
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZnO decorated laser-induced graphene produced by direct laser scribing†
Joana
Rodrigues
 *,
Julia
Zanoni
,
Guilherme
Gaspar
,
António J. S.
Fernandes
,
Alexandre F.
Carvalho
,
Nuno F.
Santos
,
Teresa
Monteiro
and
Florinda M.
Costa
*,
Julia
Zanoni
,
Guilherme
Gaspar
,
António J. S.
Fernandes
,
Alexandre F.
Carvalho
,
Nuno F.
Santos
,
Teresa
Monteiro
and
Florinda M.
Costa
Departamento de Física & I3N, Universidade de Aveiro, Campus Universitário de Santiago, 3810-193 Aveiro, Portugal. E-mail: joana.catarina@ua.pt; Tel: +351 234247261
First published on 12th July 2019
Abstract
A scalable laser scribing approach to produce zinc oxide (ZnO) decorated laser-induced graphene (LIG) in a unique laser-processing step was developed by irradiating a polyimide sheet covered with a Zn/ZnO precursor with a CO2 laser (10.6 μm) under ambient conditions. The laser scribing parameters revealed a strong impact on the surface morphology of the formed LIG, on ZnO microparticles' formation and distribution, as well as on the physical properties of the fashioned composites. The ZnO microparticles were seen to be randomly distributed along the LIG surface, with the amount and dimensions depending on the used laser processing conditions. Besides the synthesis conditions, the use of different precursors also resulted in distinct ZnO growth's yields and morphologies. Raman spectroscopy revealed the existence of both wurtzite-ZnO and sp2 carbon in the majority of the produced samples. Broad emission bands in the visible range and the typical ZnO near band edge (NBE) emission were detected by photoluminescence studies. The spectral shape of the luminescence signal was seen to be extremely sensitive to the employed processing parameters and precursors, highlighting their influence on the composites' optical defect distribution. The sample produced from the ZnO-based precursor evidenced the highest luminescence signal, with a dominant NBE recombination. Electrochemical measurements pointed to the existence of charge transfer processes between LIG and the ZnO particles.
1. Introduction
Zinc oxide (ZnO) is considered one of the most explored and versatile semiconductor oxide materials in terms of its physical/chemical properties, namely the high surface-area provided by the myriad of morphologies produced at the nano/micro scale level, high crystallinity, nontoxicity, biological compatibility, and improved optical, electrical and electrochemical response. All these features make ZnO structures highly desirable for various applications including optics, optoelectronics, supercapacitors, biosensors, tissue engineering or drug delivery systems, among others.1–5The development of ZnO composites with other materials is expected to combine the properties of both components, allowing the production of functional materials with more advantageous properties than each component by itself.6,7 Amongst these composite structures, an interesting class of nanostructured carbon-based forms (e.g. graphene, carbon nanotubes-CNTs) have been employed in the fabrication of a wide range of applications, as for instance in biosensors, photovoltaics, supercapacitors or photocatalysts.8–16 Previous works by our research group17,18 already explored the synergistic properties of ZnO with carbon-based materials. Specifically, an enhancement of the ZnO photoluminescence (PL) was identified when nanostructures of this semiconductor were deposited on a forest of vertically-aligned CNTs (VACNTs),17 while ZnO/CNT buckypaper composites were seen to maintain the strong luminescent properties of the ZnO structures, exhibiting the electrical properties associated with the CNTs.18 Furthermore, an enhancement of the physical properties of ZnO and carbon-based materials when brought together has also been reported, showing interesting advantages, namely in sensing applications.19–23 For instance, Lupan et al.19 recently described the influence on ultraviolet (UV) and gas sensing properties of ZnO nanowires after combining them with CNTs. These authors observed that the addition of CNTs to the porous ZnO networks led to an enhancement of the sensing properties of ZnO, with an efficient detection of different gases, especially in the case of ammonia. Fullerene C60 hybridized ZnO tetrapods were also employed in the fabrication of gas sensing devices, revealing interesting sensing behaviour towards volatile organic compounds.24 Besides gas sensing, ZnO/carbon composites have been also used in biosensing applications for the detection of several biospecies, such as glucose,20,25 DNA26 or cancer indicators.21,27 For example, a device for detection of prostate-specific antigen (PSA) was accomplished using ZnO quantum dots/CNTs composites, abling the sensor to detect PSA concentration in serum samples as low as 0.61 pg ml−1.27 Moreover, Zhao et al.20 reported the fabrication of a glucose sensor based on ZnO-nanorods/graphene heterostructures, which showed a linear response to glucose concentrations in the range of 0.2–1.6 mM.20 A common feature in the different ZnO/carbon composites reported in the literature is the need of several steps to produce the materials that comprise the composite and the composite itself. Usually, both components are produced separately, with the desired dimensions and morphologies, and then mixed together to form the final material.15,19,28 Frequently, this approach involves surface modification of one or both constituents to establish covalent, noncovalent or electrostatic interactions between them,29,30 resulting in elaborated and time consuming processes. Another approach involves the synthesis of ZnO in the presence of the previously prepared carbon material, onto which the oxide semiconductor grows with the chosen morphology.16,17,29 This approach is less time consuming and promotes a closer link between both components, since the ZnO grows directly on the carbon-based substrate. Even so, a multi-step preparation is required to reach the desired composite. Aiming at simplifying these processes, in the present work we report on the production of ZnO decorated laser-induced graphene (LIG) in the same laser-processing step. In this process, a CO2 laser irradiates a Zn/ZnO-based layer covering a polyimide – C22H10O5N2 (Kapton®) sheet used as precursors for ZnO and LIG, respectively. The production of LIG from Kapton® by laser scribing using a CO2 laser was firstly reported by the group of Tour.31 Its formation by graphenisation is believed to be induced by the high localised temperatures (higher than 2500 °C according to laser-induced fluorescence) attained by a highly focused laser beam, giving rise to a photothermal carbonisation process.31 Additionally, some attempts have been made to improve the specific capacitance of LIG-based electrodes32 by forming composites with other materials, as is the case of MoS2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33 and metal oxides as Co3O4, MoO2, and Fe3O4,34 mostly by embedding the metal-complexes used as precursors for the metal oxide particles in the polyimide film during the polymer preparation. Laser induction is then conducted on this metal-complex-containing polyimide substrate to form the composites.34
33 and metal oxides as Co3O4, MoO2, and Fe3O4,34 mostly by embedding the metal-complexes used as precursors for the metal oxide particles in the polyimide film during the polymer preparation. Laser induction is then conducted on this metal-complex-containing polyimide substrate to form the composites.34
Following the work of Lin et al.,31 several others have been reported regarding the development, optimisation and application of LIG.32,35–46 In fact, the production of LIG by such scribing process has now been accomplished using other carbon sources besides the polyimide, as for instance cloth, paper or even food,41,43,47 as well as other laser wavelengths.48 Moreover, its application extend from microfluidic devices, sensors, optoelectronic devices, microsupercapacitors, to catalysts, among others.40,41,43,45,46,49
When compared to other techniques, the method in focus by the present report possesses key advantages. Among these, we highlight the prompt synthesis process of the composites, the ability to perform a pattern with the computer-controlled laser-scribing system, the production of flexible composite's samples and its easy scalability in a time- and cost-effective way. Moreover, since the formation of LIG and ZnO takes place simultaneously, a stronger link and interaction between the two materials is expected when compared with other approaches.
The main goal of the present work is to develop these nanostructured composites, aiming to use them as transducer elements to be further incorporated into affordable, flexible and disposable biosensors. Nevertheless, before testing the new composites on the foreseen application, an adequate knowledge of their fundamental properties is of paramount importance to understand the materials potentialities and also to be able to control/tune the desired features.
