
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of hollow ferrogadolinium nanonetworks for dual-modal MRI guided cancer chemotherapy†
Ting Tang‡
 ab,
Xiao Sun‡*c,
Xuedong Xu‡d,
Yifeng Biana,
Xiaojie Maa and
Ning Chen*a
ab,
Xiao Sun‡*c,
Xuedong Xu‡d,
Yifeng Biana,
Xiaojie Maa and
Ning Chen*a
aJiangsu Key Laboratory of Oral Diseases, Nanjing Medical University, Nanjing, 210029, China. E-mail: dentistchenning@126.com
bDepartment of Dental Implantology, Hefei Stomatology Hospital, Clinical School of Anhui Medical University, Hefei, 230001, China
cDepartment of Chemical and Biomolecular Engineering, National University of Singapore, Singapore 117585, Singapore. E-mail: xiaosun8000@163.com
dChina Shijiazhuang Pharmaceutical Group Co., Ltd., Shijiazhuang, 050038, China
First published on 18th January 2019
Abstract
The development of hollow ferrogadolinium nanonetworks has not been reported for nanomedicine application until now. In this study, we developed a hollow and porous ferrogadolinium nanonetwork structure using the one-pot solvothermal method. This nanoparticle could be simultaneously used as a T1 and T2 dual-modal magnetic resonance imaging (MRI) contrast agent. In addition, the hollow lumen and abundant pores of the nanonetworks maximized the loading capacity and conferred the nanoplatforms for suitable anticancer drug loading capacity. Using these nanonetworks, MRI and anticancer experiments were conducted in vitro and satisfactory dual-modal MRI and cancer chemotherapy results were obtained. Therefore, the nanonetworks with dual-modal MRI and drug loading abilities effectively complement the ferrogadolinium composites' library and hold great promise in nanomedicine for simultaneous cancer diagnosis and chemotherapy.
1. Introduction
Currently, magnetic resonance imaging (MRI) is one of the most powerful non-invasive imaging modalities used in the diagnosis of different cancers.1–3 However, more than half of the diseased patients require the use of an MRI contrast agent (CA) to enhance the contrast of the targeted site within the surrounding tissues and improve the resolution of the MRI images.4,5 There are two types of CAs commonly used: Gd-based compounds are often employed as T1 CAs and Fe-based magnetic nanomaterials are often utilized as T2 CAs.6–9 However, the inherent limitations of single mode CAs are often criticized, thus rendering dual-modal MRI CAs as more acceptable because the synergistic combination of T1-and T2-weighted MRI can provide more comprehensive imaging information, increase the diagnostic accuracy, and improve the overall prognosis of patients.10,11In the study of dual-modal MRI CAs, most attention has been paid to ferrogadolinium nanocomposites because Gd- and Fe-based compounds are commonly used as T1 and T2 CAs, respectively.12,13 It is worth noting that as T1 CAs are directly in contact with the magnetic core and located within the magnetic field generated by the magnetic core and opposite to the external magnetic field, T2 CAs can severely perturb T1 imaging.6,7,14 Hence, T1 CAs should be on the inside or away from the outside of the magnetic core. However, many previous studies have failed to observe this.15–17 Many interesting nanostructures of ferrogadolinium composites have been reported to reduce the interference of T2 CAs for T1 imaging, including yolk-like structures, barbell-like structures, and others.6,18 However, their synthesis process is multi-step and very complicated. Furthermore, many of these nanostructures are limited in their ability to load drugs because of their non-porous surface structures.6,7,15–23
Up until now, a hollow and porous ferrogadolinium nanonetwork (HPFN) has not been reported. In this case, Gd3+ is located inside the HPFN and has a parallel spin ordering with the same direction as the magnetic field induced by the Fe-based material, which will not disturb the T1 imaging, thus providing effective T1 imaging.7,10,24 Moreover, the porous and hollow structure is beneficial for the water molecules contacting T1 and T2 CAs as well as for efficient drug loading.
In this study, we developed a HPFN using the one-pot solvothermal method. This nanoparticle simultaneously and efficiently exerted T1 and T2 contrast ability. In addition, the hollow lumen and abundant mesopores conferred the nanonetworks a high specific surface area, which was critical for the abundant loading of anticancer drugs. Cisplatin, a commonly used anticancer drug,25,26 was selected as the model drug and loaded into the nanonetworks. Using these nanonetworks, MRI and anticancer experiments were conducted in vitro and satisfactory dual-modal MRI and cancer chemotherapy results were obtained. Therefore, the easily synthesized nanonetworks with dual-modal MRI and drug loading abilities effectively complement the ferrogadolinium nanocomposites' library and holds great promise in nanomedicine for simultaneous cancer diagnosis and chemotherapy.
