vivo by a living fluorescence imaging for small animals (IVIS Lumina Series III; PerkinElmer Inc., Waltham, MA, USA). The major organs and tumors of one mouse were collected for ex vivo fluorescence imaging. Furthermore, in order to evaluate the targeting efficiency of C-HPNs after LIFU irradiation, DiI-labeled HPNs and C-HPNs (2.5 mg mL−1, 200 μL) were injected through the tail vein of C643 tumor-bearing mice, and LIFU (1.2 W cm−2; duty cycle, 50%; pulse wave mode; 6 W, 3 min) was carried out six hours after injection. The tumor tissues and major organs were collected, sectioned, and frozen at the predetermined time post-injection. After fixation with 4% paraformaldehyde, DAPI dyeing was performed for 5 min in the dark. CLSM was applied to observe the biodistribution of DiI-labeled NPs.2.13 In vivo ADV and ultrasound imaging
Fifteen mice bearing C643 tumors (100 mm3 in volume) were randomly divided into three groups (n = 5): (1) saline solution, (2) HPNs, and (3) C-HPNs. All experiments were conducted under complete anesthesia using 1% pentobarbital. The LIFU probe was placed on the tumor surface, and ultrasound imaging in the B mode and CEUS mode was performed and images captured before and after LIFU irradiation (1.2 W cm−2; duty cycle, 50%; pulse wave mode; 6 W, 3 min) six hours after injection. The mean grayscales of both modes in the tumor area were analyzed using the DFY software.2.14 Therapeutic effect in vivo
An antitumor assay was performed on mice bearing anaplastic thyroid cancer xenografts when the subcutaneous tumor reached 100 mm3 in volume. In total, the tumor-bearing mice were divided randomly into 10 groups (n = 5 per group): the saline control group (control) and the groups administered free 10-HCPT solution (10-HCPT), C-PNs, HPNs, C-HPNs, LIFU, 10-HCPT + LIFU, C-PNs + LIFU, HPNs + LIFU, and C-HPNs + LIFU. Two hundred microliters of the mixture in 0.9% saline solution was injected through the tail vein at the same 10-HCPT dose (1 mg kg−1). LIFU parameters (acoustic intensity, 1.2 W cm−2; duty cycle, 50%; pulse wave mode; 6 W, 5 min) were determined with the ultrasound coupling agent filling between the LIFU probe and tumor surface six hours after injection. Five consecutive treatments were conducted every three days starting on day 20 and ending on day 37 after the injection of C643 cells. The tumor volume and body weight of each mouse were recorded every two days, and the tumor volume changes were determined from the relative tumor volume V/V0 (V0: the initial tumor volume before treatment), and the tumor growth curves were drawn simultaneously. All mice were euthanized on day 37, and the tumor masses were dissected and weighed. The tumor inhibition rate (TIR) was calculated as follows: TIR (%) = [(x − y)/x] × 100%, where x and y represent the average weight of tumors in the control and treatment groups, respectively.18 Additionally, histology and immunohistochemistry experiments were carried out. Tissue sections were stained with H&E to identify the histopathological changes. To evaluate tumor cell proliferation and apoptosis, proliferating cell nuclear antigen (PCNA) and terminal deoxynucleotidyl transferase (TdT) dUTP nick-end labeling (TUNEL) staining was performed. In addition, to evaluate the biological toxicity of each treatment group, blood samples were collected for hematological examination, biochemical examination of the liver and a renal function assay.2.15 Statistical assay
Each experiment was repeated at least three times. The mean ± standard deviation of all the data was analyzed in the tables and figures. One-way ANOVA was utilized for the inter-group comparison, and the bilateral paired t-test and the least significant difference (LSD) test were utilized for statistical evaluation. P values < 0.05 were considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).3. Results and discussion
3.1 Characterization of C-HPNs
C-HPNs were fabricated to codeliver C225/10-HCPT, where 10-HCPT and PFP were encapsulated in the core simultaneously with C225 conjugation on the surface. As shown in the photograph inset in Fig. 2A, C-HPNs produced an emulsion with a yellow appearance, while the PNs formed a white emulsion, and the nanoparticles exhibited an almost perfect spherical morphology without obvious aggregation under an optical microscope (Fig. 2A). The average particle size, PDI, and zeta potential of various formulations of NPs are listed in Table S1.