2. Experimental details
2.1 Materials synthesis
A continuous wave (CW) CO2 laser (Redsail M500) with a wavelength of 10.6 μm (maximum power output of 50 W and a beam diameter in the order of ∼100 μm) was used to produce the ZnO-decorated LIG composites (hereafter designated by ZnO/LIG). This system is fitted with a mechanically driven X–Y focusing head. Fig. 1 depicts a schematic representation of all steps involved in the preparation of the precursor materials and the synthesis process. The ZnO/LIG samples were formed by laser irradiation of a Kapton® sheet (HN500), previously covered with metallic Zn/ZnO powders embedded in a paste. This paste was used in order to promote a good adhesion of the Zn/ZnO powder to the Kapton® sheet, thus avoiding the loss of this precursor during the laser scribing process. Therefore, the Kapton® will act as the precursor for LIG, while the Zn/ZnO-based paste is the precursor for ZnO. This paste was spread onto the Kapton® sheet by dropping 1 ml in a defined area of ∼4 × 4 cm2, delimited with a scotch tape, as illustrated in Fig. 1. Different organic ligands were tested to evaluate the best approach in forming a homogeneous cover of the Kapton® surface by the precursor. First, a mixture of Zn (Sigma-Aldrich, purum) powder with polyvinyl alcohol (PVA) dispersed in water (with a concentration of 0.1 g ml−1) was tested and 1 g of the commercial Zn powder was added to 10 ml of PVA. This mixture was stirred for 1 h, followed by ultrasonic bath with the same duration, aiming to obtain a fairly homogeneous paste. The Kapton® sheet was then covered with this paste via doctor blade technique, and then left to dry in air. During the drying process, it was observed that the Zn/PVA paste (hereafter designated by Zn#1) tends to accumulate more on the borders delimited by the tape. The average thickness of the Zn#1 layer was found to be ∼6.7 μm, as shown in Fig. S1a.† However, this layer is rather inhomogeneous, as highlighted in Fig. S1b,† with some large Zn agglomerates, as well as nearly Zn-free areas. Moreover, when left to dry, the high plasticity of this layer led to its detachment from the polymer sheet in some regions (see Fig. S2†), compromising the laser writing process in such areas.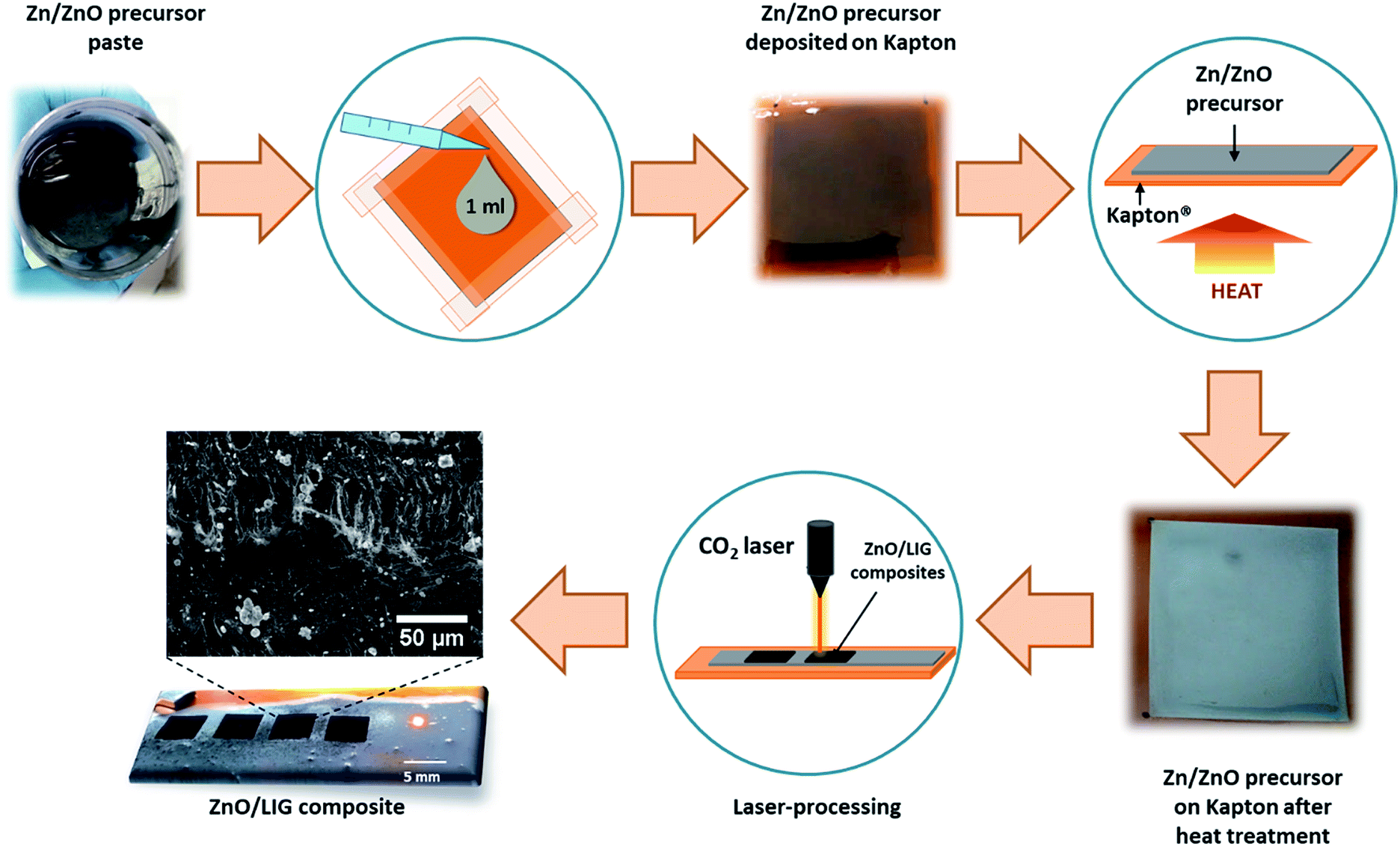 | ||
| Fig. 1 Schematic representation of all steps involved in the preparation of Zn/ZnO-covered polyimide sheet used as precursor and the laser-scribing process to form the ZnO/LIG composites. A representative SEM micrograph (processing conditions: P ∼ 24 W, v = 200 mm s−1 and d = 0.075 mm, focused conditions – set#1) was included to show the LIG structure with the presence of small ZnO particles randomly dispersed onto the surface. | ||
To overcome these drawbacks, a new Zn-based precursor paste was prepared in order to obtain a higher homogeneity in the preparation of the Zn/Kapton precursors, and subsequently in the ZnO/LIG composites. In this case, the Zn commercial powders were placed in a planetary ball mill during 1 h at 500 rpm to reduce the particle dimension and their size distribution. The milled powders (1 g) were then mixed with ethanol (2 ml), terpineol (4 g, Sigma-Aldrich) and ethyl cellulose (0.5 g, Sigma-Aldrich) to form a viscous paste (hereafter designated by Zn#2). The mixture was kept in an ultrasonic bath during 1 h and another 30 min under magnetic stirring. This procedure was repeated to inssure a homogeneous paste. The new precursor layer was then spread onto the Kapton® surface using the same method employed for the Zn#1 paste. However, in this case, an additional heating step to dry the paste was included, instead of letting the samples to dry in air. This heating step was conducted at 100 °C during ∼1 h. With this step, a more uniform cover of the polymer surface was obtained.
Additionally, another paste was prepared in the same way as the Zn#2, but now using ZnO commercial powders (AnalaR, 99.7%) instead of the Zn ones. In this case, the average thickness of the layer was found to be ∼16 μm (Fig. S1c†) and with an improved homogeneity when compared with the Zn#1 paste (Fig. S1d†). It is worth to note that a similar thickness was also found for the Zn#2 precursor paste.
A combination of different laser parameters was employed with focus on scan speed v (100–350 mm s−1), laser power output P (∼15.0–27 W) and distance between the laser head and the substrate dlaser (1.8–2.4 cm). Focused conditions corresponde to dlaser = 1.8 cm. The distance between scanning lines d was kept between 0.075 and 0.1 mm. The parameters were chosen to enable the synthesis of both LIG and ZnO while preventing the full polymer degradation by always keeping a thin unaffected Kapton® layer that provides the flexible mechanical support. Mainly, three sets of samples were considered to evaluate the influence of the different processing parameters and precursors on the properties of the composites, as summarized in Table 1. All samples were produced under ambient conditions. After their synthesis, all samples were washed with distilled water to remove the ZnO particles not attached to LIG.
| Sample's set | Precursor | Laser power (W) | Scan speed (mm s−1) | Line spacing (mm) | Distance between laser head and precursor (cm) |
|---|---|---|---|---|---|
| #1 | Zn#1 | 27 | 350 | 0.1 | 1.8 (focus) |
| 300 | 0.075 | ||||
| 24 | 350 | ||||
| 300 | |||||
| 200 | |||||
| 15 | 300 | ||||
| 200 | |||||
| 100 | |||||
| 5 | 200 | ||||
| 100 | |||||
| #2 | Zn#1 | 21 | 200 | 0.1 | 1.8 |
| 1.9 | |||||
| 2.0 | |||||
| #3 | Zn#1 | 22 | 150 | 0.1 | 2.0 |
| Zn#2 | |||||
| ZnO |
2.2 Morphological, structural, optical and electrochemical characterisation
The morphology of the produced samples was studied by scanning electron microscopy (SEM) using a TESCAN Vega3 SBH SEM microscope. Energy-dispersive X-ray spectroscopy (EDS) maps were acquired using a Bruker Xflash 410 M Silicon Drift Detector, with a 133 eV energy resolution (at MnKα) @ 100 k cps.3D reconstruction of the samples surface morphology was obtained from a S neox non-contact optical profiler from Sensofar Metrology. The instrument was operated under confocal mode, allowing measurements of smooth to rough surfaces with variable resolution, according to the microscope's objective. Three different objective magnifications were used in the present work, namely 10× , 20× and 50× , resulting in lateral resolutions of 1.29, 0.65 and 0.26 μm and vertical resolutions of 25, 8 and 3 nm, respectively. Subsequent image reconstruction and morphology profiles were assessed through SensoScan 6.2 software.
Micro-Raman spectroscopy measurements were obtained in a Horiba Jobin Yvon HR800 instrument in backscattering configuration by exciting the materials with a 442 nm line from a CW He–Cd laser (Kimmon IK Series), using a ND 0.6 neutral density filter and focusing with an objective of 50 × magnification and numerical aperture of 0.7. The spectra were acquired in the range between 150 and 3300 cm−1.
Room temperature (RT) steady-state PL measurements were performed using the 325 nm (3.81 eV) line from a CW He–Cd laser (Kimmon IK Series) with a beam spot of ∼1 mm and an excitation power density below 0.6 W cm−2. The experiments were carried out under a 90° geometry. The luminescence was measured using a dispersive system SPEX 1704 monochromator (1 m, 1200 gr mm−1) fitted with a water-cooled Hamamatsu R928 photomultiplier tube. All samples were investigated under the same excitation/detection conditions and the signal was maximised at the visible band maximum. As stated in the Introduction section, the main goal is to combine optical and electrochemical transduction in a single device, thus the luminescence outcome of the produced composites was chosen as the main criteria to be monitored for the optimisation of the processing conditions. The samples that resulted in composites with no RT measurable luminescence emission were discarded.
Electrochemical characterisation was accomplished by cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) via a Versastat 3 instrument. The CV and EIS response of the ZnO/LIG composites (from ZnO-based precursor) were measured and compared to LIG reference samples (same processing conditions as the composite samples). Measurements were performed in duplicate, using a Ag/AgCl (1 M KCl) reference electrode and a platinum wire counter electrode in N2-bubbled 10 mM phosphate buffer saline (PBS pH 7.4, from Fisher BioReagents) solutions. The ferro/ferricyanide redox couple was employed for faradaic measurements.