2. Materials and methods
2.1. Chemicals
All obtained chemicals were of analytical grade and used without further purification. Fe(acac)3, Gd(acac)3, 1,6-hexanediamine, CH3COONa, cisplatin, ethylene glycol, and fluorescein isothiocyanate (FITC) were purchased from Aladdin Co. (Shanghai, China). All other chemicals were provided by the Sinopharm Co. (Shanghai, China).2.2. Preparation of amine-functionalized HPFN
Fe(acac)3 (0.5 g), and Gd(acac)3 (0.5 g) were added into ethylene glycol (30 mL) and vigorously stirred at 80 °C for 1 h. Then, 1,6-hexanediamine (3.6 g) was added into the solution and stirred at 80 °C for an additional 0.5 h. CH3COONa (4 g) was then added into the solution and reacted for 0.5 h. The mixture was sealed in a Teflon-lined stainless-steel autoclave and heated at 200 °C for 24 h. The resulting product was precipitated by centrifugation, briefly rinsed with water and ethanol three times to effectively remove the solvent and excess 1,6-hexanediamine, and then dried at 60 °C before characterization and application.2.3. Loading cisplatin into HPFN
HPFN (30 mg) were dispersed into 10 mL of cisplatin solution (2 mg mL−1) under stirring overnight. Cisplatin-loaded HPFN nanonetworks (designated as HPFNC) were centrifuged and the supernatant was detected by inductively coupled plasma optical emission spectrometry (ICP-OES) to quantify the unloaded cisplatin. The loading capacity of cisplatin could be calculated using the equation LC (%) = [2 mg mL−1 − c(cisplatin)sup] × V/mnanoplatform × 100%. In the in vitro experiments, the HPFN and HPFNC were re-suspended in distilled water and filtered using a 0.8 µm-membrane filter.2.4. Observing the cellular uptake of HPFN in vitro
Confocal laser scanning microscopy (CLSM) was used to define the intracellular localization of fluorescein isothiocyanate (FITC)-labeled HPFN (designated as HPFN-FITC) in HeLa cells. To form HPFN-FITC conjugates, FITC was loaded into the HPFN. HeLa cells seeded in 60 mm-polystyrene plates (5 × 105 cells per dish) were treated with HPFN-FITC (24 µg mL−1) for 1 h, 3 h, and 6 h, and then stained with NucRed Live 647 ReadyProbes Reagent (Invitrogen, USA) for 15 min at 37 °C in the dark. The cells were washed twice with PBS (pH 7.4) and fixed with 4% paraformaldehyde for 30 min. The cells were then visualized using a Leica SP8 confocal microscope with 100/63× oil-immersion objective lens.2.5. Measuring MRI in vitro
The longitudinal (T1) and transverse (T2) relaxation time of HPFN was detected using a 9.4 T MRI scanner. The T1 and T2 relaxation rates were measured using the inversion recovery method and a multi-echo spin echo sequence, respectively. The imaging parameters were as follows: TR = 6000 ms; TE = 5.6 ms; slice thickness = 1.0 mm; matrix = 96 × 96. Relaxivity (r1 and r2) was determined using a linear fit of the relaxation time as a function of the amount of Gd or Fe ions determined by ICP-OES.2.6. Cytotoxicity assays
