
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePinostrobin inhibits proliferation and induces apoptosis in cancer stem-like cells through a reactive oxygen species-dependent mechanism†
Alka Jadaun‡
,
Sapna Sharma‡,
Radha Verma and
Aparna Dixit *
*
Gene Regulation Laboratory, School of Biotechnology, Jawaharlal Nehru University, New Delhi-110067, India. E-mail: adixit7@gmail.com; adix2100@mail.jnu.ac.in
First published on 16th April 2019
Abstract
Current treatments and targeted therapies for malignancies are limited due to their severe toxicity and the development of resistance against such treatments, which leads to relapse. Past evidence has indicated that a number of plant-derived dietary agents possess biological activity against highly tumorigenic and resistant cell populations associated with cancer relapse. These subpopulations, termed cancer stem-like cells (CSCs), have been targeted with plant-derived dietary flavonoids. The present study was undertaken to assess the anti-proliferative potential of pinostrobin, a dietary flavonoid, against CSCs. Sphere-forming cells were developed from HeLa cell lines using specific culture conditions. The existence of a CSC population was confirmed by the morphological examination and analysis of surface markers using confocal microscopy and flow cytometry. The effect of pinostrobin on the cell viability of the CSC population, evaluated through MTT reduction assays and the expression levels of surface markers (CD44+ and CD24+), was studied through various biological assays. HeLa-derived CSCs showed higher CD44+ and lower CD24+ expression. Pinostrobin inhibited the self-renewal capacity and sphere formation efficiency of CSCs in a dose-dependent manner. Increased ROS production, and decreased mitochondrial membrane potential and CD44+ expression indicated that pinostrobin promoted ROS-mediated apoptosis in CSCs. These results thus demonstrate the therapeutic potential and effectiveness of pinostrobin in the chemoprevention and relapse of cancer by targeting the CSC population. Thus, pinostrobin, in combination with currently available chemo and radiation therapies, could possibly be used as a safe strategy to alleviate adverse treatment effects, together with enhancing the efficacy.
Introduction
In India, the prevalence of cervical cancer is the highest among all the cancer cases in females. It is the most common cancer among 15–44 aged females.1 Compared to the urban population, the rural population is at a higher risk of developing cervical cancer. Despite great advances in targeted therapies and major steps toward customised therapy for cancer patients, there has yet not been a significant increase in survival rate for many individuals. Target cancer therapies show limited efficacy, due to the specific criteria of individuals. It has been observed in many patient cohorts that drug efficacy imbalance often arises due to the development of drug resistance through acquiring genetic and epigenetic alterations.2 Survival rates can be increased, if the disease is diagnosed at an early stage, with improved treatment strategies.Despite the availability of several therapies, a number of patients develop drug resistance and face tumour relapse, significantly contributing to a poor survival rate.3 It is now well established that cancer relapse and progression is influenced by a cell population within the cancerous tissue, termed as cancer stem-like cells or cancer stem cells (CSCs).4–6 These CSCs play a major role in determining tumor aggressiveness, heterogeneity, drug resistance and recurrence of the disease.5,7,8 Generally, conventional and targeted therapy target only mature, proliferating tumor cells without affecting the CSC population.9,10 Targeting the CSC population would possibly help in controlling disease progression and thus enhance the survival rate of cancer patients. If treatment results in the elimination of CSCs, it helps to reduce tumor growth and prevent relapse. Since CSCs adapt to different survival mechanisms, it is important to identify a molecule that could simultaneously modulate a number of cellular pathways/mechanisms.
Natural products such as curcumin, berberine and resveratrol have been reported to interact with a number of cellular molecules, controlling the proliferation of the CSC population.11 These natural products play a crucial role in targeting the CSC population and its surface markers, and modulate critical signalling pathways that are essential for its maintenance and survival.12,13 Pinostrobin (PN), extracted from several medicinal plants, belongs to the flavanone family of phytochemicals. The anti-leukemic and anti-cancer activities of PN against different types of carcinomas have been reported.13–15 We have previously reported the cytotoxic activity of PN in cervical cancer cells, without any cytotoxic effects on normal HEK293 cells, and demonstrated that it induced ROS-mediated cell death.16 In addition, we also demonstrated the direct interaction of PN with topoisomerase I, an enzyme involved in DNA replication, and inhibited its activity.17 PN from different sources has been reported to regulate a variety of cellular systems that are necessary for normal cellular function,18 by possibly affecting multiple cellular and molecular processes. Since it has shown anti-proliferative activity in cancer cells, it is hypothesized that it may have the potential to inhibit the proliferation of CSCs and induce cell death. In this study, we investigated if PN is indeed able to target a HeLa-CSC population and affect their proliferation. We further investigated the possible mechanism underlying PN-induced cell death in HeLa-CSCs.
Materials and methods
Reagents
Analytical grade chemicals were used in the present study. Pinostrobin (PN), doxorubicin (DX), N-acetyl cysteine (NAC) and fluorescent dyes used in the study such as 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine iodide (JC-1), 2′,7′-dichlorofluorescin diacetate (DCFH-DA), hydroethidine (HE), Annexin V-FITC, propidium iodide (PI), verapamil, Hoechst 33342 dye and 10-N-nonyl acridine orange (NAO) were purchased from Sigma-Aldrich Chemical Co., USA. Epidermal growth factor and basic fibroblast growth factor (bFGF) were obtained from Invitrogen, USA. All buffers/solutions used in the study were prepared in Milli-Q water. PN stock solution (20 mM) was prepared in triple solvent (TS, dimethyl formamide![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetonitrile:dimethyl sulfoxide, 1:1:1).19 Triple solvent was used as a vehicle control (VC). We included DX as a reference control in all of the experiments as it has been shown to inhibit the cell proliferation of CSCs derived from human colon carcinoma HT29 cells20 and ovarian carcinoma SKOV3 cells.21
:acetonitrile:dimethyl sulfoxide, 1:1:1).19 Triple solvent was used as a vehicle control (VC). We included DX as a reference control in all of the experiments as it has been shown to inhibit the cell proliferation of CSCs derived from human colon carcinoma HT29 cells20 and ovarian carcinoma SKOV3 cells.21
Cell culture
HeLa cells (human cervical cancer cell line) were obtained from the National Centre for Cell Science (NCCS), Pune (India), cultured and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% foetal bovine serum (FBS), 100 U ml−1 of penicillin and 100 μg ml−1 of streptomycin (Biological Industries, Israel) at 37 °C in a 5% CO2 incubator with a 95% air atmosphere, essentially as described previously.16Identification of the side population
