
Facile synthesis of aquo-cisplatin arsenite multidrug nanocomposites for overcoming drug resistance and efficient combination therapy†
Jingyu
Xin‡
a,
Ke
Zhang‡
b,
Jiaqi
Huang
a,
Xiangjie
Luo
a,
Xuanqing
Gong
a,
Zhaoxuan
Yang
a,
Hongyu
Lin
a,
Hong
Shan
b and
Jinhao
Gao
 *a
*a
aState Key Laboratory of Physical Chemistry of Solid Surfaces, The Key Laboratory for Chemical Biology of Fujian Province, and Department of Chemical Biology, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: jhgao@xmu.edu.cn
bDepartment of Interventional Medicine, Guangdong Provincial Engineering Research Center of Molecular Imaging, The Fifth Affiliated Hospital, Sun Yat-sen University, Zhuhai, 519000, China
First published on 8th November 2018
Abstract
Cisplatin (CDDP) and arsenic trioxide (ATO), two representative inorganic anticancer drugs, have been successful in the treatment against several kinds of malignancies. However, combination therapy with these two drugs in clinical application suffers from poor pharmacokinetics, serious side effects, and drug resistance of the tumor. Herein, we report a carrier-free aquo-cisplatin arsenite multidrug nanocomposite loaded with cisplatin and arsenic trioxide prodrugs simultaneously. This nanocomposite achieves a high loading capacity and pH-dependent controlled release of the drugs. Because of these features, this nanocomposite shows better in vitro toxicity against various carcinoma cell lines than either the single drug or free drug combination, promotes the synergistic effect of cisplatin and arsenic trioxide, and significantly inhibits the growth of tumors in vivo. Furthermore, cisplatin and arsenic trioxide in this nanocomposite can realize a coordination of both enhanced DNA damage and DNA repair interference within cisplatin-resistant cells, which results in overcoming the drug resistance effectively. Gene expression profiles demonstrate the reduced expression of proto-oncogenes and DNA damage repair related genes MYC, MET, and MSH2, along with the increase of tumor suppressor genes PTEN, VHL, and FAS after the nanocomposite treatment. This type of multidrug nanocomposite offers an alternative and promising strategy for combination therapy and overcoming drug resistance.
1. Introduction
Cancer is one of the most common and hazardous diseases all over the world, especially in China.1 Due to the escalating drug resistance, various combination therapies with different anticancer drugs have been developed for cancer treatment.2,3 Among the assorted anticancer drugs in combination therapies, arsenic trioxide (ATO), a traditional Chinese medicine, has been used as a front-line anticancer drug for the treatment of acute promyelocytic leukemia (APL) as approved by the Food and Drug Administration (FDA).4,5 In addition to leukemia, several studies demonstrated that ATO exhibited anticancer activity against certain solid tumors.6,7 Cisplatin (CDDP), which is also an FDA-approved inorganic anticancer drug popular in current combination therapies, shows the therapeutic effect against various malignancies.8,9 Previous studies reported that CDDP showed synergistic effects with ATO in combination therapy in vitro.10,11 Hence ATO and CDDP, as two antitumor drugs, can be used to demonstrate the resurgence of inorganic drugs.12,13 Nevertheless, combination therapy in clinical application still faces many challenges due to the limitations that arise from the intrinsic characteristics of these drugs and tumors.14 These limitations include poor and inconsistent pharmacokinetic properties, unbearable and dose-limiting side effects caused by the drugs,15,16 and drug resistance, especially cisplatin resistance which can be acquired by tumors rapidly during the therapeutic process.17,18 Therefore, it is crucial to develop a new strategy for the effective combination therapy and for overcoming the drug resistance.With the advancement of nanotechnology, researchers have greatly extended the applications of nanomaterials to the biomedical field.19–23 A variety of nanodrugs with different compositions and structures have emerged.24–27 Some characteristics of these nanoplatforms, such as high drug loading capacity, controlled drug release, a prolonged circulation time in the blood, and passive tumor targeting via the EPR effect, have improved the anticancer efficacy of many traditional drugs.28,29 Many efforts have been dedicated to the nanoformulation of ATO and CDDP. Arsenite ions, the major form of free ATO in aqueous solution, could precipitate with transition metal ions to form nanocomposites.30–33 This has been exploited by previous studies to load ATO and CDDP into liposomes, which could improve the anticancer efficacy of ATO and CDDP toward solid tumors.34 However, the approaches to load drugs involve complicated fabrication, which results in low loading efficiency, high time consumption, and an unsatisfying therapeutic effect. Carrier-free self-delivery nanocomposites of drugs, which mean that drugs are formulated at the nanoscale to realize the delivery and release by themselves without the aid of carriers,35–37 have recently emerged as an encouraging strategy for the construction of next-generation nanodrugs.38 Apart from the advantages of nanoplatforms, the self-delivery nanocomposite is also free of complex manipulation involving nanocarrier handling and possible toxicity and immunogenicity induced by nanocarriers,39 which makes it a promising approach to address the challenges in combination therapy.40
In this study, we developed a novel carrier-free aquo-cisplatin arsenite (denoted as PtAs) multidrug nanocomposite with a feasible and efficient loading of CDDP and ATO prodrugs. The PtAs dual-inorganic-drug nanocomposite featured improved stability in the physiological environment, controlled release of drugs in an acidic environment, enhanced anticancer efficacy, and a promoted synergistic effect of CDDP and ATO against various cancer cells in vitro. Further exploration revealed that this nanocomposite could induce massive DNA damage, and inhibit the activity of PARP-1, which is closely related to the DNA damage repair in cisplatin-resistant tumor cells.41,42 As a result, this nanocomposite showed strong cytotoxicity against cisplatin-resistant ovarian tumor cells and could overcome cisplatin resistance effectively. The improvement in pharmacokinetics and enhancement in suppressing tumor growth in vivo indicate that this novel carrier-free multidrug nanocomposite is a promising strategy in clinical combination therapy.
2. Experimental
2.1. Materials
Cisplatin, sodium arsenite, and silver acetate were purchased from Alfa Aesar. Polyoxyethylene (5) nonylphenyl ether (Igepal CO-520), polyallyamine hydrochloride (PAH, Mw 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500), dextran sulfate (DXS, Mw 10000), and nitric acid (65–68%) were purchased from Sigma-Aldrich. Cyclohexane and ethanol were purchased from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China). All chemicals were used as received without further purification.
500), dextran sulfate (DXS, Mw 10000), and nitric acid (65–68%) were purchased from Sigma-Aldrich. Cyclohexane and ethanol were purchased from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China). All chemicals were used as received without further purification.