HeLa cells at a density of 5 × 104 cells per m2 were incubated in a humidified incubator at 37 °C for 24 h. The adherent HeLa cells were then treated with different concentrations of cisplatin and HPFNC in the growth medium at 37 °C in 96-well plates for 24 h and 48 h. Following this, MTT (5 mg mL−1, 10 µL) was added to each well and incubated for an additional 4 h. The supernatant was removed, and 100 µL of DMSO was added to completely dissolve the formazan crystals by vigorously shaking the cell culture plate for 10 min until no particulate matter was visible. The absorbance in each well was measured at 570 nm using a microplate reader (Biotek, USA). The biocompatibility of the HPFN was also tested by following the above process, except that cisplatin was replaced with HPFN.2.7. Characterization
The general morphology and microstructure of the sample were observed under a scanning electron microscope (SEM; Hitachi, Ltd., Tokyo, Japan) and a H-800 transmission electron microscope (TEM; Hitachi, Ltd., Tokyo, Japan). The nitrogen adsorption–desorption isotherm of the sample was measured using a porosimetry analyzer (Tristar II, 3020M, Micromeritics, USA). The structure and interaction of the sample was characterized using an X-ray diffractometer (XRD) (TTR-III, Rigaku Co., Japan) and an X-ray photoelectron spectroscope (XPS) (Thermo ESCALAB 250XI, USA), respectively. The composition of the sample was determined using a Thermo Nicolet iS10 Fourier transform infrared (FT-IR) spectrometer (Thermo Electron, Co., USA), and the spectral data were processed using the software of the spectrometer (OMNIC). The magnetic behavior of the sample was measured by a superconducting quantum interference device magnetometer (Bruker Co., Germany).3. Results and discussion
3.1. Synthesis and characterization of HPFN
As shown in Scheme 1, we successfully synthesized a multifunctional HPFN nanomaterial using a simple solvothermal method. Gd3+ could localize anywhere within the nanonetworks, and it had a parallel spin ordering with the same direction as the magnetic field induced by the Fe-based material. At this point, the Fe-based material did not disturb Gd3+; instead, it further enhanced the T1 imaging. In addition, the porous and hollow structure with a large specific surface area was beneficial for the water molecules contacting T1 and T2 CAs as well as for sufficient loading of the drugs. In this case, HPFN could be used for simultaneous cancer diagnosis and chemotherapy.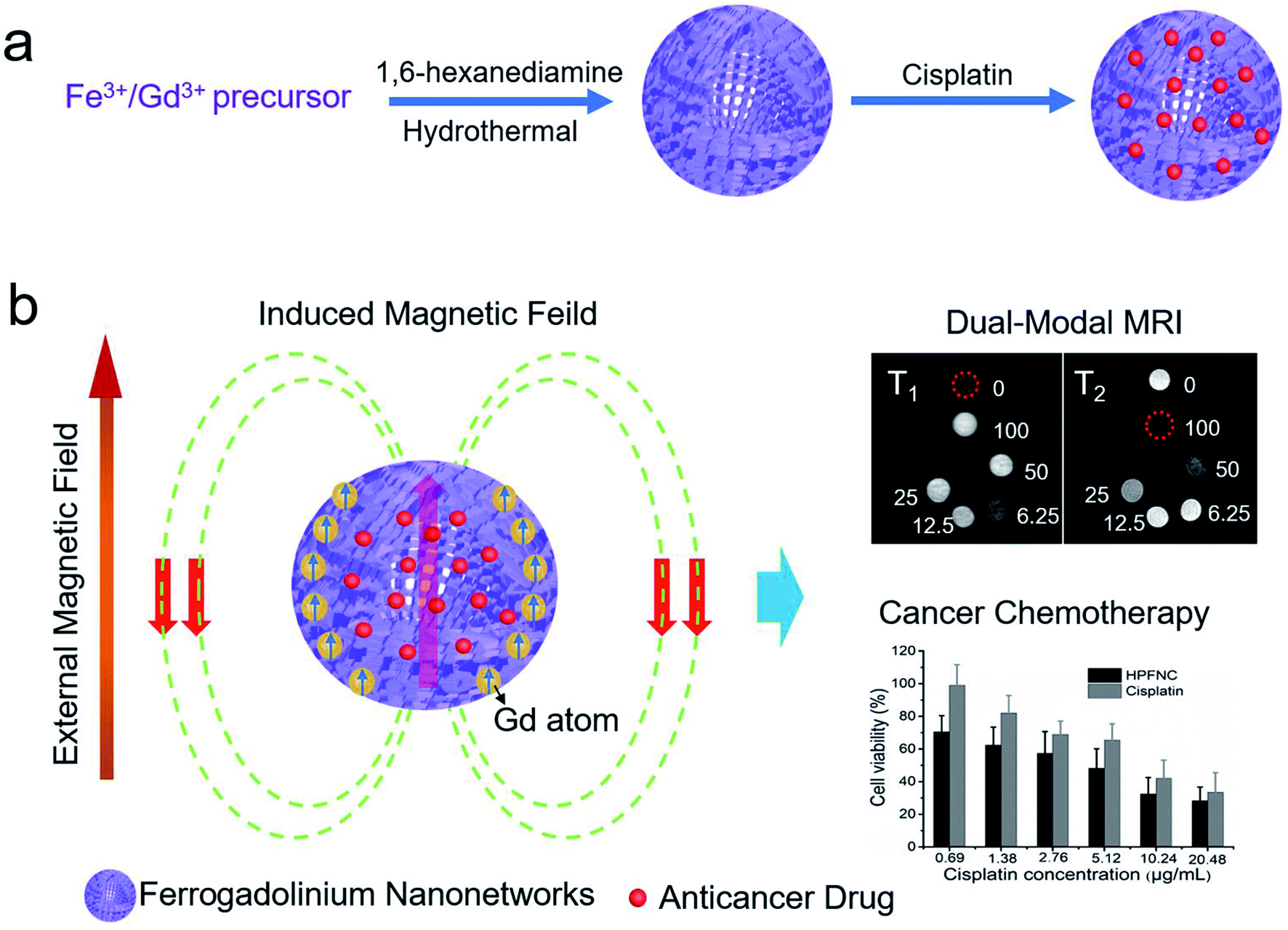 | ||
| Scheme 1 Schematic of (a) the synthesis for ferrogadolinium nanonetworks and (b) their applications in dual-modal MRI guided cancer chemotherapy. | ||
As shown in Fig. 1a and b, the TEM micrographs display that the HPFN adopted a hollow and porous nanonetwork architecture with homogeneous dispersion. It is well known that good dispersibility of the nanoparticles is important for the nanomedical applications. The nanonetworks could be well dispersed both in water and PBS, and the hydrodynamic size analysis of the HPFN also showed that the nanonetworks possessed homogeneous size distribution. These results were consistent with the observation from TEM images. XRD peaks appearing in the HPFN display broad and low-intensity peaks, which are likely to be indexed to spinel Fe3O4 (JCPDS no. 19-0629) and low-crystallinity Gd2O3 (JCPDS no. 12-0797) (Fig. 1c), confirming that the nanomaterial was composed of ferrogadolinium oxide. As previously reported, many modifiers with active groups such as amino or carboxylate groups could control the formation of nanomaterials and improve the colloidal stability and biocompatibility of the prepared nanomaterials during the