† The particle size and zeta potential of HPNs were (204.3 ± 3.84) nm and (−2.2 ± 0.77) mV, respectively. After modification with C225, the particle size and zeta potential of the obtained C-HPNs were 241.7 ± 4.04 nm and −3.28 ± 0.91 mV, respectively (Fig. 2C). The larger size further indicated the successful conjugation of C225. Additionally, as shown in Fig. S1,† the TEM images of C-HPNs further confirmed the spherical morphology of a typical shell–core structure with a uniform shell. The diameter was approximately 235 nm, which is consistent with the DLS results (Fig. 2B). Moreover, negligible variation in the particle size was observed at 37 °C for 90 min (Fig. S2†), and C-HPNs could maintain a relatively stable nano-size for 7 days at 4 °C storage, indicating excellent stability (Fig. 2D). The nanoscale size of C-HPNs allowed them to pass through the tumor vascular endothelial spaces via the EPR effect,16 and the negative charge enabled the nanoparticles to repel each other, preventing aggregation in the suspension and thereby ensuring the uniform dispersion of nanoparticles. As shown in Fig. 3D, C-HPNs exhibited small UV absorption peaks of 10-HCPT at 390 nm in the UV-vis absorption spectrum compared with the bare PNs, indicating the successful loading of 10-HCPT. Moreover, after functionalization with C225, the absorption spectrum of C-HPNs did not exhibit any significant change, indicating that polymer nanocarriers could be an appropriate vehicle to deliver 10-HCPT. Nevertheless, the small UV absorption peaks of C-HPNs might be the results of the fact that the drugs were encapsulated into the nanoparticles, and PLGA may also have an effect on the absorption peaks of 10-HCPT.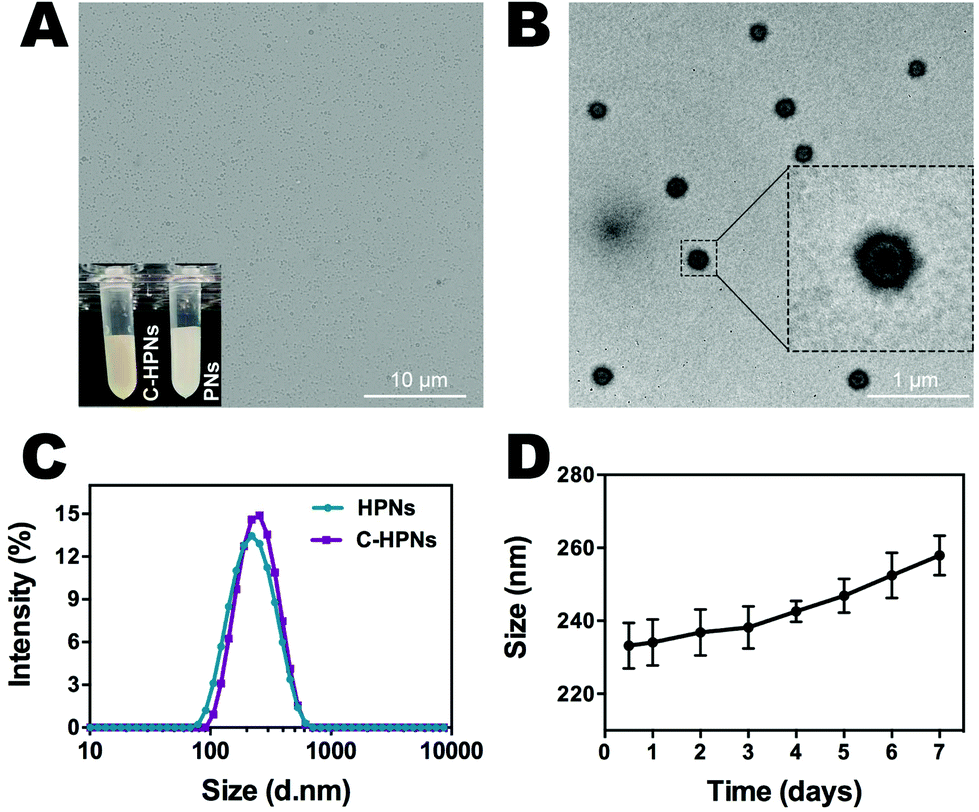 | ||
| Fig. 2 Characterization of C-HPNs. (A) Optical microscopy images of C-HPNs; digital photographs of C-HPNs and PNs are shown in the inset of (A). (B) TEM images of C-HPNs. (C) Size distribution of C-HPNs and HPNs. (D) Size changes of C-HPNs at 4 °C after long-term storage (the data are shown as mean ± SD, n = 3 per group). | ||
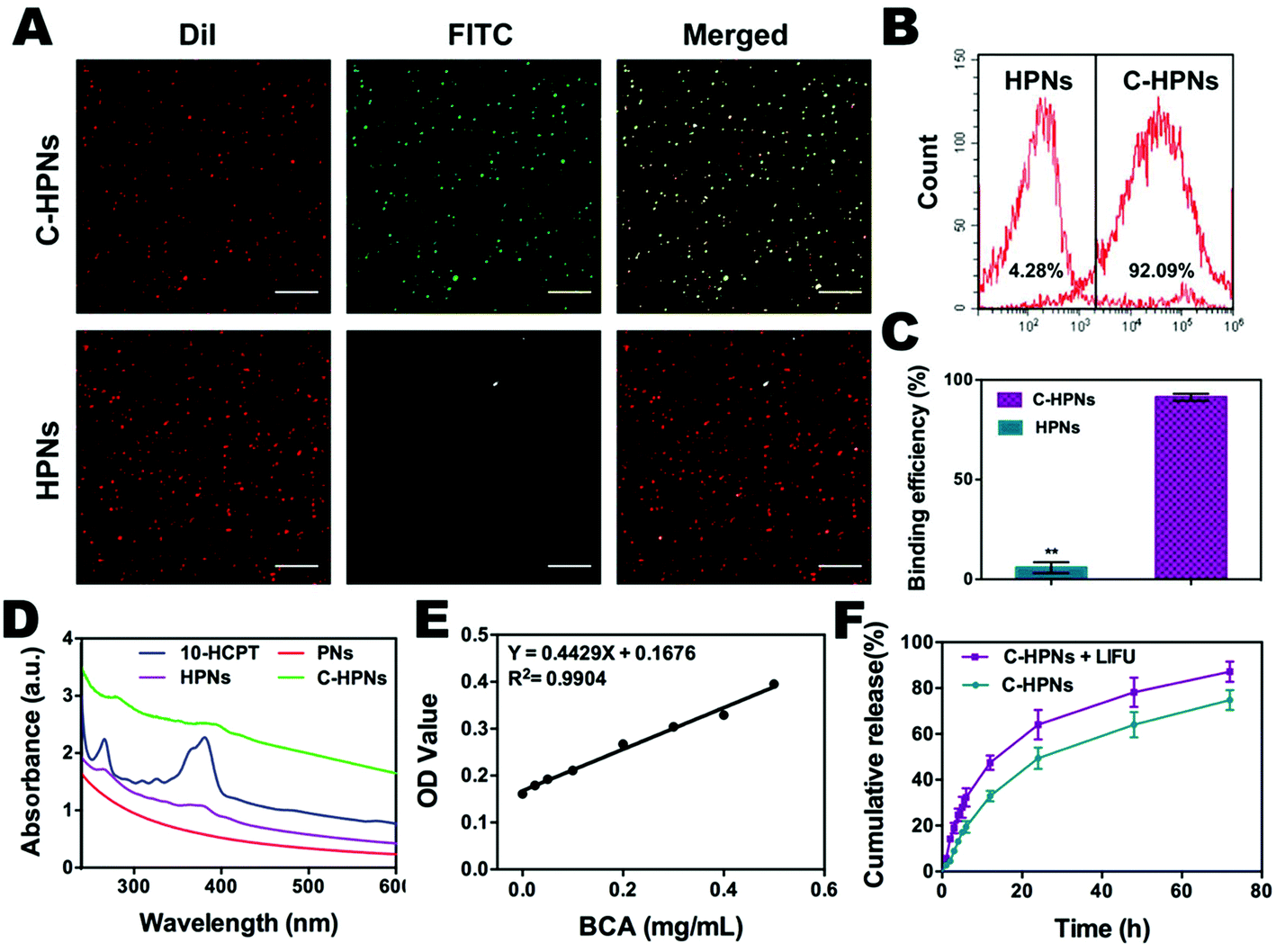 | ||
| Fig. 3 (A) CLSM image showing preferential binding between FITC-labeled secondary antibody and DiI-labeled C-HPNs and HPNs. The scale bar is 10 μm. (B) Flow-cytometry results and (C) histogram showed the average binding efficiency of different NPs (**p < 0.01). (D) Identification of C-HPNs by UV-vis spectroscopy. C-HPNs showed the typical UV absorption peaks of 10-HCPT, indicating that 10-HCPT was loaded successfully. (E) The concentration–absorbance standard curve of bicinchoninic acid (BCA) protein. (F) In vitro drug release profiles of C-HPNs and C-HPNs + LIFU (the data are shown as mean ± SD, n = 3 per group). | ||
A thermotropic phase transition experiment was carried out to further verify the encapsulation of PFP and the phase-transformation temperature. Fig. S3† shows that no obvious microbubbles appeared at 37 °C, and then a small amount of nanoparticles expanded gradually after the temperature reached 40 °C. The volumes of numerous nanoparticles gradually enlarged and transformed into larger, micron-sized bubbles at 45 °C. Moreover, almost all nanoparticles enlarged to form microbubbles when the temperature reached 50 °C, and some microbubbles started to collapse. Only a few microbubbles could be observed when the temperature was increased to 55 °C. However, no obvious changes were observed in the saline and C–HN groups. The above results indicated that PFP was successfully encapsulated into the NPs and that PFP plays a key role in the phase-changeable process of NPs. It is worth mentioning that the nanoparticles were still stable without the generation of obvious microbubbles at physiological body temperature. PFP can convert from the liquid phase to the gas phase at physiological body temperature due to its low boiling point (29 °C).63 This paradoxical phenomenon may be attributed to the coating polymer material and the increased Laplace pressure that elevated the threshold of the phase transition temperature of PFP inside the nanoparticles, enabling the nanoparticles to remain stable and rendering them unable to evaporate spontaneously at body temperature.64–67 As the temperature rose, polymer nanoparticles with PFP cores could convert into microbubbles, and the adjacent microbubbles could fuse with each other to form larger microbubbles, which was consistent with previous studies.68 The C-HPNs could circulate steadily in vivo due to their stable nanosize, which facilitated passive aggregation at the tumor site via the EPR effect.51,69
3.2 Assessment of binding efficiency between C225 and HPNs
In this study, an FITC-labeled secondary antibody was used as an indirect fluorescent marker to indicate the binding between C225 and PLGA nanoparticles (NPs). Significant yellow fluorescence signals were observed in the combined channel for the C-HPNs (Fig. 3A). In contrast, the HPNs showed distinct red fluorescence signals. FCM revealed that the mean fluorescence intensities of the C-HPNs and HPNs were approximately 92.09% and 4.28%, respectively (Fig. 3B and C), confirming the high conjugation between the second antibody and C-HPNs. This result indirectly demonstrated that C225 was successfully attached to the polymer nanoparticles. Moreover, the BCA assay demonstrated that the concentration of C225 binding on the surface of NPs was approximately 0.36 mg mL−1 (Fig. 3E).3.3 Drug loading and LIFU-triggered 10-HCPT release in vitro