2.2. Preparation of aquo-cisplatin
150 mg of cisPt (cis-(NH3)2PtCl2) was mixed with 167 mg of silver acetate in 10 mL of deionized water. The mixture was stirred at 55 °C in the dark overnight. Then the resulting mixture was filtered through a 0.2 μm membrane to remove the white AgCl precipitate. The pale yellow filtrate was stored at 4 °C in the dark.2.3. Synthesis of carrier-free aquo-cisplatin arsenite (PtAs) nanocomposites
PtAs nanocomposites were synthesized via a reverse microemulsion approach using a cyclohexane/Igepal CO-520/water system. 25 mL of cyclohexane was mixed with 10 mL of Igepal CO-520, and 1630 μL of 0.05 M aquo-cisplatin acetate was added to the cyclohexane–Igepal CO-520 mixture to form microemulsions. Then 1630 μL of 0.1 M sodium arsenite was added to the microemulsions. The microemulsions were stirred at room temperature for 24 h. PtAs nanoparticles were collected by ethanol precipitation and stored in deionized water at 4 °C.2.4. Surface modification of PtAs nanocomposites
PtAs nanocomposites were modified with PAH and DXS polymers according to a layer-by-layer assembly method. 2 mL of PtAs nanocomposites (500 μg mL−1) were added to 10 mL of PBS solution containing 200 μg mL−1 PAH. The resulting mixture was stirred at room temperature for 4 h. The intermediate product was collected by ultrafiltration. After that, 10 mL of PBS solution containing 200 μg mL−1 DXS was added to 1 mL of the concentrated intermediate product. The resulting mixture was reacted for 4 h at room temperature. The final product, denoted as PtAs@PAH-DXS nanodrugs, was collected by ultrafiltration.2.5. Characterization
Transmission electron microscopy (TEM) images were obtained on a JEM-2100 microscope and a Tecnai F30 microscope. EDS elemental analysis was performed on a Tecnai F30 microscope. Elemental quantitative analysis of Pt and As in samples was carried out by inductively coupled plasma atomic emission spectroscopy (ICP-AES) or inductively coupled plasma mass spectroscopy (ICP-MS). Hydrodynamic diameter and zeta potential measurements were performed on a Malvern Zetasizer nano ZS instrument.2.6. Evaluation of drug release in vitro
PtAs nanocomposites and PtAs@PAH-DXS nanocomposites were dispersed in 1× PBS (pH 7.4) or 0.01 M citrate buffer (pH 5.4) at 37 °C. After that, 300 μL of the solution was removed and subjected to centrifugation to collect the supernatant at selected time points. The concentration of Pt and As in the supernatant was analyzed by ICP-MS.2.7. Cell culture
All cells (SMMC-7721, HepG2, H22, COC-1 WT and COC-1 DDP) were purchased from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). HepG2 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM medium) supplemented with 10% fetal bovine serum (FBS, HyClone) and antibiotics (100 mg mL−1 streptomycin and 100 U mL−1 penicillin). The SMMC-7721, H22, COC-1 WT and COC-1 DDP cells were cultured in Roswell Park Memorial Institute 1640 medium (RPMI-1640 medium) supplemented with 10% FBS and antibiotics. All cells were cultured under a humidified atmosphere of 5% CO2 at 37 °C.2.8. Cytotoxicity evaluation
Cytotoxicity was evaluated via the CCK-8 assays. Cells were seeded into a 96-well plate at a density of 5 × 103 cells per well in culture media and incubated for 24 h. Then the cells were incubated with CDDP, ATO, CDDP + ATO, or PtAs@PAH-DXS nanocomposites at a series of Pt and As concentrations for 48 h. Each experiment was performed in triplicate. After that, the culture media were replaced with fresh culture media containing 0.5 mg mL−1 of CCK-8 and the cells were incubated for another 4 h. Finally, the absorbances at 492 nm of each well were measured using a MultiSkan FC microplate reader (Thermo scientific).2.9. Synergistic effect evaluation
The combination indexes (CIs) were put forward to evaluate the synergistic effect. The CI equation is based on the multiple drug-effect equation of the Chou–Talalay method. For certain cell viability, CI values were calculated according to the following equation:| CI = D1/(Dx)1 + D2/(Dx)2 |
2.10. Cellular uptake evaluation
After treatment with different drug formulations (CDDP, ATO, and PtAs@PAH-DXS nanocomposites) of the same Pt or As concentration (500 ng mL−1 Pt and 200 ng mL−1 As for SMMC-7721 cells and 1 μg mL−1 Pt and 400 ng mL−1 As for COC-1 cells) for 12 h, the cells were collected and washed three times with PBS to remove the remnant drugs. To measure the amount of Pt and As inside the cells, the cells were lysed completely in 500 μL of nitric acid (65–68%) and the lysate was analyzed by ICP-MS.2.11. In vivo study
All animal procedures were in accordance with the National Institute of Health Guidelines for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee of Xiamen University. Male BALB/c mice (6 weeks old, 18–22 g) were purchased from the Laboratory Animal Center of Xiamen University. The murine tumor model was established by injecting H22 cells into subcutaneous tissues. When the diameter of H22 tumors reached approximately 0.5 cm, PBS, CDDP, ATO, or PtAs@PAH-DXS nanocomposites were intravenously injected into the mice at a dose of 2 mg Pt and 0.8 mg As per kg on days 0, 3, and 5. Tumor growth and weights of the mice were monitored. The volume of the tumor was calculated using the equation: VTumor = (a2 × b)/2 (a and b represent the maximum and minimum diameter of the tumor, respectively). In vivo biodistribution analysis was conducted as follows: tissues of specific organs were excised from the mice at 24 h after injection, and completely lysed with a mixture of concentrated nitric acid and hydrogen peroxide. The lysate was analyzed by ICP-MS. For pharmacokinetic analysis, 10 μL of blood was sampled from the tail at selected time points after injection. Then the blood samples were lysed and analyzed by ICP-MS as well.2.12. Western blotting analysis
After treatment, the cells were collected and mixed with RIPA buffer containing 1% protease inhibitor cocktail at 4 °C for 30 min. After centrifugation, the supernatant was added to loading buffer of equal volume and the resulting mixture was heated at 100 °C for 5 min. Proteins were loaded on polyacrylamide gel and underwent electrophoresis in stacking gel (5%) for 20 min (80 V) and in separated gel (8%) for 60 min (120 V). Proteins were subsequently transferred to a 0.45 μm PVDF membrane (Millipore). After blocking with TBS buffer containing 5% skimmed milk powder (Oxoid) for 30 min, the membrane was incubated in TBS buffers containing 5% skimmed milk powder and primary antibodies against PARP-1 (1:1000, abcam), PAR (1:2000, abcam), γH2AX (1:1000, abcam), and β-tubulin (1:5000, abcam) overnight at 4 °C, and washed with TBS buffer 5 times between each incubation. After that, the membrane was further incubated in TBS buffer containing 5% skimmed milk powder and secondary antibodies (1:5000, goat anti-rabbit or goat anti-mouse, abcam) for 1 h, and washed with TBS buffer 5 times. Protein bands were visualized using WesternBright ECL (Advansta) and captured with an imaging system (GE Healthcare Bio-sciences AB). The expression level of β-tubulin was used as a control to normalize the expression of target proteins.
2.13. RNA sequencing analysis
After treatment with different drug formulations (CDDP, ATO, and PtAs@PAH-DXS nanocomposites) of the same Pt or As concentration (5 μM Pt and 5.25 μM As for SMMC-7721 cells) for 12 h, the cells were collected and washed three times with PBS, then Trizol was added to protect and store the RNA. RNA sequencing was done by Majorbio Company.2.14. Statistical analysis
Statistical analysis was performed using Student's t-test for unpaired data and a p value less than 0.05 was accepted as an indicator of a statistically significant difference compared to controls.3. Results and discussion
3.1. Preparation and characterization of PtAs@PAH-DXS nanodrugs
The PtAs@PAH-DXS nanodrugs were prepared by reverse microemulsion synthesis followed by layer-by-layer assembly with polymers. Carrier-free PtAs nanocomposites were synthesized by precipitation of arsenite ions and aquo-cisplatin ions within water nanodroplets in reverse microemulsion (Fig. 1A and Fig. S1†). This synthesis method is straightforward and feasible for scale up. Then PtAs nanocomposites were coated with polyallyamine hydrochloride (PAH) and dextran sulfate (DXS) sequentially to form PtAs@PAH-DXS nanodrugs (Fig. 1B). To confirm the coating of polycations and polyanions, we monitored the changes of the hydrodynamic size and surface zeta potential of nanoparticles during the process of coating. Dynamic light scattering (DLS) analysis showed that the average hydrodynamic size of PtAs nanocomposites was 22.8 ± 2.1 nm with a narrow distribution (Fig. 2A). With two layers of polymer coating on the surface, the hydrodynamic size of the nanodrugs increased significantly to 38.8 ± 1.6 nm (Fig. 2A). The zeta potential of the nanoparticles during the process of coating, which depends on the surface charge of nanoparticles, changed substantially. Finally, the PtAs@PAH-DXS nanodrugs showed a negative surface zeta potential at −46.2 ± 4.79 mV due to the coating of dextran (Fig. 2B). Typical transmission electron microscopy (TEM) images indicated that the PtAs nanocomposites were uniform and monodisperse with an average diameter of 18.0 ± 1.5 nm (Fig. S2†) and the PtAs@PAH-DXS nanodrugs were also monodisperse with an average diameter of 23.4 ± 1.8 nm (Fig. 2C). The difference between the hydrodynamic size by DLS analysis and the diameter measured from the TEM images was attributed to the hydrated layers of polymer coatings. EDS mapping of the nanodrugs indicated the colocalization of CDDP and ATO with a high loading efficiency (Fig. 2D and Fig. S2†). ICP-AES showed that the molar ratio of platinum (Pt) and arsenic (As) in the nanodrugs was about 1:1.05, suggesting that the main product of precipitation was an aquo-cisplatin arsenite coordination complex (Fig. S1†). We also monitored the change in the particle size of PtAs@PAH-DXS in DI water by DLS for two weeks. Little change in the particle size suggested that the nanodrugs are stable in water (Fig. S3 and Table S1†).