reaction processes.27 The mechanism could be attributed to the fact that these charged groups can coordinate with metal cations in solution and affect the nucleation and mass transfer process of the nanocrystals, thus resulting in the formation of controllable nanostructures. Similarly, in our case, two –NH2 groups exist in the head and tail of each 1,6-hexanediamine molecule with five C–C bonds in the middle. As shown in Fig. S1,† during the solvothermal process, in the first 1 h, Fe(acac)3 and Gd(acac)3 were continuously degraded to release iron and gadolinium cations, which could easily be coordinated by 1,6-hexanediamine, probably forming large molecular networks by the linking of positively charged iron and gadolinium cation-coordinated 1,6-hexanediamine molecules.27 At the same time, the iron and gadolinium cations in the formed large molecular networks are slowly hydrated to form Fe(OH)3 and Gd(OH)3, and reduced by ethylene glycol to form Fe3O4 or undergo pyrolysis to form Gd2O3 at high temperature and pressure condition.28 During this process, the formed amorphous solid spheres grow continuously and start crystallizing at the surface of the spheres because the surface is in full contact with the solution.29 Compared with the large and well-crystallized particles on the exteriors of the spheres, the inner amorphous crystallites have higher surface energy, which provides the driving force for the Ostwald ripening. Therefore, the inner crystallites tend to dissolve, resulting in interior void space and porous structure.29
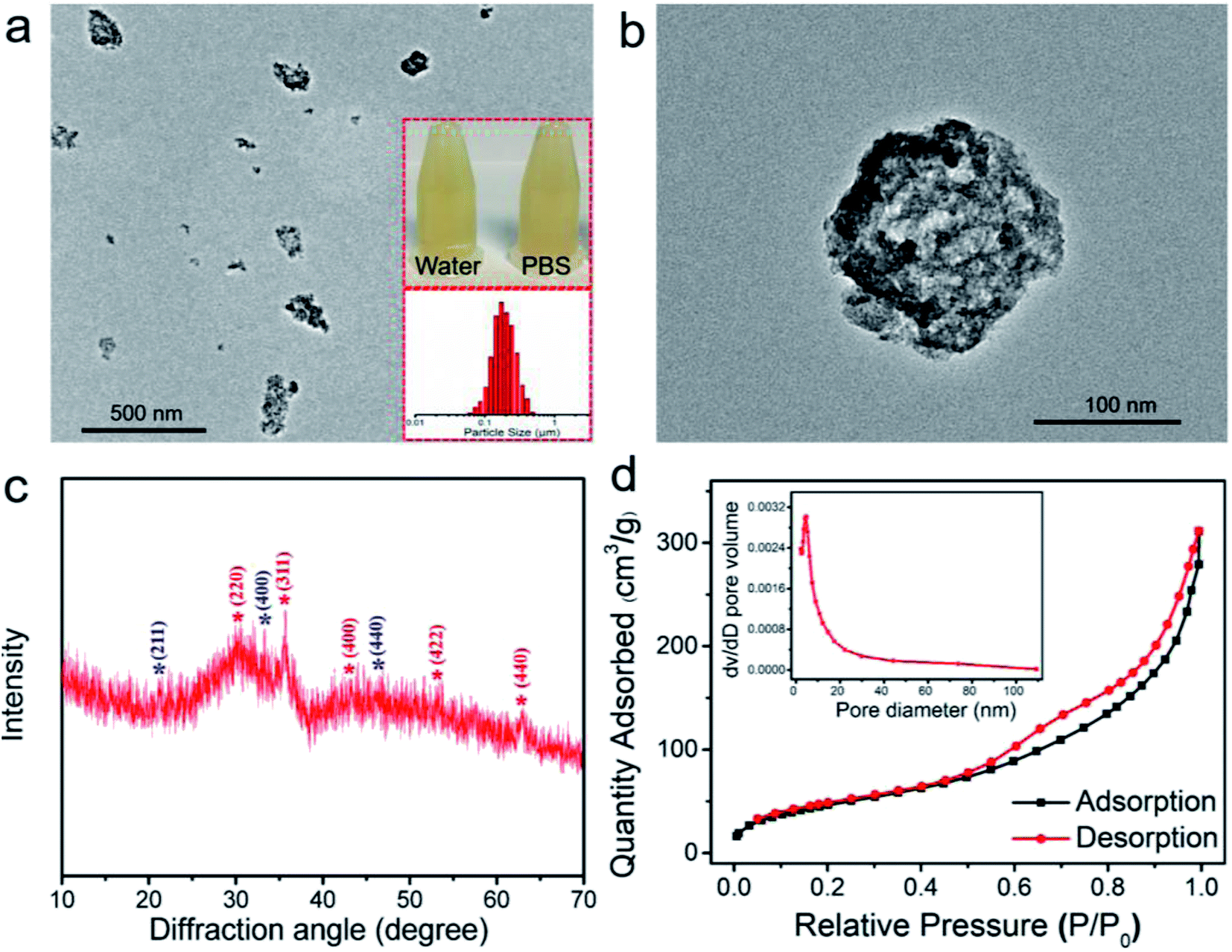 | ||
| Fig. 1 (a) Low magnification and (b) high magnification TEM images of HPFN. Inset: HPFN (0.5 mg mL−1) dispersed in water and DMEM medium. Hydrodynamic size distribution of HPFN. (c) XRD pattern, and (d) N2 adsorption/desorption isotherms of HPFN. | ||
Subsequently, the specific surface area and porosity of HPFN were analyzed using nitrogen adsorption–desorption isotherm curves (Fig. 1d). The data showed that the HPFN had a high specific surface area (175.3 m2 g−1) and a uniform pore size distribution (predominantly 4.6 nm). The isotherm curve showed that the typical hysteresis behavior at 0.5–0.8 P/P0 was attributed to the delay in nitrogen evaporation from the hollow-space of lumen, which was blocked by the surrounding mesopores. This finding demonstrated that HPFN is an ideal drug carrier.
XPS analysis of the samples (Fig. 2a) confirmed the presence of Fe, Gd, O, C, and N elements, indicating the existence of ferrogadolinium oxide and 1,6-hexanediamine in this nanocomposite. After loading cisplatin, a new peak at 73.3 eV (Pt 4f) appeared, indicating that cisplatin was successfully loaded into HPFN to form HPFNC. Moreover, the N 1s peak shifted from 399 eV (Fig. 2b) to 400.4 eV (Fig. 2c), confirming that cisplatin was loaded into HPFN through coordination between cisplatin and the NH2 group of HPFN. The drug loading amount was calculated as 7.9%. Subsequently, the composition of HPFN was confirmed using the FT-IR spectrum (Fig. 2d). The peaks at 519 cm−1, 588 cm−1, 1512 cm−1, and 1080 cm−1 were respectively assigned to Gd–O stretching vibration, Fe–O stretching vibration, N–H bending vibration, and C–N stretching vibration (Fig. 2d), confirming the existence of Gd2O3, Fe3O4, and 1,6-hexanediamine in HPFN.
