
Controllable fabrication of α-Ag2WO4 nanorod-clusters with superior simulated sunlight photocatalytic performance†
Bing-Yu
Wang
a,
Guo-Ying
Zhang
 *a,
Guan-Wei
Cui
b,
Yan-Yan
Xu
*a,
Yue
Liu
a and
Chun-Yan
Xing
a
*a,
Guan-Wei
Cui
b,
Yan-Yan
Xu
*a,
Yue
Liu
a and
Chun-Yan
Xing
a
aTianjin Key Laboratory of Structure and Performance for Functional Molecules; Key Laboratory of Inorganic-Organic Hybrid Functional Material Chemistry, Ministry of Education; College of Chemistry, Tianjin Normal University, Tianjin 300387, P. R. China. E-mail: hxxyzgy@tjnu.edu.cn; hxxyxyy@tjnu.edu.cn; Fax: +86 22 23766532; Tel: +86 22 23766532
bCollege of Chemistry, Chemical Engineering and Materials Science, Shandong Normal University, Jinan 250014, P. R. China
First published on 19th November 2018
Abstract
α-Ag2WO4 clusters comprising an assembly of nanorods were controllably fabricated via a simple ion exchange method at room temperature. The phase, morphology, microstructure, optical absorption, photoluminescence, photoelectrochemical properties and photocatalytic behavior of the products were systematically explored. α-Ag2WO4 nanorod-clusters of high purity are achieved at a medium pH of 9.8 provided by adjusting the molar ratio of AgNO3 and Na2WO4 to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4, and the sample exhibits superior photocatalytic activity for the degradation of organic pollutants. Its rate constant k is as high as 21.8 fold in comparison with that of the sample obtained with stoichiometric raw materials at a ratio of 1:0.5. The boosted photoactivity of α-Ag2WO4 clusters can be well accounted for by the broadened light harvesting and accelerated charge separation, which are proved by the red-shifted light absorption, higher photocurrent and a smaller Nyquist impedance radius. Based on the detected active species and the band edge positions, a possible migration mechanism of photoinduced e−/h+ pairs on the surface of α-Ag2WO4 clusters was proposed. This work provides some new insights into the rational design and synthesis of photocatalysts with a deep understanding of the relationship among the experimental parameters, the microstructure and properties to acquire a more desired photoactivity.
:4, and the sample exhibits superior photocatalytic activity for the degradation of organic pollutants. Its rate constant k is as high as 21.8 fold in comparison with that of the sample obtained with stoichiometric raw materials at a ratio of 1:0.5. The boosted photoactivity of α-Ag2WO4 clusters can be well accounted for by the broadened light harvesting and accelerated charge separation, which are proved by the red-shifted light absorption, higher photocurrent and a smaller Nyquist impedance radius. Based on the detected active species and the band edge positions, a possible migration mechanism of photoinduced e−/h+ pairs on the surface of α-Ag2WO4 clusters was proposed. This work provides some new insights into the rational design and synthesis of photocatalysts with a deep understanding of the relationship among the experimental parameters, the microstructure and properties to acquire a more desired photoactivity.
1. Introduction
Over the past few decades, energy crisis and the presence of toxic organic pollutants in the environment have posed serious threats to the ecosystem and human health.1–3 Compared with traditional methods such as electrochemical degradation,4 bio-degradation,5 and sorption,6 semiconductor photocatalytic technology has been widely recognized by scientific researchers to eliminate environmental pollutants due to its convenient operation, high efficiency, utilization of solar energy and environmental friendliness.7–9 The key and challenging factor of this technology is the exploration of effective photocatalytic materials with visible-light response and high degradation efficiency.Among the various semiconductors in this regard, silver-containing complex oxides including Ag2CO3,10,11 Ag2CrO4,12 AgVO3,13 and Ag3PO414,15 have aroused growing interest as efficient photocatalysts under the irradiation of sunlight. For Ag-based transition metal oxide semiconductors, the top of the valence band (VB) consists of unique hybridized Ag 4d and O 2p orbitals, which can lift the top position of the VB and narrow down the bandgap.16 The bottom of the conductance band (CB) consisting of delocalized s and/or p orbitals displays significant dispersity so that it possesses high migration efficiency of photogenerated electrons.17 As one of the Ag-based compound members, Ag2WO4 also exhibits good photocatalytic activity for the degradation of organic pollutants and antibacterial activity.18,19 In general, the Ag2WO4 crystal family covers three types of crystalline structures, i.e. an alpha (α) phase with an orthorhombic-type, a beta (β) phase with a hexagonal-type and a gamma (γ) phase with a cubic-type. Among these polymorphs, α-Ag2WO4 is thermodynamically the most stable phase20 and presents promising application as an alternative photocatalyst.
However, the light absorption range of α-Ag2WO4 is relatively narrow due to its intrinsic large band gap, and the recombination rate of photoinduced electrons and holes is usually fast in the single system. In order to enhance the photoactivity of α-Ag2WO4, many efforts have been devoted to constructing Ag2WO4-based hybrids with other compounds such as oxides Fe3O421 and WO3,22 sulfide Ag2S,23 halides AgCl24 and AgI,25 noble metal Ag,26 graphitic carbon nitride (g-C3N4),27 and Zn–Cr28 layered double hydroxides. The fabrication of composite systems usually involves multiple steps and is uneconomical, which is not beneficial for their production on a large scale. What's more, the introduction of another material would bring defects in the α-Ag2WO4 crystal, which could become the combination center of photogenerated e−/h+ pairs. By far, there are few reports of investigation of the mono-system of the α-Ag2WO4 photocatalyst without the addition of other compounds to extend the range of light-harvesting or inhibit the recombination of photoinduced carriers.29 Compared with the laser-assisted electronic reconstruction29 and surfactant-mediated morphology control,30 it should be more significant and necessary to explore the efficient α-Ag2WO4 photocatalyst from the viewpoint of solution chemistry for large scale production. It is known that in a homogeneous solution, the kinetics of nucleation and particle growth may be well adjusted by the parameters of the precursor solution such as the raw material concentration, medium acidity,31,32 and so on. The parameters would influence the submicroscopic structure of a photocatalyst and further affect its photocatalytic behavior.
In the present work, we report a facile chemical precipitation method to prepare α-Ag2WO4 nanorod-clusters by tuning the original molar ratio of AgNO3 and Na2WO4. The sample exhibits obviously superior photocatalytic activity towards the degradation of organics with a rate constant k enhanced 21.8 fold compared with that synthesized with stoichiometric raw materials. The broadened light harvesting and accelerated charge migration account well for the boosted photocatalytic activity. The active oxidation species are ascertained using scavengers and electron spin resonance techniques, based on which a possible migration mechanism of photoinduced carriers on the surface of the α-Ag2WO4 cluster is proposed. This work provides a better understanding of the relationship among the preparation, microstructure and activity of the efficient α-Ag2WO4 photocatalyst from solution chemistry.
2. Experimental section
2.1 Preparation
All the reagents were of analytical grade and used as received without further purification. The α-Ag2WO4 sample was synthesized by a facile ion-exchange method at room temperature. In a typical synthesis, 4.0 mmol of Na2WO4 was dissolved in 20.0 mL of deionized water to form a clear solution. Then 20.0 mL of AgNO3 solution (1.0 mmol) was added dropwise into the above solution under vigorous agitation, which was kept stirring for 1.5 h. Finally, the product was collected by centrifugation, washed with deionized water and absolute ethanol several times, and then dried at 60 °C in air, which was denoted as AWO-1:4 according to the molar ratio of AgNO3 and Na2WO4. By adjusting the dosage of Na2WO4, a series of α-Ag2WO4 samples were prepared, keeping the other conditions unchanged (Table S1†).
2.2 Characterization
The crystal phase and composition of the samples were characterized using the X-ray diffraction (XRD) technique on a Bruker D8-Advance diffractometer with Cu Kα radiation. X-ray photoelectron spectroscopy (XPS) measurement was performed on a Thermo Fisher X-ray photoelectron spectrometer using Al Kα radiation. The morphology and microstructures of the products were examined with a field-emission scanning electron microscope (FESEM, FEI, NOVA Nano SEM 230) and a high-resolution transmission electron microscope (HRTEM, FEI, Tecnai G2F20). Photoluminescence (PL) spectra were recorded on an F-4500 fluorescence spectrophotometer. The N2 adsorption–desorption isotherms were collected at liquid nitrogen temperature using a Micromeritics ASAP2020 surface area and porosity analyzer. The specific surface area was calculated from the multipoint adsorption data of the N2 adsorption isotherms by using Brunauer–Emmett–Teller (BET) theory. The pore size distribution was determined by the Barrett–Joyner–Halenda (BJH) method. The UV-vis diffuse reflectance spectra (DRS) were recorded on a JASCO V-550/V-570 UV-vis spectrophotometer fitted with an integrating sphere accessory. The electron spin resonance (ESR) measurements were performed on a JEOL JES-FA200 ESR spectrometer with a xenon lamp as the simulated sunlight source.2.3 Photoelectrochemical measurements
Transient photocurrent measurements and electrochemical impedance spectroscopy (EIS) were conducted using a Versa START4-200 electrochemical workstation and operated in a standard three-electrode configuration system. Typically, a Ag2WO4 suspension was obtained by dispersing 40 mg of the photocatalyst into 4.0 mL of N,N-dimethylformamide (DMF). Then the working electrode was prepared via drop-casting 0.5 mL of the Ag2WO4 suspension onto an indium tin oxide (ITO) glass (1 cm × 2 cm) and dried at 60 °C under vacuum for 1 h. A Pt wire and an Ag/AgCl electrode were used as a counter electrode and a reference electrode, respectively. The electrolyte was 0.5 M of Na2SO4 aqueous solution. A 300 W Xe lamp was employed for simulated solar irradiation.2.4 Photocatalytic degradation experiments
The photocatalytic activities of the as-prepared samples were evaluated for the degradation of RhB with a 500 W Xe lamp at room temperature. In brief, 5.0 mg of the photocatalyst was placed in a quartz tube containing 10.0 mL of 10−5 M RhB solution to form a suspension. Before illumination, the suspension was magnetically stirred in the dark for 30 min to establish an adsorption–desorption equilibrium. Then, at given time intervals, one quartz tube was taken out and centrifuged immediately to remove the photocatalysts. The UV-vis absorption spectra of the supernatant were measured using a Shimadzu 2550 UV-vis spectrophotometer. Photocatalytic degradation to methylene blue (MB), crystal violet (CV) and methyl orange (MO) followed the same procedure. Phenol was monitored using an HPLC instrument of the Agilent 1100 series equipped with an Agilent Zorbax Eclipse XDB-C18 column (150 mm × 4.6 mm, 5 μm). Methanol and water with a volume ratio of 30:70 were used as the mobile phase at a flow rate of 1 mL min−1 and the injection volume was 20 μL.