The EE and LE of C-HPNs were 73.76 ± 12.1% and 6.39 ± 1.49%, respectively. These values indicated that NPs could serve as an appropriate vehicle to efficiently encapsulate 10-HCPT. As we expected, the release rates of C-HPNs with LIFU irradiation were over 3–4-fold higher than those without LIFU irradiation at 1 h and 2 h. Moreover, substantially higher 10-HCPT release rates of C-HPNs with LIFU irradiation were detected (approximately 87.21% vs. 74.85%) up to 72 h (Fig. 3F). The LIFU-responsive enhanced release profile might be attributed to the ADV effect, which increased the diffusion of the drug from the interior of the NPs into the surrounding medium after the microbubbles collapsed.3.4 In vitro ADV and ultrasound imaging of C-HPNs
To detect whether the phase shift of C-HPNs was stimulated by LIFU in vitro, we evaluated the adequate ultrasound power and duration of LIFU. As shown in Fig. 4A, anechoic and non-contrast-enhanced signals of the C-HPNs were observed before LIFU irradiation. With a duration of 1 min to 4 min, minimal ultrasound signals were obtained in both modes at power values of 4 W and 5 W, indicating that the power was not high enough to trigger the phase transformation due to the stiffness of the polymer shell. Nevertheless, increasing the power to 6 W resulted in gradually stronger ultrasound signals in either mode as the duration increased from 1 min to 3 min. The signals peaked at 3 min, and then began to reduce at 4 min. This phenomenon indicated that the C-HPNs could convert into microbubbles triggered by LIFU at a certain power, and the declining ultrasound signals at 4 min may be attributed to the instability of microbubbles when their volume expanded to a certain extent. However, enhanced signals were observed at 1 min with a LIFU power of 7 W and then gradually declined as the irradiation time lengthened, suggesting that a higher power could generate multiple microbubbles within a short time, but the generated microbubbles collapsed quickly due to the higher power as the duration lengthened. The DYF software further verified that the average grayscale values were the strongest at a power of 6 W and a duration of 3 min (p < 0.05) (Fig. 4B and C). In addition, saline solution and C-HNs without PFP were irradiated with LIFU at the same parameters, and barely visible contrast signals appeared, indicating that the gasification of PFP was the key to enhance ultrasound imaging. The above results further indicated the excellent ultrasound imaging properties of C-HPNs, and the power and duration of LIFU were crucial to triggering the phase transition. Therefore, adequate parameters for inducing ADV with a LIFU power of 6 W and a duration of 3 min were chosen for subsequent research.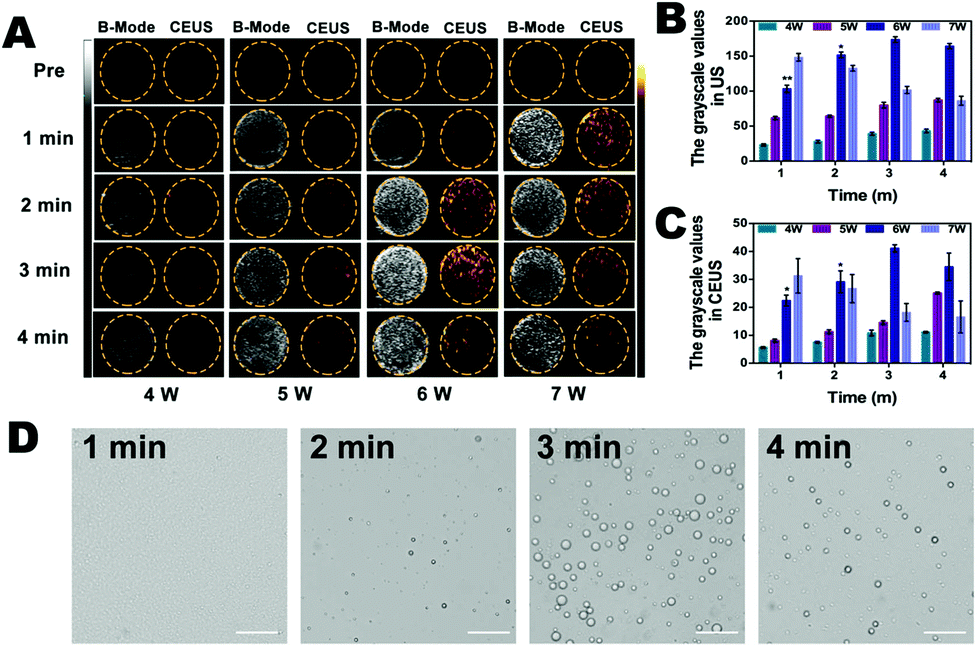 | ||
| Fig. 4 (A) B-mode and CEUS images of C-HPNs before and after LIFU irradiation under different conditions (4–7 W, 1–4 min), (B) the grayscale values in US, and (C) the grayscale values in CEUS after being irradiated with LIFU (*p < 0.05, **p < 0.01). (D) Optical images of C-HPNs after LIFU irradiation (6 W, 1–4 min; scale bar = 10 μm) (the data are shown as mean ± SD, n = 3 per group). | ||
In addition, the optical microscopy results were consistent with the ultrasound imaging results: large amounts of microbubbles were observed after LIFU irradiation and peaked at 6 W with a duration of 3 min (Fig. 4D). As shown in Fig. S4,† the average temperature was 30.3 °C after LIFU exposure at a power of 6 W and a duration of 3 min, suggesting that the adequate ultrasound power and duration of LIFU we selected could not achieve the temperature required to induce the thermal phase transition. Several reports have demonstrated that the liquid core of the phase changeable nanodroplets can vaporize to the gas phase upon activation by ultrasound energy, called acoustic droplet vaporization (ADV).70–72 It was the mechanical oscillation of ADV that generated the microbubbles.
3.5 In vitro cell experiments
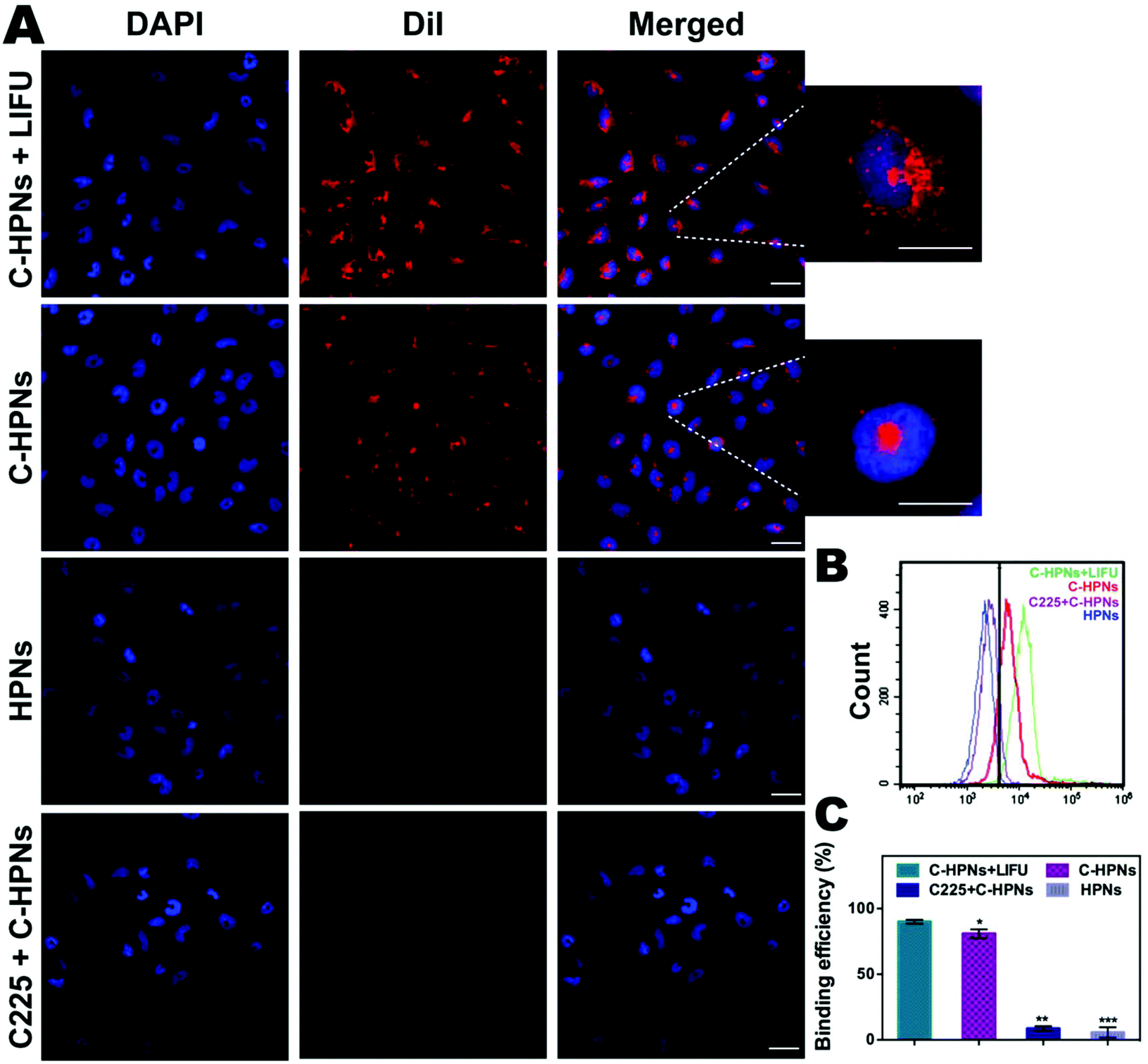 | ||
| Fig. 5 Targeting efficiency in vitro. (A) CLSM images of C643 cells after different treatments, and the intracellular uptake by C643 cells was significantly promoted by LIFU irradiation (blue fluorescence represents staining by DAPI, and red fluorescence represents nanoparticles stained with DiI; scale bar = 10 μm). (B) Flow-cytometry results and (C) histogram show the binding efficiency of different groups (the data are shown as mean ± SD, n = 3 per group, *p < 0.05, **p < 0.01, ***p < 0.001). | ||
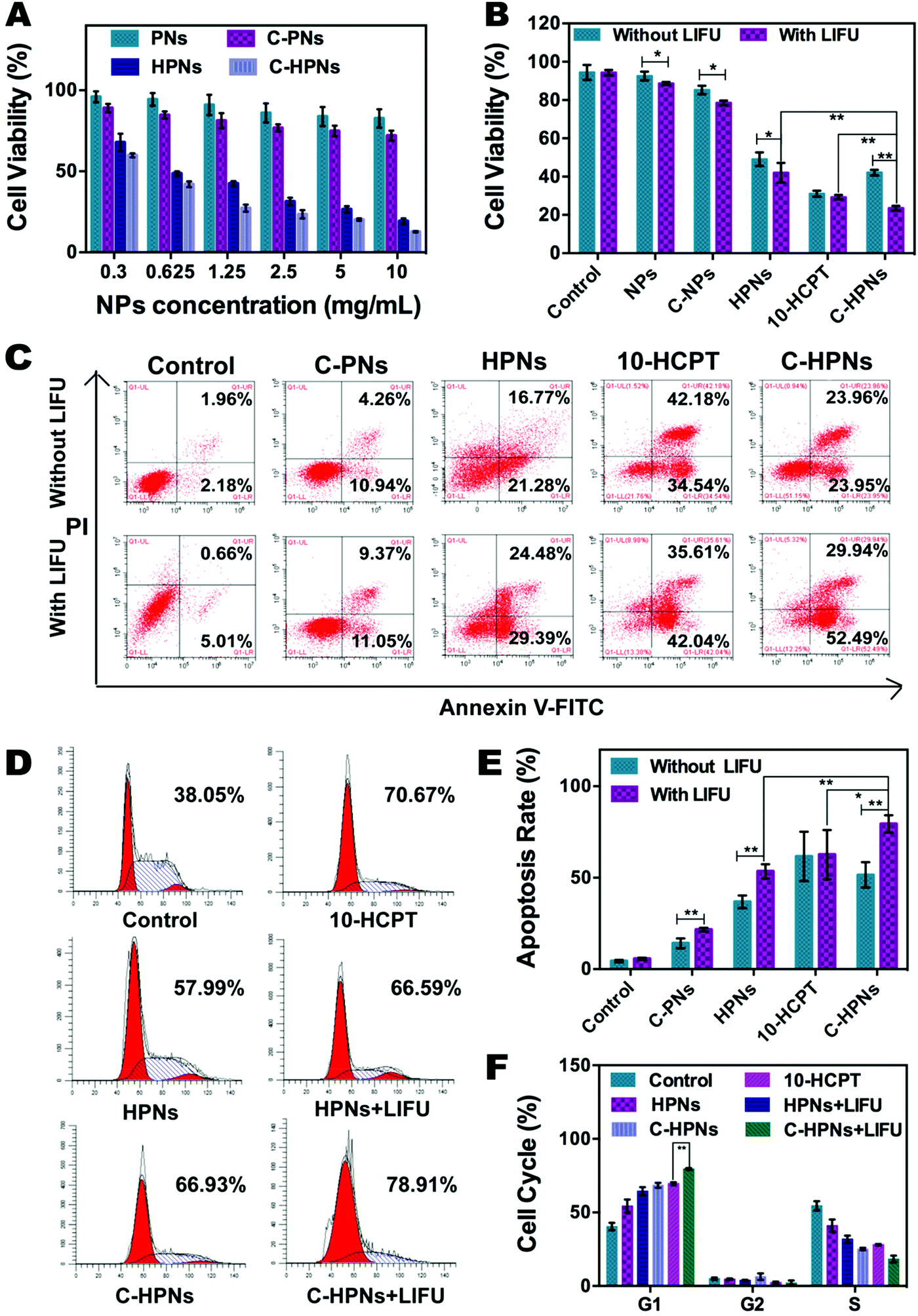 | ||
| Fig. 6 Anti-tumor efficacy in vitro. (A) Cell viability assay of C643 cells incubated with different formulations of NPs for 24 h. (B) Cell viability assay of free 10-HCPT, C-PNs, HPNs, and C-HPNs incubated with C643 cells with or without LIFU irradiation (6 W, 3 min). (C) Flow analysis of apoptosis. The number indicates the percentage of cells in the late and early apoptosis stage, respectively. (D) Cell cycle test for C643 cells with different treatments for 24 h. The number represents the percentage of cells in the G1 stage. (E) Quantitative analysis of apoptosis. (F) Quantitative analysis of cells in the G1, G2, and S phase, respectively (the data are shown as mean ± SD, n = 3 per group, *p < 0.05, **p < 0.01). | ||