3. Results and discussion
3.1 Microscopical analyses
In order to obtain the desired ZnO/LIG composites, several laser-writing conditions and ZnO precursors were tested . At the first stage (set#1), the main goal was to use a wider range of parameters to assess the general tendency of their influence on the composites synthesis. For this purpose, the Zn#1 precursor was employed and the synthesis was performed at focused conditions (dlaser = 1.8 cm), as described in the experimental section. Moreover, LIG reference samples were also produced with the same processing parameters as the composite ones. It was verified that the threshold conditions for the composite formation were different from those required for LIG production, especially regarding the applied laser powers. Due to the Zn/PVA paste covering Kapton®, higher laser powers were needed to form the ZnO/LIG structures. Indeed, when only the polymer is present, powers of ∼5 W have shown to be enough to start the graphenisation process (even for scanning speeds of 350 mm s−1), in line with what was reported by other authors.31,35 On the other hand, when the Zn-based precursor is present, a higher threshold power in the range of ∼10–15 W was necessary to initiate the composite formation, since lower powers were not enough to promote the photothermal carbonisation of the Kapton® sheet. This can be explained by photon absorption and/or dissipation by the Zn/PVA paste and partial reflection of the incident photons on the metallic Zn particles. Both effects lead to an attenuation of the laser power that reaches the polymer, with a consequent reduction of the attained local temperature for similar applied powers.Fig. 2 shows SEM micrographs of selected ZnO/LIG composites prepared in set#1. As it can be seen, the choice of laser-writing parameters has a significant effect on the formed structures. Although in the studied cases it was possible to observe that the laser irradiated region led to the formation of a carbon-based material that protrudes from the Kapton® substrate, distinct features arise from the different parameters combination. For instance, it was observed that higher laser powers (up to ∼27 W) promote the formation of a large amount of randomly oriented carbon fibres (Fig. 2a and b). Nonetheless, it was seen that the occurrence of these fibres could be controlled by an adequate combination of laser power and scanning speed. A comparison between images (a) and (b) and then between (b) and (c) of Fig. 2 allows to clearly identify that, while keeping the same line spacing, a reduction of the laser power leads to a reduction of the size/amount of the carbon fibres for the same scan rate (350 mm s−1). In a similar way, and for the same applied power (∼24 W), the amount of fibres was reduced by decreasing the scan speed from 350 mm s−1 to 200 mm s−1. As expected, these two parameters were seen to be deeply correlated, given that as the laser power increases, higher scan rates are also required to avoid partial or full degradation of the polymer sheet. Thus, an adequate tuning of the synthesis parameters enables the promotion of different surface morphologies in the produced carbon-based materials. Recently, Duy et al.42 reported a detailed work regarding the formation of these laser-induced graphene fibres (LIGF). They consider several scribing parameters in the formation of such fibres, namely the wavelength of the used laser, fluence and laser image density (dots per length unit), which determines the overlap of subsequent laser pulses used to form the image. Overlapping was found crucial to suppress the formation of the fibres. In our setup, this superposition can be determined by the laser scanned lines distance and laser power. An increase in power broadens the spot above the graphenisation threshold. Thus, the regions with a higher amount of fibres correspond to a lower overlap of the laser path.
 | ||
| Fig. 2 SEM micrographs of the ZnO/LIG composites with different laser processing conditions from set#1. The yellow circles and arrow highlight the presence of ZnO particles with different sizes. (a) P ∼ 27 W, v = 350 mm s−1; (b) P ∼ 24 W, v = 350 mm s−1; (c) P ∼ 24 W, v = 200 mm s−1; and (d) P ∼ 15 W, v = 100 mm s−1. | ||
By adjusting the processing parameters in order to reduce the formation of these carbon fibres, a 3D foam-like LIG structure was visible (Fig. 2c and d). This foam exhibits large pores with an irregular shape and diameters in the range of one to a few tens of micrometres, which are attributed to the rapid liberation of gases produced during the photothermal process.31,35 Besides the large-size pores, LIG also displays a porous structure with much smaller pores' diameters (microns to sub micrometres) than the previous ones (Fig. 2d). According to Luo et al.,35 these porous regions are also due to the release of the mentioned pyrolysis gases, being more pronounced as the laser power increases, resulting in higher thicknesses and heights. In fact, in a typical polyimide carbonisation process, two main steps are present: pyrolysis and carbonisation itself.50,51 In the first step, which occurs between 500–650 °C, there is a breakage of the carbonyl groups in the imide part, leading to an abrupt release of CO and CO2 gases, accompanied by a pronounced weight loss of the polymer. In the second step, which occurs over a temperature range from 800 °C to more than 1000 °C (for a Kapton® substrate only), mostly H2 is released.50,51 After the release of these molecules, the aromatic compounds can be rearranged to form few-layer graphene nanostructures.31,32 This pyrolysis/carbonisation proceeds at a very fast rate due to the rapid laser heating inherent to the laser-writing process.35
Additionally, it was also observed that with the increase of the scan speed (>350 mm s−1) the uniformity of the samples was reduced, showing alternating regions of high and low porosity, likely to be due to the non-uniform exposition of the precursors to the laser.
Simultaneously with the LIG formation, the production of randomly distributed spherical-shaped particles also takes place on LIG's surface, depicting a wide range of micrometre-scale sizes. EDS mapping revealed their correspondence to ZnO (Fig. 3), which was further corroborated by Raman measurements (section 3.2). A closer inspection of the SEM images (insets in Fig. 2 and 3) showed that the larger microparticles are in fact comprised by aggregates of much smaller ones, especially when higher laser powers are applied. Scrutinising the sample produced with the highest power employed in this study (∼27 W – Fig. 2a), it was evident that, besides the large aggregates of microparticles (diameters in the range of a few micrometers – arrow in inset of Fig. 2a and S3a†), smaller particles can be found along with the carbon fibres (e.g., circles in the inset of Fig. 2a). On the other hand, in the case of the sample synthesised with ∼15 W (lowest applied power for the composites' formation), the ZnO aggregates tend to be scarcer, the particles are smaller and with a more irregular shape, as highlighted in the inset of Fig. 2d. This can be attributed to the lower temperature expected for such powers, since the formation of these ZnO microparticles is promoted by the thermal oxidation of the Zn powder in air. The local heating induced by the CO2 laser beam leads to the Zn transformation into ZnO, as the temperatures attained in this process are much higher than the melting point of bulk metallic Zn (420 °C52). Therefore, one can assume that partial melting followed by evaporation of Zn should occur during the laser-writing process. The reaction with oxygen (present in the surrounding atmosphere) with the outer surface of the previously formed Zn droplets originates the ZnO nuclei from which the particles start to grow.53 Thus, Zn acts as both reactant and catalyst for the ZnO growth, as reported by other authors.54 For a constant oxygen content, the size of the ZnO structures is strongly dependent on the local temperature and Zn partial vapour pressure. Both high temperature and high Zn availability lead to a rapid growth into large structures, whereas the opposite results in slow nucleation/growth rates.55 It may explain why less and smaller microparticles/aggregates are observed in the case of the samples processed at ∼15 W. A lower scan rate is expected to enable a higher exposure of the Zn precursor to the laser beam (heating source), providing more energy (at a fixed laser power) to initiate the ZnO nucleation and growth, and possibly leading to the production of a higher amount of ZnO particles. Since this Zn precursor is comprised by a dispersion of the metal particles in PVA, it is also worth to mention that, at such temperatures, the organic material is expected to fully decompose during the laser processing (two stage decomposition at ∼240 °C and 450 °C56,57), being absent from the laser-processed areas.
 | ||
| Fig. 3 SEM image of a sample produced with ∼24 W, v = 350 mm s−1, d = 0.075 mm and focused conditions (left) and EDS mapping of the correspondent region (right). | ||
Finally, another aspect to take into account in the composite formation is the gas liberation during the graphenisation process. It is expected that the outgassing during carbonisation of LIG may spread to the newly formed ZnO particles and remaining metallic Zn to the peripheral areas that will be further irradiated. The interaction of these gases with the particles under formation can be responsible for the presence of different chemical groups at the ZnO surface, which may give rise to distinct optical properties, as will be discussed later on. Furthermore, the way the gases are released (depending on the applied scribing conditions) may also influence how the ZnO particles are distributed on the LIG surface. For example, the quick “bombing” effect (as denominated by Luo et al.35) experienced by applying high laser powers may contribute to the “expulsion” of some ZnO particles out of the surface of LIG. When the ZnO/LIG composites samples are compared to the ones only containing LIG, one should bear in mind that the precursor layer/paste is likely to absorb/retain part of the formed gases, which will change the local environment at the polymer surface and therefore contribute to the differences observed in the topographic/morphological structures.
Considering now the influence of the focus conditions, it was verified that increasing the distance between the laser head and the precursor (set#2) to 2 cm, which corresponds to slightly defocused conditions, results in the formation of a composite with better luminescent properties (see section 3.3), namely higher PL intensity and, consequently, higher signal to noise ratio. Moreover, comparing samples prepared with the same scan speed and similar laser powers (Fig. 2c and 4 top left), one clearly sees a further reduction of the carbon fibres formation, as well as a more uniform surface morphology and porosity of LIG. A likely explanation for this noticeable change is related with the increase in the laser beam spot and more homogeneous radial distribution of the power. As reported by Chyan et al.,47 this will result in multiple lasing at a given location for the same processing speed and in one single laser pass, which leads to a less fibrous morphology and to a more sheet-like structure of LIG. It is worth to mention that for values of dlaser > 2.2 cm no composites could be formed. Regarding the ZnO formation itself, no significant changes were identified for the amount, shape and size of the microparticles. Therefore, the results suggest that using defocused conditions may promote an enhancement in the crystallinity of the formed ZnO (see Raman section), but not in their production yield. Similarly to what was discussed in the case of the scan speed, laser beam defocusing results in a longer and milder exposure of the Zn precursor and the formed ZnO particles to the beam for the same processing time, thus leading to better structural and optical properties.
 | ||
| Fig. 4 SEM images of samples from set#3, using different precursors and similar processing conditions. | ||
Changing the precursor from Zn#1 to Zn#2 (Fig. 4 top left and right, respectively), and focusing on samples from set#3, results in changes in the surface morphology of the composites, especially concerning the porous distribution of LIG. The sample produced with the Zn#2 precursor reveals the presence of larger pores than the ones identified in the Zn#1 sample, dispersed in a mesh of much smaller pores. The differences can be attributed to different laser interaction/absorption by the organic compounds that comprise the paste (PVA vs. a mixture of ethanol, terpineol and ethyl cellulose). Additionally, the higher homogeneity of the Zn particles distribution using the Zn#2 precursor, as well as its better adhesion to the Kapton® sheet, may also contribute to the observed differences. Nevertheless, the ZnO particles seem very similar in these two cases (see also Fig. S3a and b†), as attested by their size and shape, and corroborated by the Raman (section 3.2) and PL (section 3.3) results.