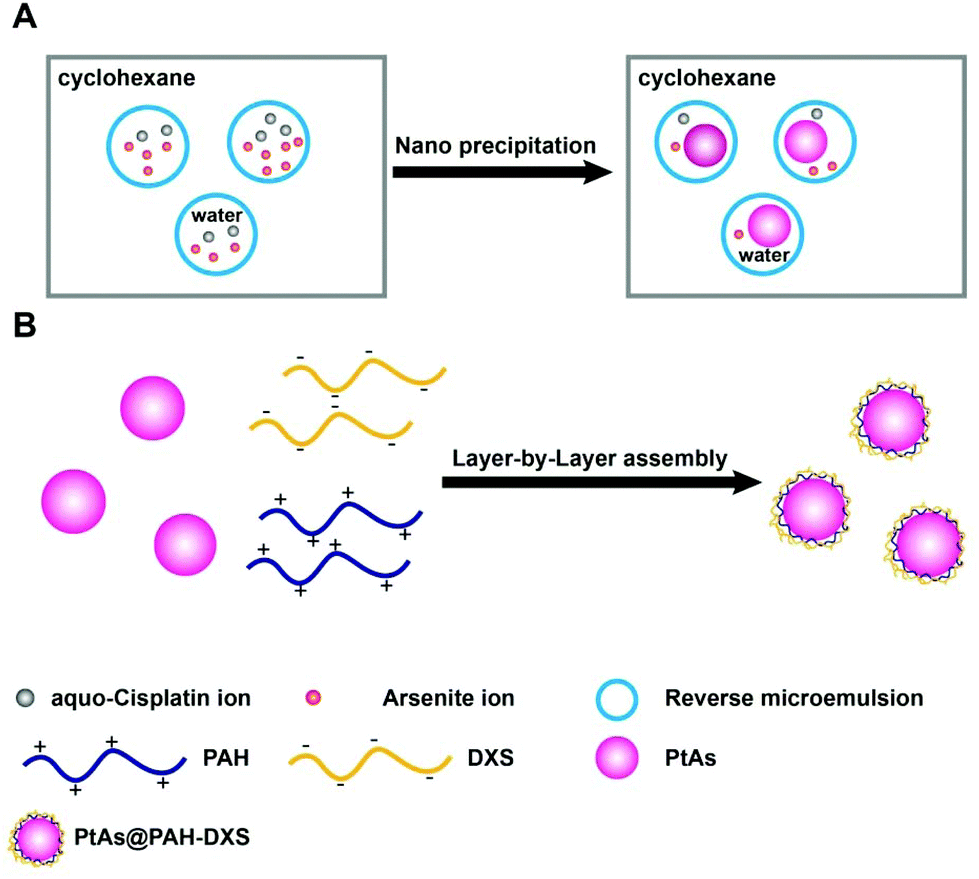 | ||
| Fig. 1 Schematic illustration of the formation of (A) PtAs NPs and (B) PtAs@PAH-DXS nanodrugs by the reverse microemulsion method followed by layer-by-layer assembly with polymers. | ||
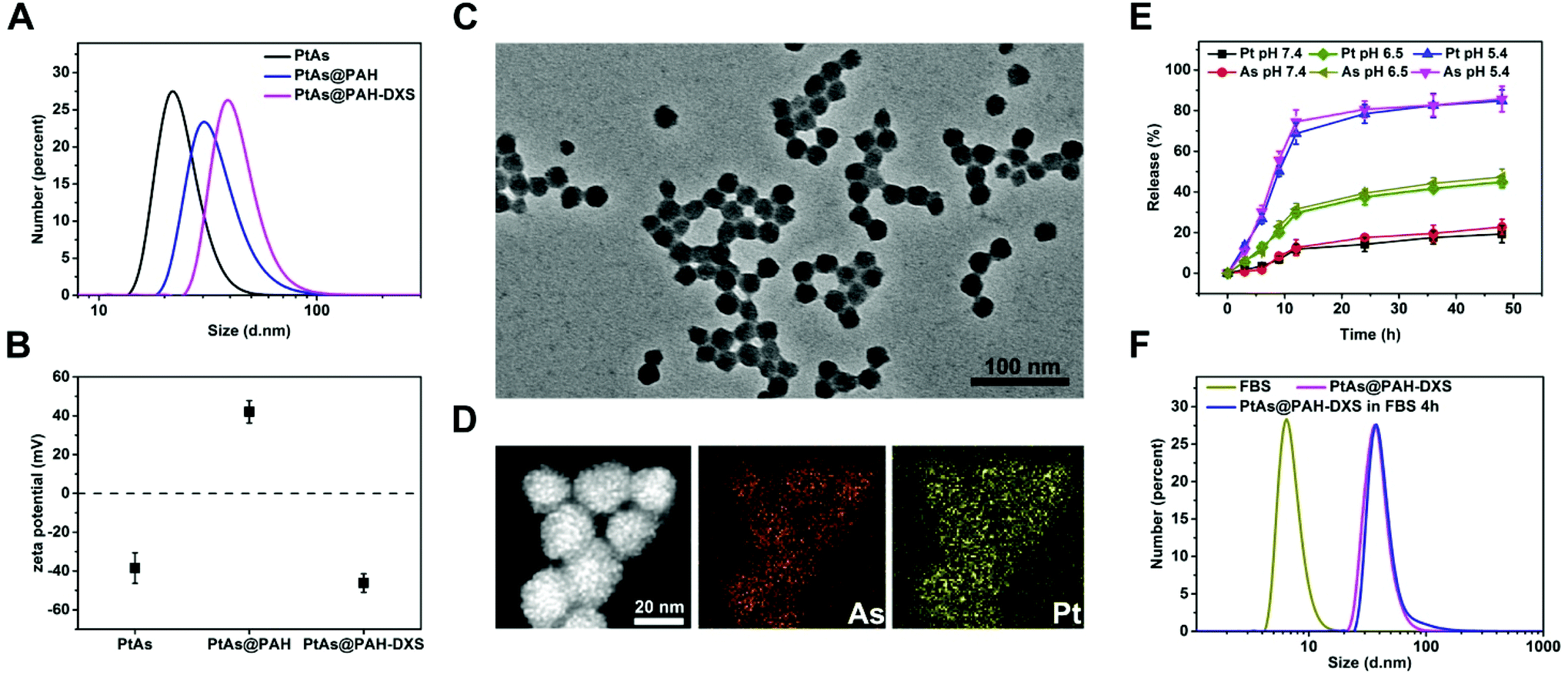 | ||
| Fig. 2 (A) Particle size distributions of PtAs, PtAs@PAH, and PtAs@PAH-DXS nanoparticles. (B) Zeta-potential analysis of PtAs, PtAs@PAH and PtAs@PAH-DXS nanoparticles in PBS. (C) A typical TEM image of PtAs@PAH-DXS nanodrugs. (D) STEM and EDS mapping images of PtAs@PAH-DXS nanodrugs. (E) Releasing profiles of PtAs@PAH-DXS nanodrugs at pH 5.4, 6.5 and 7.4 (n = 3 per group). (F) Particle size distribution of PtAs@PAH-DXS nanodrugs after incubation with 20% (v/v) fetal bovine serum (FBS) in PBS for 4 h. | ||
Due to excessive anaerobic metabolism of malignant cells, the microenvironment of tumors is mildly acidic compared to the normal physiological environment.43 Therefore, the difference in pH between the tumor microenvironment and physiological environment, as well as the difference between some intracellular compartments (such as endosomes and lysosomes) and the cytoplasm, has been exploited to achieve controlled drug release from nanoplatforms. We assessed the releasing profiles of PtAs nanocomposites and PtAs@PAH-DXS nanodrugs in phosphate buffer (PBS) (pH 7.4), PBS (pH 6.5) and citrate buffer (pH 5.4) at 37 °C. The release of CDDP and ATO was monitored by ICP-MS elemental analysis. About 78% of Pt and 81% of As were released from PtAs@PAH-DXS nanodrugs within 24 h at pH 5.4, and about 38% of Pt and 39% of As were released within 24 h at pH 6.5, while less than 20% of Pt and As were released within 24 h at pH 7.4 (Fig. 2E). These results indicated an unapparent release from nanodrugs at pH 7.4 and a considerable release in an acidic environment. In contrast, PtAs nanocomposites were less stable at pH 7.4 and half of Pt and As were released within 24 h (Fig. S4†). The enhanced stability of PtAs@PAH-DXS nanodrugs in a near neutral environment was attributed to the surface coating of polymers. All these results indicated that the PtAs@PAH-DXS nanodrugs could selectively release two kinds of anticancer drugs simultaneously and controllably in the acidic microenvironment of tumors or cellular endosomes. More importantly, the continuous release process could last for more than 40 h, which provides the feasibility for long-time therapy.