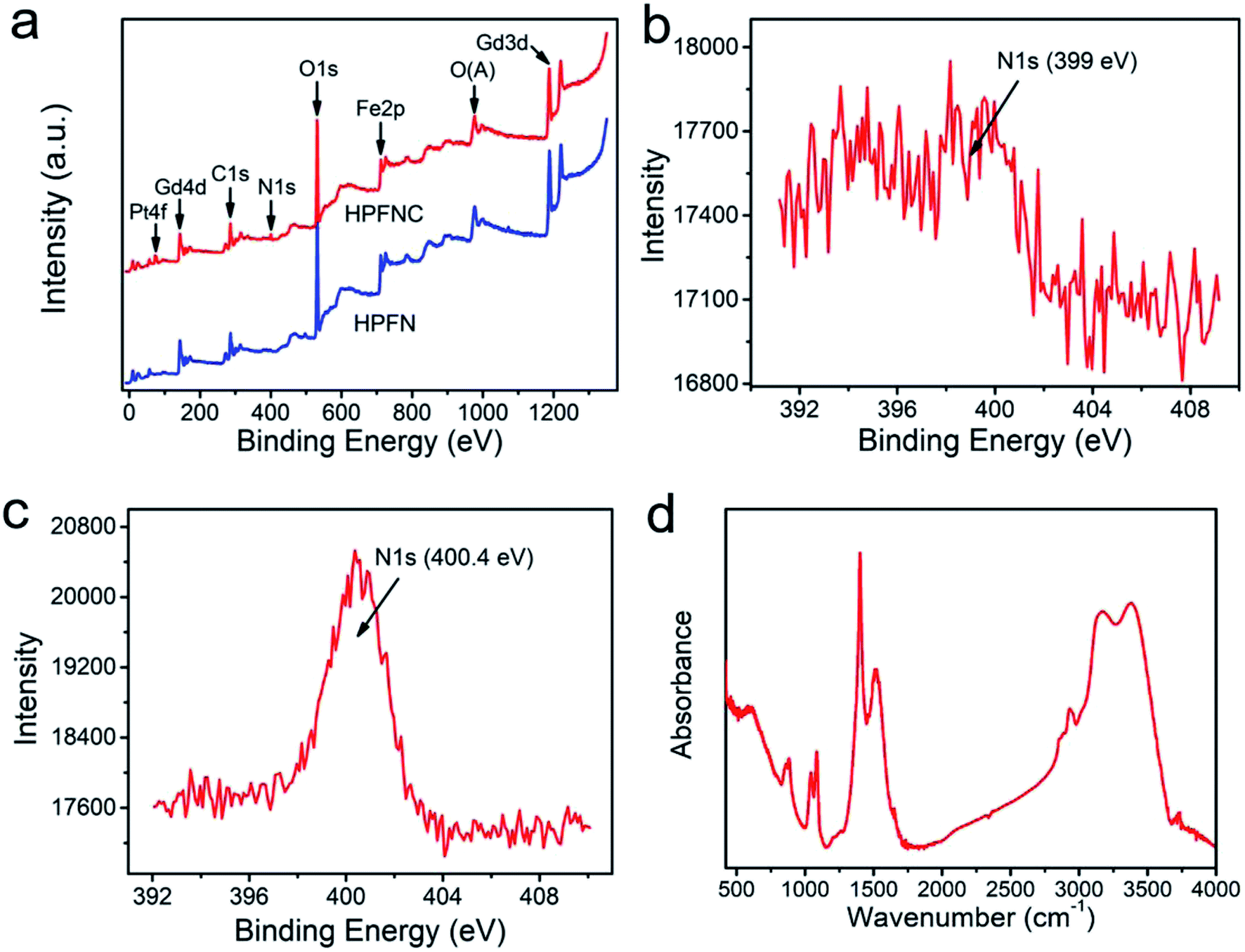 | ||
| Fig. 2 (a) Full XPS spectra and N 1s peaks of (b) HPFN, and (c) HPFNC. (d) FT-IR spectrum of HPFN. | ||
Field-dependent magnetization curves showed that the saturated magnetization value of HPFN was 9.6 emu g−1 at 300 K (Fig. 3a). In addition, the magnetization of HPFN continuously increased along with the increase in the applied magnetic field at 3 K, which may have been due to the enhanced spin canting effect of Gd3+ in HPFN.30,31 In addition, the hysteresis at 3 K indicated that the nanonetworks possessed superparamagnetism. Standard zero-field cooling (ZFC) and field cooling (FC) measurements of HPFN displayed similar curves compared to those of paramagnetic Gd2O3 at a low temperature, but gradually became similar to the curves of superparamagentic Fe3O4 with an increase in temperature (Fig. 3b). These magnetic properties of HPFN were similar to those previously reported for ferrogadolinium oxide composites,6 indicating that the nanonetwork was composed of ferrogadolinium oxide composite.