3. Results and discussion
3.1 Influence of preparation parameters
The phase, crystallinity and purity of the series of the as-obtained α-Ag2WO4 samples with different molar ratios of AgNO3 and Na2WO4 were determined by XRD. Fig. 1a shows the typical diffraction pattern of the product prepared with a stoichiometric reactant ratio of 1:0.5. Most of the diffraction peaks can be indexed to the orthorhombic phase α-Ag2WO4 with cell constants of a = 10.82 Å, b = 12.02 Å and c = 5.90 Å (JCPDS 34-0061). But it is also seen that some impurity with reflection peaks at 2θ of 26.9°, 35.6°, 44.7° and 53.0° as marked in diamond is appeared, which can be assigned to Ag2W2O7 (JCPDS 75-1506) with triclinic structures. That is, the AWO-1:0.5 sample is a mixed phase of α-Ag2WO4 and Ag2W2O7. The repeated experiment gives the same result to eliminate the doubt of any unexpected factors (Fig. S1†). It is interesting to note that when the Na2WO4 dosage is adjusted to be in excess or insufficient than the stoichiometric ratio, all of the diffraction peaks of the samples, including AWO-1:0.25, AWO-1:2, AWO-1:4 and AWO-1:5, can be readily indexed to the orthorhombic phase α-Ag2WO4. No peaks of any other phases or impurities are detected, indicating the high purity of these products. The sharp and well-defined diffraction peaks indicate a good degree of crystallinity of the samples synthesized at ambient temperature. The above analysis shows that the phase purity of α-Ag2WO4 can be influenced by the dosage of Na2WO4.
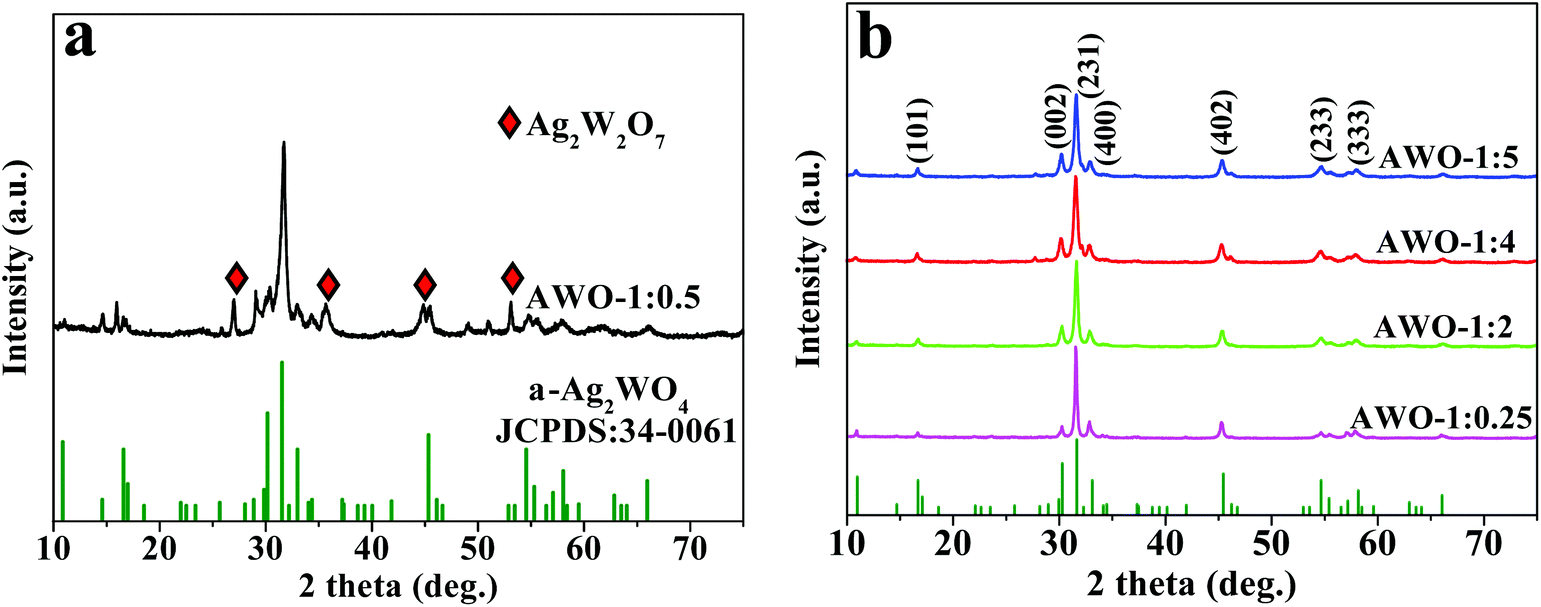 | ||
| Fig. 1 XRD patterns of the Ag2WO4 samples prepared with different molar ratios of AgNO3 and Na2WO4 (the JCPDS standard is shown at the bottom). | ||
To investigate the influence of the reactant molar ratio on the morphology of the products, SEM images of a series of α-Ag2WO4 samples were collected and the results are shown in Fig. 2. When insufficient Na2WO4 with a reactant molar ratio of 1:0.25 is added to the precursor, the sample presents long microrods with average sizes of 5 μm length and 270 nm width (Fig. 2a). The product synthesized at a stoichiometric molar ratio of 1:0.5 (Fig. 2b) consists of a mixture of thicker and longer microrods and tiny-sized nanorods. When the molar ratio is higher than the stoichiometric ratio from 1:2 to 1:5 (Fig. 2c–e), all the α-Ag2WO4 samples are made up of small-sized nanorods. Interestingly, AWO-1:4 exhibits well-defined 3D clusters (Fig. 2d) which are constructed from 1D nanorods of 200–400 nm length and 40–45 nm diameter. The SEM results demonstrate that the reactant molar ratio not only influences the phase purity, but also greatly affects the morphology and size of α-Ag2WO4. Compared with the randomly stacked nanorods in the samples of AWO-1:2 and AWO-1:5, the hierarchical AWO-1:4 may have potential application as a photocatalyst owing to its multiple reflections and porous cluster structure.
 | ||
| Fig. 2 SEM images of the samples obtained with different molar ratios of AgNO3 and Na2WO4: (a) AWO-1:0.25, (b) AWO-1:0.5, (c) AWO-1:2, (d) AWO-1:4, and (e) AWO-1:5. | ||
The influence of Na2WO4 may be attributed to its hydrolysis effect, which would affect the medium acidity. As shown in Table S2,† the pH of the precursor solution gradually increases from 7.49 to 9.83 with the increasing Na2WO4 amount. Based on a previous report,33 the initial pH value of the reactive solution plays an important role in the formation of the product microstructures due to the adjustment of the nucleation and the crystal growth rate. In order to further verify the influence of medium acidity, we changed the pH of the precursor using diluted NaOH or HNO3 solutions based on AWO-1:4. As indicated in Fig. S2,† at a lower pH less than 8.11, α-Ag2WO4 bulk or nanoparticles are obtained and the product is amorphous with weak crystallinity while a higher pH more than 9.00 gives a rod-shaped morphology with good crystallinity. In particular, the precursor without any acidity adjustment corresponding to a pH value of 9.81 gives a perfect 3D structure of nanorod-clusters. Therefore, an appropriate molar ratio of AgNO3 and Na2WO4 is essential to acquire well-crystallized α-Ag2WO4 clusters.