Sphere forming cells (SFCs) were also recognised as the side population (SP).19 For identification and validation of the SP in the parent HeLa cell population, Hoechst 33342 staining in the presence or absence of verapamil, a calcium channel antagonist, was performed. The adherent parent HeLa cells were trypsinised (0.25%) and washed twice with 1× PBS. The cells (1 × 104/well) were incubated in pre-warmed DMEM (containing 2% fetal bovine serum (FBS), 1 mM HEPES, and 5 μg ml−1 Hoechst 33342) for 90 min at 37 °C in the dark on a rocking platform shaker with slow agitation. The CSCs (1 × 104 cells per well) were generated as described above and cultured in SFM. To obtain appropriate sized spheres, 100 μl of fresh SFM was added after every 48 h for 8 days. The cells were washed twice with pre-cooled 1× PBS and centrifuged at 2000 rpm at 4 °C. The cells were then incubated with verapamil (50 μM) at 37 °C for 30 min after the addition of Hoechst 33342 stain, to check if the fluorescent efflux from the SP cells present in the HeLa population was blocked by verapamil. The cells were counterstained with propidium iodide (PI) (2 mg ml−1) in the dark. Acquisition was performed using a FACS Calibur™ (Beckon Dickinson Immuno cytometry system, San Jose, USA) operated in dual wavelength mode, by measuring the fluorescence of the Hoechst 33342 dye and PI at 460 and 580 nm, respectively. The results were analysed by FCS Express.v5 Flow Cytometry data analysis software. A small number of cells that were capable to efflux Hoechst stain with low fluorescing were identified as the SP.In vitro culture and passage of sphere forming cells
SFCs were generated from HeLa cells using the method developed by López et al.22 HeLa cells (1 × 106 cells per ml) were cultured in 2 ml of serum-free media (SFM) supplemented with epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF), (20 ng ml−1 each) and bovine serum albumin (BSA) (0.4%) in a 35 mm culture dish at 37 °C in a 5% CO2 atmosphere. The culture medium was replenished every 48 h after SFC generation for the next 8 days. After generation of 70–80% spheres, the SFCs were collected by gentle centrifugation (2500 rpm, 5 min). Each sphere was separated using 500 μl of EDTA (1.0 mM) and collected using a Pasteur pipette followed by gentle shaking at 2500 rpm for 5 min to achieve single-cell suspension. These SFCs were recognized as cancer stem-like cells (CSCs).23Morphological assessment of CSCs
The growth of the cells was monitored on days 1, 3, 6, 9, and 12. On day 12, the spheres were detached from the surface of the flask and floated in suspension as clumps/aggregates. The CSCs bodies thus collected, were visualized under a bright field microscope (Leica, DMIL, Germany), as described by Gu et al.19 Parent HeLa cells (1 × 104 cells per well), included as a control, were cultured in complete DMEM at 37 °C in a 5% CO2 incubator.Identification of the CSC population
:100) (Invitrogen, USA) for 30 min at RT. The cells were then washed once with 1× PBS and the morphology of the stained CSCs and parent HeLa cells was examined under an Olympus Fluoview FV1000 confocal laser-scanning microscope at 40× magnification using a 1.4NA oil objective lens at the Advanced Instrument Research Facility (AIRF), JNU, New Delhi (India). The size of the acquired images was 512 × 512 pixels (0.414 μm per pixel).Quantification of the CD24+/CD44+ expression levels
Changes in the expression levels of CD24+/CD44+ in the CSC population and HeLa cells induced by different treatments, i.e. PN, DX, vehicle and in the untreated CSCs population, were quantified using a FACS Calibur™ at 488 nm (ex). The green and red fluorescence intensities were visualized in the FL1 and FL2 channels, respectively and analysed using FCS Express.v5 Flow Cytometry data analysis software.Regeneration of the parent HeLa cells from the CSCs
Regeneration of the parent HeLa cells from the CSCs was carried out using the method of Gu et al. with minor modifications.19 CSCs (1 × 104 cells per well) were cultured in constituted DMEM in a 35 mm culture dish at 37 °C in a humidified 5% CO2 incubator for 12 days and the morphology of the generated HeLa cells was assessed under a bright field microscope (Leica, DMIL, Germany).Cell proliferation assay
CSCs were cultured (1 × 104 cell per well) in SFM, (as described above). Cells were seeded separately in 96 well plates and cultured at 37 °C in a 5% CO2 incubator. The cytotoxic effect of PN on the CSCs was evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) reduction assay.21 CSCs were treated with different concentrations of PN and DX, as indicated in the legends to the figures, and corresponding volume of vehicle as a control for 24 and 48 h. MTT assay was also performed in the PN-treated cells in the presence or absence of 5 mM NAC, an ROS-scavenger.24 After treatment, MTT (0.5 mg ml−1) was added to each well and incubated for the next 4 h in the dark at 37 °C in a CO2 incubator. The medium was removed and formazan crystals were dissolved in 100 μl of dimethyl sulfoxide (DMSO) with gentle shaking. The amount of formazan crystals was measured at 570 nm using a TECAN ELISA reader (Tecan Sunrise™). The data are expressed as cytotoxicity (%) with respect to vehicle treated cells.Sphere-forming efficiency (SFE) assay
The effect of PN (50 and 100 μM) on the sphere forming efficiency (SFE) of sphere forming cells was investigated. Subconfluent sphere forming cells (1 × 104 cells per well) were dissociated using 1× EDTA (1.0 mM) to achieve a single cell suspension. Cells (1 × 104 cells per well) were seeded in a 6-well culture dish and cultured in SFM with the addition of 25 μl per well of fresh SFM after every 48 h. The numbers of spheres generated in each well were examined under a bright field microscope, as described by López et al.22Flow cytometric analysis
000 events and analysis was performed by FCS Express.v5 Flow Cytometry data analysis software.Assessment of the mitochondrial membrane potential (ΔΨm)
The effect of PN on the mitochondrial membrane potential, a crucial event in caspase-dependent apoptosis, was measured as previously described.16 CSCs (1 × 104 cells per well) were treated with PN or DX for 48 h. The cells were then stained with JC-1 (1 μg ml−1) for 20 min at 37 °C in the dark and washed once with 1× PBS after staining. Fluorescence of JC-1 aggregates (red), indicative of healthy cells with functional mitochondria, and of monomers (green), indicative of apoptotic or unhealthy cells with collapsed mitochondria were measured at 590 nm with excitation at 580 nm and 527 nm with excitation at 510 nm, respectively, using a FACS Calibur™. On average 10000 events were recorded and analysed by FCS Express.v5 Flow cytometry data analysis software.
Annexin-V FITC staining
CSCs (1 × 104 cells per well) were treated with PN in the presence or absence of NAC or DX for 48 h to determine apoptotic population. After the treatment, CSCs were collected by centrifugation at 2000 rpm for 5 min at RT and washed once with 1× PBS. The cells were fixed in ice cold 4% paraformaldehyde (PFA) for 20 min, washed once in 1× PBS and suspended in 500 μl of 1× Annexin binding buffer [10× buffer composition: 0.1 M HEPES/NaOH (pH 7.4), 1.4 M NaCl, and 25 mM CaCl2], stained with Annexin V-FITC reagent (5 μl; 0.019 mg ml−1) and incubated for 20 min at 37 °C in the dark.28 Stained cells were washed once with 1× PBS. Fluorescence (10000 events) was monitored using the FL1 channel by a FACS Calibur™ and analysed using FCS Express.v5 Flow Cytometry data analysis software.
PI staining
CSCs cells (1 × 104 cells per well) were treated with PN or DX for 48 h to determine late apoptosis cell death in the total population. Vehicle treated cells were included as controls. Cells were fixed with 4% ice cold PFA for 20 min, washed with 1× PBS once and vortexed to remove clumping. The cells were suspended in 500 μl of a PI master mix [prepared in 1× PBS buffer, DNase-free RNAse (100 μg ml−1), PI (40 μg ml−1) and 0.1% Triton × 100]29 and incubated for 30 min at 37 °C in the dark. Fluorescence was measured using a FACS Calibur™ at a 485 nm excitation wavelength with 10000 events, and analysed by FCS Express.v5 Flow Cytometry data analysis software.
Statistical analysis
GraphPad software (Chicago, USA) was used for statistical analyses. The levels of significance were calculated using a two-way analysis of variance (ANOVA) test (Dunnett's multiple comparison test). A ‘p’ value of less than 0.05 (p < 0.05) was considered as statistically significant with respect to the vehicle-treated group. The results are expressed as mean ± SD from at least three independent experiments, performed in triplicate.Results
Establishing the CSC population from the parent HeLa cells
In order to establish the CSC population from the HeLa cells, it was necessary to observe the presence of the side population in the HeLa cells. The CSC population comprises signature surface molecules such as ABC transporter (Ca2+ channel), which is extensively engaged in efflux action. The existence of a highly tumorigenic SP in parent HeLa cells was established by Hoechst dye efflux assay in the presence or absence of verapamil, a Ca2+ channel blocker. Verapamil inhibited the efflux of the dye from the SP. Approximately, a 1.86% SP (low efflux fluorescence) among the total number of HeLa cells was observed in the absence of verapamil. However, the presence of verapamil significantly reduced the low efflux fluorescence population to 0.64% (Fig. 1A). Thus, the reduction in SP in the presence of verapamil confirmed the presence of a SP in the parent HeLa cells. The CSC population generated from parent HeLa cells was first established and optimized in the presence of SFM and growth factors. The CSCs are known to continuously differentiate and make a cluster of heterogeneous mature cells. The side population present in the HeLa cells was significantly transformed into a compact cluster of cancer sphere cells (Fig. 1B). Microscopic examination of the cells showed that the formation of clusters of cancer sphere cells was initiated within 3 days and a progressive increase in the size of the spheres was observed with time (Fig. 1B). As is evident from Fig. 1B, the HeLa-CSCs continuously proliferated, made large number of clusters and differentiated into non-adherent spherical stem-like clusters. The CSCs generated from HeLa cells were also confirmed by expression analysis of cell surface markers on the CSC population (Fig. 1C). Furthermore, quantified high CD44+ expression (94.55%) and relatively lower CD24+ expression (28.94%) were observed on HeLa-sphere forming cells, whereas the parent HeLa cells showed low CD44+ (60.33%) and high CD24+ (93.60%) expression (Fig. 1D). These results confirmed the authenticity of the CSC cultures generated from the HeLa cells.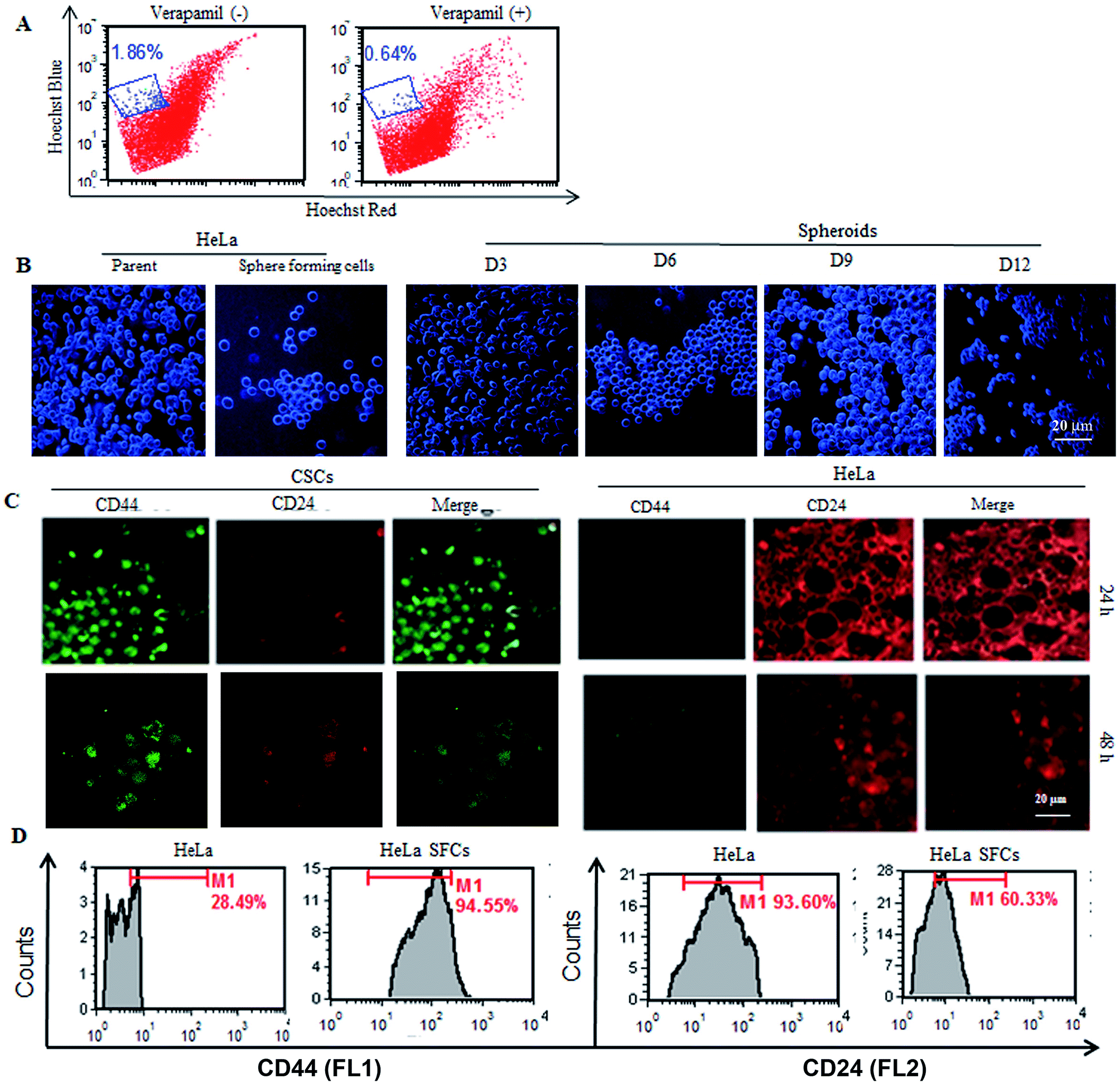 | ||
| Fig. 1 Identification and validation of CSCs from HeLa cells. (A) Establishing the presence of the side population (SP) in HeLa cells. (B) Comparative morphological examination of sphere forming cells (SFC)/cancer stem-like cells (CSCs) and parent HeLa cells. Sphere forming cells derived from adherent populations of HeLa cells at different time intervals. (C) Expression analysis of cell surface markers (CD24+ and CD44+) in CSCs and HeLa cells after 48 h of seeding, visualized under a confocal microscope (40×). (D) Quantitative analysis of CD44+ and CD24+ expression on HeLa cells and CSCs by flow cytometry. | ||
Regeneration of the HeLa cells from SFCs and spheroids
The authenticity of the CSCs was further confirmed by checking their ability to generate HeLa cells. The self-renewal ability of SFCs/CSCs and spheroids to regenerate parent HeLa cells was evaluated by growing them in constituted DMEM. The SFCs adhered to the surface of the culture flask, exhibiting a similar morphology to that of the parent HeLa cells within 5 days. Furthermore, the self-renewal activity of the SFCs was also validated by the formation of spheroids. Spheroids were formed as a cluster of heterogeneous cells after long culturing of SFCs. When spheroids of 3 and 5 days were cultured in constituted DMEM, the cells quickly adopted that same morphology as that of the parent HeLa cells within 4 days (Fig. S1†). Thus, these results indicated that both SFCs and spheroids derived from them adopted parent HeLa cell morphology.PN decreases cell viability and modulates side population
The effect of PN on the cell viability of CSCs was assessed by MTT assay. As shown in Fig. 2A, PN treatment significantly inhibited CSC population cell viability in a dose-dependent manner at concentrations starting from 12.5 μM (Fig. 2A). DX treatment, included as a reference control, also resulted in decreased cell viability of the CSCs. Based on the cell viability data of the PN-treated CSCs, 50 μM of PN was used for subsequent experiments. To establish if PN affected the side population in the HeLa cells, the effect of PN treatment on the SP in the HeLa cells in the presence or absence of verapamil was also investigated. PN (50 μM & 100 μM) significantly reduced the SP to 1%, and 0.44% in comparison to that observed in vehicle treated cells (∼1.42%) in the absence of verapamil. In the presence of verapamil, the percentage of SP was further reduced to 0.42% and 0.19% at 50 and 100 μM of PN, respectively, as compared to the vehicle treated cells (0.55%) (Fig. 2C). Thus, PN was competent in reducing the SP number, either in the absence or presence of verapamil in a dose-dependent manner. | ||
| Fig. 2 Impact of PN on the CSC population and side population. MTT reduction assay to determine the cytotoxicity of PN in the CSC population at 24 h (A) and 48 h (B) post treatment. The data represent the mean ± SD of three independent experiments, performed in triplicate. Two-way ANOVA (Dunnett's multiple comparisons) was performed to calculate the statistical difference among all of the treated groups with respect to the vehicle treated group. * = p ≤ 0.05, ** = p ≤ 0.01, and **** = p ≤ 0.0001. (C) The effect of PN on the SP in the presence and absence of verapamil analyzed using Hoechst 33342 staining by flow cytometry. VC, vehicle control; PN, pinostrobin; DX, doxorubicin. | ||
PN alters the expression of CSC cell surface markers
Cancer stem cells proficiently express unique surface markers, including CD44+, CD24+ and ATP binding cassette (ABC) transporters. A reduction in the number of viable CSCs by PN, as evident from MTT assay, was confirmed by the analysis of the expression of cell surface markers on the CSC population. A higher expression of CD44+ with negligible expression of CD24+ is characteristic of CSCs, whereas HeLa cells express higher CD24+ with minimal CD44+. Confocal microscopy of the treated cells clearly showed a decrease in the green fluorescence of FITC in the PN-treated CSCs, indicating decreased expression of CD44+ when compared to vehicle-treated CSCs (Fig. 3A). Likewise, a decrease in the red fluorescence of the PE stain indicated a decreased expression of CD24+ in comparison to vehicle treated HeLa cells (Fig. 3A). The change in the expression of the surface markers on the CSCs and HeLa cells was also observed after 24 h with evident disintegration of the spheroids in the DX treated cells (Fig. S2†). However, after 48 h, the change was prominent, as evident from the increased fluorescence on PN exposure. Thus, PN adversely affected both the HeLa cells and its CSCs.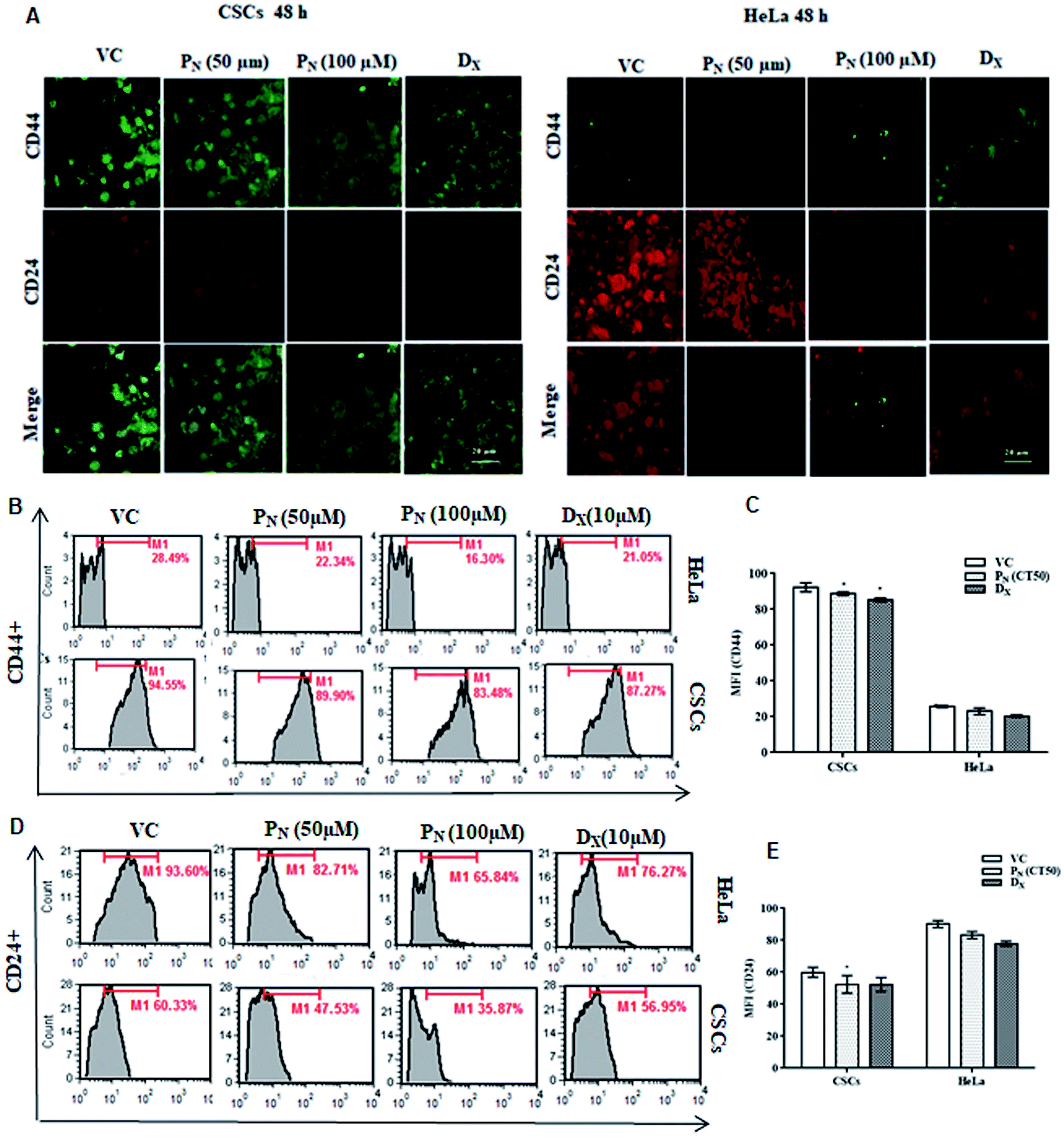 | ||
| Fig. 3 Effect of PN on CD44+ and CD24+ expression in CSCs and HeLa cells. (A) HeLa cells and CSCs, treated with the vehicle and different concentrations of PN and DX for 48 h, were stained with FITC-conjugated anti-CD44 and PE-conjugated anti-CD24 antibodies and visualized (40×) under a confocal microscopy. (B and D) Flow cytometric analysis of CD44+ & CD24+ expression in PN-treated (50 μM, 100 μM) HeLa-CSCs at 48 h post-treatment. (C and E) Mean fluorescence intensity of CD44+ and CD24+ HeLa cells and CSCs treated with PN. VC, vehicle control; PN, pinostrobin; DX, doxorubicin. | ||
Quantitative analysis of CD44+ and CD24+ on the CSCs and HeLa cells by flow cytometry further confirmed the microscopic results. As evident from Fig. 3, PN (100 μM) reduced CD44+ in both the CSCs and HeLa cells to 83.48 and 16.30% compared to vehicle treated CSCs (94.55%) and HeLa cells (28.49%) (Fig. 3B). A decrease in the CD44+ CSC population was observed in PN treated CSCs (Fig. 3C). The vehicle treated CSCs showed 60.33% CD24+ cells, whereas in PN-treated (100 μM) cells, their expression was reduced to 35.87%. Likewise, a decrease in the expression of CD44+ and CD24+ was noted in PN-treated HeLa cells. PN exposure reduced the expression levels of CD44+ and CD24+ to 16.30 and 65.84%, respectively in HeLa cells in comparison to vehicle treated cells (CD44+ and CD24+, 28.49% and 93.60%, respectively) after 48 h (Fig. 3D and E). The expression of CD44+and CD24+ was also found to be lower in DX-CSCs and HeLa cells treated with DX for 48 h, respectively.
PN inhibits the sphere formation efficiency (SFE) of CSCs and alters the morphology of the spheres
Since CSCs form spheres that contain tumorigenic cells, it was of interest to know if PN affected the sphere formation. For this, the effect of PN on the sphere formation efficiency of the HeLa-CSCs was analysed at different time intervals. As evident from Fig. 4, there was an increase in the number of cell clusters/spheroids with time in the untreated/vehicle treated cells. PN-treatment of CSCs resulted in a relatively lower number of cell clusters and decreased the sphere and spheroid formation when compared to the vehicle control. Likewise, DX-treated CSCs also showed a relatively lower number of compact spheroids as compared to the vehicle treated group and the spheroids appeared to be disintegrated into smaller individual spheres. Thus, the sphere formation efficiency was significantly reduced in PN-treated CSCs when compared to vehicle treated cells. Morphological examination of the spheres showed a time-dependent increase in the size of the spheres and that they formed symmetric prototypical spheroids that indicated the self-renewal function of normal stem cells, in vehicle treated cells. On the other hand, in the PN-treated CSCs as well as DX-treated CSCs, a loss in the compact spheroid structure was observed. PN-treatment resulted in complete disruption of the morphology of a single sphere with its outer cell membrane significantly disintegrated at 48 h. DX-treated spheres also showed disintegration of the membrane integrity. Vehicle-treated CSCs showed intact spheres similar to those seen in untreated CSCs (Fig. 4). | ||
| Fig. 4 Effect of PN on sphere formation efficiency. The morphology and number of spheres were examined microscopically (20×) at different time points after PN treatment. VC, vehicle control; PN, pinostrobin; DX, doxorubicin. | ||
PN enhances ROS generation
Reactive oxygen species (ROS) production was analysed in PN treated CSCs by DCFH-DA (Fig. 5A). PN-treated (50 μM) CSCs exhibited higher fluorescence events of DCF (57.43 + 3.54%) in comparison to vehicle-treated CSCs (19.26 + 0.21%). Higher DCF fluorescence in PN-treated cells is an indicator of ROS production in CSCs. The site of ROS production was analysed using 10-nonyl acridine orange (NAO) dye. PN-treated CSCs showed lower binding affinity of NAO with cardiolipin (27.71 + 4.40%) when compared to vehicle-treated CSCs (64.23 + 2.0%) at 48 h of treatment (Fig. 5B). The consequences of ROS production in PN-treated CSCs were analysed using HE. Fig. 5C shows the red fluorescence of ethidium bromide that binds to DNA of damaged cells. PN-treated CSCs showed presence of DNA fragmentation indicated by greater red fluorescence events (40.59 + 6.15%). DNA fragmentation was found to increase with time and increasing dose of PN. | ||
| Fig. 5 Effect of PN on ROS production, cardiolipin oxidation and DNA damage. (A) Flow cytometric analysis of the ROS production using DCFH staining in the PN-treated CSC population. (B) Analysis of cardiolipin oxidation using 10-nonyl acridine orange (NAO) staining by flow cytometry in the PN treated CSC population. (C) Flow cytometric analysis showing oxidation of hydroethidine (HE) by ROS in PN treated CSCs. The figures show the representative dot plots for each analysis. The bar diagrams on the right show the mean fluorescence intensity of the respective fluoroprobe in the CSCs subjected to different treatments. Significance (p value) levels were calculated with respect to vehicle treated CSCs (VC) using two-way ANOVA (Dunnett's multiple comparisons) * = p ≤ 0.05, ** = p ≤ 0.01, *** = p ≤ 0.001 and **** = p ≤ 0.0001. VC, vehicle control; PN, pinostrobin; DX, doxorubicin. | ||
PN treatment promotes apoptotic cell population and depolarization of the mitochondrial membrane potential (ΔΨm)
Cell membrane integrity is an indicator of a healthy cell. In apoptotic cells, phosphatidylserine, an inner cell membrane protein, is externalized from the inner side to the outer side of the cells, to which Annexin V is able to bind. In order to see if PN affects the cell membrane integrity, FITC-Annexin V staining was carried out. PN-treated cells showed greater phosphatidylserine exclusion in the form of Annexin V-FITC fluorescence events in CSCs (61.34 ± 3.37%) indicating early apoptotic cell population, in comparison to vehicle (∼10%) treated cells at 48 h (Fig. 6A). Thus, externalization of phosphatidylserine was significantly increased in PN-treated cells in a time dependent manner, indicating disruption of membrane integrity.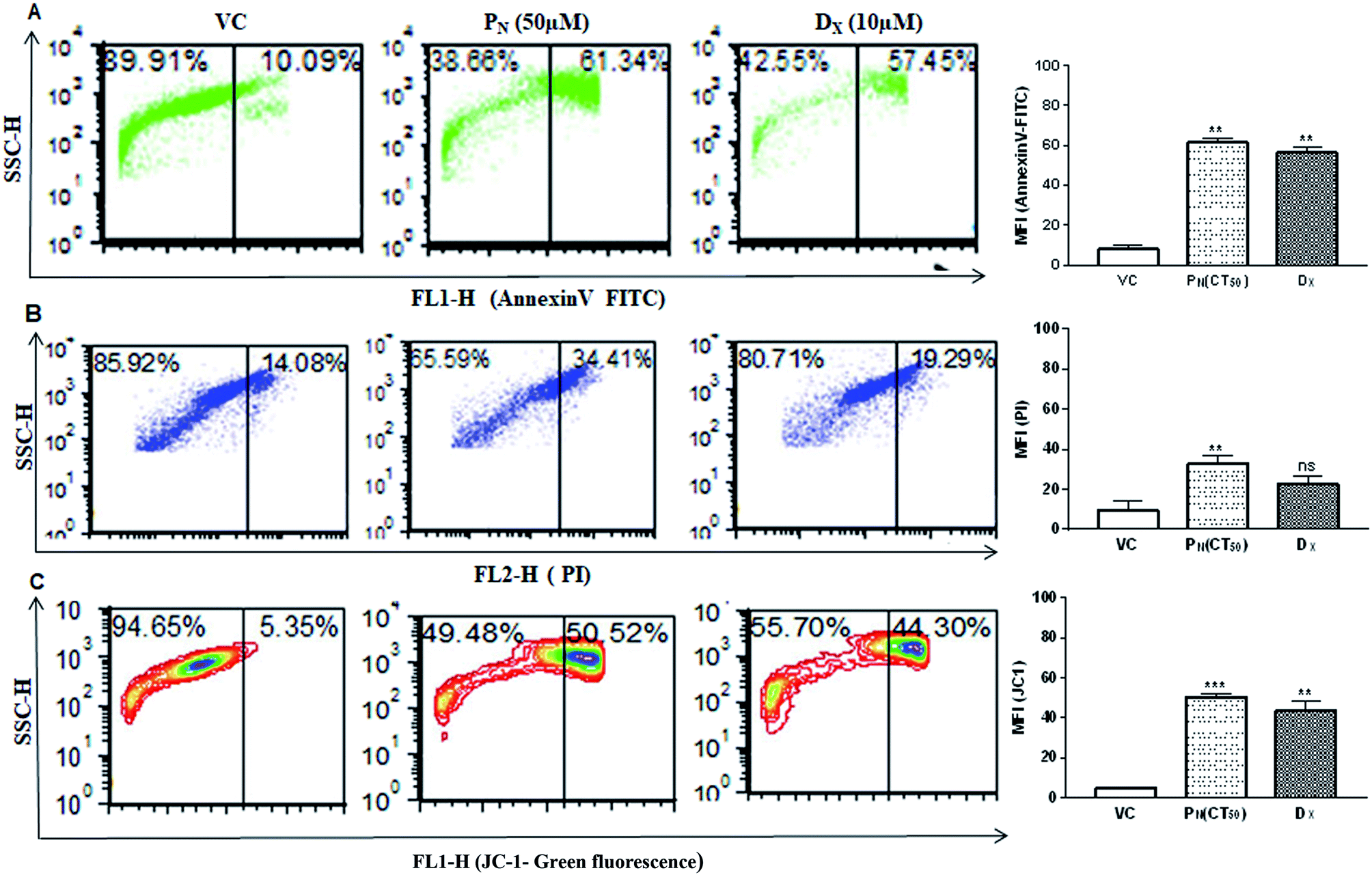 | ||
| Fig. 6 Measurement of apoptotic/necrotic cell populations and change in the mitochondrial membrane potential in PN-treated CSCs. (A) Annexin-V-FITC staining to determine apoptotic cell population in PN treated CSCs population. (B) Flow cytometric analysis of PI stained CSCs to determine late apoptotic cell population. (C) Flow cytometric analysis of the change in the mitochondrial membrane potential (ΔΨm) in the PN treated CSCs population using JC-1 probes. The bar diagrams on the right show the mean fluorescence intensity of the respective fluoroprobe in the CSCs subjected to different treatments. Significant difference (p value) was calculated using two-way ANOVA (Dunnett's multiple comparisons with respect to the vehicle treated group). * = p ≤ 0.05, ** = p ≤ 0.01, *** = p ≤ 0.001 and **** = p ≤ 0.0001. VC, vehicle control; PN, pinostrobin; DX, doxorubicin. | ||
DNA fragmentation was analysed by PI staining to determine the late apoptotic cell population. CSCs exposure to PN resulted in an increase in PI-stained nuclei also with red fluorescence events at 34.41 ± 3.72%, compared to vehicle treated cells with PI fluorescence events at 14.08 ± 4.43% (Fig. 6B). The greater number of stained nuclei in PN-treated CSCs indicated the disintegration of nuclear membrane. Integrity of the mitochondrial membrane is absolutely necessary for the normal metabolic functions of cells. Hence, the mitochondrial membrane potential was analysed using a cationic carbocyanine JC-1 probe in vehicle-treated and PN treated CSCs. Fig. 6C shows that PN-treated CSCs showed a higher number of JC-1 monomer fluorescence events (50.52 ± 1.71) in comparison to that observed in vehicle treated cells (fluorescence events ∼ 5%) at 48 h. DX, used as a positive control, also resulted in the formation of JC-1 monomer fluorescence events (∼44%) in CSCs. The vehicle-treated cells showed high ΔΨm with intense red fluorescence and exhibited fewer JC-1 monomers with intact membranes.
NAC rescues PN-induced ROS-mediated apoptosis in CSCs
To confirm the role of ROS in PN-induced cell death, the ROS scavenger NAC was used. For this, the CSCs were incubated with 5 mM NAC for 5 h prior to the addition of 50 μM PN and allowed to grow for a further 48 h. As is evident from the figure, pre-treatment with NAC resulted in ∼30% reduction in cytotoxicity (Fig. 7A) when compared to the cells treated with PN alone (∼60%). Since PN treatment resulted in increased ROS production, ultimately leading to cell death, ROS production in the cells pre-treated with NAC was also estimated. Fig. 7B and C clearly show a 53.8% reduction in the ROS levels assessed by DCFH-DA staining in the cells pre-treated with NAC in comparison to those treated with PN alone. A reduction in cell death accompanied by decreased ROS levels in the NAC-pretreated cells clearly indicate that NAC was able to rescue the CSCs from the ill effects of ROS generated due to PN treatment. Apoptosis assay using FITC conjugated Annexin V also confirmed that NAC inhibited PN-induced apoptosis in CSCs (Fig. 7D and E). Pre-treatment with NAC resulted in a significant decrease in the apoptotic cell population (∼20%) when compared to the PN-treated cells alone (52%). No significant changes in the necrotic population in the NAC pre-treated group were observed in comparison with the PN-treated group. These data validated that PN inhibited cell proliferation and caused ROS-mediated apoptosis in the CSC population.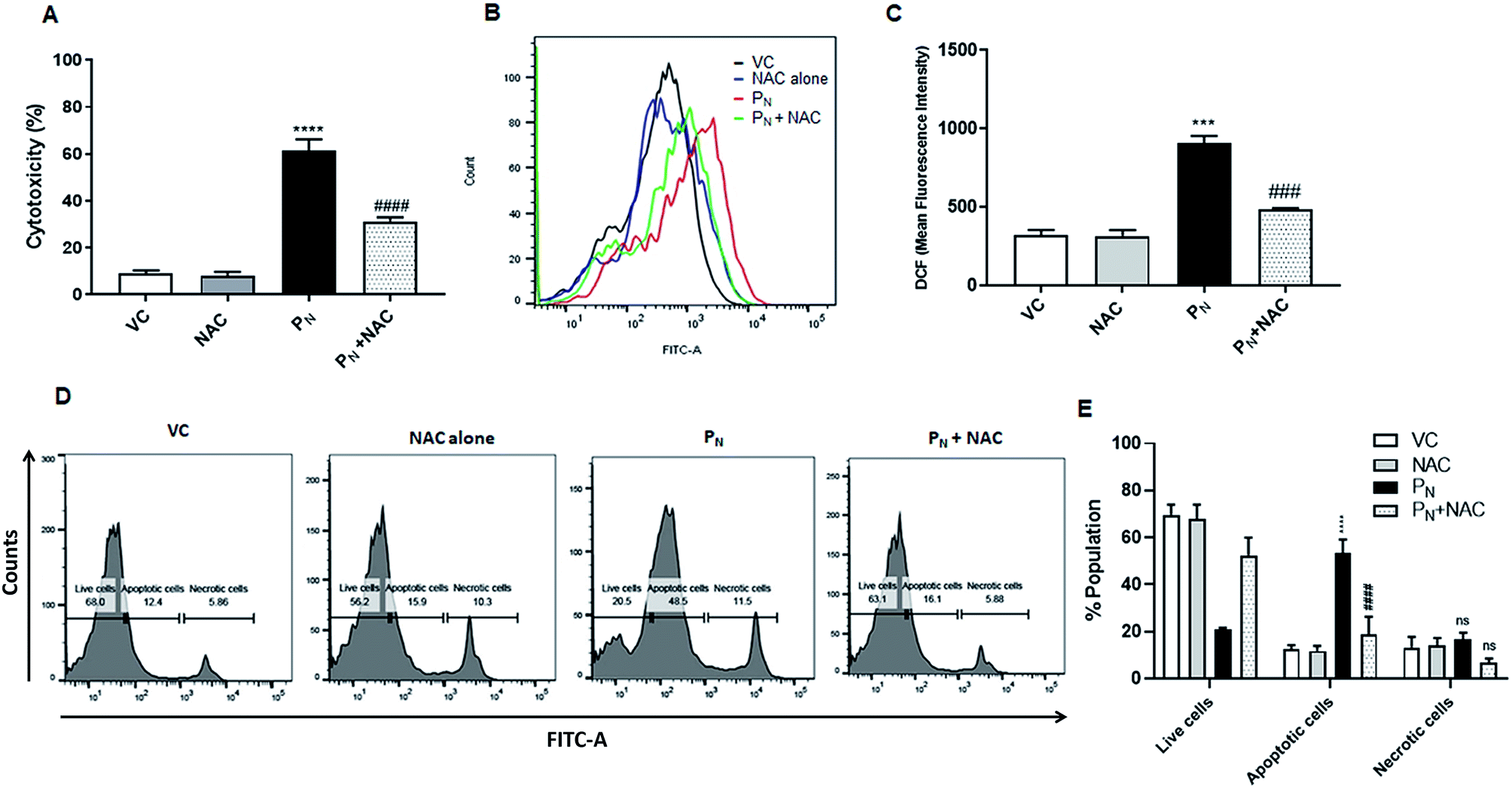 | ||
| Fig. 7 Role of ROS in apoptosis induced by PN. CSCs were pre-incubated with NAC (5 mM) for 5 h, followed by treatment with PN (50 μM) for 48 h. (A) Cell toxicity (%) was measured by MTT assay. (B) Overlay histogram showing ROS generation in PN-treated and NAC-treated groups. (C) Bar diagram representing the effect of NAC on PN-induced ROS generation. (D and E) NAC reduced apoptosis resulting in a reduction of the PN-treated CSC population using an Annexin-V-FITC apoptosis detection kit (Biovision), as per manufacturer's instructions. All of the FACS experiments were carried out on a BD FACSAria™ III cell sorter (BD Biosciences). Ordinary one-way ANOVA (Dunnett's multiple comparisons test) was performed to calculate the statistical difference (p ≤ 0.05). (*) compared with vehicle control cells, whereas # represents the significance compared to PN-treated cells. NS: non-significant, VC: vehicle control, PN: pinostrobin, NAC: n-acetyl cysteine. | ||
Discussion
Despite the availability of various therapies such as chemotherapy, radiotherapy and surgical intervention for the treatment of cancer, the incidence of relapse and subsequent mortality due to various cancers have been reported to increase. The primary cause of this recurrence has been attributed to the SP of cancer cells recognised as the CSC population, which are often resistant to existing therapies. Therefore, it is hypothesized that successful treatment of cancer does not only depend on the main tumor mass, but also on highly resistant CSCs. The existence of CSCs suggests that tumor development and metastasis are driven by the SP of parent cancer cells, which are responsible for tumor initiation, growth and recurrence. Therefore, the targeting of CSCs is gaining attention in devising new treatment strategies for cancer. Indeed, combination therapies that target both stem cells and tumor cells have been proven to be more effective in the treatment of breast cancer.29Traditional medicines systems including Unani, Ayurveda and Siddha have used formulations derived from plants and marine sources for the treatment of different ailments with minimal toxicity. Indeed, a number of chemotherapeutic agents such as taxol and vinblastine, currently used for the treatment of cancer, are derived from plants.12 The use of molecules/compounds derived from dietary substances that are part of the daily diet for the prevention and treatment of cancer would possibly provide an alternate strategy without the adverse effects associated with currently available therapies. With respect to targeting cancer cells, a number of compounds have shown selective toxicity to cancer stem cells in in vivo models.30 A few of these compounds are present in our daily diet, including curcumin, resveratrol, epigallocatechin gallate (EGCG), vitamin A and its precursors, isothiocyanate–sulforaphane, and various other flavonoids.30–32 Curcumin and EGCG have been reported to downregulate the self-renewal ability of CSCs by modulating associated pathways in an in vitro model.33 Similarly, resveratrol has also been reported to significantly reduce the nuclear localisation of β-catenin, important for normal signalling in CSC maintenance.34 Dietary agents have been reported to overcome CSC-dependent drug resistance.35 Treatment with plant-derived quercetin showed greater inhibition of tumor growth, indicating the influence on drug resistant cells in comparison to control mice, suggesting a lower proportion of chemotherapy-resistant CSCs.30 Some flavonoids show synergistic action in different cancer cells, whereas some show specific toxicity against certain types of cells.36–38 Thus, there is a need to search for molecules that have anti-cancer activity for a variety of cancer cells together with the ability to inhibit CSC proliferation.
PN, a flavonoid present in a number of dietary compounds, has been shown to inhibit the growth and proliferation of a variety of cancer cells.13–16 Previous findings from our laboratory have demonstrated the anticancer potential of PN through the inhibition of topoisomerase I activity, control of cell proliferation, and oxidative-stress induced cell death in HeLa cells.16,17 PN efficiently induced apoptosis in HeLa cells through ROS-mediated activation of extrinsic and intrinsic signalling pathways, as well as ROS-mediated mitochondrial damage.16 Stem cells contain own signature molecules like ABC membrane transporter proteins that efflux certain lipophilic drugs and are recognized as the SP.39 A few ABC membrane transporter proteins act as Ca2+ channels, increasing the cellular uptake of Ca2+ and activating Ca2+ dependent proteases (e.g. calpain) and endonuclease(s), which leads to DNA fragmentation and apoptosis.40,41 We first demonstrated the existence of highly tumorigenic CSCs or side population in parental HeLa cells by Hoechst staining in the presence of verapamil, a Ca2+ channel blocker, which would prevent the efflux of Hoechst dye from the SP.
Our results confirmed that the HeLa cells used in the study did contain a SP or CSCs. After confirming the presence of the SP or CSCs in the parental HeLa cells, sphere forming cells (SFCs) or CSCs were successfully established from HeLa cells in vitro to assess the effect of PN on their self-renewal properties. Compact clusters of cancer sphere cells were non-adherent and spherical with a morphology different from that of the parent HeLa cells. A progressive increase in their diameter clearly indicated their high proliferative and self-renewal ability. Similar uniformity in sphere formation and maturation has also been reported in CSCs derived from breast cancer epithelial cell lines including MCF-7 and MDA-MD-231, and various sarcoma and brain tumors.42–44 PN treatment showed significant cytotoxicity towards CSCs in a dose and time-dependent manner. This is evident from the reduction in the number and size of the spheroids and overall decrease in sphere formation efficiency. Our results suggest that HeLa cells contain a PN-sensitive CSC population. Our results are in agreement with those of Tang et al.,45 who showed that EGCG, another flavonoid, inhibited the growth of CSCs derived from human prostate cancer cells (PC-3 and LNCaP) in a dose-dependent manner. Likewise, a significant reduction in tumor volume in a MDA-MB-231 mammosphere model with a CD44high/CD24low phenotype was observed after curcumin administration.30
Like the CSCs derived from human colon carcinoma HT29 cells20 and ovarian carcinoma SKOV3 cells,21 DX also inhibited cell proliferation of the CSCs derived from HeLa cells in the present study. Previous studies have shown that CSC subpopulations can be identified, isolated from various different tumors by exploiting cell-surface markers such as CD44+ and CD24+ in ovarian and mammospheres from breast cancer cell lines.50 These specific signature molecules or cell surface markers have been correlated with disease determination.46 The CD24+ population is involved in the formation of sphere-like bodies, cell proliferation, differentiation, promotion of tumour growth, and increases resistance towards chemotherapeutic drugs, whereas the CD44+ population is engaged in cancer promotion, metastasis and cancer cell growth.47 Gu and his colleagues reported HeLa-SFC/CSC populations that expressed CD44high/CD24low cell-surface markers and showed drug resistance.19 A reduction in the CSC population upon PN-treatment was therefore confirmed by analysis of CSC-specific cell surface markers on HeLa-CSCs. As expected, the HeLa-CSCs showed high CD44+ and low CD24+ expression, whereas the parental HeLa cells showed lower CD44+ and higher CD24+ expression.19 Thus, both the morphological examination and expression analysis of the cell surface markers confirmed the identity of the HeLa-CSCs and their stem-like properties. PN-treatment resulted in a significant decline in the expression of CD44+ and CD24+ in HeLa cells as well as HeLa-CSCs, although the extent of reduction was more in CSCs than that observed in parent HeLa cells.48 These data thus demonstrate the potential of PN to inhibit the self-renewal activity of CSCs. The decreased expression of CSC-specific markers endorses cell death in PN-treated cells and a decrease in the high CD44+ population would inhibit tumor growth, metastasis and cause relapse.47 Phytochemicals capable of altering the expression of both CD44+ and CD24+ have been identified as anti-metastatic agents.49 We further explored the possible effects of PN exposure on the cytotoxicity, morphology, and expression levels of surface markers among the CSC population by analysing the changes in the CD24+ and CD44+ expression levels. Our previous findings clearly revealed that in parent HeLa cells,16 PN was found to be very promotive towards modulating the cellular integrity and mitochondrial membrane potential, DNA fragmentation and ROS production. A reduction in the cytotoxicity, ROS production and apoptotic cell population by PN in cells pre-treated with NAC, a ROS scavenger, further confirmed that generation of ROS is the key mechanism by which PN inhibits cell proliferation and induces cell death in CSCs. In the present study, the results show enhanced production of ROS, DNA fragmentation, and depolarized mitochondrial membrane potential in the PN-treated CSCs, which could lead them to cell death through an apoptotic pathway.
Conclusions
Overall, through the generation of in vitro CSC cultures from HeLa cancer cells, we demonstrated that PN has the ability to inhibit CSC proliferation and spheroid formation, ultimately leading the CSCs to apoptotic cell death. These data clearly demonstrate that treatment with plant derived flavonoids such as PN, which targets both cancer cells16 and cancer stem cells (present study), is likely to prove a promising therapeutic strategy, by reducing relapse and recurrence. The individual and combined effects of chemotherapies and flavonoids have been tested in various cancers. The anti-CSC activity of a combination of sulforaphane and dietary-derived flavonoid quercetin has been examined in MIA-PaCa2 pancreatic cancer cells (high proportion of CSCs) using a xenograft model. Although both of these agents individually inhibited the growth of tumors, the combination led to complete eradication.47 Similarly, PN or other such compounds could possibly be used in combination with existing therapies for improved outcome.In our study, the mechanism of PN-induced cell death is probably associated with an increase in ROS generation and depolarization of the mitochondrial membrane potential in CSCs, which leads to downregulation of expressions of stem cell markers and regeneration capacity. However, further studies using in vivo models would confirm the potential of PN for cancer treatment, either alone or in combination with existing therapies.
Conflicts of interest
The authors declare that no conflicting interests exist.Abbreviation
| BSA | Bovine serum albumin |
| CSCs | Cancer stem-like cells |
| DMEM | Dulbecco's modified Eagle's medium |
| EDTA | Ethylenediaminetetraacetic acid |
| FACS | Fluorescence-activated cell sorting |
| FBS | Fetal bovine serum |
| MMP | Mitochondrial membrane potential |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PBS | Phosphate buffer saline |
| PN | Pinostrobin |
| ROS | Reactive oxygen species |
| SP | Side population |
Acknowledgements
The authors are thankful for the financial support from the Department of Biotechnology, New Delhi, India, for the DBT-BUILDER grant (BT/PR5006/INF/22/153/2012) and the PURSE grant from the Department of Science and Technology [SR/PURSE/Phase2/11(C) 2015], New Delhi, India to the Jawaharlal Nehru University, New Delhi, India. The University Grant Commission (UGC) is acknowledged for providing a research fellowship to AJ and SS, respectively. The Advanced Instrumentation Research Facility (AIRF), Jawaharlal Nehru University, New Delhi is acknowledged for providing the FACS and confocal microscopy facilities.References
- A. Sreedevi, R. Javed and A. Dinesh, Int. J. Women's Health, 2015, 7, 405–414 Search PubMed.
- I. Dagogo-Jack and A. T. Shaw, Nat. Rev. Clin. Oncol., 2018, 15, 81–94 CrossRef CAS PubMed.
- B. Bao, A. Ahmad, A. S. Azmi, S. Ali and F. H. Sarkar, Curr. Protoc. Pharmacol., 2013, 61, 14.25.1–14.25.14 CrossRef PubMed.
- G. Lee, R. R. Hall 3rd and A. U. Ahmed, J. Stem Cell Res. Ther., 2016, 6(10), 363 CrossRef PubMed.
- J. C. Chang, Medicine, 2016, 95, S20–S25 CrossRef CAS PubMed.
- K. Chen, Y. H. Huang and J. L. Chen, Acta Pharmacol. Sin., 2013, 34, 732–740 CrossRef CAS PubMed.
- A. Flemming, Nat. Rev. Drug Discovery, 2015, 14, 165 CrossRef PubMed.
- Z. A. Rasheed and W. Matsui, J. Gastroenterol. Hepatol., 2012, 27(suppl. 2), 15–18 CrossRef CAS PubMed.
- T. Wang, S. Shigdar, M. P. Gantier, Y. Hou, L. Wang, Y. Li, H. A. Shamaileh, W. Yin, S. F. Zhou, X. Zhao and W. Duan, Oncotarget, 2015, 6, 44191–44206 Search PubMed.
- N. Y. Frank, T. Schatton and M. H. Frank, J. Clin. Invest., 2010, 120, 41–50 CrossRef CAS PubMed.
- J. A. McCubrey, K. Lertpiriyapong, L. S. Steelman, S. L. Abrams, L. V. Yang, R. M. Murata, P. L. Rosalen, A. Scalisi, L. M. Neri, L. Cocco, S. Ratti, A. M. Martelli, P. Laidler, J. Dulinska-Litewka, D. Rakus, A. Gizak, P. Lombardi, F. Nicoletti, S. Candido, M. Libra, G. Montalto and M. Cervello, Aging, 2017, 9, 1477–1536 CAS.
- G. M. Cragg and J. M. Pezzuto, Med. Princ. Pract., 2016, 25(suppl. 2), 41–59 CrossRef PubMed.
- H. D. Smolarz, E. Mendyk, A. Bogucka-Kocka and J. Kocki, Z. Naturforsch., C: J. Biosci., 2006, 61, 64–68 CAS.
- Sukardiman, A. Darwanto, M. Tanjung and M. O. Darmadi, Clin. Hemorheol. Microcirc., 2000, 23, 185–190 CAS.
- J. C. Le Bail, L. Aubourg and G. Habrioux, Cancer Lett., 2000, 156, 37–44 CrossRef CAS PubMed.
- A. Jaudan, S. Sharma, S. N. A. Malek and A. Dixit, PLoS One, 2018, 13, e0191523 CrossRef PubMed.
- A. Jadaun, N. Subbarao and A. Dixit, J. Photochem. Photobiol., B, 2017, 167, 299–308 CrossRef CAS.
- J. W. Fahey and K. K. Stephenson, J. Agric. Food Chem., 2002, 50, 7472–7476 CrossRef CAS PubMed.
- W. Gu, E. Yeo, N. McMillan and C. Yu, Cancer Gene Ther., 2011, 18, 897–905 CrossRef CAS PubMed.
- S. Atashpour, S. Fouladdel, T. K. Movahhed, E. Barzegar, M. H. Ghahremani, S. N. Ostad and E. Azizi, Iran. J. Basic Med. Sci., 2015, 18, 635–643 Search PubMed.
- A. Pouyafar, M. Z. Heydarabad, S. B. Aghdam, M. Khaksar, A. Azimi, R. Rahbarghazi and M. Talebi, 2019, DOI:10.1002/jcb.28129, Epub ahead of print.
- J. López, A. Poitevin, V. Mendoza-Martinez, C. Perez-Plasencia and A. Garcia-Carranca, BMC Cancer, 2012, 12, 48 CrossRef PubMed.
- M. A. Goodell, K. Brose, G. Paradis, A. S. Conner and R. C. Mulligan, J. Exp. Med., 1996, 183, 1797–1806 CrossRef CAS PubMed.
- M. Bhardwaj, N. H. Kim, S. Paul, R. Jakhar, J. Han and S. C. Kang, PLoS One, 2016, 26, e0154525, DOI:10.1371/journal.pone.0154525.
- L. U. Ling, K. B. Tan, H. Lin and G. N. Chiu, Cell Death Dis., 2011, 2, e129 CrossRef PubMed.
- E. C. Dietze, L. E. Caldwell, S. L. Grupin, M. Mancini and V. L. Seewaldt, J. Biol. Chem., 2001, 276, 5384–5394 CrossRef CAS PubMed.
- C. Bucana, I. Saiki and R. Nayar, J. Histochem. Cytochem., 1986, 34, 1109–1115 CrossRef CAS PubMed.
- S. S. Fard, M. Jeddi-Tehrani, M. M. Akhondi, M. Hashemi and A. M. Ardekani, Avicenna J. Med. Biotechnol., 2010, 2, 53–61 Search PubMed.
- T. Wang, R. Narayanaswamy, H. Ren and V. P. Torchilin, Cancer Biol. Ther., 2016, 17, 698–707 CrossRef CAS PubMed.
- S. Khan, A. Karmokar, L. Howells, A. L. Thomas, R. Bayliss, A. Gescher and K. Brown, Mol. Nutr. Food Res., 2016, 60, 1295–1309 CrossRef CAS PubMed.
- P. B. Gupta, T. T. Onder, G. Jiang, K. Tao, C. Kuperwasser, R. A. Weinberg and E. S. Lander, Cell, 2009, 138, 645–659 CrossRef CAS PubMed.
- M. Ahmed, K. Chaudhari, R. Babaei-Jadidi, L. V. Dekker and A. Shams Nateri, Stem Cells, 2017, 35, 839–850 CrossRef PubMed.
- S. S. Chung and J. V. Vadgama, Anticancer Res., 2015, 35, 39–46 CAS.
- R. S. Tarapore, I. A. Siddiqui and H. Mukhtar, Carcinogenesis, 2012, 33, 483–491 CrossRef CAS PubMed.
- S. F. Chen, S. Nieh, S. W. Jao, C. L. Liu, C. H. Wu, Y. C. Chang, C. Y. Yang and Y. S. Lin, PLoS One, 2012, 7, e49275 CrossRef CAS PubMed.
- P. R. Pandey, H. Okuda, M. Watabe, S. K. Pai, W. Liu, A. Kobayashi, F. Xing, K. Fukuda, S. Hirota, T. Sugai, G. Wakabayashi, K. Koeda, M. Kashiwaba, K. Suzuki, T. Chiba, M. Endo, T. Fujioka, S. Tanji, Y. Y. Mo, D. Cao, A. C. Wilber and K. Watabe, Breast Cancer Res. Treat., 2011, 130, 387–398 CrossRef CAS PubMed.
- A. A. Gokbulut, E. Apohan and Y. Baran, Hematology, 2013, 18, 144–150 CrossRef CAS PubMed.
- A. Del Follo-Martinez, N. Banerjee, X. Li, S. Safe and S. Mertens-Talcott, Nutr. Cancer, 2013, 65, 494–504 CrossRef CAS PubMed.
- S. Orrenius, M. Ankarcrona and P. Nicotera, Adv. Neurol., 1996, 71, 137–149 CAS ; discussion 149–151.
- S. Jiang, S. C. Chow, P. Nicotera and S. Orrenius, Exp. Cell Res., 1994, 212, 84–92 CrossRef CAS PubMed.
- D. L. Holliday and V. Speirs, Breast Cancer Res., 2011, 13, 215 CrossRef PubMed.
- F. S. Dela Cruz, Front. Oncol., 2013, 3, 168 Search PubMed.
- E. Sugihara and H. Saya, Int. J. Cancer, 2013, 132, 1249–1259 CrossRef CAS.
- W. Zhao, Y. Li and X. Zhang, Cancer Transl. Med., 2017, 3, 87–95 CrossRef CAS PubMed.
- S. N. Tang, C. Singh, D. Nall, D. Meeker, S. Shankar and R. K. Srivastava, J. Mol. Signaling, 2010, 5, 14 CrossRef PubMed.
- J. Manuel Iglesias, I. Beloqui, F. Garcia-Garcia, O. Leis, A. Vazquez-Martin, A. Eguiara, S. Cufi, A. Pavon, J. A. Menendez, J. Dopazo and A. G. Martin, PLoS One, 2013, 8, e77281 CrossRef PubMed.
- A. Z. Ayob and T. S. Ramasamy, J. Biomed. Sci., 2018, 25, 20 CrossRef.
- D. Wang, B. Upadhyaya, Y. Liu, D. Knudsen and M. Dey, BMC Cancer, 2014, 14, 591 CrossRef.
- P. R. Dandawate, D. Subramaniam, R. A. Jensen and S. Anant, Semin. Cancer Biol., 2016, 40–41, 192–208 CrossRef CAS PubMed.
- M. Appari, K. R. Babu, A. Kaczorowski, W. Gross and I. Herr, Int. J. Oncol., 2014, 45, 1391–1400 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08380k |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2019 |