Most nanodrugs injected intravenously often face the problem that nanodrugs absorb various serum proteins on the surfaces nonspecifically and form a “protein corona” in the physiological environment,44 which would cause nanodrugs to be recognized and taken up by phagocytic cells in the mononuclear phagocyte system (MPS) and ultimately accumulated into the liver and spleen.45 The phenomenon decreases the tumor uptake and the anticancer efficacy of drugs. To assess the nonspecific absorption of proteins on the surface of PtAs@PAH-DXS nanodrugs, we monitored the changes of hydrodynamic diameter by DLS analysis. The nanodrugs were incubated with PBS solution containing 20% (v/v) fetal bovine serum (FBS) for 4 h at 37 °C. PBS solution with an equal concentration of FBS was used as a control. It was noticed that the hydrodynamic diameter of PtAs@PAH-DXS nanodrugs increased slightly after incubation with the FBS solution (Fig. 2F and Table S1†). These results suggested that the surface coating with dextran could prevent the nonspecific absorption of serum proteins. This indicates a prolonged circulation time, improved in vivo stability and biocompatibility, which would be beneficial for passive tumor targeting by the enhanced permeability and retention (EPR) effect.
3.2. In vitro anticancer efficacy of PtAs@PAH-DXS nanodrugs
To evaluate the anticancer efficacy of PtAs@PAH-DXS nanodrugs and the synergistic effect of CDDP and ATO in vitro, free ATO, free CDDP, a combination of free CDDP and ATO (CDDP + ATO), and PtAs@PAH-DXS nanodrugs were incubated with human or murine hepatocellular carcinoma cell lines (SMMC-7721, HepG2, and H22) and human cisplatin-resistant or wild ovarian carcinoma cell lines (COC-1 DDP andCOC-1 WT) at different Pt and As doses for 48 h (Fig. 3). The molar ratio of Pt and As in the combination treatment (CDDP + ATO) was fixed at 1:1.05, consistent with the molar ratio of Pt and As in the PtAs@PAH-DXS nanodrugs. Half maximal inhibitory concentrations (IC50) in terms of Pt and As were calculated according to the cytotoxicity results. The results showed that the PtAs@PAH-DXS nanodrugs exhibited an apparently lower IC50 than free ATO, free CDDP, and the combination of free CDDP and ATO against all these cell lines (Fig. 3), and further indicated that the nanodrugs significantly increased the combination therapeutic efficacy and overcame the cisplatin-resistance.
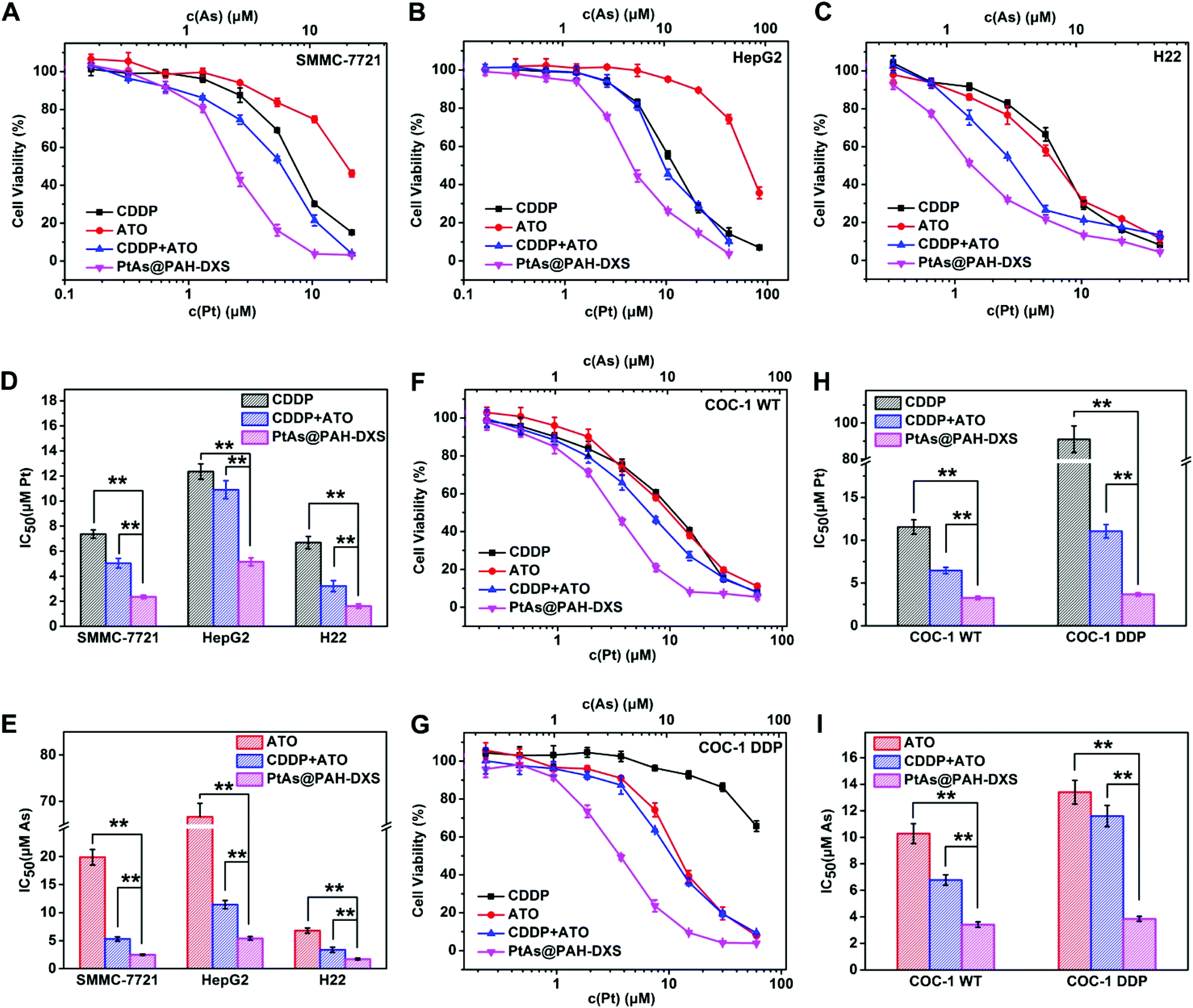 | ||
| Fig. 3 In vitro cytotoxicity assays. CCK-8 assay of liver cancer cell lines (A) SMMC-7721, (B) HepG2, and (C) H22 cells treated with free CDDP, free ATO, a combination of free CDDP and ATO (CDDP + ATO), or PtAs@PAH-DXS for 48 h (n = 5 per group). Comparison of IC50 values (48 h) in terms of (D) Pt and (E) As for free CDDP, free ATO, a combination of free CDDP and ATO (CDDP + ATO), or PtAs@PAH-DXS for liver cancer cells (**p < 0.01). CCK-8 assay of (F) COC-1 WT cells and (G) cisplatin-resistant COC-1 DDP cells treated with different drug formulations for 48 h (n = 5 per group). Comparison of IC50 values (48 h) in terms of (H) Pt and (I) As for different drug formulations for COC-1 WT and COC-1 DDP cells (**p < 0.01). | ||
Then we further analyzed the synergistic effect of CDDP and ATO according to the equation of the Chou–Talalay method.46 The calculated combination index (CI) quantitatively indicates the interactions of two drugs: CI < 1 means synergism, while CI = 1 and CI > 1 stand for the additive effect and antagonism, respectively. A smaller CI means a stronger synergistic effect. We compared the CI values for PtAs@PAH-DXS nanodrugs and the combination of free CDDP and ATO for the above-mentioned cell lines according to the cytotoxicity results. The CI values for the combination of free CDDP and ATO at low cell viability were close to 1 or greater than 1, while the CI values for PtAs@PAH-DXS nanodrugs were much smaller than 1 (Fig. 4A, B, and Fig. S5†), suggesting that loading CDDP and ATO into nanodrugs could promote the synergistic effect significantly for these two drugs.