Comprehensive consideration of the in vitro experiment results clearly indicated that polymer nanoparticles could serve as an outstanding vehicle for PFP and 10-HCPT, and the conjugation of C225 afforded the target capability to improve cell recognition and endocytosis, as well as to improve the therapeutic effect of 10-HCPT. In addition, LIFU facilitated 10-HCPT release on demand, maximizing the inhibitory effects on cell proliferation. It may be a result of the increased permeability of the cell membrane induced by the cavitation and UTMD effects74 and the LIFU-responsive enhanced release of 10-HCPT at the target site, which considerably enhanced the inhibitory impact on cell proliferation.
3.6 Biosafety evaluation of C-HPNs
The biosafety of C-HPNs was evaluated before the in vivo experiments. H&E staining of the major organs (heart, liver, spleen, lung and kidney) at day 1, day 7, and day 15 post-injection revealed no acute or chronic pathological damage at a dose of 5 mg mL−1, which was nearly five-fold higher than the therapeutic dose (Fig. S5†). Liver biochemical markers (ALT, AST, and ALB), kidney functional markers (BUN and CREA), and complete blood counts (RBC, HB, and WBC) showed no obvious difference among all the mice (Fig. S6†). These results highlight the excellent biocompatibility of C-HPNs in vivo without severe side effects in the short-term and with relatively long-term effects on the mice. Thus, the C-HPNs that we fabricated possess the potential to be an appropriate nanocarrier for in vivo applications.3.7 In vivo fluorescence imaging in xenograft tumors
Fluorescence imaging was applied at predetermined time points to evaluate the targeting efficacy and biodistribution of C-HPNs in vivo. Compared with the low fluorescence signal of the nontargeted group at each time point, a significant accumulating fluorescence signal of the targeted group appeared at the tumor site and peaked at 6 h at values 5.1-fold higher than those of the nontargeted group ((4.54 ± 1.58) × 108vs. (8.92 ± 0.95) × 107 (ps−1 cm−2 sr−1)/(μW cm−2), respectively) (Fig. 7A). After 24 h, the fluorescence intensities of the removed tumors and major organs were analyzed ex vivo (Fig. 7B), and the fluorescence intensity of the tumors in the targeted group was still 1.8-fold higher than that in the nontargeted group ((3.4 ± 0.49) × 108vs. (1.89 ± 0.29) × 108 (ps−1 cm−2 sr−1)/(μW cm−2), respectively). There was almost no difference in the fluorescence intensities in the major organs between the targeted and nontargeted groups (Fig. 7C). Meanwhile, significantly stronger red fluorescence signals were observed in the tumor cryosections of the targeted group at 6 h under CLSM after the ultrathin sectioning of tumor tissues compared with the fewer red signals in the nontargeted group. It is worth noting that the red fluorescence signals were remarkably improved in the tumor cryosections of the C-HPN + LIFU group than that of the C-HPN group after LIFU irradiation (Fig. 7D). The fluorescence signal distribution in the major organs did not show a significant variance in either group but mainly accumulated in the liver and spleen (Fig. 7E). The low fluorescence signals of the nontargeted group at the tumor site may be a result of the EPR effect, which facilitates passive aggregation in the tumor tissues, whereas the stronger fluorescence signal of the targeted group was mainly a result of the C225-mediated endocytosis mechanism. Moreover, C-HPNs might overcome the biological barriers of tumors with LIFU assistance, and the accumulation of C-HPNs at the target sites was promoted by the mechanical forces from the shear stream and liquid jets after the microbubble oscillation, cavitation, and destruction.75,76 During the process of the acoustic microbubble oscillation and collapse by the ultrasound-targeted microbubble destruction (UTMD) effect, the cell membrane could be disrupted and the permeability could be improved, which allowed more efficient accumulation of C-HPNs at the target sites.77 This finding further verified that C225 could specifically drive the nanocarriers to tumor tissues with the LIFU assistant for a longer period of time, preventing them from quickly re-entering systemic circulation and thereby enabling extravascular diagnosis and effective agent-assisted antitumor therapy.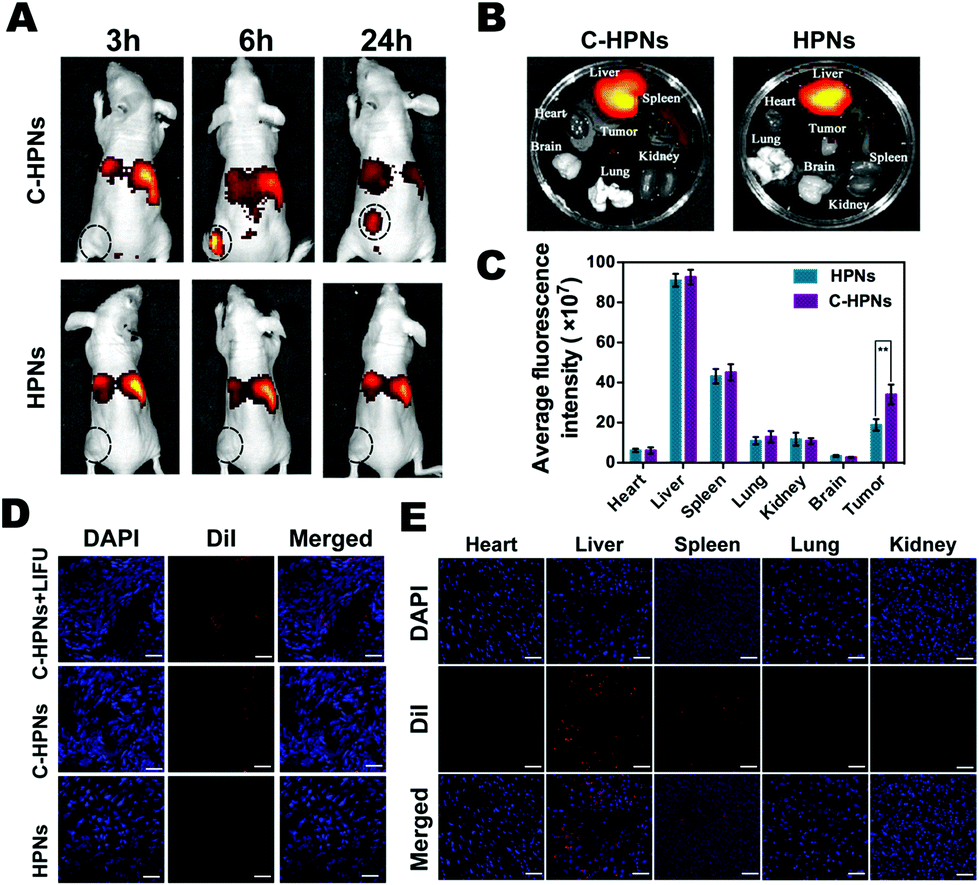 | ||