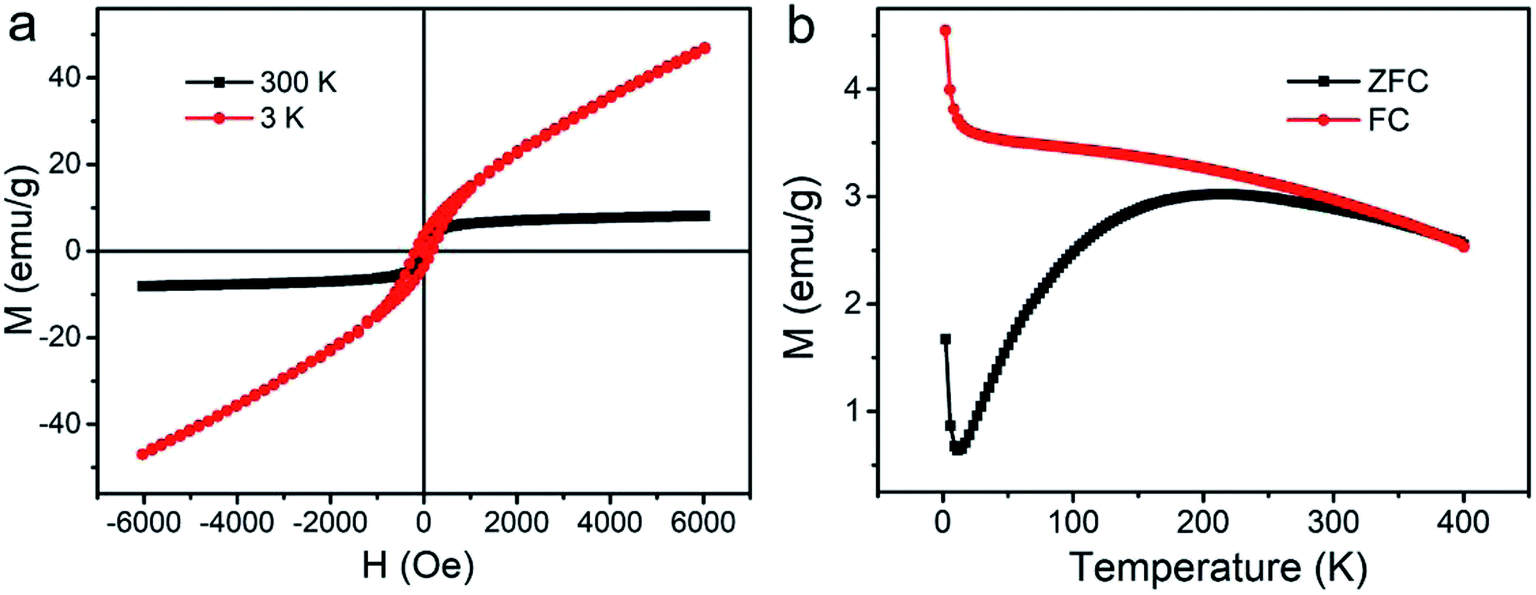 | ||
| Fig. 3 M − H and M–T curves of HPFN. | ||
3.2. Observation of uptake by HeLa cells
For the CLSM experiment, after the HeLa cells were incubated with HPFN-FITC for different times, the fluorescence of the cells displayed a time-dependent enhancement (Fig. 4). In the initial 1 h, weak green fluorescence was observed within the cytomembrane, indicating that only a few HPFN-FITC particles were localized within the cytomembrane of the HeLa cells. With the increase in incubation time, the fluorescence within the cytomembrane gradually increased, and weak fluorescence was also found in the cytoplasm. After 6 h of incubation, strong fluorescence distributed uniformly within the cells, suggesting that increasing number of HPFN-FITC particles crossed the cell membrane and entered the cytoplasm. These results confirmed that the HPFN could be effectively internalized by the HeLa cells, and can enhance the localized dose in HeLa cells.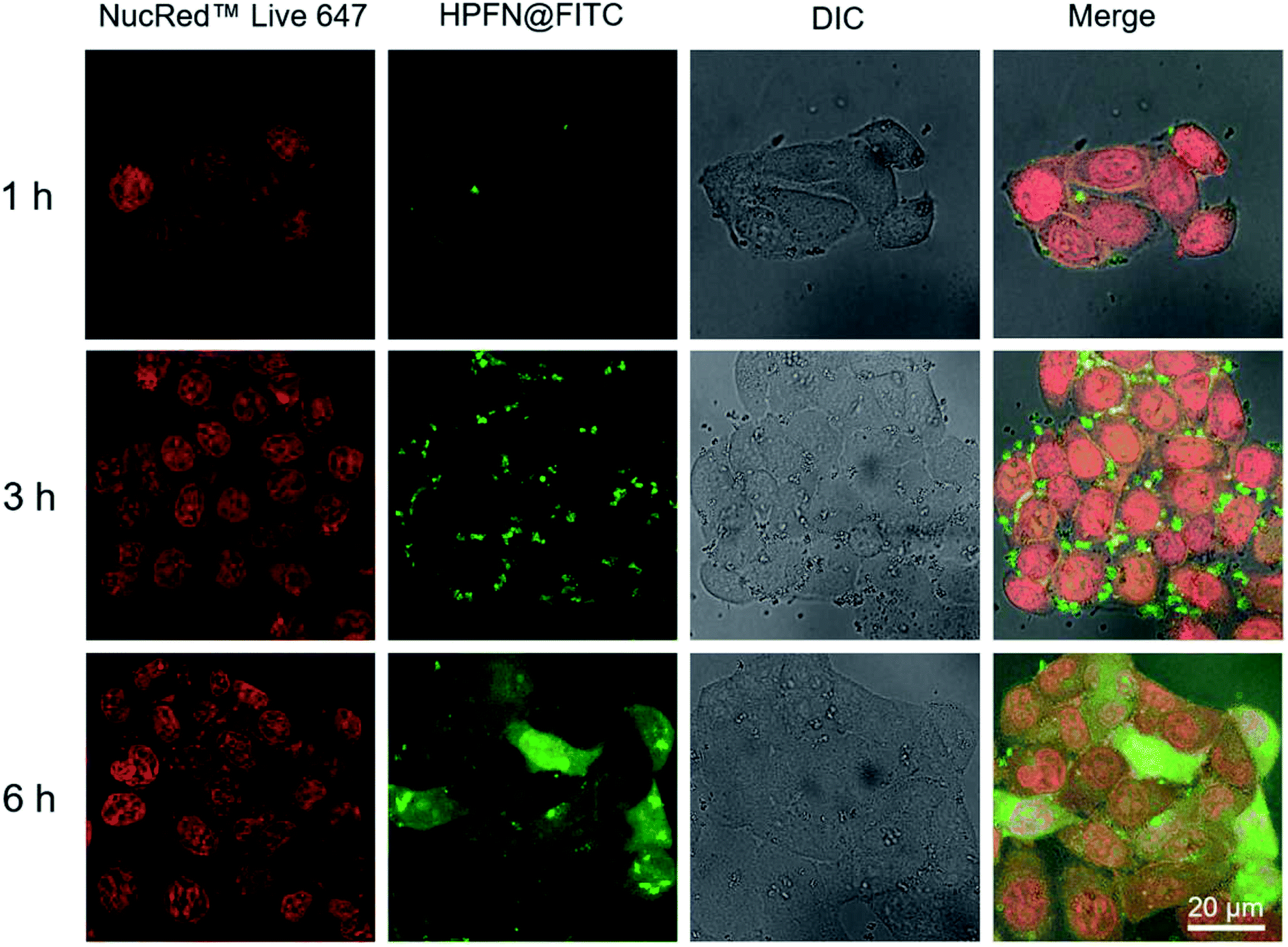 | ||
| Fig. 4 (a) CLSM images of HeLa cells treated with HPFN-FITC for different indicated times. For each panel, the images from left to right show cell nuclei stained by NucRed Live 647 ReadyProbes Reagent (red), FITC fluorescence in cells (green), bright field, and a merged image of the three left-most images, respectively. All images had the same scale bar of 20 µm. | ||
3.3. T1–T2 dual-modal MRI Performance