3.2 Composition and submicroscopic structure of AWO-1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :4
:4
The XPS technique was applied to further validate the surface compositions of the AWO-1:0.5 and AWO-1:4 samples (Fig. 3). The four sets of major peaks in the survey XPS spectrum (Fig. 3a) can be assigned to C 1s, Ag 3d, W 4f and O 1s, respectively. The unexpected C 1s peak is attributed to the adventitious hydrocarbon from the XPS instrument. Ag 3d is located at 374 eV and 368 eV with an obvious symmetry, which are indexed to be Ag 3d3/2 and Ag 3d5/2 binding energies of Ag+, respectively.34 The XPS peak of W 4f is found at 37.1 eV and 35.0 eV (Fig. 3c), which could be attributed to W6+ in WO42− or W2O72− ions. The O 1s spectrum shown in Fig. 3d can be fitted into two peaks at 530.6 eV and 531.9 eV, which correspond to the bulk O2− in the crystal lattice and –OH species, respectively. The percentage of hydroxy oxygen calculated from its peak area is 30.8% for AWO-1:4 and 14.6% for AWO-1:0.5, respectively. It is reported that the surface hydroxyl can react with photoinduced holes to form hydroxyl radicals (˙OH);35 so the higher number of hydroxyl groups in AWO-1:4 is expected to produce more oxidative ˙OH in the subsequent photocatalysis.
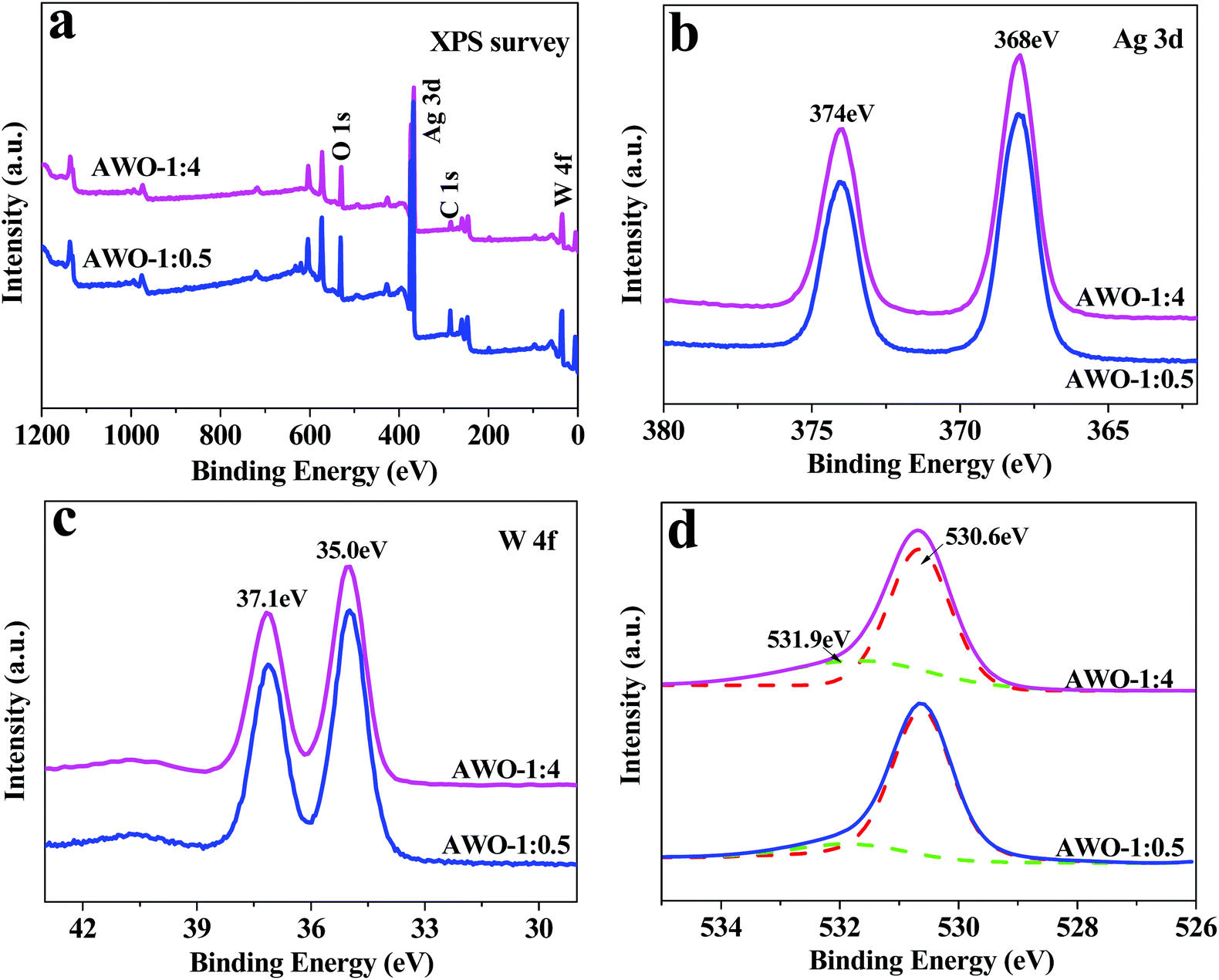 | ||
| Fig. 3 XPS spectra of the AWO-1:4 and AWO-1:0.5 samples: (a) the survey spectrum, and (b–d) the high resolution XPS spectra of Ag 3d, W 4f and O 1s, respectively. | ||
The morphology and microstructure of the typical AWO-1:4 sample were further identified using SEM and TEM. Fig. 4a presents the panoramic SEM image, which shows that the AWO-1:4 product is composed of 3D clusters assembled by tiny 1D nanorods. The enlarged SEM image shown in Fig. 4b indicates that the well-defined nanorods are 200–400 nm in length and 40–45 nm in diameter. The STEM mapping analysis shown in Fig. 4c and the EDX result shown in Fig. S3† display that the Ag, O and W elements spread over the entire area of the samples. TEM analysis (Fig. 4d) further showed the rod-shaped morphology and the size of the product, corresponding well to the SEM result. Fig. 4e shows the HRTEM image recorded from the squared area indicated in Fig. 4d. Clear lattice fringes can be observed, indicating the good crystallinity of the product. The d-spacing of 2.8 Å measured from the fringes corresponds to the (231) plane of α-Ag2WO4, whereas the d-spacing of 2.7 Å matches well with the (400) plane of α-Ag2WO4. The well-defined diffraction spots of the corresponding selected area electron diffraction (SAED) pattern can be indexed to the (400), (231) and (002) reflections, further confirming the crystal nature of the α-Ag2WO4 nanorod (Fig. 4f).
 | ||
| Fig. 4 SEM and TEM images of the typical AWO-1:4 sample: (a) panoramic SEM image, (b) magnified SEM image, (c) STEM mapping analysis, (d) TEM image of a single nanorod, (e) HRTEM image of the labelled square in d, and (f) the corresponding SAED pattern of the nanorod. | ||
3.3 BET and optical properties
Fig. 5a shows N2 adsorption–desorption isotherms and BJH pore-size distribution plots of AWO-1:4 and AWO-1:0.5, respectively. Both the samples exhibit type IV isotherms (BDDT classification) with type H3 hysteresis loops. The maximum adsorption quantity of AWO-1:4 reaches up to 16.2 m3 g−1, which is about 5.6 fold that of the AWO-1:0.5 sample (2.9 m3 g−1). The result is well interpreted from the higher BET surface area of 8.8 m2 g−1 for the AWO-1:4 sample than that of 1.7 m2 g−1 for AWO-1:0.5. Compared with the narrow pore size distribution and little pore area of AWO-1:0.5, the AWO-1:4 exhibits a broad pore size distribution from 1.7 to 4.9 nm with an obviously higher pore area. The enhanced surface area and pores of AWO-1:4 can be ascribed to the thinner width of the nanorods and their self-assembly into the 3D cluster structures, which would provide more catalytic sites and transport paths for reactant molecules.