| Fig. 7 In vivo targeting efficiency of DiR-labeled C-HPNs to C643 xenograft-bearing nude mice. (A) In vivo fluorescence images of the nude mice after intravenous injection of DiR-labeled C-HPNs or DiR-labeled HPNs at 3, 6, and 24 h, respectively. (B) Ex vivo fluorescence images of tumor and major organs at 24 h after the administration of DiR-labeled C-HPNs or DiR-labeled HPNs. (C) Semi-quantitative bio-distribution of DiR-labeled C-HPNs or DiR-labeled HPNs in mice determined from the average FL intensities of organs and tumor (**p < 0.01). (D) CLSM images of tumor sections for the experimental groups (nuclei were stained with DAPI, scale bar = 10 μm). (E) Distribution of DiI-labeled C-HPNs in major organs (scale bar = 20 μm). n = 3 per group. | ||
3.8 In vivo ADV and ultrasound imaging
Based on the targeted accumulation ability of C-HPNs in the tumor tissues, we speculated that the phase-changeable NPs could serve as ultrasound agents to enhance ultrasound imaging and diagnose lesions. Regular low or anechoic and no contrast-enhanced signals were observed in all groups after the administration of different treatments before LIFU irradiation (Fig. 8A). LIFU was carried out in all groups six hours after the administration of different treatments, and the time points were in accordance with those of in vivo fluorescence imaging. Compared to the preimages, significantly stronger spot-like echo signals gradually accumulated at the tumor sites in the targeted group in both modes, whereas no obvious changes were observed in the saline group, and only minimal signals appeared in the nontargeted group. These results suggested that C225 facilitated targeting accumulation in the tumor tissues and when phase-changeable NPs underwent ADV at the tumor site triggered by LIFU, large amounts of microbubbles were generated, resulting in enhanced ultrasound imaging. However, the insufficient ADV of the HPNs could not efficiently enhance ultrasound imaging due to the lack of C225-mediated targeting ability. In addition, obvious enhancement was not observed in the C-HPNs without LIFU irradiation, indicating that C-HPNs could not enhance the ultrasound imaging by themselves in vivo. These results indicated that C-HPNs were suitable as ultrasound imaging agents and effective nanocarriers in vivo due to their relative stability. The mean grayscale values assayed using the DFY software in both modes for the C-HPNs were considerably higher than those obtained for the nontargeted and saline control groups (peaking at approximately 51, 15, and 5 in the B mode and 36, 8, and 3 in the CEUS mode, respectively) (Fig. 8B and C). The above data were consistent with the ultrasound imaging results, further confirming the feasibility of the targeted ultrasound imaging of C-HPNs under LIFU irradiation, and local LIFU irradiation could increase the accuracy of the phase-changeable NPs.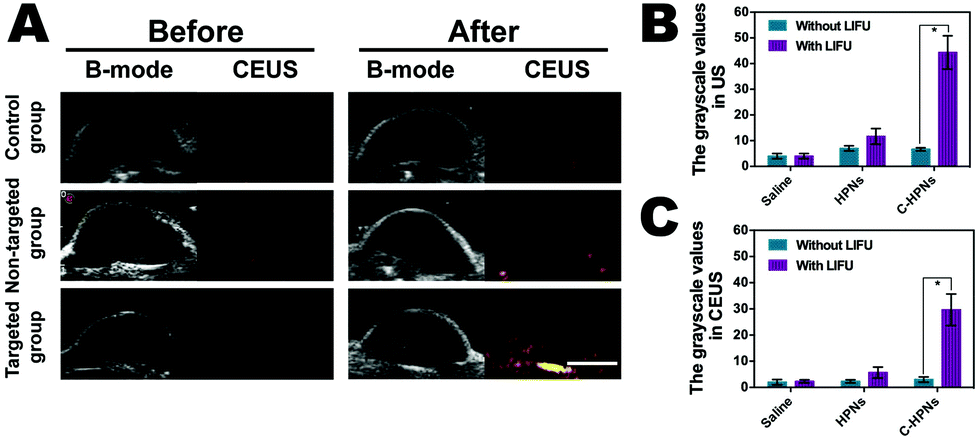 | ||
| Fig. 8 In vivo ADV with LIFU sonication. (A) Ultrasound imaging of mice tumors in B-mode and CEUS mode before and after different treatments. The grayscale values in B-mode (B) and CEUS mode (C) of mice tumors before and after LIFU irradiation for the HPNs, C-HPNs, and controls (scale bar = 5 mm, *p < 0.05), respectively; n = 3 per group. | ||
3.9 Therapeutic effect in vivo
Motivated by the remarkable therapeutic efficacy of the combination of C-HPNs and LIFU in vitro, the antitumor efficacy in vivo was investigated in subcutaneous C643 tumor models. Digital photographs of different groups of mice were taken to show the treatment effect (Fig. 9A). The therapeutic efficacy was assessed by monitoring changes in the tumor volume (Fig. 9B), quantity, and TIR of each group. As shown in Table S2,† the TIR in several groups increased as follows: control < LIFU < C-PNs < C-PNs + LIFU < HPNs < HPNs + LIFU < 10-HCPT < 10-HCPT + LIFU < C-HPNs < C-HPNs + LIFU. The tumors in the control group grew the fastest, and no significant reduction in the tumor volume was observed in the LIFU group, indicating that the LIFU irradiation doses were reliable in vivo. The epidermal growth factor (EGFR) is a well-known appropriate target for the detection and therapy of tumor cells. However, no therapeutic effect was observed because no antitumor drugs were encapsulated in the C-PNs. The C-HPNs exhibited a significantly stronger antitumor effect than the HPNs (53.36% ± 4.3% vs. 30.61% ± 8.0%), indicating that C-HPNs take advantage of EGFR-mediated active targeting, allowing high accumulation in the tumor tissues with sustained 10-HCPT release in situ. However, the accumulation of HPNs in the tumor site relied only on the presence of vessel fenestrations and vascular leaking, and insufficient drug release from HPNs in the tumor site limited the therapeutic effect. Notably, the therapeutic efficacy of free 10-HCPT was inferior to that of C-HPNs, which was inconsistent with the antiproliferation results in vitro. This inconsistency may be the result of the poor solubility and rapid hydrolysis of free 10-HCPT in vivo; only a few drugs accumulated in the tumor region due to the systemic administration, and thus, the therapeutic effect of free 10-HCPT was not satisfactory and could only be fully displayed in vivo. Furthermore, encapsulating 10-HCPT in the NPs enhanced the stability of 10-HCPT, increasing the concentration of 10-HCPT at the tumor site due to the active targeting capability of C-HPNs and thereby augmenting and prolonging the action of 10-HCPT in vivo. Furthermore, the inhibition rate was unexpectedly elevated from 53.36% ± 4.3% to 81.24% ± 4.0% when C-HPNs were administered simultaneously with LIFU, resulting in a significantly higher TIR. These results showed that LIFU irradiation could further improve the therapeutic effect of subcutaneous anaplastic thyroid cancer in nude mice.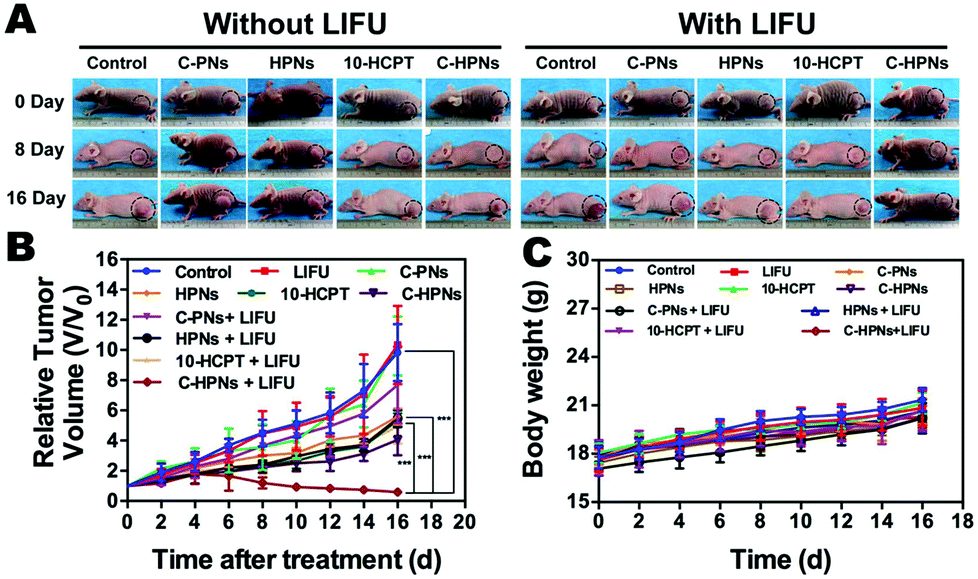 | ||