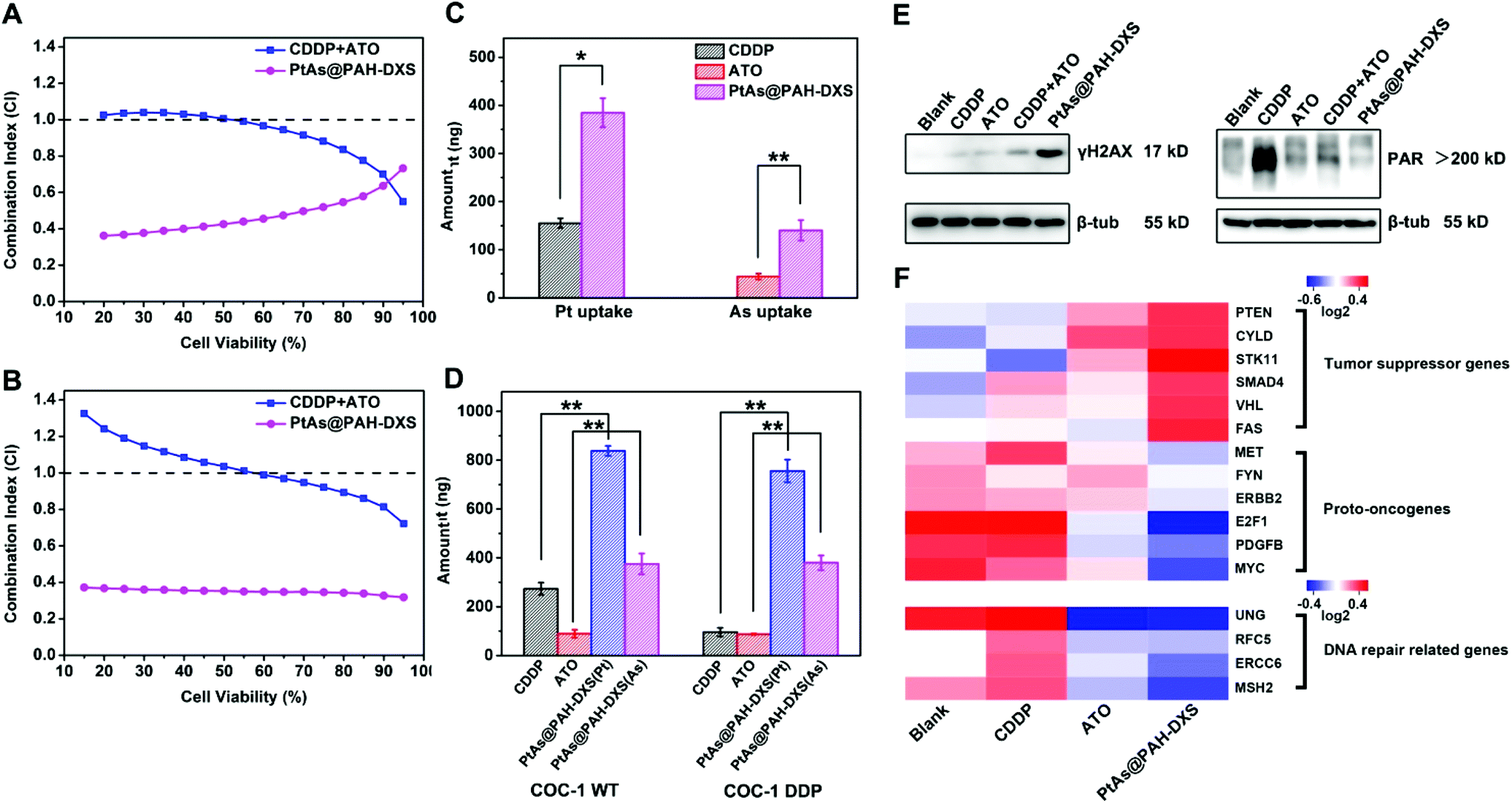 | ||
| Fig. 4 Mechanisms of the synergistic effect and overcoming drug resistance. Comparison of the combination index of CDDP and ATO for (A) SMMC-7721 and (B) COC-1 DDP cells after treatment with a combination of free CDDP and ATO (CDDP + ATO) or PtAs@PAH-DXS for 48 h. Comparison of cellular uptake of CDDP and ATO in (C) SMMC-7721 cells and (D) COC-1 WT and COC-1 DDP cells treated with free CDDP, ATO, or PtAs@PAH-DXS for 12 h (n = 3 per group, *p < 0.05, **p < 0.01). (E) The expression levels of γH2AX (left) and PAR (right) were detected by western blotting. Cisplatin resistant COC-1 DDP cells were treated with different drug formulations (5 μM CDDP, 5.25 μM ATO, 5 μM CDDP + 5.25 μM ATO, or 5 μM PtAs@PAH-DXS in terms of Pt) for 12 h. (F) Heatmaps of the transcriptional levels of certain DNA damage repair related genes, proto-oncogenes, and tumor suppressor genes in SMMC-7721 cells treated with different drug formulations (5 μM CDDP, 5.25 μM ATO, or 5 μM PtAs@PAH-DXS in terms of Pt) for 12 h. Red indicates high levels, whereas blue indicates low levels. | ||
After that, we incubated SMMC-7721, COC-1 WT and COC-1 DDP cells with free ATO, CDDP, and PtAs@PAH-DXS nanodrugs at the same dose for 12 h and analyzed the cellular uptake of drugs quantitatively by ICP-MS to study the behavior of free drugs and nanodrugs during cellular internalization and intracellular delivery (Fig. 4C and D). We noted that free ATO-treated or free CDDP-treated cells only took up a small amount of ATO or CDDP. The cellular uptake of free ATO and CDDP needs the assistance of membrane transport proteins.47 Overexpression of efflux proteins or reduced expression of transport proteins on the membrane,48,49 which partially accounts for the drug resistance of cancer cells, significantly decreases the effective concentrations of the drug inside cells. The nanodrugs avoid the problems for cellular internalization and intracellular delivery of free drugs due to the different ways of cellular uptake like endocytosis.50 Therefore, the amount of ATO and CDDP inside the cells treated with PtAs@PAH-DXS nanodrugs increased significantly compared to the cells treated with free-CDDP and the cells treated with free-ATO, suggesting that PtAs@PAH-DXS nanodrugs enhanced effective accumulation of ATO and CDDP in the cells substantially, resulting in the improvement of anticancer efficacy.