Since the yield of ZnO particles production was relatively low for all the applied processing conditions, a new precursor paste was tested using ZnO commercial powder instead of metallic Zn. Fig. 4 bottom depicts the SEM image of the resultant composite. As seen in this figure, a larger amount of ZnO particles is present in this sample, when considering the same laser processing conditions and similar amount of precursor paste. The ZnO structures tend to accumulate more in the protruded regions of the sample. The higher concentration of ZnO particles in these regions may also be accounted by the "bombing" effect mentioned earlier. A fast gas release from the polyimide graphenisation may shove the ZnO to those regions. Additionally, the formed particles do not present the same spherical-like shape as the ones produced from the Zn pastes. Instead, a more irregular morphology is observed: not only do the particles appear to have smaller dimensions (Fig. S3c†) but also a propensity to form agglomerates. The higher yield observed in this case may be related to the different interaction between the laser and the ZnO powder particles vs. the metallic Zn particles. As previously mentioned, metallic Zn can reflect part of the incident photons, thus reducing the number of photons absorbed. On the other hand, the used wavelength is well absorbed by the ZnO powders, as corroborated by other growth techniques using a similar heating source.58 Laser incidence promotes the thermal decomposition of ZnO, leading to the formation of vapours containing the precursors materials. Zn will react with oxygen from both the atmosphere and the precursor powder, leading to the formation of ZnO structures that will further be deposited on the surface of the freshly formed LIG.
Fig. 5 depicts 3D optical profilometer images of the selected samples. Their respective surface topographic profiles taken at the central position of the images (blue dashed-lines in the 3D pictures) are also displayed. Moreover, a reference sample corresponding only to LIG was analysed (Fig. S4a†) in order to compare its surface profile and texture aspect ratio with the ZnO/LIG composite samples for the same laser-writing conditions (specifically the one depicted on Fig. 5a). It was observed that all the samples present alternating valley and hill regions (better identified in Fig. S2a, c† and 5c), which correpond to the protruded regions mentioned in the SEM analysis. The thickness of these hills roughly increases for higher laser powers, as it was also reported by Luo et al.35 The topographical data is obtained from the corresponding 3D reconstruction of the several individual 2D images, where each individual point corresponds to a certain height value. Given that the surface of the samples comprises a fibre-like structure, it is likely to measure discrete points. As such, a continuous line was only added to the topographical data for the sake of clarity. The texture aspect ratio (Str) parameter was used to spatially evaluate the uniformity of the surface texture of these samples, where a strong directionality of surface structure corresponds to a Str closer to zero (wavy-like) while an Str closer to unit indicates a more disordered texture arrangement.59
 | ||
| Fig. 5 Optical profilometer images acquired with 20 × magnification and surface profile (taken at the centre of the images – marked line). (a–c) ZnO/LIG composites with different laser processing conditions. The colour scale was defined from blue to red, corresponding to the minimum and maximum of the Z values, respectively, and for each individual sample (peak-to-valley full colour scale). (d) Texture aspect ratio (Str) values for the different analysed samples. | ||
As it was observed in the SEM micrographs (Fig. 2), the samples prepared with higher power and scanning speed evidence a higher amount of misoriented carbon fibres. In addition, a reduction of the speed from 350 to 200 mm s−1 revealed a strong impact on the surface topography of the ZnO/LIG samples, leading to a more ordered carbon structure. Str values (Fig. 5d) show an increase with laser power and scan speed, therefore revealing more disordered features formed at the surface. By comparing the samples with and without the presence of the Zn precursor paste and laser-processed under the same conditions, remarkable differences emerge. A more regular surface is observed in the case of the LIG reference sample (denominated Ref LIG in Fig. S4a†), with a clear alternation between valleys and hills. In fact, and based on previous experiments, these hills are formed by several arranged fibres that arise from the overlapping of part of the laser spot under consecutive scan lines. As previously described, the spot affected zone exceeds the predefined laser line spacing (0.075 mm) and parts of the sample are thus irradiated twice, i.e. on peripheries of the scanned lines. In line with what has been observed for the samples prepared with Zn#1 (please consider P ∼ 24 W and v = 200 mm s−1 for comparison), the sample processed from Zn#2 (Fig. S4d†) evidence a high disorder surface. In addition, and from a wider point of view, this sample seems to present larger agglomerates of Zn/ZnO at the surface, compared to his counter-part based on Zn#1 precursor, and showing also agglomerates of similar dimensions to the ones found in the sample processed under P ∼ 15 W and v = 100 mm s−1. This indicates that the Zn#2 precursor and the conjugation of laser defocusing along with lower scan rate may result in the formation of larger pores and ZnO agglomerates. Its texture aspect ratio is comparable with the one obtained for the sample prepared with Zn#1 and processed under similar conditions, which shows that the laser power sets most of the samples' surface disorder rather than a reduction of the scan rate. Regarding the sample produced from the ZnO-based paste, a much more textured surface is observed, with a clear alternation between hill and valley, similar to what was observed in set#1 for the sample processed with lower scan speed (Fig. S4c†). Moreover, a low value for the texture aspect ratio was also attained (Fig. 5d), even lower than the one obtained for the sample processed under P ∼ 15 W and v = 100 mm s−1 (Zn#1) and for the sample processed under the same laser conditions but using the Zn#2 precursor. This shows the importance of the light absorption by the precursor's layer to the morphology of the ZnO/LIG composite, as well as the dispersion of ZnO particles on the surface of LIG.
3.2 Raman spectroscopy
Representative micro-Raman spectra acquired on different areas of interest of the ZnO/LIG samples are shown in Fig. 6. By taking advantage of the contrast between the different regions, it was possible to characterise areas with a higher amount of ZnO particles (“whiter” regions) and others dominated by the LIG features (“darker” regions). Probing the samples in ZnO-rich regions resulted in the observation of the characteristic Raman active vibrational modes of the ZnO wurtzite crystalline structure, A1, E1 and E2, as well as their overtones and combined modes. The “darker” regions exhibited the vibrational modes typically associated with sp2 coordinated carbon, proving that LIG is indeed present in all the samples. As a matter of fact, the vibrational modes of LIG resembles the ones typically observed in reduced graphene oxide (rGO)60 The presence of the well-defined ZnO vibrational modes in the analysed samples attests the crystallinity of the produced particles. Nevertheless, clear differences are observed between the relative intensities of the different Raman modes, as well as on their full width at half maximum (FWHM). The differences observed in the relative intensity of the vibrational modes may also arise from polarisation effects caused by the different orientations between the ZnO structures and the incidence wavevector of the laser beam.61–64 Additionally, the high intensity of the mode observed in the 2LO region (2A1(LO), 2E1(LO)) when compared to the first order phonons can be justified by the resonant excitation conditions, as previously reported in ZnO bulk samples.65 Regarding the FWHM, typically sharper peaks are associated with a high degree of crystallinity of the samples, while broader peaks may be accounted by amorphous phases or higher density of defects. For instance, in the case of the samples produced from the Zn-based precursors, the existence of non-stoichiometric ZnOx phases or residual metallic Zn with some spontaneously formed native oxide may contribute for the broadening of the Raman lines. In fact, the spectrum acquired on the Zn powders included in Fig. 6 shows the presence of such native oxide already in the precursor powders, although with broader features than the ones observed for the processed samples. Therefore, the observation of a broad A1(LO) mode may indeed result from the contribution of Zn particles that were not fully oxidised during the laser writing process. | ||
| Fig. 6 Raman spectra for selected samples produced with different laser-scribing conditions, obtained in backscattering configuration by exciting the samples with 442 nm laser line. All spectra were acquired using a ND 0.6 neutral density filter in the excitation. The red lines correspond to regions with higher ZnO content, while the dark grey lines correspond to regions where LIG predominates. For the samples processed with higher scan speed, the blue lines displayed denote intermediate regions between ZnO-rich and LIG-rich areas. A spectrum of the Zn powders used as precursor was also added for comparison (light blue line). | ||
As a general trend, defocused conditions seem to lead to sharper ZnO vibrational modes (see, for instance Ehigh2 mode) and for the same applied power lower scan rates exhibits sharper peaks, suggesting an improved crystallinity of the produced ZnO particles in such cases. Regarding the vibrational modes recorded for LIG, the D-band, frequently associated to the sp2 coordinated defective/disordered carbon phases, appears at ∼1363–1369 cm−1, while the G-band, related to C–C bond stretching in sp2, can be identified at ∼1577–1586 cm−1.66–68 The ratio between the peak intensity of these modes, ID/IG, can be used to measure the relative defect content in the sp2 carbon lattice.67,68 In the present case, ratios between 0.11 and 0.60 were obtained, showing a tendency to increase for higher scan speeds. High values of the ID/IG indicate the presence of a significant amount of defects, with possibly part of those being originated by oxygen-containing functional groups, suggesting some degree of oxidation in the samples prepared with higher laser power.68 Moreover, it was observed that higher ID/IG values were obtained for samples with a higher amount of carbon fibres (higher laser powers). This can be inferred, for instance, from the sample produced with P ∼ 15 W, where one can clearly note that when carbon fibres are substantially absent the D-band intensity is extremely low (ID/IG ∼ 0.11). Nevertheless, it is also important to take into account the contribution of the high density of graphene's edges for the intensity of the D band, since they behave as defective regions, thus activating the D mode.67 Besides, the FWHM of these modes is in the order of 52–59 cm−1 and 66–80 cm−1 for G-band and D-band, respectively, which constitutes another indication of the presence of a substantial density of defects and is concomitant with the presence of oxygen-related ones.69,70
In fact, the increase in the ID/IG ratio seems to have the same tendency as the Str value (excluding the sample prepared under the highest laser power), suggesting a correspondence between the presence of a higher amount of carbon fibres (and thus a higher surface disorder) and the presence of defects. The 2D band appears between ∼2724–2740 cm−1, originating from second order zone-boundary phonons.31,71 Therefore, the presence of a single and symmetric 2D-band (especially noticeable in the sample prepared with P ∼ 15 W), together with the G-band in the presented spectra, clearly indicates the formation of a graphene-based material.47 The FWHM of the 2D-band ranges from ∼97 to 104 cm−1 in all the analysed samples, while I2D < IG, which are characteristics typically associated to the formation of multi-layer graphene. Furthermore, additional and less intense modes are associated with the D + D′′, D + D′ and 2D′ modes. These multi-phonon processes are commonly indicative of defects in graphene-based materials.71
It is also important to mention that the absence of LIG Raman features observed when the beam was focused on the ZnO rich regions may be accounted by the limited penetration depth of the laser beam, probing essentially the ZnO particles and not the LIG underneath. Furthermore, since it was possible to identify broad and overlapped modes associated with the carbon phases, the presence of amorphous carbon in such spectra must be also considered, likely present in the interface between the ZnO particles and the LIG foam. This suggests that when a ZnO particle is formed, a small amount of amorphous carbon can also be produced in its vicinity due to the laser interaction with LIG/Kapton® and/or the organic compounds present in the precursor paste. When the sample is probed in a nearby region but away from the ZnO, the spectra display the well-defined LIG characteristic Raman modes. Additionally, intermediate regions were also probed in some samples, as displayed in Fig. 6 for the ones processed under higher scan speeds, evidencing the presence of both ZnO and carbon-related modes. These regions correspond to the transition areas between the “whiter” ZnO-rich regions and the “darker” LIG-rich ones.