As shown in Fig. 5, with the increase in concentrations of HPFN, T1-and T2-weighted MR images gradually brightened and darkened, respectively, indicating that the HPFN had a dose-dependent MRI contrast ability. The r1 value of the HPFN was 6.63 s−1 mM−1, which was calculated as the ratio of the proton relaxation (1/T1) to the Gd3+ ion concentration. The r2 value of HPFN was 244.07 s−1 mM−1, which was calculated as the ratio of the 1/T2 to the Fe3+ ion concentration. These values were higher than the r1 and r2 values of the commercially available Gd or Fe-based contrast agents.32,33 On one hand, the high r1 and r2 values may be attributed to the porous and hollow structure of HPFN, which was beneficial for the water freely contacting Gd3+ ions or being close to Fe3O4 and thus, good T1 and T2 imaging was obtained. On the other hand, the Fe-based material inside the HPFN would not disturb the Gd3+ ions.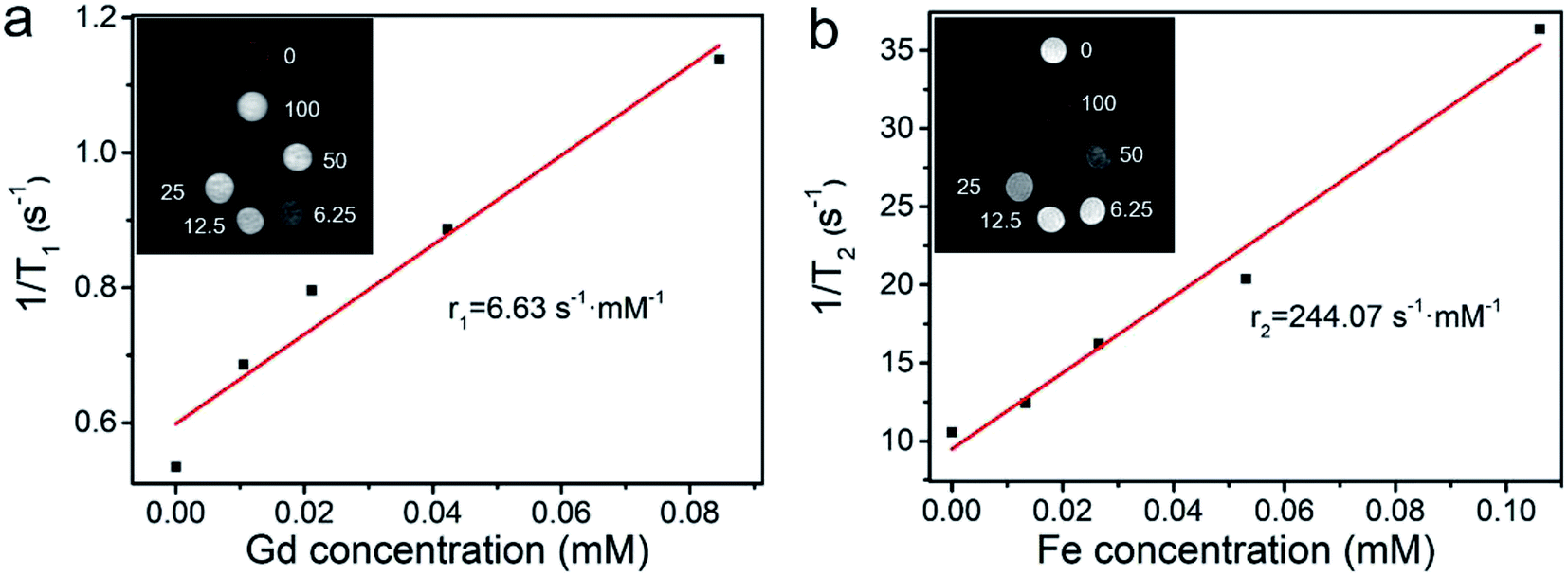 | ||
| Fig. 5 (a) T1 and (b) T2 relaxivity and MR images of HPFN at different concentrations. | ||
3.4. Investigation of biocompatibility and antitumor effect of the nanosystem
The release behavior of the drugs from HPFNC under different pH conditions was explored before it was used for the antitumor experiment (Fig. S2†). The result indicated that the cisplatin release was pH-dependent. Cisplatin had low drug release rate at pH 7.4. However, the drug release rate could be promoted under acidic condition. This was because the coordination effect between –NH2 and cisplatin could be easily destroyed under acidic conditions.28 We know that good biocompatibility of nanomaterials is important for biomedical applications. As shown in Fig. 6, no significant decrease