3.3. Possible mechanisms of the synergistic effect and overcoming drug resistance
Platinum-based drugs like cisplatin induce the apoptosis of tumor cells generally through breaking the DNA double-strands and damaging the structure of DNA in tumor cells directly.51 When DNA double-strands are broken, H2AX, a member of the histone family which functions in packaging and organizing eukaryotic DNA into chromatin, is phosphorylated rapidly near the breaking sites to form γH2AX.52,53 While the DNA double-strand breaks are repaired, the γH2AX is dephosphorylated and returns to original H2AX, thus the γH2AX is a potential biomarker of DNA damage.54 Tumor cells often acquire cisplatin resistance by improving the ability of DNA damage repair.18 PARP-1, a type of polymerase, plays an important role in DNA damage repair. It catalyzes the synthesis of poly (ADP-ribose) (PAR) at the location of DNA damage to recruit related damage repair factors, and hence facilitates DNA damage repair.55 Utilizing PARP-1 inhibitors is an efficient strategy to overcome drug resistance. Recently, several PARP-1 inhibitors have been evaluated in clinical combination therapy trials.56,57 ATO, which is a potential PARP-1 inhibitor, has been reported to inhibit the activity of PARP-1 and interfere with DNA damage repair.58,59 Western blotting analysis revealed a significantly increased γH2AX expression in PtAs@PAH-DXS-treated COC-1 DDP cells compared to CDDP-treated cells (Fig. 4E, left), indicating that PtAs@PAH-DXS could induce massive DNA damage and result in enhanced cytotoxicity to cisplatin-resistant cells. Then the PAR expression in COC-1 DDP cells was analyzed (Fig. 4E, right). CDDP-treated cells showed a large amount of PAR synthesis, indicating that cisplatin-resistant cells could detect DNA damage and initiate DNA damage repair in a short time. In comparison, PtAs@PAH-DXS-treated cells showed much less PAR synthesis within the same time frame, indicating that ATO in nanodrugs substantially inhibited PARP-1 activities, resulting in the blockade of DNA damage repair, enhancing the DNA damage and the apoptosis of tumor cells. Therefore, we concluded that the inhibition of DNA damage repair could account for the synergistic effect of CDDP and ATO and the surmounting of drug-resistance by nanodrugs.RNA sequencing analysis showed that the transcriptions of proto-oncogenes and DNA damage repair related genes like MYC, MET, and MSH260,61 were downregulated by PtAs@PAH-DXS nanodrugs, while the transcriptions of tumor suppressor genes like PTEN, FAS, and VHL62 were upregulated by PtAs@PAH-DXS nanodrugs in SMMC-7721 cells compared to CDDP and ATO groups (Fig. 4F). These results suggested that the nanodrugs could enhance the therapeutic effect and overcome drug resistance through regulating the transcriptional levels of related genes. Based on the above results, we plan to investigate the efficacy of overcoming drug resistance by PtAs@PAH-DXS nanodrugs in vivo in the future.
3.4. In vivo study of PtAs@PAH-DXS nanodrugs
Male BALB/c mice bearing subcutaneously H22 tumors were used to examine the behavior and the anticancer efficacy of PtAs@PAH-DXS nanodrugs in vivo. Blood samples were collected at selected time points after injection and subjected to ICP-MS analysis to determine the concentration of platinum in the blood (Fig. 5A and B). We noticed that the blood circulation half-life of PtAs@PAH-DXS nanodrugs (40 min in terms of Pt and 44 min in terms of As) was longer than that of CDDP (21 min) and ATO (21 min), suggesting that the blood circulation half-life of drugs was substantially prolonged. In addition, the concentration of PtAs@PAH-DXS in the blood was higher than 10% ID per gram even at 6 h after injection. A long blood circulation half-life is essential for efficient passive tumor targeting.63 Then, we measured the biodistribution of ATO and CDDP in mice at 24 h after injection. The tumor uptake of PtAs@PAH-DXS nanodrugs and free CDDP, in terms of Pt, was 2.9 ± 0.28% and 0.7 ± 0.33% ID per gram, as detected by ICP-MS, respectively, while the tumor uptake of PtAs@PAH-DXS nanodrugs and free ATO in terms of As was 3.2 ± 0.20% and 0.4 ± 0.17% ID per gram, respectively (Fig. 5C and D). These results suggested that the nanodrugs achieved much better accumulation in tumors than free drugs, which is consistent with the results of cell experiments. We also noted that a large portion of the nanodrugs was accumulated in the liver and spleen which could be ascribed to the clearance by MPS and RES. In addition, the majority of free CDDP and ATO was removed from the body, probably via rapid renal clearance, which is one of the major obstacles for successful tumor therapy.64 The results of pharmacokinetic analysis and biodistribution study further demonstrated that these nanodrugs with a longer blood circulation half-life could enhance the accumulation of drugs at tumor sites due to the EPR effect.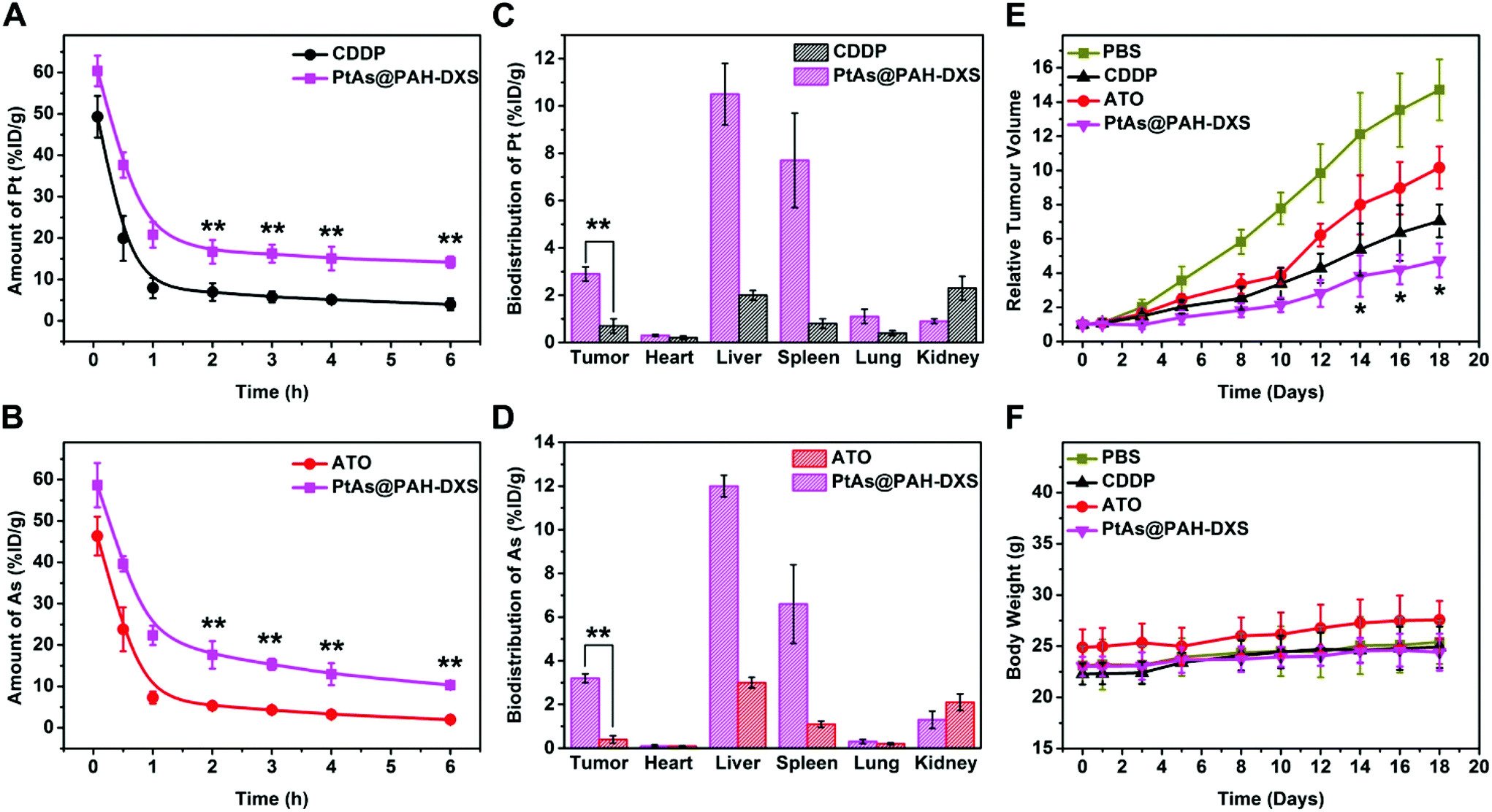 | ||
| Fig. 5 In vivo study of PtAs@PAH-DXS nanodrugs. Blood circulation curves of (A) Pt and (B) As after intravenous injection of free ATO, CDDP, or PtAs@PAH-DXS (n = 4 per group, **p < 0.01). Biodistribution of (C) Pt and (D) As at 24 h after intravenous injection of free ATO, CDDP, or PtAs@PAH-DXS (n = 4 per group, **p < 0.01). (E) Growth curves of tumors after intravenous injection of PBS, free ATO, free CDDP, or PtAs@PAH-DXS at a dose of 2 mg Pt and 0.8 mg As per kg body weight (*p < 0.05 compared to PBS, ATO, and CDDP groups, n = 5 per group). (F) Body weight curves of mice during the treatment with PBS, free ATO, free CDDP, or PtAs@PAH-DXS (n = 5 per group). | ||