| Fig. 9 Anti-tumor therapy effects in vivo. (A) Photographs of C643-tumor-bearing mice after treatments with control, C-PNs, HPNs, free 10-HCPT, and C-HPNs with or without LIFU irradiation for 0, 8, and 16 days, respectively. (B) Tumor growth curves of ten groups after various treatments (***p < 0.001). (C) Body weight curves of C643-tumor-bearing mice subjected to different treatments; n = 5 per group. | ||
At the same time, H&E staining of tumor sections showed that larger numbers of cells were destroyed in the C-HPN + LIFU group than those in the other groups, indicating the significant therapeutic efficacy of the combination therapy strategy (Fig. 10A). Moreover, compared with the other groups, large numbers of apoptotic cells (green fluorescence) were observed in the C-HPN + LIFU group, whereas only a limited proliferation (red fluorescence) appeared; these results followed the same trend as the H&E staining results (Fig. 10B). The 10-HCPT group and the 10-HCPT + LIFU group had the lowest leucocyte number but the highest ALT and BUN in comparison with the other groups (Table S3†). In addition, there was no statistically significant difference in the body weight among all groups during the treatment course (Fig. 9C).
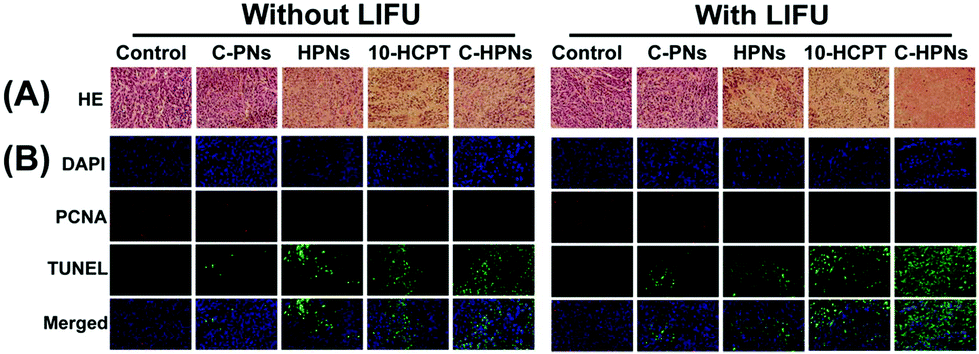 | ||
| Fig. 10 Anti-tumor efficiency observed by pathological examination of tumor tissue after different treatments. Histological assays of tumors with H&E (A), PCNA, and TUNEL staining (B). The nucleus shows red or green fluorescence representing PCNA-positive or TUNEL-positive cells, and the blue fluorescence represents the negative cells (400× magnification); n = 5 per group. | ||
The above results clearly indicated that the combination of C-HPNs and LIFU achieved a remarkable synergistic therapeutic effect against subcutaneous anaplastic thyroid cancer in nude mice highlighting the safety of ADV-mediated targeted tumor therapy at the same time. This treatment strategy is a preferable approach for anaplastic thyroid cancer, remarkably enhancing the therapeutic capability without obvious side effects.
4. Conclusions
In this work, C-HPNs were fabricated by integrating PFP, 10-HCPT and C225 into a single polymer nanocarrier, which could be triggered by LIFU. The biocompatible C-HPNs exhibit the active EGFR-targeting and LIFU-responsive features for both contrast ultrasound imaging and synergistic chemotherapy of ATC. The combination of LIFU and C-HPNs led to remarkable inhibition of tumor growth in nude mice without severe adverse side effects, as investigated both in vitro and in vivo. In conclusion, our study demonstrated that C-HPNs, triggered by LIFU, provided the co-delivery of C225/10-HCPT to achieve precisely targeted diagnosis and synergistic chemotherapy against ATC without obvious system toxicity. Thus, the excellent biosafety and noninvasiveness of these nanoparticles guarantee their further clinical translation and may provide a promising theranostic approach for ATC.Conflicts of interest
The authors have stated that there are no competing interests.Acknowledgements
The authors thank Chongqing Key Laboratory of Ultrasound Molecular for providing the laboratory. The authors are grateful to Dr Zhangluxi Liu, Yuan Guo and Jing Shao for assistance in technical support with C225 conjugation. This work was financially supported by the National Natural Science Foundation of China (Grant No. 31630026, 81630047, and 81471713), the Ministry of Science and Technology Foundation of China (Grant No. 3R216P843430) and Health Projects of Jilin Province (Grant No. SCZSY201618).References
- S. D. Perrault, C. Walkey, T. Jennings, H. C. Fischer and W. C. Chan, Nano Lett., 2009, 9, 1909–1915 CrossRef CAS PubMed.
- E. Molinaro, C. Romei, A. Biagini, E. Sabini, L. Agate, S. Mazzeo, G. Materazzi, S. Sellari-Franceschini, A. Ribechini, L. Torregrossa, F. Basolo, P. Vitti and R. Elisei, Nat. Rev. Endocrinol., 2017, 13, 644–660 CrossRef CAS PubMed.
- L. D. Green, L. Mack and J. L. Pasieka, J. Surg. Oncol., 2006, 94, 725–736 CrossRef PubMed.
- M. Zhou, Y. Chen, M. Adachi, X. Wen, B. Erwin, O. Mawlawi, S. Y. Lai and C. Li, Biomaterials, 2015, 57, 41–49 CrossRef CAS PubMed.
- T. D. Singh, S. Y. Jeong, S. W. Lee, J. H. Ha, I. K. Lee, S. H. Kim, J. Kim, S. J. Cho, B. C. Ahn, J. Lee and Y. H. Jeon, J. Nucl. Med., 2015, 56, 1690–1696 CrossRef CAS PubMed.