3.3 Photoluminescence
As stated in the experimental part, the PL emission was the main property monitored during the optimisation process of the composites' fabrication. As such, all the produced samples were analysed by PL spectroscopy at RT by exciting them with a 325 nm (3.81 eV) laser beam, which corresponds to an energy above the bandgap of ZnO (Eg ∼ 3.3 eV @ RT4). Fig. 7 depicts the main results obtained for the different sets of conditions tested (see Table 1). Fig. 7a displays the PL spectra for selected samples belonging to the first set (set#1). In this case, all the analysed ZnO/LIG composites revealed the presence of a broad visible band dominating the spectra, together with the typical near band edge (NBE) emission in the UV region, although with a lower intensity when compared to the visible band. The visible bands identified in these samples present small differences regarding their peak positions, being dependent on the used processing condition. Moreover, the relative intensity between the UV and visible emissions also changes with the synthesis parameters. In all the cases, the broad band extends from the green to the yellow and orange–red spectral range, likely corresponding to an overlap of difference recombination channels. However, these channels present different contributions to the overall broad emission band depending on the processing conditions, even though with a predominance in the orange–red spectral region for all the studied samples. These differences result in a slight redshift of the peak position of the broad band when higher scan rates and laser power were used. The sample prepared with P ∼ 15 W and v = 100 mm s−1 exhibits the maximum of the band at ∼597 nm, while for the sample prepared with P ∼ 27 W, the peak position shifted to ∼625 nm. These results show that the formation of the defects originating the orange–red emission is promoted/enhanced when higher laser powers and scan rates are applied. It is worth noting that PL measurements were also performed in the LIG reference samples produced with the same laser-scribing parameters as those of the composites; however, no luminescence signal was detected. The sample prepared with P ∼ 24 W and v = 200 mm s−1 was the one that exhibited the highest intensity, followed by the one produced at P ∼ 15 W and v = 100 mm s−1. This seems to indicate that a lower scan speed is beneficial for an improved PL signal, having a predominant effect over the laser power. Moreover, the fact that the highest scan speed used (350 mm s−1) led to the formation of a considerable amount of carbon fibres, which scattered part of the incident laser beam, may also contribute to the poor PL signal collection. Thus, a proper combination between laser power and scan speed should be met, as low scan speeds compromise the use of higher laser powers, which could result in a damaged sample. These results are in line with what was observed by Raman spectroscopy regarding the broadening of the peaks. | ||
| Fig. 7 RT PL spectra obtained by exciting the samples with the 325 nm laser line: (a) selected samples from set#1, where the line spacing (0.075 mm) and the focused conditions were kept and the Zn#1 precursor was used; (b) samples from set#2, where only the distance between laser head and precursors was changed, keeping the remain processing parameters constant (d = 0.1 mm; v = 200 mm s−1; P ∼ 21 W) and using Zn#1 precursor; (c) samples from set#3 comparing the different ZnO precursors and prepared under the same conditions (d = 0.1 mm; v = 150 mm s−1; P ∼ 22 W, dlaser = 2 cm). A spectrum acquired for the Zn powders was also included for comparison. (d) Comparison between the RT PL spectra of the sample processed by direct-laser writing using the ZnO-based precursor and the LAFD-produced ZnO tetrapods (TPs) and nanoparticles (NPs), as well as other ZnO/carbon composites (ZnO/VACNTs and buckypapers (BPs) formed with a mixture of ZnO TPs and CNTs), produced using the LAFD-ZnO structures. The spectra are vertically displaced for clarity. | ||
Regarding the NBE recombination, the emission in all the analysed cases of set#1 is peaked at ∼378 nm (∼3.28 eV), corresponding to the free exciton energy position. The broadening of the peak suggests an overlap of other optical centres usually expected in this region, namely transitions associated to surface-related defects, as the one identified at ∼392 nm (∼3.16 eV) for the samples produced by laser assisted flow deposition (LAFD).58,72
The PL results for defocus conditions (set#2) are depicted in Fig. 7b. It is clearly seen that the focus conditions strongly influence the spectral shape and peak position of the PL emission. Once again, the broad visible bands are likely composed by several emitting centres, whose relative intensity is seen to change with the employed focus conditions. The sample produced with a distance of 2.0 cm was the one that revealed the highest overall PL intensity and, therefore, this was the value chosen to carrying out the next set of tests (set#3).
The last set of samples (set#3) was produced keeping all the processing parameters fixed and only changing the precursors for the ZnO formation (Fig. 7c). No significant changes were observed between Zn#1 and Zn#2, which correspond to the Zn-based pastes. Similarly to what was observed in the previous sets of samples, the spectra of these samples are dominated by a broad band peaked in the orange–red spectral region. Conversely, when the precursor based on ZnO was used, the NBE became the dominant emission, even though a visible broad band is also present in the same spectral range as the previous ones, but exhibiting a higher contribution from the green component(s). This spectral shape resembles the one typically obtained when the LAFD-ZnO is used to form composites (see Fig. 7d). Indeed, those ZnO precursor powders were the same as the ones used here, while, in the case of the Zn-based precursors, the same Zn metallic powder was used, thus, resulting in the appearance of similar defect-related centres in the PL spectra. Hence, the precursors nature seems to have a strong effect on the PL emission, even taking into account that, in the all the cases, the Zn/ZnO powders are embedded in an organic-based layer. The use of different precursor powders is likely to lead to the presence of distinct trace contaminants in the samples, which can give rise to different defects centres and/or distribution of the defects. In fact, it is known that the zinc sources have a strong effect on the properties of the ZnO structures.73,74 For instance, Yu et al.75 studied three zinc sources (pure metallic zinc, crystalline ZnO, and ZnCO3 powders) and concluded that different ZnO structures were formed for each precursor. Moreover, the samples also revealed distinct luminescence features. The authors attributed the observed differences to the yield and constituents/contaminants of the corresponding zinc vapour that is formed during the synthesis process. In the present case, a higher contribution from the deep level defects seems to be promoted by the use of Zn-based precursors. It is important to take into account that, as suggested by the broader peaks in the Raman spectra, the use of Zn as precursor may result in the presence of some non-stoichiometric ZnOx phases and/or lattice disorder, as well as some residual Zn, with a native oxide on its surface, which may also play a role in the resultant PL features. In fact, as seen by the PL spectrum recorded in the unprocessed Zn powders, the NBE recombination and the visible broad band could be identified even prior laser processing. In this case, a higher NBE/visible band intensity was observed when compared to the processed samples using these powders as precursor. Moreover, the visible broad band presents a higher contribution from the red component, being peaked at ∼625 nm (∼1.98 eV). These emissions are likely due to the spontaneously formed native oxide already identified by Raman spectroscopy. The differences in the relative intensity of the emission prior and after the laser processing may be related with charge transfer processes between ZnO and LIG. The decrease in the NBE relative intensity in the presence of LIG could be explained considering interfacial charge transfer between ZnO and LIG, as reported in other works concerning ZnO/rGO composites.76,77 According to Kavitha et al.,76 when the semiconductor is excited with UV light, the photogenerated electrons are likely to transfer to rGO, since the energy position of the conduction band of ZnO is higher than that of graphene, resulting in a reduction in the recombination of electrons and holes, and thus lower NBE signal. Actually, this seems to be a common feature in other ZnO/graphene-based composites.76–79 However, care must be taken when comparing literature reports on this kind of composites since significant differences may arise depending not only on the type of ZnO structures used, but also on the graphene synthesis method and its oxidation state.28,76–83 In order to highlight the differences that are frequently encountered in distinct ZnO structures, as well as when they are incorporated into carbon-based composites, Fig. 7d depicts a comparison between the PL spectra of the sample produced by direct-laser writing using the ZnO-based paste and LAFD-produced ZnO structures, as well as other ZnO/carbon composites prepared with the LAFD-ZnO structures. As can be seen, the luminescence features display distinct spectral shapes, even when comparing the samples composed only by ZnO. It is known that the PL signal is strongly depend on several factors, especially on the morphology of the samples and their growth/synthesis methods and conditions (as is evident in the here presented examples of tetrapods and nanoparticles both produced by the LAFD technique).7,58,84 Therefore, a comparison between the PL features of different ZnO/carbon-based composites is not straightforward and needs to take into account several factors. Notwithstanding, the observation of broad luminescence bands peaked in the visible region seems to be a common trend in the present ZnO/carbon composites, as well as in the ones reported in the literature. Actually, such bands are very common in ZnO structures and have been attributed to the presence of several types of defects in this semiconductor.85–88 Although extensively studied, the nature of these defects remains unclear, accentuated by the fact that different defect centres are known to originate PL emissions in the same spectral regions and even with similar spectral shapes.4,85 The most common defect-related emission is the well-known green luminescence (GL), with a maximum in the range of 2.3–2.5 eV (∼495–540 nm). This band has been associated to defects that range from intrinsic ones, as is the case of oxygen/zinc vacancies (VO/VZn), ZnO antisites, interstitial Zn atoms, transitions from Zn interstitials to Zn vacancies, to extrinsic, namely Cu ions.86,89 Nevertheless, there are also some significant evidences that surface-related defects may give rise to a broad luminescence in this spectral region.85,90 On the other hand, yellow (YL) and orange–red (RL) bands are less common than the GL and, therefore, less discussed in the literature, although often observed,85,87 as in the case of the present ZnO/LIG composites. Literature reports regarding the nature of defects emitting in the yellow and orange–red spectral regions have proposed that these emissions may arise from similar deep levels but with different initial states, for instance, between the ZnO conduction band or shallow donors and deep acceptors.85,88 It has been suggested that the orange–red emission is related with excess oxygen, particularly with interstitial oxygen (Oi) defects.85,87 Typically, intrinsic defects are the most pointed ones for the origin