Based on the results that PtAs@PAH-DXS nanodrugs exhibited better anticancer efficacy than traditional chemotherapeutic agents in vitro and favorable biodistribution and pharmacokinetics properties in vivo, PtAs@PAH-DXS nanodrugs were evaluated for therapy against HCC solid tumors in vivo (Fig. 5E). The mice bearing subcutaneous H22 tumors were injected intravenously with PBS, free ATO, free CDDP, or PtAs@PAH-DXS nanodrugs (2 mg Pt per kg body weight for free CDDP and PtAs@PAH-DXS, and 0.8 mg As per kg body weight for free ATO) on days 0, 3, and 5. The tumors in the PBS group grew very rapidly. The mean volume of subcutaneous tumors in this group increased approximately 14 fold by day 18, while the mean subcutaneous tumor volume of the free ATO treated group increased about 10 fold, and that of the CDDP treated group increased 7 fold at day 18. The treatment with PtAs@PAH-DXS nanodrugs resulted in the minimal growth of tumors within the same period, in comparison with the other groups. The body weights of the nanodrug-treated mice increased slightly during the treatment, suggesting that the nanodrugs did not cause significant side effects (Fig. 5F). In addition, representative sections of the major organs from mice treated with PBS, ATO, CDDP or PtAs@PAH-DXS nanodrugs were collected at 24 h after injection. The sections were stained with hematoxylin and eosin (Fig. S6†). No evident lesions were found after administration with nanodrugs, indicating the minimal systemic toxicity and excellent in vivo biocompatibility. These therapeutic benefits were ascribed to the continuous drug release process and the effective passive tumor targeting of nanodrugs. These results demonstrated the potential of PtAs@PAH-DXS as an effective combination therapy for solid tumors.
4. Conclusion
In summary, we developed a new carrier-free aquo-cisplatin arsenite nanocomposite which had a high capacity for simultaneous loading of CDDP and ATO prodrugs. The synthesis of the PtAs@PAH-DXS nanodrugs was straightforward and facile. These nanodrugs featured pH-responsive controlled release, high stability in the physiological environment, and improved in vivo biodistribution and pharmacokinetics. Owing to these advantages, the nanodrugs could induce massive DNA damage, substantially enhance the in vitro and in vivo therapeutic effect for HCC, and effectively overcome the drug resistance. In addition, mechanistic exploration of the combination therapy with CDDP and ATO in nanodrugs revealed that the inhibitory activity of ATO toward PARP-1, one of the DNA damage repair related enzymes, was the key to the synergistic effect and overcoming drug resistance. We believe that this novel aquo-cisplatin arsenite multidrug nanocomposite holds great promise for overcoming drug resistance of tumors and combination therapy in clinical application.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21771148, 21602186, 21521004, 81430041, 81620108017, and 81771879) and the Fundamental Research Funds for the Central Universities (20720170020, 20720170088, and 20720180033).References
- W. Chen, R. Zheng, P. D. Baade, S. Zhang, H. Zeng, F. Bray, A. Jemal, X. Q. Yu and J. He, CA-Cancer J. Clin., 2016, 66, 115–132 CrossRef PubMed.
- J. Jia, F. Zhu, X. Ma, Z. Cao, Z. W. Cao, Y. Li, Y. X. Li and Y. Z. Chen, Nat. Rev. Drug Discovery, 2009, 8, 111–128 CrossRef CAS PubMed.
- G. V. Long, Z. Eroglu, J. Infante, S. Patel, A. Daud, D. B. Johnson, R. Gonzalez, R. Kefford, O. Hamid, L. Schuchter, J. Cebon, W. Sharfman, R. McWilliams, M. Sznol, S. Redhu, E. Gasal, B. Mookerjee, J. Weber and K. T. Flaherty, J. Clin. Oncol., 2018, 36, 667–673 CrossRef CAS PubMed.
- J. Zhu, Z. Chen, V. Lallemand-Breitenbach and H. de The, Nat. Rev. Cancer, 2002, 2, 705–713 CrossRef CAS PubMed.
- M. Hoonjan, V. Jadhav and P. Bhatt, J. Biol. Inorg. Chem., 2018, 23, 313–329 CrossRef CAS PubMed.
- B. Tan, J. F. Huang, Q. Wei, H. Zhang and R. Z. Ni, World J. Gastroenterol., 2005, 11, 5938–5943 CrossRef CAS PubMed.
- C. C. Lin, C. Hsu, C. H. Hsu, W. L. Hsu, A. L. Cheng and C. H. Yang, Invest. New Drugs, 2007, 25, 77–84 CrossRef CAS PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- W.-Z. Zhong, Q. Wang, W.-M. Mao, S.-T. Xu, L. Wu, Y. Shen and Y.-Y. Liu, Lancet Oncol., 2018, 19, 139–148 CrossRef CAS PubMed.
- N. Zhang, Z. M. Wu, E. McGowan, J. Shi, Z. B. Hong, C. W. Ding, P. Xia and W. Di, Cancer Sci., 2009, 100, 2459–2464 CrossRef CAS PubMed.
- C. W. Helm and J. C. States, J. Ovarian Res., 2009, 2, 2–8 CrossRef PubMed.
- I. Romero-Canelon and P. J. Sadler, Inorg. Chem., 2013, 52, 12276–12291 CrossRef CAS PubMed.
- P. J. Dilda and P. J. Hogg, Cancer Treat. Rev., 2007, 33, 542–564 CrossRef CAS PubMed.
- J. A. Kemp, M. S. Shim, C. Y. Heo and Y. J. Kwon, Adv. Drug Delivery Rev., 2016, 98, 3–18 CrossRef CAS PubMed.
- N. Pabla and Z. Dong, Kidney Int., 2008, 73, 994–1007 CrossRef CAS PubMed.
- R. Oun, Y. E. Moussa and N. J. Wheate, Dalton Trans., 2018, 47, 6645–6653 RSC.
- F. H. Igney and P. H. Krammer, Nat. Rev. Cancer, 2002, 2, 277–288 CrossRef CAS PubMed.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- Z. H. Zhao, Z. J. Zhou, J. F. Bao, Z. Y. Wang, J. Hu, X. Q. Chi, K. Y. Ni, R. F. Wang, X. Y. Chen, Z. Chen and J. H. Gao, Nat. Commun., 2013, 4, 2266 CrossRef PubMed.