- R. C. Smallridge and J. A. Copland, Clin. Oncol. (R. Coll. Radiol.), 2010, 22, 486–497 CrossRef CAS PubMed.
- K. B. Ain, Thyroid, 1998, 8, 715–726 CrossRef CAS PubMed.
- N. Prasongsook, A. Kumar, A. V. Chintakuntlawar, R. L. Foote, J. Kasperbauer, J. Molina, Y. Garces, D. Ma, M. A. N. Wittich, J. Rubin, R. Richardson, J. Morris, I. Hay, V. Fatourechi, B. McIver, M. Ryder, G. Thompson, C. Grant, M. Richards, T. J. Sebo, M. Rivera, V. Suman, S. M. Jenkins, R. C. Smallridge and K. C. Bible, J. Clin. Endocrinol. Metab., 2017, 102, 4506–4514 CrossRef PubMed.
- Y. Liu, V. Gunda, X. Zhu, X. Xu, J. Wu, D. Askhatova, O. C. Farokhzad, S. Parangi and J. Shi, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7750–7755 CrossRef CAS PubMed.
- G. E. Lombardo, V. Maggisano, M. Celano, D. Cosco, C. Mignogna, F. Baldan, S. M. Lepore, L. Allegri, S. Moretti, C. Durante, G. Damante, M. Fresta, D. Russo, S. Bulotta and E. Puxeddu, Mol. Cancer Ther., 2018, 17, 1187–1195 CrossRef CAS PubMed.
- Q. Feng, Y. Zhang, W. Zhang, X. Shan, Y. Yuan, H. Zhang, L. Hou and Z. Zhang, Acta Biomater., 2016, 38, 129–142 CrossRef CAS PubMed.
- S. Rizzitelli, P. Giustetto, J. C. Cutrin, D. Delli Castelli, C. Boffa, M. Ruzza, V. Menchise, F. Molinari, S. Aime and E. Terreno, J. Controlled Release, 2015, 202, 21–30 CrossRef CAS PubMed.
- A. Bystritsky, A. S. Korb, P. K. Douglas, M. S. Cohen, W. P. Melega, A. P. Mulgaonkar, A. DeSalles, B. K. Min and S. S. Yoo, Brain Stimul., 2011, 4, 125–136 CrossRef PubMed.
- L. Zhu, H. Zhao, Z. Zhou, Y. Xia, Z. Wang, H. Ran, P. Li and J. Ren, Nano Lett., 2018, 18, 1831–1841 CrossRef CAS PubMed.
- Y. Cao, Y. Chen, T. Yu, Y. Guo, F. Liu, Y. Yao, P. Li, D. Wang, Z. Wang, Y. Chen and H. Ran, Theranostics, 2018, 8, 1327–1339 CrossRef PubMed.
- J. Liu, T. Shang, F. Wang, Y. Cao, L. Hao, J. Ren, H. Ran, Z. Wang, P. Li and Z. Du, Int. J. Nanomed., 2017, 12, 911–923 CrossRef CAS PubMed.
- Y. Gong, Z. Wang, G. Dong, Y. Sun, X. Wang, Y. Rong, M. Li, D. Wang and H. Ran, Drug Delivery, 2016, 23, 2280–2289 CAS.
- H. Zhao, M. Wu, L. Zhu, Y. Tian, M. Wu, Y. Li, L. Deng, W. Jiang, W. Shen, Z. Wang, Z. Mei, P. Li, H. Ran, Z. Zhou and J. Ren, Theranostics, 2018, 8, 1892–1910 CrossRef PubMed.
- N. Abdullah-Al-Nahain, J. E. Lee, I. In, H. Lee, K. D. Lee, J. H. Jeong and S. Y. Park, Mol. Pharm., 2013, 10, 3736–3744 CrossRef PubMed.
- N. Zhou, Z. Hao, X. Zhao, S. Maharjan, S. Zhu, Y. Song, B. Yang and L. Lu, Nanoscale, 2015, 7, 15635–15642 RSC.
- L. Deng, X. Cai, D. Sheng, Y. Yang, E. M. Strohm, Z. Wang, H. Ran, D. Wang, Y. Zheng, P. Li, T. Shang, Y. Ling, F. Wang and Y. Sun, Theranostics, 2017, 7, 4410–4423 CrossRef CAS PubMed.
- Y. H. Bae and K. Park, J. Controlled Release, 2011, 153, 198–205 CrossRef CAS PubMed.
- Y. Liu, M. Ji, M. K. Wong, K. I. Joo and P. Wang, BioMed Res. Int., 2013, 2013, 378380 Search PubMed.
- Z. Yu, R. M. Schmaltz, T. C. Bozeman, R. Paul, M. J. Rishel, K. S. Tsosie and S. M. Hecht, J. Am. Chem. Soc., 2013, 135, 2883–2886 CrossRef CAS PubMed.
- L. Zhang, Y. Zhang, A. Mehta, M. Boufraqech, S. Davis, J. Wang, Z. Tian, Z. Yu, M. B. Boxer, J. A. Kiefer, J. A. Copland, R. C. Smallridge, Z. Li, M. Shen and E. Kebebew, Oncotarget, 2015, 6, 9073–9085 Search PubMed.
- C. Ensinger, G. Spizzo, P. Moser, I. Tschoerner, R. Prommegger, M. Gabriel, G. Mikuz and K. W. Schmid, Ann. N. Y. Acad. Sci., 2004, 1030, 69–77 CrossRef CAS PubMed.
- J. Janković, S. Tatić, V. Božić, V. Živaljević, D. Cvejić and S. Paskaš, Hum. Pathol., 2017, 61, 164–172 CrossRef PubMed.
- M. Garg, D. Kanojia, R. Okamoto, S. Jain, V. Madan, W. Chien, A. Sampath, L. W. Ding, M. Xuan, J. W. Said, N. B. Doan, L. Z. Liu, H. Yang, S. Gery, G. D. Braunstein and H. P. Koeffler, J. Clin. Endocrinol. Metab., 2014, 99, E62–E72 CrossRef PubMed.
- B. A. Schiff, A. B. McMurphy, S. A. Jasser, M. N. Younes, D. Doan, O. G. Yigitbasi, S. Kim, G. Zhou, M. Mandal, B. N. Bekele, F. C. Holsinger, S. I. Sherman, S. C. Yeung, A. K. El-Naggar and J. N. Myers, Clin. Cancer Res., 2004, 10, 8594–8602 CrossRef CAS PubMed.
- Y. Nobuhara, N. Onoda, Y. Yamashita, M. Yamasaki, K. Ogisawa, T. Takashima, T. Ishikawa and K. Hirakawa, Br. J. Cancer, 2005, 92, 1110–1116 CrossRef CAS PubMed.
- B. Gabler, T. Aicher, P. Heiss and R. Senekowitsch-Schmidtke, Anticancer Res., 1997, 17, 3157–3159 CAS.
- J. D. Bergström, B. Westermark and N. E. Heldin, Exp. Cell Res., 2000, 259, 293–299 CrossRef PubMed.
- S. M. Thomas and J. R. Grandis, Cancer Treat. Rev., 2004, 30, 255–268 CrossRef CAS PubMed.
- M. J. Park, Y. S. Kim, J. Yang, W. C. Sun, H. Park, S. Y. Chae, M. S. Namgung and K. S. Choi, Ultrasound Med. Biol., 2013, 39, 292–299 CrossRef PubMed.
- P. Bossi, C. Bergamini, M. Siano, M. Cossu Rocca, A. P. Sponghini, F. Favales, M. Giannoccaro, E. Marchesi, B. Cortelazzi, F. Perrone, S. Pilotti, L. D. Locati, L. Licitra, S. Canevari and L. De Cecco, Clin. Cancer Res., 2016, 22, 3961–3970 CrossRef CAS PubMed.
- T. Yungan, A. Anne, S. Christian, M. Laurent, K. Cedric, M. Philippe, B. Etienne, K. Marie-Christine, L. Michel, C. Thierry, A. Marc, P. Yoann, J. Eric, G. Bernard, Z. Ayman, T. Jean-Marc, L. Brigitte, C. Alexandre, R. Séverine, H. Ali, M. Emanuelle, B. Christian, M. Nicolas, C. Alexandre, O. Nathalie, C. Odile, S. Xu Shan and B. Jean, J. Clin. Oncol., 2018, 36, 3084–3090 CrossRef PubMed.
- A. K. Berger, S. Lücke, U. Abel, G. M. Haag, C. Grüllich, A. Stange, M. Dietrich, L. Apostolidis, A. Freitag, C. Trierweiler, C. von Gall, J. Ose, F. Giesel, T. F. Weber, F. Lordick, U. Haberkorn and D. Jäger, Br. J. Cancer, 2018, 119, 170–175 CrossRef CAS PubMed.