of this emission.85,91–95 For instance, Alvi et al.96 proposed that the RL is due to the recombination from zinc interstitial to oxygen interstitial defect levels in ZnO. On the other hand, positron annihilation spectroscopy measured in ZnO bulk samples suggests that the band peaked at 2.1 eV (∼590 nm) correlates with the zinc vacancy density.97 Previous works on ZnO structures prepared by the hydrothermal method84,98 have suggested that surface mediated processes (dependent of the synthesis/growth methods) could also contribute to luminescent features in this spectral region. Taking into account the fastness of the laser scribing process, which takes only few seconds per laser stride, such structural defects are likely to be present in here reported samples and their contribution in the observed emission should be considered. In the case of the YL, oxygen interstitial defects are the most commonly proposed origin, although the contribution of some impurities (e.g. Li doping/contamination) have also been suggested.4 In fact, Djurišić et al.87 reported that both YL and RL were reduced upon annealing in an argon atmosphere, which seems to indicate their correlation with the presence of excess oxygen. However, subsequent annealing in air led to an increase of the RL but not the YL. Indeed, the later has also been related with the presence of Zn(OH)2 at the surface of the ZnO nanostructures, leading to a weak UV and a strong broad yellow emission.85,87 This could be a possibility in the present case, since the gases that are generated during the thermal graphenisation of Kapton®, as well as the thermal degradation of the organic compounds that comprise the ZnO precursor pastes, are likely to influence the ZnO growth and its bulk and surface defect distribution. In this case, Zn(OH)2 and/or –OH groups can be formed at the surface of ZnO, due either to the polyimide (C22H10O5N2) or the organic compounds that composed the ZnO precursor, resulting in the presence of recombination processes in the yellow spectral region.85 Notwithstanding, as distinct optical centres can be overlapped in the same spectral region and frequently different types of defects are present in the same sample, these multiple recombination channels in the same spectral region give rise to broad emissions bands, which makes it rather difficult to assess the origin of the observed emissions.87,99,100
The ratio between the NBE recombination and the deep level emission is frequently used as an indication of the optical quality of the samples. Typically, higher NBE/deep level indicates a high optical quality of the produced ZnO structures. In addition, this also constitutes a sign of the good structural quality of the samples, since the observation of the free exciton recombination suggests a lower defect concentration. Therefore, the ZnO formed from the Zn-based precursors is expected to have a higher concentration of defect centres, leading to a higher intensity of the visible bands regarding the NBE emission. Moreover, even after improving the processing conditions of the samples prepared with the Zn-based pastes, the PL signal still presented a rather low intensity when compared to the ZnO/LIG composites prepared from the ZnO paste. This can be partially justified by the higher amount of ZnO particles on the surface of LIG in the latter case, as seen in the SEM image of Fig. 4 (bottom), as well as by the higher crystalline quality of the produced samples, as seem to be suggested by the Raman spectra (Fig. 6). In this case, the produced composites revealed a PL outcome fairly reproducible, homogenous along the sample and with a good intensity of the signal.
In summary, the laser processing conditions and the used precursors show a noteworthy influence in the defect concentration/distribution of the resultant composites, although some common features were found. Indeed, similar optical centres are expected to be present in the analysed samples, resulting in the broad emission bands. It is also interesting to highlight that, despite the fact that for all the tested conditions, the LIG content predominates over the ZnO one (even when the ZnO-based precursor was used), the characteristic PL emission of ZnO could be identified at RT and with the visible emission being observed with the naked eye upon ultraviolet laser excitation. Nevertheless, an intense signal is desired for further sensing applications, therefore the sample prepared with the ZnO-based precursor was considered the most adequate for device application purposes and for further electrochemical characterisation.
3.4 Electrochemical characterisation
As mentioned in the experimental section, the electrochemical characterisation was accomplished by performing EIS and CV measurements in the selected samples. In order to get some insight concerning the charge transfer process between ZnO particles and LIG, the selected ZnO/LIG sample and electrodes composed only by LIG prepared under the same laser-writing conditions were analysed. Nonetheless, it is important to bear in mind that, since there is no additional ZnO precursor layer on top of the polyimide sheet, the laser power that reaches the polymer is expected to be somewhat higher than in the case of the composite. Even so, it is fair to assume that this change has no significant influence in the resulted analysis, allowing to compare the data for both samples.The CVs in PBS of both LIG and ZnO/LIG electrodes (Fig. 8) denote a symmetric, quasi-rectangular shape, even at relatively high scan rates of 100 mV s−1, which is a good indication of their capacitive nature. Capacitive currents and the area inside the CVs are enhanced for the ZnO/LIG electrode, which clearly underline the augmented capacitance due to the ZnO particles. This correlates well with that observed in several reports in the literature.101,102
 | ||
| Fig. 8 CVs of LIG reference (black lines) and ZnO/LIG composite (red lines) at 100 mV s−1. The measuring solutions are PBS only (dashed lines) and PBS with 1 mM ferrocyanide (solid lines). | ||
When 1 mM ferrocyanide is added to the electrolyte, well-defined waves of redox activity of diffusing species appear in both CVs (solid lines in Fig. 8). In fact, the CV of the LIG sample resembles that of a reversible redox pair. The anodic-to-cathodic peak separation is only 46 mV, which is below the theoretical minimum of ∼57 mV103 for a reversible one-electron redox process based on diffusing species. This could be due to partial and weak adsorption of ferro/ferricyanide ions to LIG surface, which is known to influence the peak position, whereas for strongly adsorbed species no peak-to-peak separation is often observed. Also, there are no additional peaks, which would appear in the case of strong adsorption, arising from the different activation energies for oxidation of strongly adsorbed and diffusing ferrocyanide ions. Moreover, the fact that the adsorption is weak and reversible is confirmed by the absence of the faradaic peaks after rinsing and re-measuring the CV response in PBS buffer only. Noticeably, the peak-to-peak separation is increased to 68 mV for the ZnO/LIG sample, accompanied by lower intensity faradaic peak currents. This indicates that charge transfer between LIG and ZnO microparticles occurs and the ZnO phase participates in the faradaic process, also inducing some degree of irreversibility to it.
The EIS Nyquist and Bode plots of the same samples are shown in Fig. 9. The spectra of both sample types are well-fitted by the mixed kinetic-diffusion equivalent circuit shown in Fig. 9, where Rs is the series ohmic resistance, RCT the charge transfer resistance and CPE 1 and CPE 2 are constant phase elements modelling the (non-ideal) double layer capacitance and ferro/ferricyanide diffusion, respectively. The plateaus of the impedance Bode plot were used to initialise the values of the fittings.
 | ||
| Fig. 9 EIS Nyquist plot of LIG reference (solid squares) and ZnO/LIG composite (open circles) using an AC perturbation of 5 mV upon the open circuit potential. The measuring solution is 1 mM ferro/ferricyanide in PBS. Insets: corresponding Bode plots and the equivalent circuit used to model the cell are also shown. | ||
The LIG reference shows only a slight modulation due to RCT at higher frequencies, before diffusion takes over the impedimetric response at lower frequencies. This resistance appears in the form of a small, depressed semi-circle in the Nyquist plot (plateaus and peaks in the Bode impedance and phase plots, respectively). On the contrary, that modulation is much clearer for the ZnO/LIG. A four-fold increase in RCT from ∼30 to ∼120 Ω is inferred when ZnO particles are present, in line with the findings from the CV measurements.
4. Conclusions
In this work we have successfully synthesised ZnO decorated LIG samples via a direct-laser scribing approach, using a CO2 laser as the irradiation source and a commercial polymer (Kapton®) sheet covered with ZnO precursor for simultaneous production of ZnO and LIG. The ZnO/LIG composites produced through this method and using Zn-based precursors comprise spherical-shaped aggregates of ZnO microparticles randomly distributed at the LIG surface, while the ones produced from a ZnO-based precursor paste exhibit a more irregular shape. Moreover, even using the same amount of powder in the precursor paste, different precursors lead to different yields of the ZnO particles' production, with the ZnO-based precursor presenting the highest yield. Different combinations of processing parameters were employed and it was observed that the surface morphology was strongly dependent on the chosen conditions. For instance, it was seen that the highest laser powers and scanning speeds resulted in a higher amount of ZnO-decorated randomly oriented carbon fibres, while for lower values of both parameters (P ≤ 24 W and v ≤ 200 mm s−1) it led to a reduction in the formation of the carbon fibres and a highly porous 3D foam-like LIG structure was obtained. Raman spectroscopy revealed the crystallinity of the formed ZnO particles, whose RT luminescence is dominated by broad visible bands in the case of the samples obtained from Zn#1 and Zn#2 precursors, also exhibiting the presence of the NBE emission in the UV, even when low laser powers were employed. The NBE/visible emission ratio was altered when the ZnO-based precursor was used, evidencing a higher NBE intensity.Electrochemical characterisation revealed an increase in the capacitance of the composite samples, when compared to the LIG reference, due to the presence of the ZnO particles. Moreover, both CV and EIS measurements point to the presence of charge transfer processes between LIG and ZnO microparticles.
This study has indeed shown the possibility of producing ZnO/LIG composites that can be easily patterned in a time- and cost-effective scalable approach. The simplicity of the fabrication process and the interesting structural, optical and electrochemical properties that result from the combination of ZnO with graphene makes ZnO/LIG composites good candidates to be employed in flexible and miniaturised devices in a wide range of applications, namely in the sensing field.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from FEDER funds through the COMPETE 2020 Programme and National Funds through FCT – Portuguese Foundation for Science and Technology under the projects UID/CTM/50025/2019 and POCI-01-0145-FEDER-028755. A. F. Carvalho acknowledges the PhD grant DAEPHYS–FCT PD/BD/114063/2015, and the funding from “Programa de Estímulo à Investigação 2016” from Fundação Calouste Gulbenkian. Acknowledgements are also due to Doctor Filipe Oliveira from CICECO for the access of the optical profiler equipment.Notes and references
- S. S. Bhat, A. Qurashi and F. A. Khanday, TrAC, Trends Anal. Chem., 2017, 86, 1–13 CrossRef CAS
.
- L. Zheng, Y. Wan, P. Qi, Y. Sun, D. Zhang and L. Yu, Talanta, 2017, 167, 600–606 CrossRef CAS PubMed
- X. Dong, Y. Cao, J. Wang, M. B. Chan-Park, L. Wang, W. Huang and P. Chen, RSC Adv., 2012, 2, 4364 RSC
- U. Özgür, Y. I. Alivov, C. Liu, A. Teke, M. A. Reshchikov, S. Doğan, V. Avrutin, S.-J. Cho and H. Morkoç, J. Appl. Phys., 2005, 98, 041301 CrossRef
- Y. K. Mishra and R. Adelung, Mater. Today, 2017, 21, 631–651 CrossRef
- O. Lupan, V. Postica, J. Gröttrup, A. K. Mishra, N. H. de Leeuw, J. F. C. Carreira, J. Rodrigues, N. Ben Sedrine, M. R. Correia, T. Monteiro, V. Cretu, I. Tiginyanu, D. Smazna, Y. K. Mishra and R. Adelung, ACS Appl. Mater. Interfaces, 2017, 9, 4084–4099 CrossRef CAS PubMed
- V. Postica, J. Gröttrup, R. Adelung, O. Lupan, A. K. Mishra, N. H. de Leeuw, N. Ababii, J. F. C. Carreira, J. Rodrigues, N. Ben Sedrine, M. R. Correia, T. Monteiro, V. Sontea and Y. K. Mishra, Adv. Funct. Mater., 2017, 27, 1604676 CrossRef
- J. Rodrigues, S. M. C. Miranda, A. J. S. Fernandes, E. Nogales, L. C. Alves, E. Alves, G. Tourbot, T. Auzelle, B. Daudin, B. Méndez, T. Trindade, K. Lorenz, F. M. Costa and T. Monteiro, Phys. Status Solidi C, 2013, 10, 667–672 CrossRef CAS
- B. K. Gupta, V. Grover, G. Gupta and V. Shanker, Nanotechnology, 2010, 21, 475701 CrossRef PubMed
- I. Sameera, R. Bhatia and V. Prasad, Phys. B, 2010, 405, 1709–1714 CrossRef CAS
- T. Kavitha, A. I. Gopalan, K.-P. Lee and S.-Y. Park, Carbon, 2012, 50, 2994–3000 CrossRef CAS
- N. A. Noor Azmy, A. A. A. Bakar, N. Arsad, S. Idris, A. R. Mohmad and A. Abdul Hamid, Appl. Surf. Sci., 2017, 392, 1134–1143 CrossRef CAS
- G.-Y. Zeng, K.-S. Nian and K.-Y. Lee, Diamond Relat. Mater., 2010, 19, 1457–1460 CrossRef CAS
- J. Chen, C. Li, D. W. Zhao, W. Lei, Y. Zhang, M. T. Cole, D. P. Chu, B. P. Wang, Y. P. Cui, X. W. Sun and W. I. Milne, Electrochem. Commun., 2010, 12, 1432–1435 CrossRef CAS
- H. Moussa, E. Girot, K. Mozet, H. Alem, G. Medjahdi and R. Schneider, Appl. Catal., B, 2016, 185, 11–21 CrossRef CAS
- X. Li, Z. Wang, Y. Qiu, Q. Pan and P. Hu, J. Alloys Compd., 2015, 620, 31–37 CrossRef CAS
- J. Rodrigues, D. Mata, A. J. S. Fernandes, M. A. Neto, R. F. Silva, T. Monteiro and F. M. Costa, Acta Mater., 2012, 60, 5143–5150 CrossRef CAS
- J. Rodrigues, D. Mata, A. Pimentel, D. Nunes, R. Martins, E. Fortunato, A. J. Neves, T. Monteiro and F. M. Costa, J. Mater. Sci. Eng. B, 2015, 195, 38–44 CrossRef CAS
- O. Lupan, F. Schütt, V. Postica, D. Smazna, Y. K. Mishra and R. Adelung, Sci. Rep., 2017, 7, 14715 CrossRef PubMed
- Y. Zhao, W. Li, L. Pan, D. Zhai, Y. Wang and L. Li, Sci. Rep., 2016, 6, 32327 CrossRef CAS PubMed
- F. Liu, Y. Zhang, J. Yu, S. Wang, S. Ge and X. Song, Biosens. Bioelectron., 2014, 51, 413–420 CrossRef CAS PubMed
- S. Kumar, W. Ahlawat, R. Kumar and N. Dilbaghi, Biosens. Bioelectron., 2015, 70, 498–503 CrossRef CAS PubMed
- P. Norouzi, H. Ganjali, B. Larijani, M. R. Ganjali, F. Faridbod and H. A. Zamani, Int. J. Electrochem. Sci., 2011, 6, 5189–5199 CAS
- D. Smazna, J. Rodrigues, S. Shree, V. Postica, G. Neubüser, A. F. Martins, N. Ben Sedrine, N. K. Jena, L. Siebert, F. Schütt, O. Lupan, R. Ahuja, M. R. P. Correia, T. Monteiro, L. Kienle, Y. Yang, Y. K. Mishra and R. Adelung, Nanoscale, 2018, 10, 10050–10062 RSC
- S. Palanisamy and S. Cheemalapati, Int. J. Electrochem. Sci., 2012, 7, 8394–8407 CAS
- S. S. Low, M. T. T. Tan, H.-S. Loh, P. S. Khiew and W. S. Chiu, Anal. Chim. Acta, 2016, 903, 131–141 CrossRef CAS PubMed
- F. Liu, W. Deng, Y. Zhang, S. Ge, J. Yu and X. Song, Anal. Chim. Acta, 2014, 818, 46–53 CrossRef CAS PubMed
- E. Rokhsat and O. Akhavan, Appl. Surf. Sci., 2016, 371, 590–595 CrossRef CAS
- D. Eder, Chem. Rev., 2010, 110, 1348–1385 CrossRef CAS PubMed
- M.-M. Lu, W.-Q. Cao, H.-L. Shi, X.-Y. Fang, J. Yang, Z.-L. Hou, H.-B. Jin, W.-Z. Wang, J. Yuan and M.-S. Cao, J. Mater. Chem. A, 2014, 2, 10540 RSC
- J. Lin, Z. Peng, Y. Liu, F. Ruiz-Zepeda, R. Ye, E. L. G. Samuel, M. J. Yacaman, B. I. Yakobson and J. M. Tour, Nat. Commun., 2014, 5, 5714 CrossRef CAS PubMed
- A. Lamberti, F. Perrucci, M. Caprioli, M. Serrapede, M. Fontana, S. Bianco, S. Ferrero and E. Tresso, Nanotechnology, 2017, 28, 174002 CrossRef PubMed
- F. Clerici, M. Fontana, S. Bianco, M. Serrapede, F. Perrucci, S. Ferrero, E. Tresso and A. Lamberti, ACS Appl. Mater. Interfaces, 2016, 8, 10459–10465 CrossRef CAS PubMed
- R. Ye, Z. Peng, T. Wang, Y. Xu, J. Zhang, Y. Li, L. G. Nilewski, J. Lin and J. M. Tour, ACS Nano, 2015, 9, 9244–9251 CrossRef CAS PubMed
- S. Luo, P. T. Hoang and T. Liu, Carbon, 2016, 96, 522–531 CrossRef CAS
- Y. Li, D. X. Luong, J. Zhang, Y. R. Tarkunde, C. Kittrell, F. Sargunaraj, Y. Ji, C. J. Arnusch and J. M. Tour, Adv. Mater., 2017, 29, 1700496 CrossRef PubMed
- L. Li, J. Zhang, Z. Peng, Y. Li, C. Gao, Y. Ji, R. Ye, N. D. Kim, Q. Zhong, Y. Yang, H. Fei, G. Ruan and J. M. Tour, Adv. Mater., 2016, 28, 838–845 CrossRef CAS PubMed
- S. P. Singh, Y. Li, A. Be'er, Y. Oren, J. M. Tour and C. J. Arnusch, ACS Appl. Mater. Interfaces, 2017, 9, 18238–18247 CrossRef CAS PubMed
- M. Bayati, H. Peng, H. Deng, J. Lin and M. Fidalgo, MRS Adv., 2017, 2, 2489–2495 CrossRef CAS
- J. Zhang, M. Ren, L. Wang, Y. Li, B. I. Yakobson and J. M. Tour, Adv. Mater., 2018, 30, 1707319 CrossRef PubMed
- R. Ye, D. K. James and J. M. Tour, Acc. Chem. Res., 2018, 51, 1609–1620 CrossRef CAS PubMed
- L. X. Duy, Z. Peng, Y. Li, J. Zhang, Y. Ji and J. M. Tour, Carbon, 2018, 126, 472–479 CrossRef CAS
- R. Ye, D. K. James and J. M. Tour, Adv. Mater., 2018, 30, 1803621 Search PubMed
- D. X. Luong, A. K. Subramanian, G. A. L. Silva, J. Yoon, S. Cofer, K. Yang, P. S. Owuor, T. Wang, Z. Wang, J. Lou, P. M. Ajayan and J. M. Tour, Adv. Mater., 2018, 30, 1707416 CrossRef PubMed
- W. Song, J. Zhu, B. Gan, S. Zhao, H. Wang, C. Li and J. Wang, Small, 2018, 14, 1702249 CrossRef PubMed
- F. Wang, K. Wang, B. Zheng, X. Dong, X. Mei, J. Lv, W. Duan and W. Wang, Mater. Technol., 2018, 33, 340–356 CrossRef CAS
- Y. Chyan, R. Ye, Y. Li, S. P. Singh, C. J. Arnusch and J. M. Tour, ACS Nano, 2018, 12, 2176–2183 CrossRef CAS PubMed
- A. F. Carvalho, A. J. S. Fernandes, C. Leitão, J. Deuermeier, A. C. Marques, R. Martins, E. Fortunato and F. M. Costa, Adv. Funct. Mater., 2018, 28, 1805271 CrossRef
- J. Zhang, M. Ren, Y. Li and J. M. Tour, ACS Energy Lett., 2018, 3, 677–683 CrossRef CAS
- M. Inagaki, S. Harada, T. Sato, T. Nakajima, Y. Horino and K. Morita, Carbon, 1989, 27, 253–257 CrossRef CAS
- M. Inagaki, N. Ohta and Y. Hishiyama, Carbon, 2013, 61, 1–21 CrossRef CAS
- Z. W. Li, W. Gao and R. J. Reeves, Surf. Coat. Technol., 2005, 198, 319–323 CrossRef CAS
- A. Sekar, S. H. Kim, A. Umar and Y. B. Hahn, J. Cryst. Growth, 2005, 277, 471–478 CrossRef CAS
- H. J. Fan, R. Scholz, F. M. Kolb and M. Zacharias, Appl. Phys. Lett., 2004, 85, 4142–4144 CrossRef CAS
- Y. Qiu and S. Yang, Adv. Funct. Mater., 2007, 17, 1345–1352 CrossRef CAS
- Y. Tsuchiya and K. Sumi, J. Polym. Sci. A-1 Polym. Chem., 1969, 7, 3151–3158 CrossRef CAS
- Z. Peng and L. X. Kong, Polym. Degrad. Stab., 2007, 92, 1061–1071 CrossRef CAS
- J. Rodrigues, A. J. Fernandes, T. Monteiro and F. Costa, CrystEngComm, 2019, 21, 1071–1090 RSC
- Development of a basis for 3D surface roughness standards, Webpage from Cent. Ultra Precis. Technol. Univ. Huddersf., 1998.