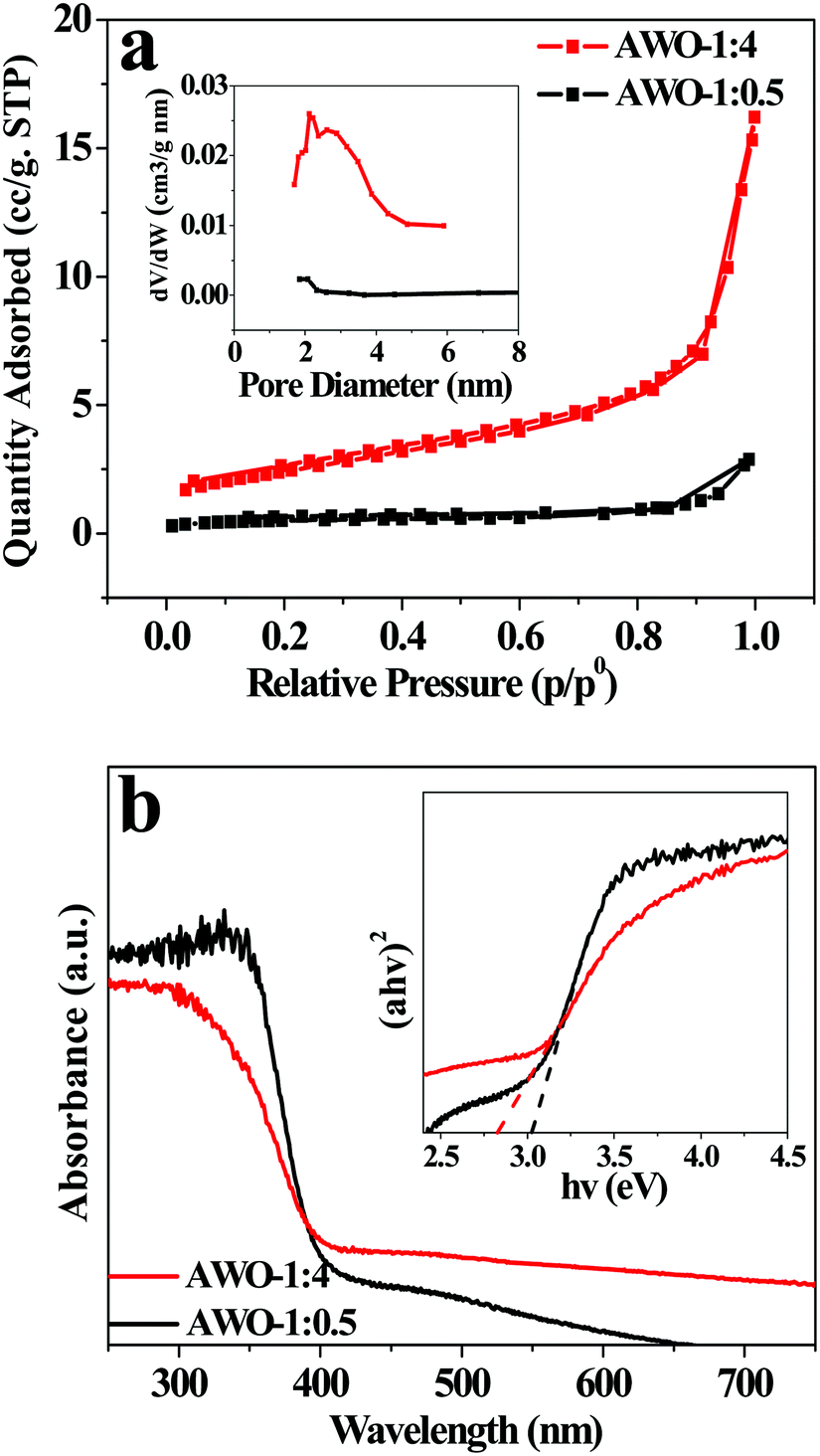 | ||
| Fig. 5 (a) N2 adsorption–desorption isotherm with a BJH pore-size distribution plot in the inset. (b) UV-vis DRS spectra and the inset shows an (αhv)2versus hv curve. | ||
The optical properties of AWO-1:0.5 and AWO-1:4 were studied using UV-vis DRS and the results are shown in Fig. 5b. The absorption curve of AWO-1:0.5 is steep with an absorption threshold at ca. 409 nm. In comparison, AWO-1:4 exhibits a red-shifted absorption edge to visible light of 438 nm. The band gap (Eg) of the two samples is determined by fitting absorption data to the direct transition equation36αhν = A(hν − Eg)1/2, where α is the absorption coefficient, hν is the photon energy, A is a proportionality constant and Eg is the optical band gap. By plotting (αhν)2 as a function of photon energy and extrapolating the linear portion of the curve to absorption equal to zero, the optical band gap values were estimated to be 3.03 eV and 2.83 eV for AWO-1:0.5 and AWO-1:4, respectively. The narrowed Eg of AWO-1:4 may arise from the greater amount of disorder in the tiny constructed α-Ag2WO4 nanorod-clusters30 which indicates more efficient utilization of sunlight.
3.4 Photocatalytic performance
The photocatalytic activities of α-Ag2WO4 samples were evaluated by the photocatalytic degradation of RhB dye under simulated sunlight irradiation (Fig. 6). As shown in Fig. 6a, the self-photolysis of the RhB molecules without any photocatalysts and dye adsorption in the dark can both be negligible. The photocatalytic degradation efficiencies over AWO-1:0.25 and AWO-1:0.5 are rather low. With the increased molar ratio of Na2WO4 to AgNO3, the photoactivity of the obtained sample is obviously enhanced. In particular, AWO-1:4 exhibits the most excellent photocatalytic activity, over which the RhB absorption at 553 nm cannot be monitored after light irradiation for 50 min. However, there is still 74.1% of RhB left after illumination for the same time over AWO-1:0.5. On further increasing the molar ratio up to AWO-1:5, the photocatalytic activity is slightly decreased. The corresponding pseudo-first-order kinetic plots and apparent rate constants k are shown in Fig. 6b. The k value over AWO-1:4 is calculated to be 0.08480 min−1, magnified 21.8 times compared with that of AWO-1:0.5 (0.00389 min−1).
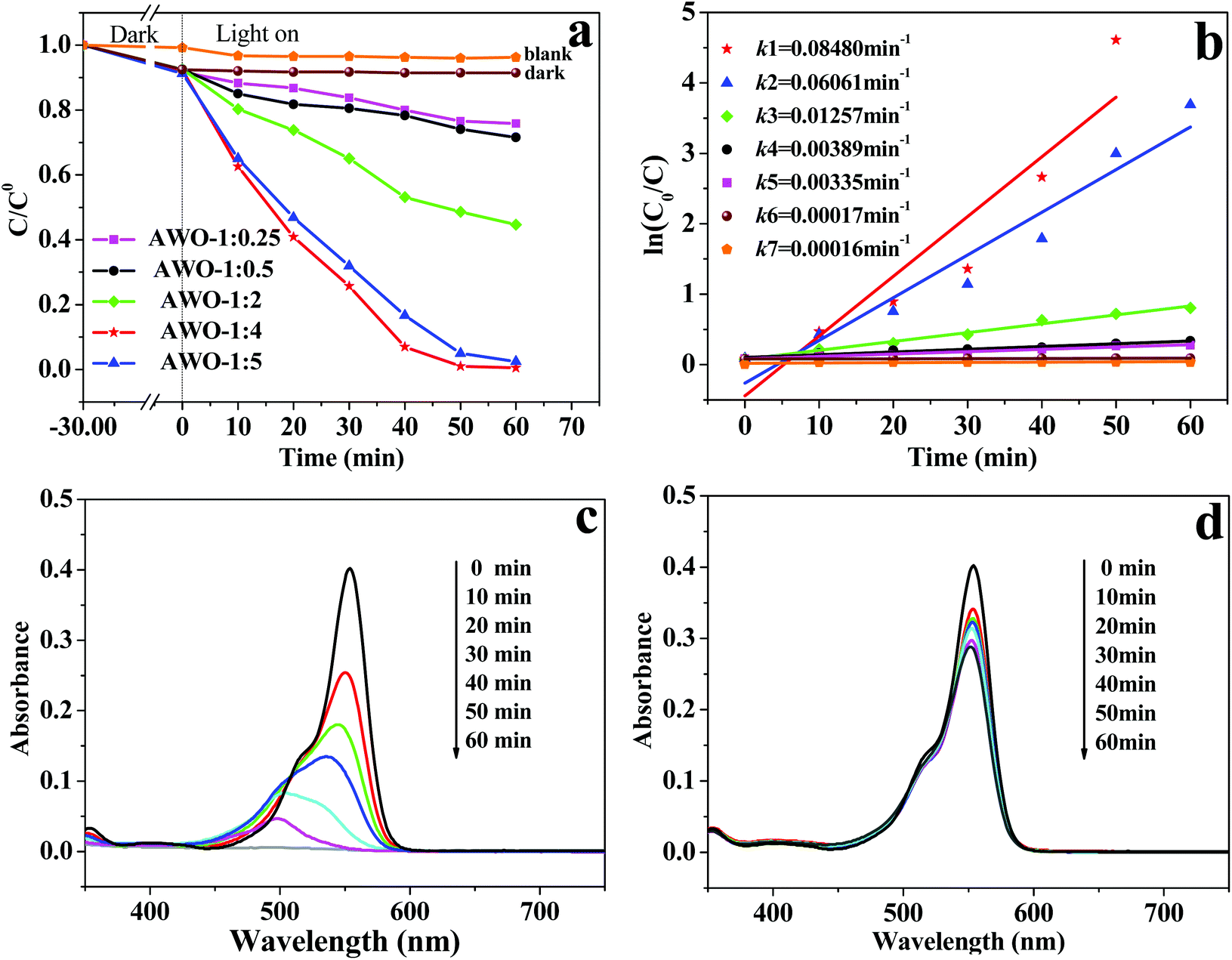 | ||
| Fig. 6 (a) Photocatalytic degradation of RhB over a series of Ag2WO4 samples and (b) corresponding pseudo first-order plots. (c and d) Temporal evolution of RhB absorption spectra over AWO-1:4 and AWO-1:0.5, respectively. | ||
More vivid temporal evolution of the RhB spectral change over AWO-1:4 and AWO-1:0.5 is shown in Fig. 6c and d, respectively. The absorption of RhB quickly decays with the exposure time over the AWO-1:4 photocatalyst and it takes 60 min to achieve complete discoloration. However, the degradation of RhB over AWO-1:0.5 is extremely slow and only 28.4% of RhB was discolorized under the same conditions. The stepwise blue-shift of the major absorption peak from 553 to 498 nm shown in Fig. 6c indicates that the photosensitized de-ethylation is also involved in the degradation process of RhB.37 It is reported that the molar absorptivity of the fully de-ethylated RhB at 498 nm by photosensitization should be maintained at about 70.0%.38 The rapid decline of the peak intensity at 498 nm to only 11.7% in the presence of AWO-1:4 indicates that it is the photocatalysis rather than photosensitization which predominates the degradation of RhB. Extended photocatalytic degradation to dyes of MB, CV and MO demonstrates similar better removal efficiencies over AWO-1:4 (Fig. S4†). The photocatalytic experiment was further applied to colorless phenol (2.4 ppm) under simulated sunlight irradiation. As shown in Fig. S5,† AWO-1:4 exhibits an improved rate constant k by a factor of 4.36 than that of AWO-1:0.5, convincingly displaying the superior structure of α-Ag2WO4 nanorod-clusters.