- L. Yang, L. Ma, J. Xin, A. Li, C. Sun, R. Wei, B. W. Ren, Z. Chen, H. Lin and J. Gao, Chem. Mater., 2017, 29, 3038–3047 CrossRef CAS.
- C. M. Hartshorn, M. S. Bradbury, G. M. Lanza, A. E. Nel, J. Rao, A. Z. Wang, U. B. Wiesner, L. Yang and P. Grodzinski, ACS Nano, 2018, 12, 24–43 CrossRef CAS PubMed.
- L. Yang, Z. Wang, L. Ma, A. Li, J. Xin, R. Wei, H. Lin, R. Wang, Z. Chen and J. Gao, ACS Nano, 2018, 12, 4605–4614 CrossRef CAS PubMed.
- M. H. Li, Z. Luo and Y. L. Zhao, Chem. Mater., 2018, 30, 25–53 CrossRef CAS.
- K. Ulbrich, K. Hola, V. Subr, A. Bakandritsos, J. Tucek and R. Zboril, Chem. Rev., 2016, 116, 5338–5431 CrossRef CAS PubMed.
- Z. Zhao, D. Huang, Z. Yin, X. Chi, X. Wang and J. Gao, J. Mater. Chem., 2012, 22, 15717–15725 RSC.
- Z. H. Zhao, H. Zhang, X. Q. Chi, H. Li, Z. Y. Yin, D. T. Huang, X. M. Wang and J. H. Gao, J. Mater. Chem. B, 2014, 2, 6313–6323 RSC.
- Y. Zhang, F. Wang, C. Liu, Z. Wang, L. Kang, Y. Huang, K. Dong, J. Ren and X. Qu, ACS Nano, 2018, 12, 651–661 CrossRef CAS PubMed.
- J. J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed.
- M. Zhou, X. Zhang, Y. Yang, Z. Liu, B. Tian, J. Jie and X. Zhang, Biomaterials, 2013, 34, 8960–8967 CrossRef CAS PubMed.
- H. M. Chen, R. C. MacDonald, S. Y. Li, N. L. Krett, S. T. Rosen and T. V. O'Halloran, J. Am. Chem. Soc., 2006, 128, 13348–13349 CrossRef CAS PubMed.
- Z. H. Zhao, X. M. Wang, Z. J. Zhang, H. Zhang, H. Y. Liu, X. L. Zhu, H. Li, X. Q. Chi, Z. Y. Yin and J. H. Gao, ACS Nano, 2015, 9, 2749–2759 CrossRef CAS PubMed.
- Z. Zhang, H. Liu, H. Zhou, X. Zhu, Z. Zhao, X. Chi, H. Shan and J. Gao, Nanoscale, 2016, 8, 4373–4380 RSC.
- S. M. Lee, O. S. Lee, T. V. O'Halloran, G. C. Schatz and S. T. Nguyen, ACS Nano, 2011, 5, 3961–3969 CrossRef CAS PubMed.
- H. Chen, S. Pazicni, N. L. Krett, R. W. Ahn, J. E. Penner-Hahn, S. T. Rosen and T. V. O'Halloran, Angew. Chem., Int. Ed., 2009, 48, 9295–9299 CrossRef CAS PubMed.
- S. Y. Qin, A. Q. Zhang, S. X. Cheng, L. Rong and X. Z. Zhang, Biomaterials, 2017, 112, 234–247 CrossRef CAS PubMed.
- L. Fan, B. Zhang, A. Xu, Z. Shen, Y. Guo, R. Zhao, H. Yao and J.-W. Shao, Mol. Pharmaceutics, 2018, 15, 2466–2478 CrossRef CAS PubMed.
- T. Li, W. Xiang, F. Li and H. Xu, Biomaterials, 2018, 157, 17–25 CrossRef CAS PubMed.
- J. Zhang, Y. Li, F.-F. An, X. Zhang, X. Chen and C.-S. Lee, Nano Lett., 2015, 15, 313–318 CrossRef CAS PubMed.
- T. Sun, Y. S. Zhang, B. Pang, D. C. Hyun, M. Yang and Y. Xia, Angew. Chem., Int. Ed., 2014, 53, 12320–12364 CAS.
- W. Ma, A. G. Cheetham and H. G. Cui, Nano Today, 2016, 11, 13–30 CrossRef CAS PubMed.
- M. Rouleau, A. Patel, M. J. Hendzel, S. H. Kaufmann and G. G. Poirier, Nat. Rev. Cancer, 2010, 10, 293–301 CrossRef CAS PubMed.
- C. J. Lord and A. Ashworth, Nature, 2012, 481, 287–294 CrossRef CAS PubMed.
- O. Tredan, C. M. Galmarini, K. Patel and I. F. Tannock, J. Natl. Cancer Inst., 2007, 99, 1441–1454 CrossRef CAS PubMed.
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi, C. Reinhardt, K. Landfester, H. Schild, M. Maskos, S. K. Knauer and R. H. Stauber, Nat. Nanotechnol., 2013, 8, 772–781 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- T. C. Chou, Pharmacol. Rev., 2006, 58, 621–681 CrossRef CAS PubMed.
- S. B. Howell, R. Safaei, C. A. Larson and M. J. Sailor, Mol. Pharm., 2010, 77, 887–894 CrossRef CAS PubMed.
- L. Galluzzi, L. Senovilla, I. Vitale, J. Michels, I. Martins, O. Kepp, M. Castedo and G. Kroemer, Oncogene, 2012, 31, 1869–1883 CrossRef CAS PubMed.
- P. D. W. Eckford and F. J. Sharom, Chem. Rev., 2009, 109, 2989–3011 CrossRef CAS PubMed.
- S. Zhang, H. Gao and G. Bao, ACS Nano, 2015, 9, 8655–8671 CrossRef CAS PubMed.
- S. M. Cohen and S. J. Lippard, Prog. Nucleic Acid Res. Mol. Biol., 2001, 67, 93–130 CAS.
- E. P. Rogakou, D. R. Pilch, A. H. Orr, V. S. Ivanova and W. M. Bonner, J. Biol. Chem., 1998, 273, 5858–5868 CrossRef CAS PubMed.
- W. M. Bonner, C. E. Redon, J. S. Dickey, A. J. Nakamura, O. A. Sedelnikova, S. Solier and Y. Pommier, Nat. Rev. Cancer, 2008, 8, 957–967 CrossRef CAS PubMed.
- O. A. Martin and W. M. Bonner, Cell Cycle, 2006, 5, 2909–2913 CrossRef PubMed.
- M. Li and X. Yu, Oncogene, 2015, 34, 3349–3356 CrossRef CAS PubMed.
- N. J. Curtin and C. Szabo, Mol. Aspects Med., 2013, 34, 1217–1256 CrossRef CAS PubMed.
- X. Wang, Y. Shi, D. Huang and X. Guan, Cancer Treat. Rev., 2018, 68, 62–68 CrossRef PubMed.
- H. Liu, Z. Zhang, X. Chi, Z. Zhao, D. Huang, J. Jin and J. Gao, Sci. Rep., 2016, 6, 31009–31021 CrossRef CAS PubMed.
- I. Walter, T. Schwerdtle, C. Thuy, J. L. Parsons, G. L. Dianov and A. Hartwig, DNA Repair, 2007, 6, 61–70 CrossRef CAS PubMed.
- B. Vogelstein and K. W. Kinzler, Nat. Med., 2004, 10, 789–799 CrossRef CAS PubMed.
- F. Sanchez-Vega, M. Mina, J. Armenia, W. K. Chatila, A. Luna, K. C. La, S. Dimitriadoy and D. L. Liu, Cell, 2018, 173, 321–337.e310 CrossRef CAS PubMed.
- C. J. Sherr, Cell, 2004, 116, 235–246 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. FaroKHzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8bm01039k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |