
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of alkalinity on the synthesis of hierarchical LTA zeolite by using bridged polysilsesquioxane†
Sufang Hu ,
Guoqiang Song,
Da Xue and
Fuxiang Li*
,
Guoqiang Song,
Da Xue and
Fuxiang Li*
Research Institute of Special Chemicals, Taiyuan University of Technology, Taiyuan, 030024, China
First published on 21st January 2019
Abstract
The mechanism of formation of hierarchical LTA zeolite involved the methoxyl groups of bridged polysilsesquioxane hydrolyzing into hydroxyl groups. The covalent Si–O–Si bond between silicon hydroxyl in the above solution and other silica sources forms by dehydration, avoiding phase separation. The influence of alkalinity on the synthesis of hierarchical LTA zeolites was investigated by using bridged polysilsesquioxane. XRD patterns indicate the synthesis of sodalite at the same molar composition of the hierarchical LTA zeolites without bridged polysilsesquioxane. The characterization results from TG and DTG revealed that bridged polysilsesquioxane was successfully incorporated into the as-synthesized hierarchical LTA zeolite. N2-adsorption/desorption results proved that mesopores and the porosities of the hierarchical LTA zeolites are adjustable by a change of alkalinity. SEM images indicated that the morphologies of the hierarchical LTA zeolites changed with increasing alkalinity. The hierarchical LTA zeolites showed faster initial exchange rates of Na+ to Mg2+ compared with conventional LTA.
Introduction
Zeolites, crystalline microporous aluminosilicate materials with well-defined pores and channels, have been widely used in the fields of catalysis, adsorption and separation.1–4 However, the diffusion limitation or spatial resistance in the microporous channels of zeolites reduces the accessibility of active sites within the micropores.2,5 Fortunately, the diffusion limitation of zeolite micropores has been resolved by the introduction of mesopores in zeolites by using a mesoporogen or the preparation of nanozeolites (particles no bigger than 100 nm).6–8 Nevertheless, a high surface energy and large amounts of surface defects cause thermodynamic instability, difficult operability and lower yields of nanozeolites.In recent years, hierarchical zeolites have been successfully synthesized utilizing various mesoporogens. For instance, carbon particles, carbon nanotubes, carbon nanofibers, carbon aerogel, and 3DOM carbon have been used as hard templates to generate intracrystalline mesopores in zeolites with unique morphologies.9–12 However, the pore size distribution of the synthesized mesoporous material depends on the carbon-mesoporogen. Due to the high-price and stiffness of hard templates, the recent use of organosilane as a mesoporogen has provided a new and promising alternative for the preparation of hierarchical zeolites.13–15 Currently, the zeolite LTA with intracrystalline mesopores has been successfully synthesized by using amphiphilic organosilane.1,16 Ryoo et al.1 synthesized hierarchical zeolites by using a designed amphiphilic organosilane, which avoided phase separation. Moreover, the porosity can be adjusted through adjusting the amounts of the templates.17 However, the design of an amphiphilic organosilane is not easy to achieve or control.
With the purpose of solving the problems described above, this group has synthesized hierarchical LTA and single-crystalline core–shell zeolite LTA by using the bridged polysilsesquioxane as an amphiphilic organosilane.18,19 Bridged polysilsesquioxane, covalently linked with silica, provides a silica source that can be easily incorporated and introduces mesopores into the zeolite framework. The hierarchical LTA zeolites showed abundant porosity and interconnected mesopores. The hierarchical LTA zeolite was hydrothermally synthesized at 373 K for 10 days. Numerous studies have shown that the chemical composition in the gel is a crucial factor for controlling the crystallization pathway and physicochemical properties of the crystalline end products.20–23 Meanwhile, the crystallization mechanism of LTA zeolite involves distinct classical and nonclassical pathways.24–32 The unique nonclassical pathway at low temperature (T < 40 °C) involves the formation of 3D gel-like islands (rough growth).23,24 The crystal growth of LTA zeolite at higher temperatures enables the solute to bind at all possible sites, allowing for the rapid formation of nuclei within the growth solution.24
The Si/Al ratio (1) of zeolite A is lower, thus zeolite A has the largest cation exchange capacity among all zeolites. Zeolite A has ion exchange capacity for Ca2+ and Mg2+ to soften water, which has a wide range of applications, such as acting as a detergent assistant, sewage treatment agent, or in the adsorption and separation industry. Zeolite A, as a detergent assistant, has a high affinity for Ca2+ and a large adsorption capacity, which can enhance the decontamination efficiency of detergents. However, the poor exchange capacity for Mg2+ of zeolite A, especially at room temperature, results in the incomplete removal of Mg2+ from water. This is due to the fact that Mg2+ has a larger charge density and exists as a hydrated ion in water. The large hydration radius of Mg2+ makes it difficult to diffuse into the pores of zeolite A, which greatly limits its application. Mesoporous LTA zeolite can greatly accelerate the Mg2+ exchange rate of zeolite A, thereby improving its application in the detergent industry. The mesoporous LTA zeolite changes the diffusion path of Mg2+, and accelerates the diffusion and exchange rate of Mg2+.
Herein, the work further discusses the influence of alkalinity on the synthesis of hierarchical LTA zeolites. The mechanism of hierarchical LTA zeolite formation was speculated. The effect of alkalinity in the gel on the crystallinity of mesoporous LTA was discussed through the characterization method of XRD. The effect of alkalinity on the pore structure and morphology of mesoporous LTA was investigated through the characterization method of nitrogen adsorption–desorption and SEM images. The Mg2+ exchange ability of hierarchical LTA zeolite was detected by the method of ion densimetry.
Experimental
Synthesis of mesoporous LTA zeolite
The synthesis method of mesostructured LTA zeolite followed a previous article.18 The influence of alkalinity in the synthesis gel was discussed. Thus, the intracrystalline mesoporous LTA zeolite was listed as follows: Al2O3:1.17SiO2:nNa2O:198H2O:0.11BP (bridged polysilsesquioxane) (n = 2.4–6.9). The conventional LTA zeolite is named A-0. The Na-type and Ca-type zeolite is described by Na-A-0 and Ca-A-0. The mesoporous LTA zeolite is named MA-x. The letter x is equal to 1, 2, 3 and 4. The molar ratios of Na2O/SiO2 of MA-1, 2, 3 and 4 are 2.05, 3.85, 4.85 and 5.85, respectively. Also, the Na-type and Ca-type mesoporous LTA zeolite is described by Na-MA-x and Ca-MA-x.In the synthesis of mesoporous LTA zeolite, sodium aluminate and distilled water were stirred until the sodium aluminate was completely dissolved. Then, sodium hydroxide was added to the above solution and the mixture was stirred continually for 2 h. The desired amount of bridged polysilsesquioxane (C24H57O12NSi3, molecular weight 635) was dropped into a sodium silicate solution in another beaker. After the above silicate mixture was stirred for 2 h, the silicate mixture was added stepwise into the sodium aluminate mixture. Then, the final mixture was further stirred for 1 h at room temperature. Crystallization was carried out in a Teflon-coated stainless-steel autoclave for a period of time at 373 K. Then, the autoclave was quickly cooled. The final product was washed with distilled water, filtered by centrifugation, dried at 373 K overnight and calcined in air at 823 K.
Ca2+ and Mg2+ exchange of zeolite Na-A
The ion exchange with Ca2+ of the Na-type zeolite sample was conducted by magnetically stirring zeolite samples in a 0.5 mol L−1 CaCl2 solution for 2 h at room temperature. This procedure was repeated three times to achieve the maximum ion exchange.18 After that, the sample was washed with deionized water three times until there was no remaining Cl−. Then, the ion-exchanged product was dried at 373 K overnight and calcined in air at 823 K.100 mL of 0.05 mol L−1 MgCl2 solution was added to a beaker. Then, the electrodes of the ion densimeter were placed in the above beaker. 0.1 g of zeolite sample was put into the solution. The amount of ion exchange with Mg2+ of the Na-type zeolite sample was recorded over a period of time.
Characterization
Powder X-ray diffraction (XRD) patterns were obtained using a Shimadzu XRD-6000 diffractometer equipped with a Ni filter using Cu Kα (k = 0.15418 nm) radiation (40 kV, 30 mA). The degree of crystallinity of the samples was quantitatively estimated using the following method. The XRD diffraction peak area of conventional LTA zeolite between 5 and 35° was taken as 100% crystallinity. The XRD diffraction peak area of the synthesized samples in the 2θ range of 5–35° was integrated. The estimated peak area was compared with conventional LTA zeolite to obtain relative crystallinity (RC). The skeletal structure of the samples was determined by using a Shimadzu IR Affinity-1 Fourier Transform Infrared Spectrometer. The solid powder of samples was mixed with KBr at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 (mass ratio) and homogenized. Then, the samples were pressed into transparent flakes under a certain pressure. The wavenumber ranged from 4000 to 400 cm−1 and the scanning resolution was 4 cm−1. TG and DTG curves were recorded on a German NETZSCH STA409C thermogravimetric analyzer. Nitrogen adsorption–desorption isotherms were measured and analyzed at liquid nitrogen temperature (77 K) using a Quantachrome Nova2000e Surface Area & Pore Size Analyzer. Prior to the analysis, the samples were outgassed at 573 K under vacuum for 3 h. The total surface area was calculated by using the Brunauer–Emmett–Teller (BET) equation from the adsorption branch of the isotherms in the relative pressure range of p/p0 = 0.05–0.3. The micropore volume, micropore area and external surface area were calculated by using the t-plots. The pore size distribution and total volume were obtained with the DFT adsorption branch. The scanning electron microscopy (SEM) images were obtained by using a JSM-6700F instrument. Transmission electron microscopy (TEM) performed with a Philips Tecnai G2 F20 microscope.
:50 (mass ratio) and homogenized. Then, the samples were pressed into transparent flakes under a certain pressure. The wavenumber ranged from 4000 to 400 cm−1 and the scanning resolution was 4 cm−1. TG and DTG curves were recorded on a German NETZSCH STA409C thermogravimetric analyzer. Nitrogen adsorption–desorption isotherms were measured and analyzed at liquid nitrogen temperature (77 K) using a Quantachrome Nova2000e Surface Area & Pore Size Analyzer. Prior to the analysis, the samples were outgassed at 573 K under vacuum for 3 h. The total surface area was calculated by using the Brunauer–Emmett–Teller (BET) equation from the adsorption branch of the isotherms in the relative pressure range of p/p0 = 0.05–0.3. The micropore volume, micropore area and external surface area were calculated by using the t-plots. The pore size distribution and total volume were obtained with the DFT adsorption branch. The scanning electron microscopy (SEM) images were obtained by using a JSM-6700F instrument. Transmission electron microscopy (TEM) performed with a Philips Tecnai G2 F20 microscope.
Results and discussion
The mechanism of hierarchical LTA zeolite formation is shown in Scheme 1. The synthesized hierarchical LTA zeolite experienced rearrangements from amorphous nanoparticles into a crystal nucleus and finally crystalline zeolite.23 Amorphous silica particles, an essential component for the formation of viable nuclei, fully depolymerize into monomer, dimer or a low polymerization degree of silicate species33 in the alkaline solution mixture, as shown in Scheme 1A. The primary building unit (precursor species) of mainly silica nanoparticles is formed in the alkaline solution mixture. The hydrolysable methoxysilyl groups (–OCH3) at each end of bridged polysilsesquioxane are linked together by Si–C bonds, which have good chemical stability under the synthesis conditions of zeolites. The bridged polysilsesquioxane, which is introduced in the synthetic mixture of LTA zeolite, spontaneously hydrolyzes into silicon hydroxyl in the alkaline medium, as shown in Scheme S1.†1,16,34,35 Meanwhile, an intense reaction between bridged polysilsesquioxane and aluminosilicate species indicates that silicon hydroxyl species easily condensed into the crystal nucleus of zeolite in alkaline medium. Thus, Si–O–Si covalent bonds were formed and the organosilane long chains of bridged polysilsesquioxane were tightly linked to the surface of the zeolite nucleus (Scheme 1B and S1†).1,16,18 The covalently Si–O–Si bonded framework formed, avoiding phase separation between the zeolite phase and mesopore phase. The amorphous nanoparticles finally converted into crystalline LTA zeolite (Scheme 1C). The incorporation of mesoporogen bridged polysilsesquioxane into crystalline LTA zeolite can be proved by the NMR spectrum.18 Two distinct resonances at −20.05 and −57.24 ppm in the NMR spectrum of uncalcined hierarchical LTA zeolite correspond to the T2 (C2–SiO2) and T3 sites (C–SiO3), respectively, indicating that mesoporogen bridged polysilsesquioxane still remains in the mesopores of uncalcined hierarchical LTA zeolite.36,37 Besides, bridged polysilsesquioxane is linked to the crystal surface of hierarchical LTA zeolite through Si–O bonds. As shown in the partial enlarged detail of Scheme 1C, the black line signifies the mesoporogen bridged polysilsesquioxane, which is covalently linked with silica, and the hydrophobic long chain moiety of bridged polysilsesquioxane is exposed on the surface of the silica. The hierarchical LTA zeolite (Scheme 1D) contains abundant mesopores after removal of the mesoporogen. Moreover, N2-adsorption/desorption data and the TEM image proved the existence of mesopores and the porosities of the hierarchical LTA zeolite.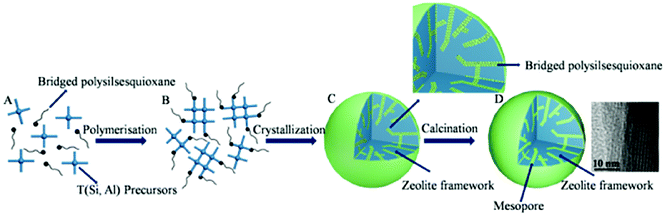 | ||
| Scheme 1 Schematic illustration for mesopore preparation in hierarchical LTA zeolite. | ||
The molar gel compositions and crystallinity of synthesized hierarchical LTA zeolites and conventional LTA are shown in Table 1. The powder X-ray diffraction (XRD) patterns of synthesized LTA zeolites with different molar ratios (n(Na2O/SiO2) = 2.05–5.85) and conventional LTA are shown in Fig. 1. The amorphous aluminosilicates completely transformed into crystalline LTA zeolite during hydrothermal treatment at 373 K. The characteristic diffraction peaks definitely belong to the characteristic diffraction peaks of the LTA topology structure.38
| Samples | Molar gel composition | RC (%) | ||
|---|---|---|---|---|
| Na2O/SiO2 | SiO2/Al2O3 | BP/Al2O3 | ||
| Na-A-0 | 2.05 | 1.17 | 0 | 100 |
| Na-MA-1 | 2.05 | 1.17 | 0.11 | 56.5 |
| Na-MA-2 | 3.85 | 1.17 | 0.11 | 84.9 |
| Na-MA-3 | 4.85 | 1.17 | 0.11 | 95.1 |
| Na-MA-4 | 5.85 | 1.17 | 0.11 | 91.1 |
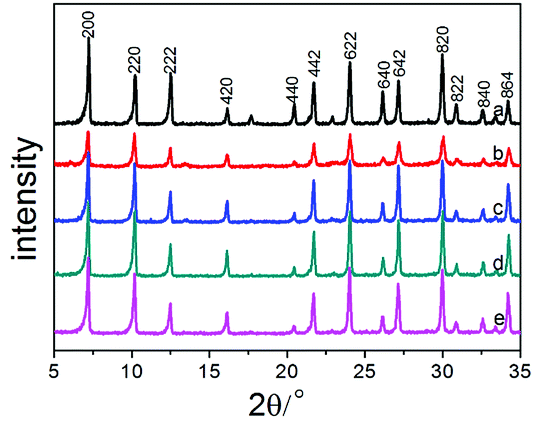 | ||
| Fig. 1 Powder X-ray diffraction patterns of Na-A and Na-MA-zeolites: (a) Na-A-0; (b) Na-MA-1; (c) Na-MA-2; (d) Na-MA-3; (e) Na-MA-4. | ||
As shown in Fig. 1, the intensity of the diffraction peaks first increases and then decreases as the amount of Na2O is increased. Comparing to the conventional LTA zeolite, the relative crystallinity (RC) also has the same tendency as the intensity of the diffraction peaks, as listed in Table 1. The relative crystallinity of zeolite LTA (MA-3) reaches 95.1%, which is close to the relative crystallinity of the conventional LTA zeolite, indicating the high crystallinity of the synthesized LTA. The crystallinity of mesoporous LTA increased along with the increase of Na2O amount. The increase of free Na+ induces the formation of nuclei in the gel, increases the number of lower-polymerized silicate anions (monomers and dimers) and accelerates the crystallization of the synthesized materials.39–42 The crystallinity of MA-3 is higher than that of MA-1 and MA-2 at the same crystallization time because of the higher amount of Na2O in the sample of MA-3. The following decrease in the crystallinity of MA-4 will be explained later.
Interestingly, for the same amount of Na2O (n(Na2O/SiO2) = 3.85) added into the gel without the template, the diffraction peaks of sodalite appeared (Fig. 2). The sample (Na-A-3-no template) contains almost pure sodalite when increasing the amount of Na2O (n(Na2O/SiO2) = 4.85) (Fig. 2). Those diffraction peaks, such as (110), (211), etc., could be assigned to the standard structural data, which indicates that the alkalinity changed the structure of the synthesized material and has a remarkable influence on the synthesis of zeolites.43 According to Greer, more aluminosilicates will dissolve into the solution when more NaOH is added, thus resulting in a lower viscosity in the gel and accelerating the crystallization of zeolite.44 The higher the alkalinity, the faster the crystallization rate of zeolite, and the easier it is to obtain the sodalite phase. Furthermore, a higher alkalinity of the system will lower the degree of polymerization of polysilicate, which is beneficial to the formation of four-membered rings during the crystallization of the silica–alumina gel. Therefore, pure sodalite was synthesized at higher alkalinity under the same crystallization conditions.
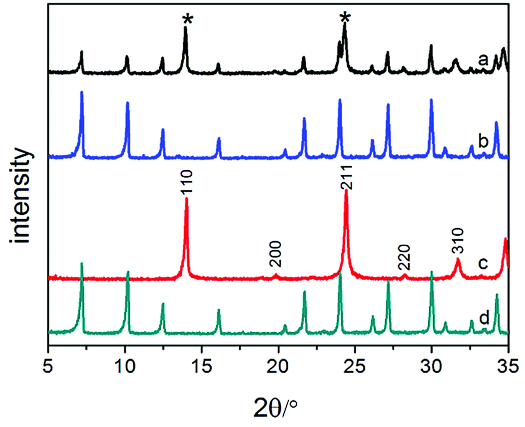 | ||
| Fig. 2 Powder X-ray diffraction patterns of Na-A and Na-MA-x zeolites: (a) Na-A-2-no template; (b) Na-MA-2; (c) Na-A-3- no template; (d) Na-MA-3. | ||
The hydrolysis degree of siloxanes on the bridged polysilsesquioxane is higher in a relatively higher alkaline environment in a gel involving a mesoporogen.33 The hydrolysis of siloxanes on the template consumes alkali in the gel and the alkalinity in the gel decreases. Consequently, pure mesoporous LTA is obtained by adding the bridged polysilsesquioxane into the solution in spite of the higher alkaline environment in the gel.
FTIR spectroscopy was performed on the mesoporous Na-MA-x (x = 2–4) zeolites synthesized in the presence of bridged polysilsesquioxane. LTA zeolite shows bands at around 468, 560, 670 and 1010 cm−1 (Fig. 3). The bands centred at 670 and 1010 cm−1 belong to the asymmetric and symmetric stretching vibrations of tetrahedra on the basis of previous studies.45,46 The band of internal bending of the tetrahedron is centered at 468 cm−1. The band at 560 cm−1 is the characteristic band of a double four ring, and it is assigned to the external vibration of Al–O and Si–O of the double four ring (4DR). The intensity of the band at 560 cm−1 signifies the intensity of structural tetrahedra. As shown in Fig. 3, the intensity of the structural tetrahedra of mesoporous zeolites Na-MA-3 and Na-MA-4 is higher than the sample Na-MA-2.
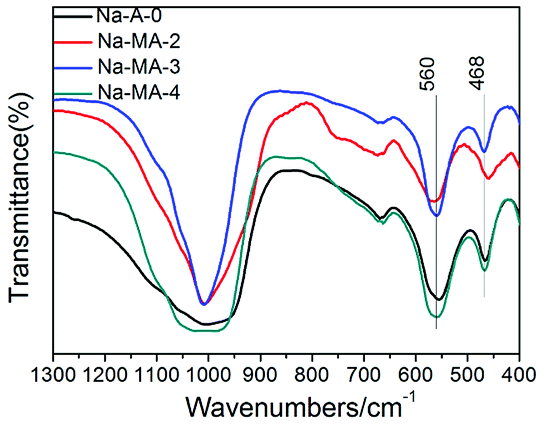 | ||
| Fig. 3 FTIR spectra of Na-A-0 and Na-MA-x samples (1300–400 cm−1). | ||
As shown in Fig. 4, the thermogravimetric curve displays the weight losses at different stages. It also provides detailed thermodynamic information through thermogravimetric (TG) analysis and differential thermogravimetry (DTG). The TG curve of the synthesized LTA sample shows that total weight loss (20.54%) occurs mainly in the following two major steps. A weight loss (14.33%) is observed within 200 °C and the amount of weight loss above 200 °C is 6.21%. There are three peaks observed in the DTG curve along with two fairly noticeable humps and a weak peak in the range of 100 °C to 700 °C. The hump around 150 °C indicates the removal of free water on the surface and intercrystalline water in the mesoporous LTA. The sharp peak at about 318 °C can be ascribed to the elimination of alkyl groups and amine groups of bridged polysilsesquioxane, while the shallow dip between 450 °C and 550 °C signifies a small fragment of residual bridged polysilsesquioxane molecules in the mesopores of LTA. The above result indicates that bridged polysilsesquioxane has successfully been incorporated into the synthesized material.9,22
 | ||
| Fig. 4 TG and DTG curves of as-prepared mesoporous LTA (Na-MA-3). | ||
The Na+ cations in the conventional Na-A zeolite occupy positions in the cages, which reduces the size of the pore apertures from 4.4 Å to less than 3.1 Å. Therefore, nitrogen molecules (kinetic radius ∼ 3.4 Å) at 77 K cannot enter the super-cage of zeolite LTA.23,45,47 The micropores of zeolite LTA could be detected by nitrogen adsorption after ion-exchange with Ca2+ of the sample. As shown in Fig. S1,† the Ca2+ exchanged samples still have the LTA topology, with higher crystallinity.
The N2 adsorption/desorption isotherms and pore size distribution (Fig. 5) of Ca-A-0 and Ca-MA-x (x = 1–4) exhibit detailed information on the difference of the pore properties. The sharp increase in the amount of N2 adsorption at P/P0 < 0.05 shows the presence of micropores, yet the gradual increase in the pressure range of P/P0 = 0.2–0.9 for Ca-MA-x suggests the existence of mesopores inside the zeolite LTA. The N2 adsorption/desorption isotherm of the conventional LTA shows no significant increase in the pressure range of P/P0 = 0.2–0.9. The distribution of pores with limited necks of uniform aperture can be observed from the shape of the isotherms and the type of the hysteresis loops.48,49 The pore size distribution, calculated by the NLDFT method (Fig. 5b), displays that the pore diameters center at 3–5 nm, which is the length of a dimer of bridged polysilsesquioxane, indicating the co-existing of mesopores and micropores in the zeolite LTA.22
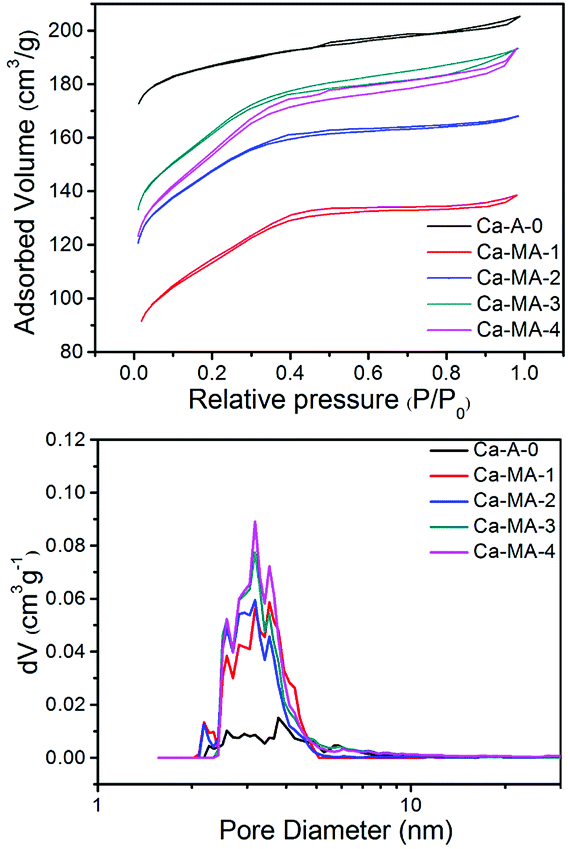 | ||
| Fig. 5 N2 adsorption/desorption isotherms at 77 K of Ca-A and Ca-MA-x samples and corresponding NLDFT pore size analysis derived from the adsorption branch. | ||
The textural properties of the synthesized materials Ca-MA-x (x = 1–4) prepared with bridged polysilsesquioxane and conventional LTA (Ca-A-0) are listed in Table 2. The textural properties of the samples include the BET surface areas, external surface areas, mesoporous volume, microporous volume, and hierarchical factor (HF). It can be seen that the amount of mesoporosity of Ca-MA-x shows an increasing trend along with the increase of the amount of alkali in Table 2. This manifests that the meso- and microporosity can be tuned and controlled by changing the amount of Na2O in the preparation of zeolites under the condition that a certain amount of templating agent has been added. The hierarchical factor (HF) is one of the most important tools to evaluate hierarchical zeolites. In the zeolite material, a higher HF signifies a larger mesopore surface area without severe damage to the micropore volume.50 As listed in Table 2, the HF of the as-synthesized zeolite LTA changes little, except Ca-MA-2 because of the existence of some relatively smaller pores (2.2 nm), which are due to monomers of bridged polysilsesquioxane.22 However, the HF of other Ca-MA-x samples indicates that the mesoporous LTA zeolites almost have the same hierarchical system.
In consequence, the synergetic effect between bridged polysilsesquioxane and a certain amount of Na2O changes the crystallinity and porosity of the mesoporous zeolite. Upon adding more NaOH into the gel, the external surface area of Ca-MA-4 increases to 287 m2 g−1. The higher hydrolysis degree of siloxanes on the bridged polysilsesquioxane in the relatively more alkaline environment results in the improvement of the effective utilization of the bridged polysilsesquioxane, thus improving the external surface area of Ca-MA-4. The total surface area of Ca-MA-4 declines slightly (565 m2 g−1), compared with the sample of Ca-MA-3 (603 m2 g−1). This can be explained by the fact that excess Na2O again dissolves the synthesized LTA zeolite and secondary crystallization occurs in the synthesis process.51,52 This is in agreement with the change of crystallinity (Table 1 and Fig. 1). Although the microporous volume and area of these samples (Ca-MA-x) are lower than those of conventional LTA (Ca-A-0), the crystallinity of these samples (Ca-MA-x) forcefully proves the synthesized high crystallinity of mesoporous LTA.
The improvement of alkalinity results in the increase of ‘‘free’’ Na+, strongly induces the formation of nuclei in the gel and greatly shortens reaction time.39,53,54 After the increase of alkalinity in the gel, the mesoporous LTA can be obtained under the condition of crystallization for 24 h at 373 K. The crystallization of mesoporous LTA is required for 168 h under the previous alkalinity (n(Na2O/SiO2) = 2.05). Meanwhile, the alkalinity increases the number of lower-polymerized silicate anions (monomers and dimers), which controls the formation of gels with a relatively higher aluminum content.40–42
The crystallization time is sharply shortened after adding more alkali into the system, which indicates that the rates of nucleation and growth of zeolite LTA significantly speed up. The improvement of alkalinity results in the decrease of viscosity in the liquid solution, which signifies that the solubility of the gel increases.20
The considerable influence of alkalinity on the crystal morphology is shown by the SEM images (Fig. 6) of the LTA samples (Na-MA-1, 2, and 3). The conventional zeolite LTA is regular cubic shape with sharp or truncated edges, based on previous investigations.18,22 The higher hydrolysis degree of siloxanes on the bridged polysilsesquioxane in a relatively higher alkaline environment results in the improvement of the linkage between aluminum and bridged polysilsesquioxane. Thus, the total silicon content linked with bridged polysilsesquioxane is reduced under the condition of higher alkalinity, which results in the relative excess of aluminum in the synthesized gel. Under such conditions, one of the silicon–oxygen bonds is distorted and shortened due to the existence of an aluminum atom on a neighboring Si site.55 The presence of aluminate ions delays the growth and nucleation rate at the points or in the directions of distortions, e.g., crystal edges and/or apexes (Fig. 6a and b).18
 | ||
| Fig. 6 SEM images of calcined hierarchical LTA zeolites synthesized by adjusting the alkalinity ((a1 and b2) Na-MA-1; (a2 and b2) Na-MA-2; (a3 and b3) Na-MA-3). | ||
A further increase of alkalinity causes partial or complete loss of crystal faces, which can be called ‘‘rounding’’ of the crystal sides and apexes (Fig. 6). The pseudo-spherical shape of mesoporous LTA (Fig. 6) illustrates that the growth rate of crystals is approximately the same in all directions under the given chemical conditions. Otherwise, it is more credible that a gradual ‘‘rounding’’ of mesoporous LTA is the reason for intensive surface nucleation in a highly supersaturated solution. Moreover, the rapid growth of the formed surface nuclei under such conditions is accompanied by a high roughness of the crystal surfaces (Fig. 6).
Conventional zeolite LTA shows a relatively slower removal ability of Mg2+ because of its higher charge density than Ca2+ and larger hydration sphere in solution. Fig. 7 shows the exchange curves of Na+ to Mg2+ at 298 K. As listed in Table 3, the Na-MA-x samples show faster initial exchange rates of Na+ to Mg2+ compared with that of Na-A-0. The exchange of 50% (defined as T50) was reached within 3 min for Na-MA-4 whereas the exchange of 50% for the sample of Na-A-0 was 36 min. Mg2+ exchange reached maximum (defined as T100) after 96 min for Na-MA-4, but more than 300 min was needed for the zeolite Na-A-0.
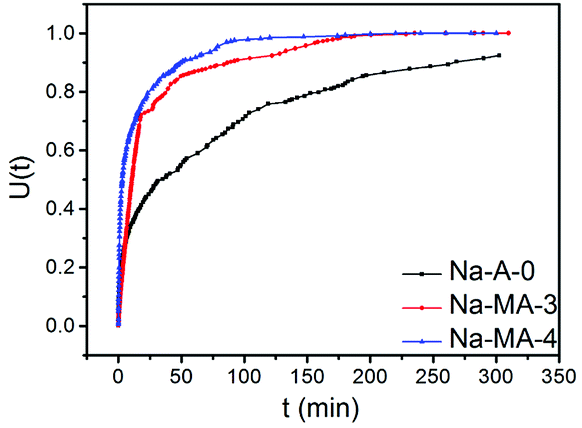 | ||
| Fig. 7 The exchange curves of Na+ to Mg2+ at 298 K for zeolite Na-A-0, Na-MA-3 and Na-MA-4. | ||
| Samples | Vmeso [cm3 g−1] | T50a (minutes) | T100b (minutes) |
|---|---|---|---|
| a T50: time of 50% exchange of Na+ to Mg2+.b T100: time of exchange of Na+ to Mg2+ to reach maximum (100 mL of a 0.05 mol L−1 MgCl2 solution was added into the beaker containing 0.1 g of zeolite sample). | |||
| Na-A-0 | 0.06 | 36 | >300 |
| Na-MA-3 | 0.16 | 12 | 176 |
| Na-MA-4 | 0.17 | 2.8 | 96 |
An important phenomenon is that the Na-MA-4 sample also shows faster initial exchange rates, 50% and maximum of Na+ to Mg2+ compared with those of Na-MA-3, as listed in Table 3. The main difference of the Na-MA-4 sample to the other samples is the higher mesopore volume than Na-MA-3. The presence of hierarchical pores in the LTA zeolites results in the distinct difference of the exchange rates of Na+ to Mg2+ for these samples, which shorten the lengths of diffusion paths and increase the approaching possibility of active sites. Therefore, Mg2+ more easily enters the exchange sites of Na+ in the zeolite particles.
Conclusions
The degree of hydrolysis of the methoxyl groups of bridged polysilsesquioxane increases under higher alkaline conditions. With the increase of alkalinity in the reaction mixture, the crystallization time sharply decreases. The increase of alkalinity causes a decrease of viscosity in the gel, thus the solubility of the gel increases faster than the dissolved zeolite. Furthermore, the mesoporosity and microporosity of hierarchical LTA zeolites are adjustable by changing the amount of Na2O. The methoxyl groups of bridged polysilsesquioxane spontaneously hydrolyze into hydroxyl groups in the silicate solution, which consumes an appropriate amount of alkali. The lower hydrolysis degree of methoxyl groups on the bridged polysilsesquioxane along with the lower alkalinity in the gel results in a decrease of the effective utilization of the bridged polysilsesquioxane. Therefore, a lower mesoporosity (0.13 cm3 g−1 and 0.10 cm3 g−1) of hierarchical LTA zeolites is obtained with the lower alkalinity in the gel. However, with a higher alkalinity in the gel, the higher hydrolysis degree of methoxyl groups on the bridged polysilsesquioxane improves the effective utilization of the bridged polysilsesquioxane, thus improving the mesoporosity (0.16 cm3 g−1 and 0.17 cm3 g−1) of Ca-MA-3 and Ca-MA-4. Besides, this study indicates that the morphology of zeolite LTA is related to the ratio of Na2O/Al2O3. The terminal SiO4 tetrahedra are distorted in the presence of aluminate ions, which constrain the growth of some crystal planes. The higher mesopore volume of these samples results in the faster exchange rates of Na+ to Mg2+ than conventional LTA. Also, the change of amount of alkali provides a kind of new tunable method of high mesopore volume in the range of a certain template amount.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 50972097).References
- M. Choi, H. S. Cho, R. Srivastava, C. Venkatesan, D. Choi and R. Ryoo, Nat. Mater., 2006, 5, 718–723 CrossRef CAS PubMed.
- Y. Tao, H. Kanoh, L. Abrams and K. Katsumi, Chem. Rev., 2006, 106, 896–910 CrossRef CAS PubMed.
- P. S. Wheatley, P. K. Allan, S. J. Teat, S. E. Ashbrook and R. E. Morris, Chem. Sci., 2010, 1, 483–487 RSC.
- M. Hartmann, Angew. Chem., Int. Ed., 2004, 43, 5880–5882 CrossRef CAS PubMed.
- Q. Q. Zhang, W. X. Ming, J. H. Ma, J. L. Zhang, P. Wang and R. F. Li, J. Mater. Chem. A, 2014, 2, 8712–8718 RSC.
- C. E. A. Kirschhock, S. P. B. Kremer, J. Vermant, G. Van Tendeloo, P. A. Jacobs and J. Martens, Chem.–Eur. J., 2005, 11, 4306–4313 CrossRef CAS PubMed.
- A. Corma, Chem. Rev., 1997, 97, 2373–2420 CrossRef CAS PubMed.
- I. Schmidt, A. Boisen, E. Gustavsson, K. Ståhl, S. Pehrson, S. Dahl, A. Carlsson and C. J. H. Jacobsen, Chem. Mater., 2001, 13, 4416–4418 CrossRef CAS.
- C. H. Christensen, I. Schmidt, A. Carlsson, K. Johannsen and H. Konrad, J. Am. Chem. Soc., 2005, 127, 8098–8102 CrossRef CAS PubMed.
- K. Selvaraj, Microporous Mesoporous Mater., 2010, 135, 82–89 CrossRef CAS.
- A. H. Janssen, I. Schmidt, C. J. H. Jacobsen, A. J. Koster and K. P. deJong, Microporous Mesoporous Mater., 2003, 65, 59–75 CrossRef CAS.
- A. Boisen, I. Schmidt, A. Carlsson, S. Dahl, M. Brorson and C. J. H. Jacobsen, Chem. Commun., 2003, 8, 958–959 RSC.
- W. Fan, M. A. Snyder, S. Kumar, P. S. Lee, W. C. Yoo, A. V. McCormick, R. L. Penn, A. Stein and M. Tsapatsis, Nat. Mater., 2008, 7, 984–991 CrossRef CAS PubMed.
- K. Egeblad, M. Kustova, S. K. Klitgaard, K. Zhu and C. H. Christensen, Microporous Mesoporous Mater., 2007, 101, 214–223 CrossRef CAS.
- H. Li, Y. Sakamoto, Z. Liu, T. Ohsuna, O. Terasaki, M. Thommes and S. Che, Microporous Mesoporous Mater., 2007, 106, 174–179 CrossRef CAS.
- R. Srivastava, M. Choi and R. Ryoo, Chem. Commun., 2006, 43, 4489–4491 RSC.
- W. J. Christopher, T. Katsuyuki and M. E. Davis, Nature, 1998, 393, 52–54 CrossRef.
- G. Song, H. Liu, F. Li, Z. Lv and J. Xue, J. Porous Mater., 2014, 21, 1101–1111 CrossRef CAS.
- G. Song, F. Li, Z. Lv and J. Xue, Mater. Lett., 2015, 161, 57–59 CrossRef CAS.
- E. I. Bassaldella, A. Kikot and J. C. Tara, Mater. Lett., 1997, 31, 83–86 CrossRef.
- T. Brar, P. France and P. G. Smirniotis, Ind. Eng. Chem. Res., 2001, 40, 1133–1139 CrossRef CAS.
- B. Bayati, A. A. Babaluo and R. Karimi, J. Eur. Ceram. Soc., 2008, 28, 2653–2657 CrossRef CAS.
- J. Addai-Mensah, J. Li, S. Rosencrance and W. Wilmarth, J. Chem. Eng. Data, 2004, 49, 1682–1687 CrossRef CAS.
- J. Grand, H. Awala and S. Mintova, CrystEngComm, 2016, 18, 650–664 RSC.
- M. Kumar, M. K. Choudhary and J. D. Rimer, Nat. Commun., 2018, 9, 2129 CrossRef PubMed.
- M. D. Oleksiak, J. A. Soltis, M. T. Conato, R. L. Penn and J. D. Rimer, Chem. Mater., 2016, 28, 4906–4916 CrossRef CAS.
- M. D. Oleksiak, K. Muraoka, M. F. Hsieh, M. T. Conato, A. Shimojima, T. Okubo, W. Chaikittisilp and J. D. Rimer, Angew. Chem., Int. Ed., 2017, 56, 13366–13371 CrossRef CAS PubMed.
- S. Mintova, J. Grand and V. Valtchev, C. R. Chim., 2016, 19, 183–191 CrossRef CAS.
- A. Sachse and J. Garcia-Martinez, Chem. Mater., 2017, 29, 3827–3853 CrossRef CAS.
- K. Li, J. Valla and J. Garcia Martinez, ChemCatChem, 2014, 6, 46–66 CrossRef CAS.
- N. Kosinov, J. Gascon, F. Kapteijn and E. J. Hensen, J. Membr. Sci., 2016, 499, 65–79 CrossRef CAS.
- H. Yamada, S. Sukenaga, K. Ohara, C. Anand, M. Ando, H. Shibata, T. Okubo and T. Wakihara, Microporous Mesoporous Mater., 2018, 271, 33–40 CrossRef CAS.
- R. K. Iler, J. Colloid Interface Sci., 1980, 75, 138–148 CrossRef CAS.
- N. Nishiyama, K. Horie and T. Asakura, J. Appl. Polym. Sci., 1987, 34, 1619–1630 CrossRef CAS.
- M. C. Brochier Salon and M. N. Belgacem, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 240–254 CrossRef CAS.
- H. Greer, P. S. Wheatley, S. E. Ashbrook, R. E. Morris and W. Zhou, J. Am. Chem. Soc., 2009, 131, 17986–17992 CrossRef CAS PubMed.
- R. R. Mukti, H. Hirahara, A. Sugawara, A. Shimojima and T. Okubo, Langmuir, 2010, 26, 2731–2735 CrossRef CAS PubMed.
- C. Baerlocher, L. B. McCusker and D. H. Olson, Atlas of zeolite framework types, Elsevier, 2007 Search PubMed.
- Z. Gabelica, N. Blom and E. G. Derouane, Appl. Catal., 1983, 5, 227–248 CrossRef CAS.
- A. V. McCormick, A. T. Bell and C. J. Radke, Zeolites, 1987, 7, 183–190 CrossRef CAS.
- T. W. Swadle, Coord. Chem. Rev., 2001, 219, 665–686 CrossRef.
- L. S. D. Glasser, Cem. Concr. Res., 1982, 3, 321–331 CrossRef.
- I. Hassan and H. D. Grundy, Can. Mineral., 1991, 29, 123–130 CAS.
- H. Greer, P. S. Wheatley, S. E. Ashbrook, R. E. Morris and W. Zhou, J. Am. Chem. Soc., 2009, 131, 17986–17992 CrossRef CAS PubMed.
- F. Hasan, R. Singh, G. Li, D. Zhao and P. A. Webley, J. Colloid Interface Sci., 2012, 382, 1–12 CrossRef CAS PubMed.
- A. Aronne, S. Esposito, C. Ferone, M. Pansini and P. Pernice, J. Mater. Chem., 2002, 12, 3039–3045 RSC.
- D. W. Breck, W. G. Eversole, R. M. Milton, T. B. Reed and T. L. Thomas, J. Am. Chem. Soc., 1956, 78, 5963–5972 CrossRef CAS.
- R. Dutartre, L. C. D. Ménorval, F. D. Renzo, D. Mcqueen, F. Fajula and P. Schulz, Microporous Mater., 1996, 6, 311–320 CrossRef CAS.
- M. R. Othman and I. S. Sahadan, Microporous Mesoporous Mater., 2006, 91, 145–150 CrossRef CAS.
- J. Pérez-Ramírez, D. Verboekend, A. Bonilla and S. Abelló, Adv. Funct. Mater., 2009, 19, 3972–3979 CrossRef.
- S. Sang, F. Chang, Z. Liu, C. He, Y. He and L. Xu, Catal. Today, 2004, 93, 729–734 CrossRef.
- S. D. Kim, S. H. Noh, K. H. Seong and W. J. Kim, Microporous Mesoporous Mater., 2004, 72, 185–192 CrossRef CAS.
- B. Subotić, J. Bronić and T. A. Jelić, Ordered Porous Solids, 2009, pp. 127–185 Search PubMed.
- L. Itani, Y. Liu, W. P. Zhang, K. N. Bozhilov, L. Delmotte and V. Valtchev, J. Am. Chem. Soc., 2009, 131, 10127–10139 CrossRef CAS PubMed.
- L. A. Bugaev, J. A. van Bokhoven, V. V. Khrapko, L. A. Avakyan and J. V. Latokha, J. Phys. Chem. B, 2009, 113, 4614–4618 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08952c |
| This journal is © The Royal Society of Chemistry 2019 |