The results display that the photoactivity of α-Ag2WO4 is greatly influenced by the preparation parameter of the Na2WO4 dosage. The above analysis shows that the Na2WO4 amount not only affects the sample purity but also changes the morphology, size, BET and optical properties of the product due to altering of the medium acidity by hydrolysis. An appropriate molar ratio of AgNO3 to Na2WO4 is essential to achieve α-Ag2WO4 nanorod-clusters with superior photoactivity. The conclusion is further confirmed by the photocatalytic activity of the pH series samples (Fig. S6†), in which the pH of 9.81 obtained from AWO-1:4 without any adjustment with an acid or base gives the best photodegradation efficiency.
The photocatalytic stability of AWO-1:4 was investigated through repeated photocatalytic experiments. As shown in Fig. 7a, no significant loss of photocatalytic activity is observed for AWO-1:4 during the five repeated experiments. The photodegradation efficiency of RhB can still reach 97.0% after 60 min illumination in the 5th cycle. An interesting phenomenon should be noted that the photocatalytic efficiencies of the 2nd to 4th runs are even more outstanding than the 1st run. It is deduced that α-Ag2WO4 is not photostable and may be partially decomposed to Ag nanoparticles under light irradiation. The Ag nanoparticles deposited on the surface of α-Ag2WO4 can serve as electron capture centers, which can restrain the recombination of photoinduced e−/h+ pairs and prevent α-Ag2WO4 from further photocorrosion.39,40 However, with further increasing recycles, the greater deposition of Ag particles would cause a light shielding effect for α-Ag2WO4 and even inhibit the transfer of holes from the VB to the interface of the photocatalyst and solution.41 So the photodegradation efficiency of AWO-1:4 shows a slight decrease in the fifth cycle. The generation of Ag is shown in Fig. 7b, in which the high resolution XPS spectra of Ag 3d after the 2nd and 5th runs can be fitted into two groups of peaks. The peaks located at around 368.1 and 374.1 eV are related to Ag+, whereas those at 369.0 and 375.0 eV imply the existence of Ag0.42 The calculated molar ratios of Ag0 to Ag+ after the 2nd and 5 th repeated photocatalysis are 8.0% and 10.1%, respectively, which coincide with the previous deduction of more Ag deposition and also correspond to the gradual appearance of the Ag (111) peak at 38.3° (Fig. S7†).
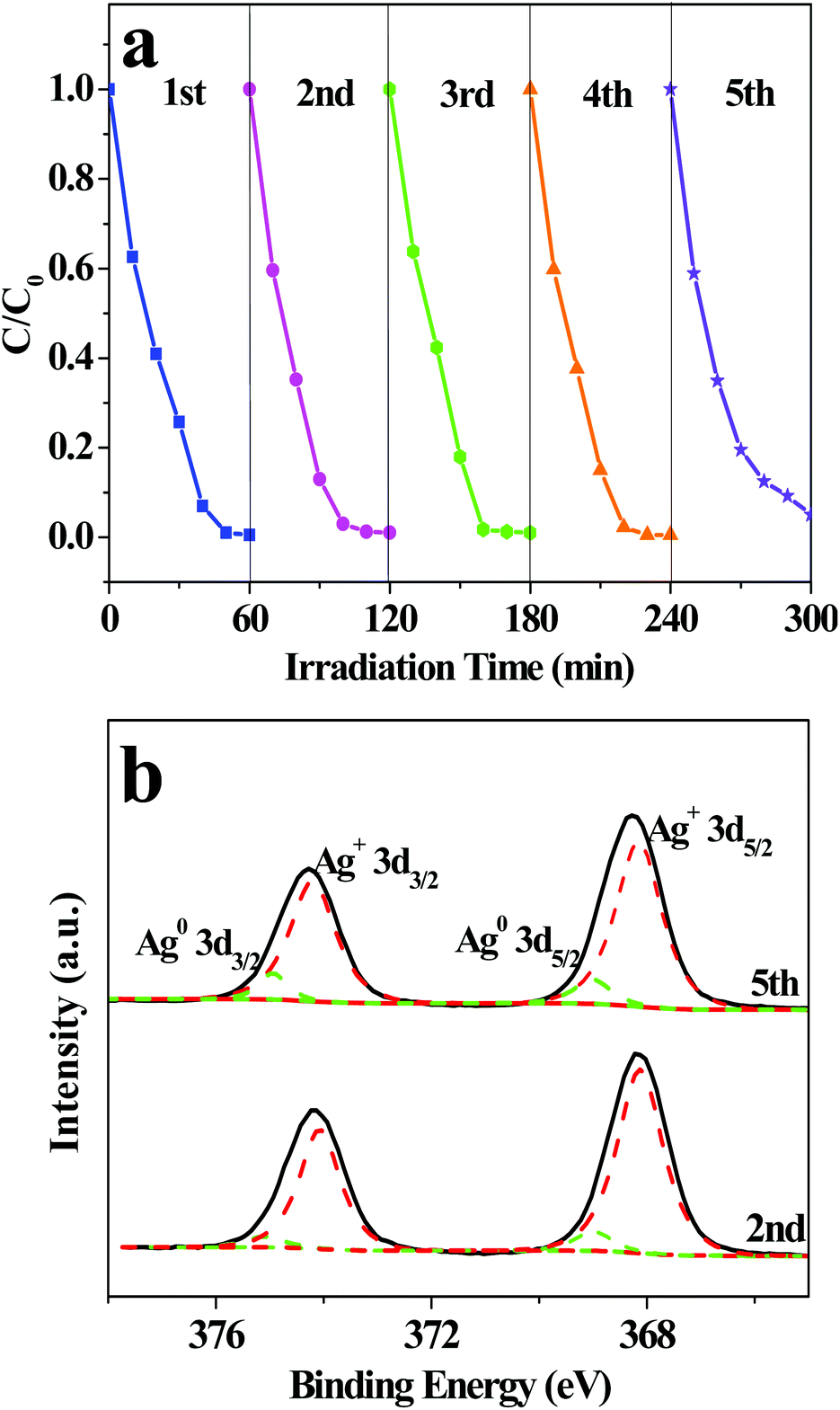 | ||
| Fig. 7 (a) Circulation runs for the degradation of RhB over AWO-1:4, and (b) XPS spectra of Ag 3d of AWO-1:4 after repeated photocatalytic experiments. | ||
3.5 PL and photoelectrochemical properties
Besides the phase, structure, morphology and light absorption, the separation and migration efficiency of photoinduced e−/h+ pairs is a crucial factor for photocatalytic activity. The PL spectra of AWO-1:4 and AWO-1:0.5 excited at 330 nm were examined to investigate the charge migration in the photocatalyst. Generally, a weaker PL intensity indicates a lower recombination of photoinduced charges, usually resulting in a higher photocatalytic activity.43 The room temperature PL spectra in Fig. 8a reveal that the emission peaks of the two samples have similar shapes with a broad spectrum. AWO-1:4 exhibits an obviously lower emission peak intensity, illustrating a restrained recombination of photogenerated carriers. Photoelectrochemical techniques of ESI and photocurrent can provide more powerful evidence to study the electron transfer behavior of a photocatalyst owing to its intrinsic band-gap structure.44 A smaller arc radius of the ESI plot and a higher photocurrent intensity indicate a faster separation and reaction rate occurring at the surface of the electrode.45Fig. 8b shows the EIS Nyquist plots of AWO-1:4 and AWO-1:0.5 without and with irradiation, respectively. The arc radii of both AWO-1:4 and AWO-1:0.5 are decreased under light irradiation, confirming the generation of photoexcited carriers. More significantly, the arc radius on the EIS Nyquist plot of AWO-1:4 is much smaller than that of AWO-1:0.5 no matter whether the sample is in the dark or under illumination. It indicates an effective separation of photogenerated carriers and a lower charge transfer resistance at the interface of the AWO-1:4 electrode and the electrolyte.46 The photocurrent responses of the photocatalysts are shown in Fig. 8c. The rapid rise and fall of the photocurrent correspond well to the switching on and off cycle of the irradiation light. AWO-1:4 exhibits a dramatically improved photocurrent intensity than AWO-1:0.5 by about 3.0 fold. The photoelectrochemical properties agree well with the PL results, further confirming the accelerated charge migration in AWO-1:4 and a resultant better photocatalytic activity.