- X. Díez-Betriu, S. Álvarez-García, C. Botas, P. Álvarez, J. Sánchez-Marcos, C. Prieto, R. Menéndez and A. de Andrés, J. Mater. Chem. C, 2013, 1, 6905 RSC
-
J. Rodrigues, A. J. S. Fernandes, D. Mata, T. Holz, R. G. Carvalho, R. Fath Allah, T. Ben, D. Gonzalez, R. F. Silva, A. F. da Cunha, M. R. Correia, L. C. Alves, K. Lorenz, A. J. Neves, F. M. Costa and T. Monteiro, in SPIE OPTO, ed. F. H. Teherani, D. C. Look and D. J. Rogers, International Society for Optics and Photonics, 2014, p. 89871F Search PubMed
- R. Cuscó, E. Alarcón-Lladó, J. Ibáñez, L. Artús, J. Jiménez, B. Wang and M. Callahan, Phys. Rev. B, 2007, 75, 165202 CrossRef
- L. Bergman, X.-B. Chen, J. Huso, J. L. Morrison and H. Hoeck, J. Appl. Phys., 2005, 98, 093507 CrossRef
- T. Sander, S. Eisermann, B. K. Meyer and P. J. Klar, Phys. Rev. B, 2012, 85, 165208 CrossRef
- J. M. Calleja and M. Cardona, Phys. Rev. B: Solid State, 1977, 16, 3753–3761 CrossRef CAS
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef CAS PubMed
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS
-
A. V. Naumov, in Graphene Oxide, John Wiley & Sons, Ltd, Chichester, UK, 2016, pp. 147–174 Search PubMed
- E. H. Martins Ferreira, M. V. O. Moutinho, F. Stavale, M. M. Lucchese, R. B. Capaz, C. A. Achete and A. Jorio, Phys. Rev. B, 2010, 82, 125429 CrossRef
- A. M. Dimiev S. Eigler, Graphene oxide: fundamentals and applications, John Wiley & Sons, Ltd, 2016 Search PubMed.
- L. G. Cançado, A. Jorio, E. H. M. Ferreira, F. Stavale, C. A. Achete, R. B. Capaz, M. V. O. Moutinho, A. Lombardo, T. S. Kulmala and A. C. Ferrari, Nano Lett., 2011, 11, 3190–3196 CrossRef PubMed
- J. Rodrigues, T. Holz, R. Fath Allah, D. Gonzalez, T. Ben, M. R. Correira, T. Monteiro and F. M. Costa, Sci. Rep., 2015, 5, 10783 CrossRef CAS PubMed
- P.-C. Chang, Z. Fan, D. Wang, W.-Y. Tseng, W.-A. Chiou, J. Hong and J. G. Lu, Chem. Mater., 2004, 16, 5133–5137 CrossRef CAS
- A. Kołodziejczak-Radzimska and T. Jesionowski, Materials, 2014, 7, 2833–2881 CrossRef PubMed
- W. D. Yu, X. M. Li, X. D. Gao, P. S. Qiu, W. X. Cheng and A. L. Ding, Appl. Phys. A, 2004, 79, 453–456 CrossRef CAS
- M. K. Kavitha, S. C. Pillai, P. Gopinath and H. John, J. Environ. Chem. Eng., 2015, 3, 1194–1199 CrossRef CAS
- Y.-C. Chen, K. Katsumata, Y.-H. Chiu, K. Okada, N. Matsushita and Y.-J. Hsu, Appl. Catal., A, 2015, 490, 1–9 CrossRef CAS
- S. Ameen, M. Shaheer Akhtar, H.-K. Seo and H. Shik Shin, Mater. Lett., 2013, 100, 261–265 CrossRef CAS
- G. Singh, A. Choudhary, D. Haranath, A. G. Joshi, N. Singh, S. Singh and R. Pasricha, Carbon, 2012, 50, 385–394 CrossRef CAS
- J. Liu, Z. Zhang, Y. Lv, J. Yan, J. Yun, W. Zhao, L. Kou and C. Zhai, Composites, Part B, 2016, 99, 366–372 CrossRef CAS
- K. Kim, S. Min Lee, Y. Seon Do, S. Il Ahn and K. Cheol Choi, J. Appl. Phys., 2013, 114, 074903 CrossRef
- G. Khurana, S. Sahoo, S. K. Barik and R. S. Katiyar, J. Alloys Compd., 2013, 578, 257–260 CrossRef CAS
- R. K. Biroju, N. Tilak, G. Rajender, S. Dhara and P. K. Giri, Nanotechnology, 2015, 26, 145601 CrossRef
- A. Pimentel, J. Rodrigues, P. Duarte, D. Nunes, F. M. Costa, T. Monteiro, R. Martins and E. Fortunato, J. Mater. Sci., 2015, 50, 5777–5787 CrossRef CAS
- A. B. Djurišić, Y. H. Leung, K. H. Tam, Y. F. Hsu, L. Ding, W. K. Ge, Y. C. Zhong, K. S. Wong, W. K. Chan, H. L. Tam, K. W. Cheah, W. M. Kwok and D. L. Phillips, Nanotechnology, 2007, 18, 095702 CrossRef
- A. F. Kohan, G. Ceder, D. Morgan and C. G. Van De Walle, Phys. Rev. B, 2000, 61, 19–27 CrossRef
- A. B. Djurišić, Y. H. Leung, K. H. Tam, L. Ding, W. K. Ge, H. Y. Chen and S. Gwo, Appl. Phys. Lett., 2006, 88, 103107 CrossRef
- M. A. Reshchikov, H. Morkoç, B. Nemeth, J. Nause, J. Xie, B. Hertog and A. Osinsky, Phys. B, 2007, 401–402, 358–361 CrossRef CAS
-
C. Jagadish and S. J. Pearton, Zinc oxide bulk, thin films and nanostructures: processing, properties and applications, Elsevier, 2006 Search PubMed
- D. Li, Y. H. Leung, A. B. Djurišić, Z. T. Liu, M. H. Xie, S. L. Shi, S. J. Xu and W. K. Chan, Appl. Phys. Lett., 2004, 85, 1601–1603 CrossRef CAS
- K. H. Tam, C. K. Cheung, Y. H. Leung, A. B. Djurišić, C. C. Ling, C. D. Beling, S. Fung, W. M. Kwok, W. K. Chan, D. L. Phillips, L. Ding and W. K. Ge, J. Phys. Chem. B, 2006, 110, 20865–20871 CrossRef CAS PubMed
- R. B. M. Cross, M. M. De Souza and E. M. S. Narayanan, Nanotechnology, 2005, 16, 2188–2192 CrossRef CAS PubMed
- S. A. Studenikin, N. Golego and M. Cocivera, J. Appl. Phys., 1998, 84, 2287 CrossRef CAS
- M. Gomi, N. Oohira, K. Ozaki and M. Koyano, Jpn. J. Appl. Phys., 2003, 42, 481–485 CrossRef CAS
- H. J. Fan, R. Scholz, F. M. Kolb, M. Zacharias, U. Gosele, F. Heyroth, C. Eisenschmidt, T. Hempel and J. Christen, Appl. Phys. A, 2004, 79, 1895–1900 CrossRef CAS
- N. H. Alvi, K. ul Hasan, O. Nur and M. Willander, Nanoscale Res. Lett., 2011, 6, 130 CrossRef CAS
- L. J. Brillson, Y. Dong, F. Tuomisto, B. G. Svensson, A. Y. Kuznetsov, D. Doutt, H. L. Mosbacker, G. Cantwell, J. Zhang, J. J. Song, Z.-Q. Fang and D. C. Look, Phys. Status Solidi, 2012, 9, 1566–1569 CrossRef CAS
- A. Pimentel, D. Nunes, P. Duarte, J. Rodrigues, F. M. Costa, T. Monteiro, R. Martins and E. Fortunato, J. Phys. Chem. C, 2014, 118, 14629–14639 CrossRef CAS
- H. Zeng, G. Duan, Y. Li, S. Yang, X. Xu and W. Cai, Adv. Funct. Mater., 2010, 20, 561–572 CrossRef CAS
- L. J. Brillson and Y. Lu, J. Appl. Phys., 2011, 109, 121301 CrossRef
- Y. Wang, X. Xiao, H. Xue and H. Pang, ChemistrySelect, 2018, 3, 550–565 CrossRef CAS
- M. Saranya, R. Ramachandran and F. Wang, Journal of Science: Advanced Materials and Devices, 2016, 1, 454–460 Search PubMed
-
A. J. Bard and L. R. Faulkner, Electrochemical methods: Fundamentals and Applications, John Wiley & Sons, Inc., 2nd edn, 2001 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8na00391b |
| This journal is © The Royal Society of Chemistry 2019 |