- S. Kim, C. N. Prichard, M. N. Younes, Y. D. Yazici, S. A. Jasser, B. N. Bekele and J. N. Myers, Clin. Cancer Res., 2006, 12, 600–607 CrossRef CAS PubMed.
- M. Burge, C. Semira, B. Lee, M. Lee, S. Kosmider, R. Wong, J. Shapiro, B. Ma, A. P. Dean, A. S. Zimet, S. A. Steel, S. W. Lok, J. Torres, M. Eastgate, H. L. Wong and P. Gibbs, Clin. Colorectal Cancer, 2018, 17, 593–599 CrossRef PubMed.
- V. G. Deepagan, B. Sarmento, D. Menon, A. Nascimento, A. Jayasree, M. Sreeranganathan, M. Koyakutty, S. V. Nair and J. Rangasamy, Nanomedicine, 2012, 7, 507–519 CrossRef CAS PubMed.
- S. Bäumer, N. Bäumer, N. Appel, L. Terheyden, J. Fremerey, S. Schelhaas, E. Wardelmann, F. Buchholz, W. E. Berdel and C. Müller-Tidow, Clin. Cancer Res., 2015, 21, 1383–1394 CrossRef PubMed.
- R. A. Beckman, L. M. Weiner and H. M. Davis, Cancer, 2007, 109, 170–179 CrossRef CAS PubMed.
- P. Krishnan, M. Rajan, S. Kumari, S. Sakinah, S. P. Priya, F. Amira, L. Danjuma, M. Pooi Ling, S. Fakurazi, P. Arulselvan, A. Higuchi, R. Arumugam, A. A. Alarfaj, M. A. Munusamy, R. A. Hamat, G. Benelli, K. Murugan and S. S. Kumar, Sci. Rep., 2017, 7, 10962 CrossRef PubMed.
- E. B. Ehlerding, P. Grodzinski, W. Cai and C. H. Liu, ACS Nano, 2018, 12, 2106–2121 CrossRef CAS PubMed.
- J. Sun, L. Sun, J. Li, J. Xu, Z. Wan, Z. Ouyang, L. Liang, S. Li and D. Zeng, Acta Biomater., 2018, 75, 312–322 CrossRef CAS PubMed.
- S. K. Hobbs, W. L. Monsky, F. Yuan, W. G. Roberts, L. Griffith, V. P. Torchilin and R. K. Jain, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4607–4612 CrossRef CAS.
- Y. Zhou, Z. Wang, Y. Chen, H. Shen, Z. Luo, A. Li, Q. Wang, H. Ran, P. Li, W. Song, Z. Yang, H. Chen, Z. Wang, G. Lu and Y. Zheng, Adv. Mater., 2013, 25, 4123–4130 CrossRef CAS PubMed.
- H. S. Min, D. G. You, S. Son, S. Jeon, J. H. Park, S. Lee, I. C. Kwon and K. Kim, Theranostics, 2015, 5, 1402–1418 CrossRef PubMed.
- M. Seo, R. Williams and N. Matsuura, Lab Chip, 2015, 15, 3581–3590 RSC.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS PubMed.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed.
- X. Zhang, Y. Zheng, Z. Wang, S. Huang, Y. Chen, W. Jiang, H. Zhang, M. Ding, Q. Li, X. Xiao, X. Luo, Z. Wang and H. Qi, Biomaterials, 2014, 35, 5148–5161 CrossRef CAS PubMed.
- H. Sah, L. A. Thoma, H. R. Desu, E. Sah and G. C. Wood, Int. J. Nanomed., 2013, 8, 747–765 CrossRef PubMed.
- T. Y. Wang, J. W. Choe, K. Pu, R. Devulapally, S. Bachawal, S. Machtaler, S. M. Chowdhury, R. Luong, L. Tian, B. Khuri-Yakub, J. Rao, R. Paulmurugan and J. K. Willmann, J. Controlled Release, 2015, 203, 99–108 CrossRef CAS PubMed.
- R. Devulapally, N. M. Sekar, T. V. Sekar, K. Foygel, T. F. Massoud, J. K. Willmann and R. Paulmurugan, ACS Nano, 2015, 9, 2290–2302 CrossRef CAS PubMed.
- S. Balthasar, K. Michaelis, N. Dinauer, H. von Briesen, J. Kreuter and K. Langer, Biomaterials, 2005, 26, 2723–2732 CrossRef CAS PubMed.
- T. Betancourt, B. Brown and L. Brannon-Peppas, Nanomedicine, 2007, 2, 219–232 CrossRef CAS PubMed.
- C. Y. Ting, C. H. Fan, H. L. Liu, C. Y. Huang, H. Y. Hsieh, T. C. Yen, K. C. Wei and C. K. Yeh, Biomaterials, 2012, 33, 704–712 CrossRef CAS PubMed.
- M. C. Cochran, J. Eisenbrey, R. O. Ouma, M. Soulen and M. A. Wheatley, Int. J. Pharm., 2011, 414, 161–170 CrossRef CAS PubMed.
- P. Li, Y. Zheng, H. Ran, J. Tan, Y. Lin, Q. Zhang, J. Ren and Z. Wang, J. Controlled Release, 2012, 162, 349–354 CrossRef CAS PubMed.
- Y. Bao, Y. Guo, X. Zhuang, D. Li, B. Cheng, S. Tan and Z. Zhang, Mol. Pharm., 2014, 11, 3196–3209 CrossRef CAS PubMed.
- L. Zhang, H. Xiao, J. Li, D. Cheng and X. Shuai, Nanoscale, 2016, 8, 12608–12617 RSC.
- K. Wilson, K. Homan and S. Emelianov, Nat. Commun., 2012, 3, 618 CrossRef PubMed.
- D. Gao, J. Gao, M. Xu, Z. Cao, L. Zhou, Y. Li, X. Xie, Q. Jiang, W. Wang and J. Liu, Mol. Pharm., 2017, 14, 984–998 CrossRef CAS PubMed.
- N. Y. Rapoport, A. M. Kennedy, J. E. Shea, C. L. Scaife and K. H. Nam, J. Controlled Release, 2009, 138, 268–276 CrossRef CAS PubMed.
- M. Guédra and F. Coulouvrat, J. Acoust. Soc. Am., 2015, 138, 3656–3667 CrossRef PubMed.
- D. Gao, M. Xu, Z. Cao, J. Gao, Y. Chen, Y. Li, Z. Yang, X. Xie, Q. Jiang, W. Wang and J. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 13524–13537 CrossRef CAS PubMed.
- Q. Sun, X. Sun, X. Ma, Z. Zhou, E. Jin, B. Zhang, Y. Shen, E. A. Van Kirk, W. J. Murdoch, J. R. Lott, T. P. Lodge, M. Radosz and Y. Zhao, Adv. Mater., 2014, 26, 7615–7621 CrossRef CAS PubMed.
- C. B. Arena, A. Novell, P. S. Sheeran, C. Puett, L. C. Moyer and P. A. Dayton, IEEE Trans. Ultrason. Ferroelectr. Freq. Control, 2015, 62, 1623–1633 Search PubMed.
- Y. J. Ho, Y. C. Chang and C. K. Yeh, Theranostics, 2015, 6, 392–403 CrossRef PubMed.
- B. A. Juliar, M. M. Bromley, A. Moncion, D. C. Jones, E. G. O'Neill, C. G. Wilson, R. T. Franceschi and M. L. Fabiilli, Adv. Healthcare Mater., 2016, 5, 1764–1774 CrossRef CAS PubMed.
- J. Schlegel, I. Peters, S. Orrenius, D. K. Miller, N. A. Thornberry, T. T. Yamin and D. W. Nicholson, J. Biol. Chem., 1996, 271, 1841–1844 CrossRef CAS PubMed.
- W. T. Chen, S. T. Kang, J. L. Lin, C. H. Wang, R. C. Chen and C. K. Yeh, Biomaterials, 2015, 53, 699–708 CrossRef CAS PubMed.
- H. Chen, W. Kreider, A. A. Brayman, M. R. Bailey and T. J. Matula, Phys. Rev. Lett., 2011, 106, 034301 CrossRef PubMed.
- C. I. De, E. Zagato, K. Braeckmans, Y. Luan, J. N. De, S. C. De Smedt and I. Lentacker, J. Controlled Release, 2015, 197, 20–28 CrossRef PubMed.
- Y. J. Ho and C. K. Yeh, Theranostics, 2017, 7, 1477–1488 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8bm00970h |
| This journal is © The Royal Society of Chemistry 2019 |