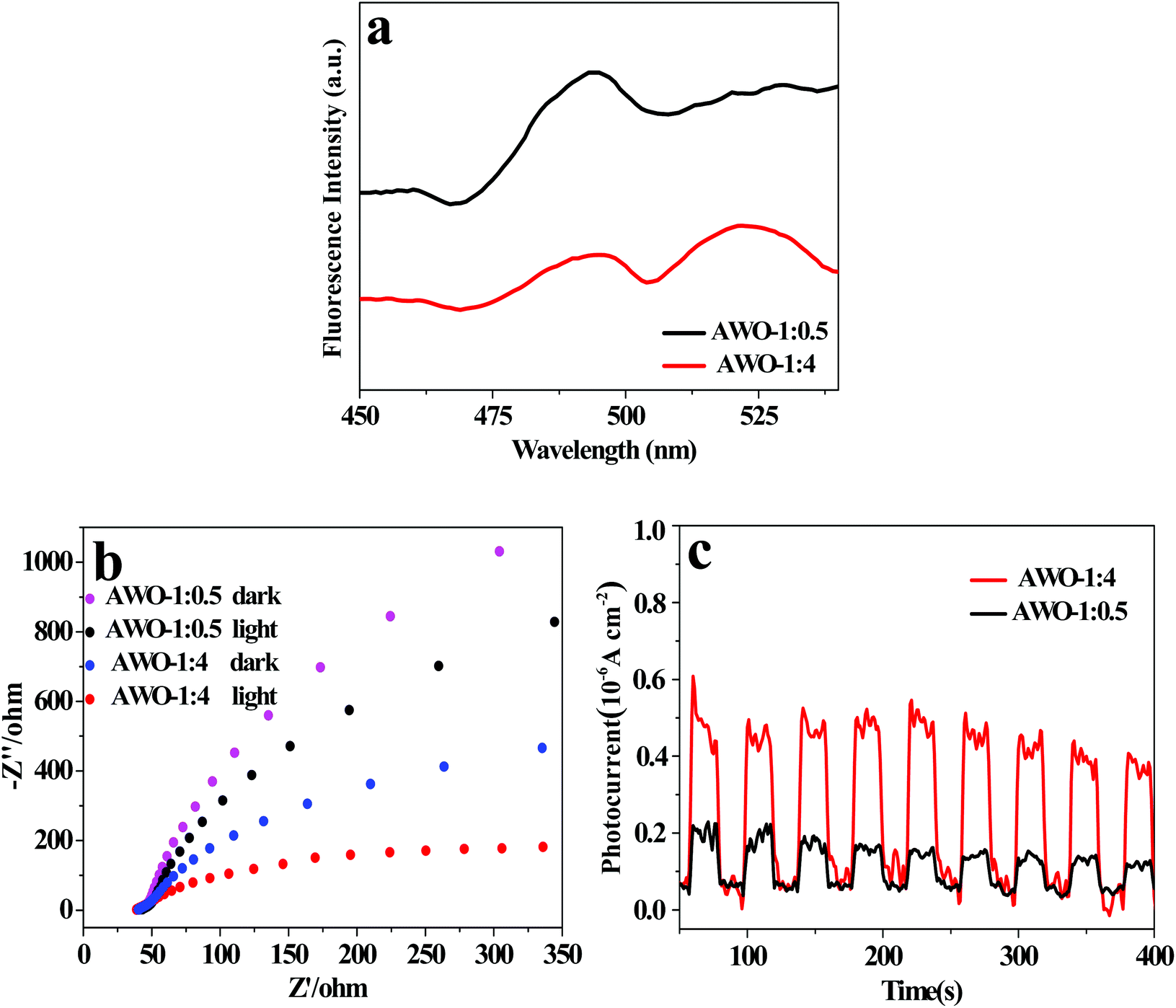 | ||
| Fig. 8 (a) PL spectra (λex = 330 nm), (b) EIS Nyquist plots and (c) photocurrent response spectra of AWO-1:4 and AWO-1:0.5, respectively. | ||
3.6 Photocatalytic mechanism
To investigate the active oxidation species in the photocatalytic degradation process of RhB over AWO-1:4, trapping experiments were performed adopting ethylene diamine tetraacetic acid (EDTA) as a hole scavenger, isopropanol (IPA) as a ˙OH scavenger and benzoquinone (BQ) as a O2˙− trapper. The results in Fig. 9a show that the introduction of BQ leads to a remarkable inhibition of the degradation efficiency from 100% to 42.0% after 50 min irradiation. The presence of EDTA also causes an obvious deactivation of the photocatalyst with the degradation efficiency reduced to 50.6%. It indicates that O2˙− and holes are important active species in the process of RhB degradation. However, the photocatalytic efficiency is inhibited by only 10.5% by the addition of IPA, demonstrating that ˙OH plays a small role in the photocatalytic degradation of RhB. The ESR spin-trap technique was further applied to detect the reactive oxidation species of O2˙− and ˙OH with 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) used as the spin trapper. As shown in Fig. 9b and c, no obvious signals can be observed for O2˙− or ˙OH radicals in the dark. However, after illumination for 3 min, the characteristic sextet peaks of DMPO-O2˙− and quartet ESR signals with a relative intensity of 1:2:2:1 for DMPO-˙OH adducts47 can be clearly discerned. The signals were both enhanced with the extension of the irradiation time to 9 min. The observation proves that O2˙− and ˙OH are both generated in the suspension of AWO-1:4 under simulated sunlight irradiation. The stronger O2˙− peaks compared with the weaker ˙OH peaks indicate that O2˙− plays a more dominant role in the photocatalytic process, which agrees well with the scavenger results. The less important role of ˙OH was further proved by the liquid PL spectra of terephthalic acid (TA) solution48 in the presence of AWO-1:4, in which only a slight increase in the PL intensity is observed with increasing illumination time.
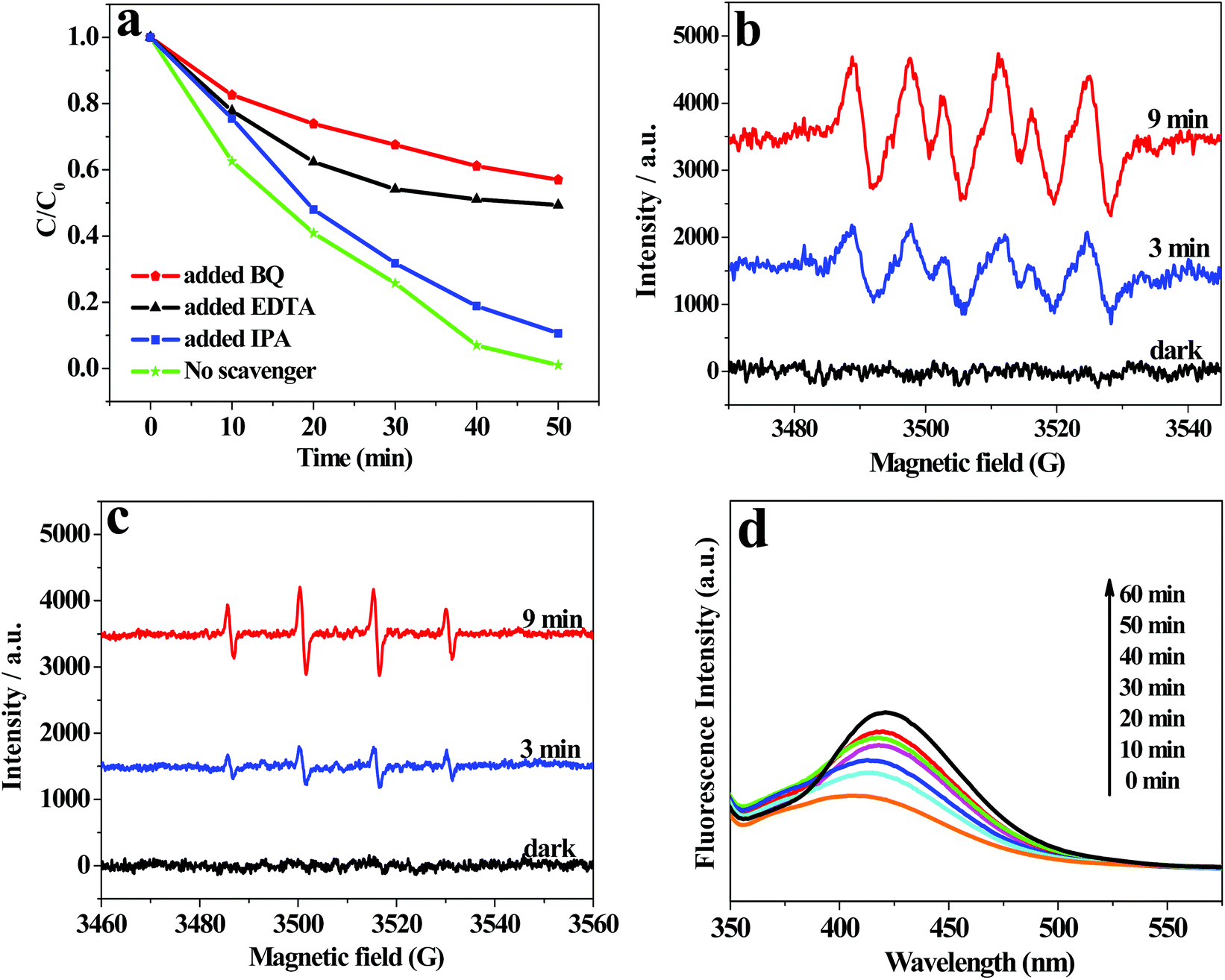 | ||
| Fig. 9 (a) Reactive species trapping experiments; ESR spectra of radical adducts trapped by DMPO with AWO-1:4 in (b) a methanol dispersion (DMPO-O2˙−) and (c) an aqueous dispersion (DMPO-˙OH), respectively; (d) PL spectral changes with the irradiation time in a 5 × 10−4 M basic solution of TA over AWO-1:4 (ex: 305 nm). | ||
The charge separation and transfer are key factors to govern the photocatalytic activity of a photocatalyst. Based on the redox potentials of the active species and the VB and CB edge positions, a mechanism of charge separation and migration at the interface of AWO-1:4 and the degradation solution can be proposed. Theoretically, the CB and VB edge positions can be determined through a widely accepted approach based on the equation 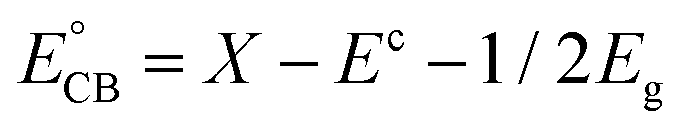 . In this equation, X is the absolute electronegativity of a semiconductor, expressed as the geometric mean of the absolute electronegativity of the constituent atoms; Ec is the energy of free electrons on the hydrogen scale (ca. 4.5 eV); and Eg is the band gap of the semiconductor. Based on the absolute electronegativity of 5.64 and the estimated Eg of 2.83 eV for AWO-1:4 in DRS spectra, the CB and VB positions of AWO-1:4 are calculated to be −0.28 V and +2.55 V, respectively. As illustrated in Fig. 10, AWO-1:4 is excited to generate CB electrons and VB holes under simulated sunlight irradiation, which would rapidly migrate to the surface of the photocatalyst and participate in the oxidation or reduction reaction. The CB potential of AWO-1:4 (−0.28 V vs. NHE) is slightly anodic than the redox potential of O2/O2˙− (−0.33 V vs. NHE); so thermodynamically the CB electrons cannot be captured by the adsorbed O2 on the AWO surface to generate O2˙− by a single-electron reduction. However, the freshly formed Ag under irradiation can act as electron traps to facilitate the separation and migration of photogenerated e−/h+ pairs due to the high Schottky barriers at the Ag/Ag2WO4 interface.49 The electrons in Ag will be further scavenged by dissolved molecular oxygen in water to form O2˙−, which is the major active species in the oxidative process. For the VB holes, the strong oxidative ability of 2.55 V enables them to directly oxidize organic pollutants, approved to be another important species by the EDTA scavenger. At the same time, the VB potential of AWO-1:4 is positive compared to the redox potential of ˙OH/OH− (2.38 V vs. NHE),50 indicating that the photoinduced holes can also oxidize the surface hydroxyl of AWO-1:4 and OH− or H2O in the degradation solution to form a few ˙OH radicals. Then with the synergic oxidation of O2˙−, h+ and ˙OH, the organic pollutants would be gradually degraded and mineralized into nontoxic inorganics.
. In this equation, X is the absolute electronegativity of a semiconductor, expressed as the geometric mean of the absolute electronegativity of the constituent atoms; Ec is the energy of free electrons on the hydrogen scale (ca. 4.5 eV); and Eg is the band gap of the semiconductor. Based on the absolute electronegativity of 5.64 and the estimated Eg of 2.83 eV for AWO-1:4 in DRS spectra, the CB and VB positions of AWO-1:4 are calculated to be −0.28 V and +2.55 V, respectively. As illustrated in Fig. 10, AWO-1:4 is excited to generate CB electrons and VB holes under simulated sunlight irradiation, which would rapidly migrate to the surface of the photocatalyst and participate in the oxidation or reduction reaction. The CB potential of AWO-1:4 (−0.28 V vs. NHE) is slightly anodic than the redox potential of O2/O2˙− (−0.33 V vs. NHE); so thermodynamically the CB electrons cannot be captured by the adsorbed O2 on the AWO surface to generate O2˙− by a single-electron reduction. However, the freshly formed Ag under irradiation can act as electron traps to facilitate the separation and migration of photogenerated e−/h+ pairs due to the high Schottky barriers at the Ag/Ag2WO4 interface.49 The electrons in Ag will be further scavenged by dissolved molecular oxygen in water to form O2˙−, which is the major active species in the oxidative process. For the VB holes, the strong oxidative ability of 2.55 V enables them to directly oxidize organic pollutants, approved to be another important species by the EDTA scavenger. At the same time, the VB potential of AWO-1:4 is positive compared to the redox potential of ˙OH/OH− (2.38 V vs. NHE),50 indicating that the photoinduced holes can also oxidize the surface hydroxyl of AWO-1:4 and OH− or H2O in the degradation solution to form a few ˙OH radicals. Then with the synergic oxidation of O2˙−, h+ and ˙OH, the organic pollutants would be gradually degraded and mineralized into nontoxic inorganics.
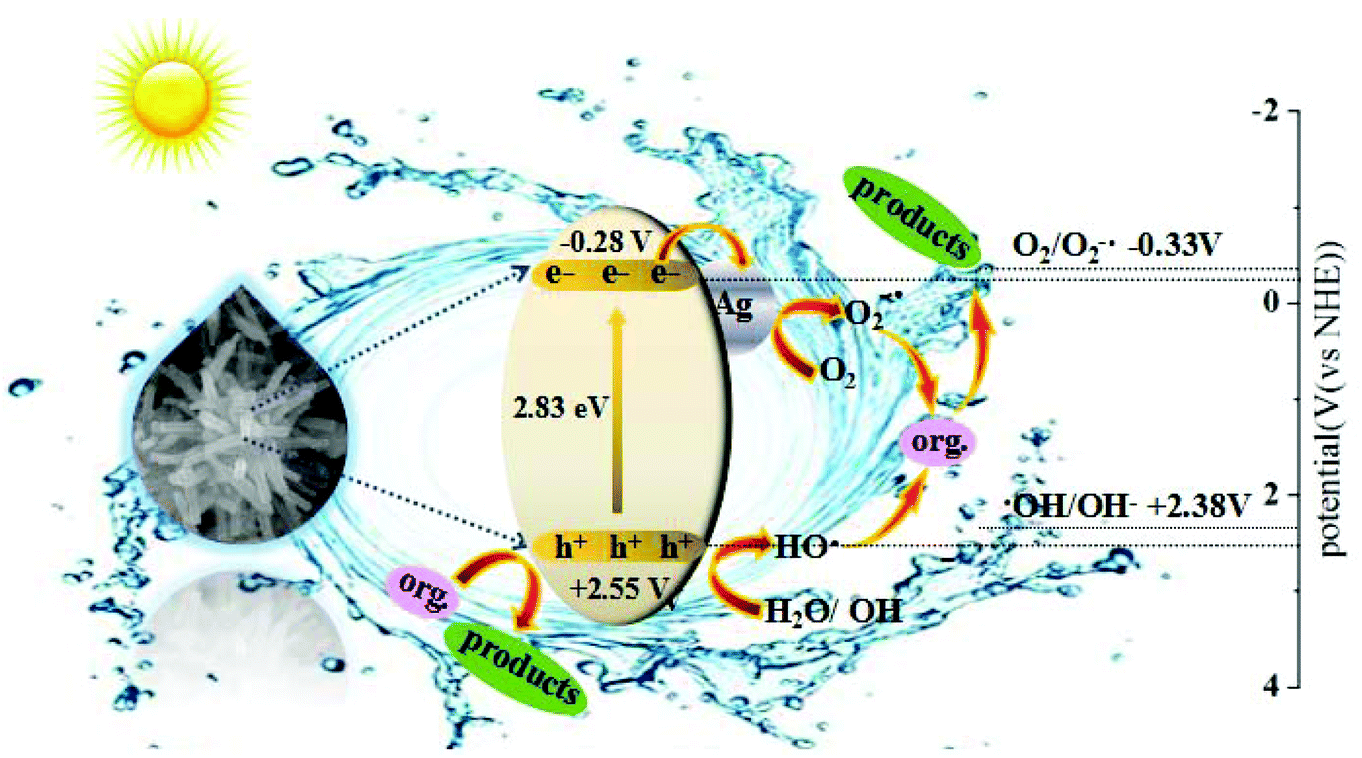 | ||
| Fig. 10 Schematic illustration of the charge separation and transfer at the interface of AWO-1:4 and degradation solution. | ||
4. Conclusions
α-Ag2WO4 nanorod-clusters were facilely constructed by a solution precipitation method at room temperature. The reactant molar ratio of AgNO3 and Na2WO4 is a key parameter to influence the sample purity, morphology, size and optical properties. At a medium pH of about 9.8 provided by a reactant molar ratio of 1:4, highly pure α-Ag2WO4 clusters constructed from nanorods are obtained. Compared with the sample prepared with a stoichiometric molar ratio of 1:0.5, the α-Ag2WO4 clusters exhibit an obviously enhanced photoactivity for the degradation of organic pollutants with a 21.8-fold improvement of the rate constant k. The boosted photocatalytic activity is attributed to the red-shifted light absorption, the increased BET surface area and especially the accelerated charge migration. Based on the active species and band edge levels, a possible migration mechanism of photoexcited e−/h+ pairs at the interface of AWO-1:4 and the degradation solution was proposed.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The project was supported by the National Natural Science Foundation of China (No. 21303122).References
- X. F. Hu, J. C. Sun, Z. F. Li, Q. Zhao, C. C. Chen and J. Chen, Angew. Chem., Int. Ed., 2016, 55, 6482–6486 CrossRef CAS PubMed.
- C. C. Wang, L. B. Wang, F. J. Li, F. Y. Cheng and J. Chen, Adv. Mater., 2017, 29, 1702212 CrossRef PubMed.
- H. H. Chen, C. E. Nanayakkara and V. H. Grassian, Chem. Rev., 2012, 112, 5919–5948 CrossRef CAS PubMed.
- M. Rahimi, A. P. Straub, F. Zhang, X. Zhu, M. Elimelech and C. A. Gorski, Energy Environ. Sci., 2017, 11, 276–285 RSC.
- C. Paredes, M. P. Bernal, J. Cegarra and A. Roig, Bioresour. Technol., 2002, 85, 1–8 CrossRef CAS PubMed.
- D. Bradshaw, T. J. Prior, E. J. Cussen, B. C. John and M. J. Rosseinsky, J. Am. Chem. Soc., 2004, 126, 6106–6114 CrossRef CAS PubMed.
- A. Kubacka, M. Fernández-García and G. Colón, Chem. Rev., 2012, 112, 1555–1614 CrossRef CAS PubMed.
- L. Cui, J. Song, A. F. Mcguire, S. Kang, X. Fang and J. Wang, ACS Nano, 2018, 12, 5551–5558 CrossRef CAS PubMed.
- J. Byun, W. Huang, D. Wang, R. Li and K. A. I. Zhang, Angew. Chem., Int. Ed., 2018, 57, 2967–2971 CrossRef CAS PubMed.
- G. P. Dai, J. G. Yu and G. Liu, J. Phys. Chem. C, 2012, 116, 15519–15524 CrossRef CAS.
- C. Dong, K. L. Wu, X. W. Wei, X. Z. Li, L. Liu, T. H. Ding, J. Wang and Y. Ye, CrystEngComm, 2014, 16, 730–736 RSC.
- D. Xu, B. Cheng, S. Cao and J. Yu, Appl. Catal., B, 2015, 164, 380–388 CrossRef CAS.
- D. P. Sahoo, S. Patnaik, D. Rath and K. M. Parida, Inorg. Chem. Front., 2018, 5, 879–896 RSC.
- Y. P. Bi, S. X. Ouyang, N. Umezawa, J. Y. Cao and J. H. Ye, J. Am. Chem. Soc., 2011, 133, 6490–6492 CrossRef CAS PubMed.
- G. Y. Zhang, X. M. Wei, X. Bai, C. M. Liu, B. Y. Wang and J. W. Liu, Inorg. Chem. Front., 2018, 5, 26–32 Search PubMed.
- Q. J. Xiang, J. G. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575–6578 CrossRef CAS PubMed.
- T. Kako, N. Kikugawa and J. Ye, Catal. Today, 2008, 131, 197–202 CrossRef CAS.
- R. A. Roca, J. C. Sczancoski, I. C. Nogueira, M. T. Fabbro, H. C. Alves and L. Gracia, Catal. Sci. Technol., 2015, 5, 4091–4107 RSC.
- R. X. Zhang, H. Y. Cui, X. F. Yang, H. Tang, H. Liu and Y. Li, Micro Nano Lett., 2012, 7, 1285–1288 CrossRef CAS.
- L. Cavalcante, M. Almeida, W. Avansi, R. Tranquilin, E. Longo, N. Batista, V. Mastelaro and S. Li, Inorg. Chem., 2012, 51, 10675–10687 CrossRef CAS PubMed.
- S. Rajamohan, V. Kumaravel, R. Muthuramalingam, S. Ayyadurai, A. Abdelwahab and B. S. Kwak, New J. Chem., 2017, 41, 11722–11730 RSC.
- F. Qiu, X. Zhu, Q. Guo, Y. Dai, J. Xu and T. Zhang, Powder Technol., 2017, 317, 287–292 CrossRef CAS.
- X. F. Wang, S. Zhan, Y. Wang, P. Wang, H. G. Yu, J. G. Yu and C. Z. Hu, J. Colloid Interface Sci., 2014, 422, 30–37 CrossRef CAS PubMed.
- H. Xu, Y. L. Cao, J. Xie, J. D. Hu, Y. Z. Li and D. Z. Jia, Mater. Res. Bull., 2018, 102, 342–352 CrossRef CAS.
- S. J. Li, S. W. Hu, W. Jiang, Y. P. Liu, Y. Liu, Y. T. Zhou, L. Y. Mo and J. S. Liu, Beilstein J. Nanotechnol., 2018, 9, 1308–1316 CrossRef CAS PubMed.
- V. M. Longo, C. C. D. Foggi, M. M. Ferrer, A. F. Gouveia, R. S. André, W. Avansi, C. E. Vergani, A. L. Machado, J. André, L. S. Cavalcante, A. C. Hernandes and E. Longo, J. Phys. Chem. A, 2014, 118, 5769–5778 CrossRef CAS PubMed.
- Y. Li, R. Jin, X. Fang, Y. Yang, M. Yang and X. Liu, J. Hazard. Mater., 2016, 313, 219–228 CrossRef CAS PubMed.
- J. Y. Zhu, H. Fan, J. C. Sun and S. Y. Ai, Sep. Purif. Technol., 2013, 120, 134–140 CrossRef CAS.
- Z. Y. Lin, J. L. Li, Z. Q. Zheng, J. H. Yan, P. Liu, C. X. Wang and G. W. Yang, ACS Nano, 2015, 9, 7256–7265 CrossRef CAS PubMed.
- N. G. Macedo, A. F. Gouveia, R. A. Roca, M. Assis, L. Gracia and J. Andrés, J. Phys. Chem. C, 2018, 122, 8667–8679 CrossRef CAS.
- A. Zhang, J. Zhang, N. Cui, X. Tie, Y. An and L. Li, J. Mol. Catal. A: Chem., 2009, 304, 28–32 CrossRef CAS.
- L. Dong, S. Guo, S. Zhu, D. Xu, L. Zhang and M. Huo, Catal. Commun., 2011, 16, 250–254 CrossRef CAS.
- X. Gao, Z. Wang, F. Fu and W. Li, Mater. Sci. Semicond. Process., 2015, 35, 197–206 CrossRef CAS.
- X. F. Wang, S. F. Li, H. G. Yu, J. G. Yu and S. W. Liu, Chem. – Eur. J., 2011, 17, 7777–7780 CrossRef CAS PubMed.
- J. J. Wang, X. N. Liu, R. H. Li, P. S. Qiao, L. P. Xiao and J. Fan, Catal. Commun., 2012, 19, 96–99 CrossRef CAS.
- A. Zeb, Z. Sun, T. Khan, M. A. Asghar, Z. Wu and L. Li, Inorg. Chem. Front., 2017, 1, 1–7 Search PubMed.
- F. Chen, J. Zhao and H. Hidaka, Int. J. Photoenergy, 2003, 5, 209–217 CrossRef CAS.
- X. F. Zhou, C. Hu, X. X. Hu, T. W. Peng and J. H. Qu, J. Phys. Chem. C, 2010, 114, 2746–2750 CrossRef CAS.
- B. Cheng, Y. Le and J. G. Yu, J. Hazard. Mater., 2010, 177, 971–977 CrossRef CAS PubMed.
- D. Xu, B. Cheng, J. Zhang, W. Wang, J. Yu and W. Ho, J. Mater. Chem. A, 2015, 3, 20153–20166 RSC.
- W. Wang, B. Cheng, J. Yu, G. Liu and W. Fan, Chem. – Asian J., 2012, 7, 1902–1908 CrossRef CAS PubMed.
- W. Teng, X. Li, Q. Zhao and G. Chen, J. Mater. Chem. A, 2013, 1, 9060–9068 RSC.
- W. J. Yue, S. K. Han, R. X. Peng, W. Shen, H. W. Geng, F. Wu, S. W. Tao and M. T. Wang, J. Mater. Chem., 2010, 20, 7570–7578 RSC.
- B. Zheng, X. Wang, C. Liu, K. Tan, Z. Xie and L. Zheng, J. Mater. Chem. A, 2013, 1, 12635–12640 RSC.
- F. Lei, Y. Sun, K. Liu, S. Gao, L. Liang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2014, 136, 6826–6829 CrossRef CAS PubMed.
- Y. Li, Y. Li, S. Ma, P. Wang, Q. Hou, J. Han and S. Zhan, J. Hazard. Mater., 2017, 338, 33–46 CrossRef CAS PubMed.
- L. Jing, Y. Xu, M. Zhang, M. Xie, H. Xu and M. He, Inorg. Chem. Front., 2018, 5, 63–72 RSC.
- Q. Xiao, Z. C. Si, J. Zhang, C. Xiao and X. K. Tan, J. Hazard. Mater., 2008, 150, 62–67 CrossRef CAS PubMed.
- Q. S. Wu, Y. Cui, L. M. Yang, G. Y. Zhang and D. Z. Gao, Sep. Purif. Technol., 2015, 142, 168–175 CrossRef CAS.
- Y. Y. Liu, Z. Y. Wang, B. B. Huang, X. Y. Zhang, X. Y. Qin and Y. Dai, J. Colloid Interface Sci., 2010, 348, 211–215 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qi01025k |
| This journal is © the Partner Organisations 2019 |