in the viability of the HeLa cells was observed after they were incubated with increasing HPFN concentrations for 48 h, indicating that HPFN had good biocompatibility. Following this, the viability of the HeLa cells incubated with free cisplatin and HPFNC was assessed. We found that cellular apoptosis induced by free cisplatin and HPFNC was strictly proportional to the dose of the anticancer drug (Fig. 7). Clearly, HPFNC displayed higher cancer cell inhibition than free cisplatin. This was because HPFN enhanced the localized drug dose in the HeLa cells. In addition, compared with 24 h of co-cultivation, 48 h of co-cultivation of the HeLa cells with either free cisplatin or HPFNC resulted in enhanced cellular apoptosis, indicating that the cellular apoptosis induced by free cisplatin and HPFNC was proportional to the co-cultivation time. This was because cisplatin needed enough time to affect DNA transcription. It is worth noting that at a high drug dose (>10.24 µg mL−1) after 48 h of co-cultivation, free cisplatin displayed a slightly higher cancer cell inhibition than HPFNC. This might be attributed to the fact that at a high-dose of HPFNC and a longer co-cultivation time, too many HPFNC particles were taken up by the cells, which inhibited the cells' uptake of more HPFNC. This was also confirmed when only a slight decrease in the viability of the HeLa cells was obtained on further increasing the dose of HPFNC.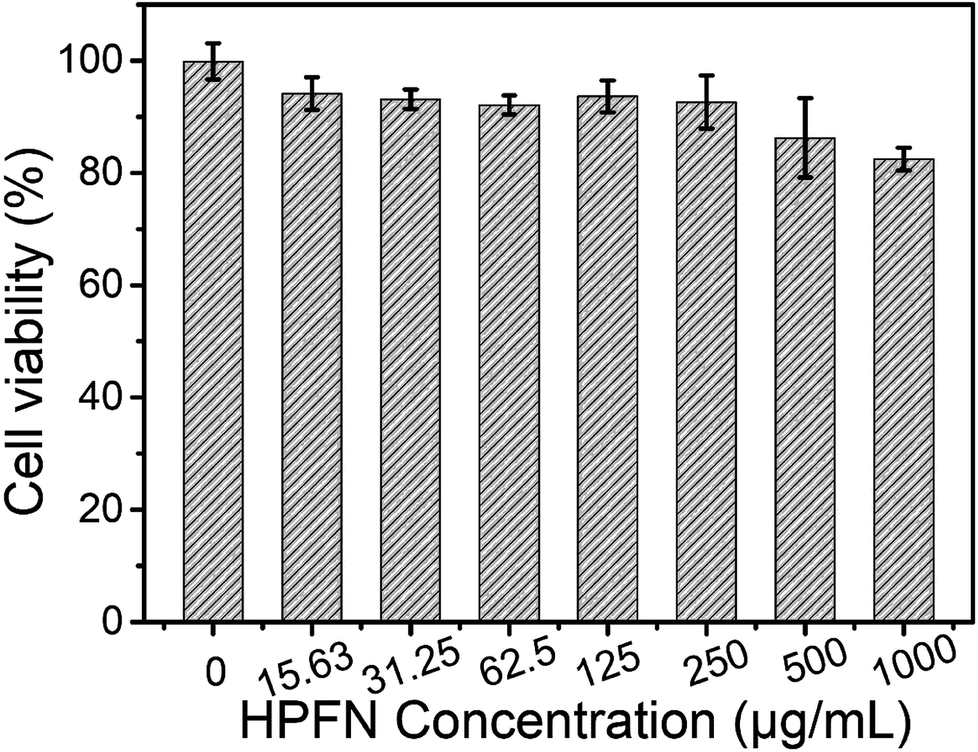 | ||
| Fig. 6 Biocompatibility of HeLa cells treated with HPFN with increasing concentrations. | ||
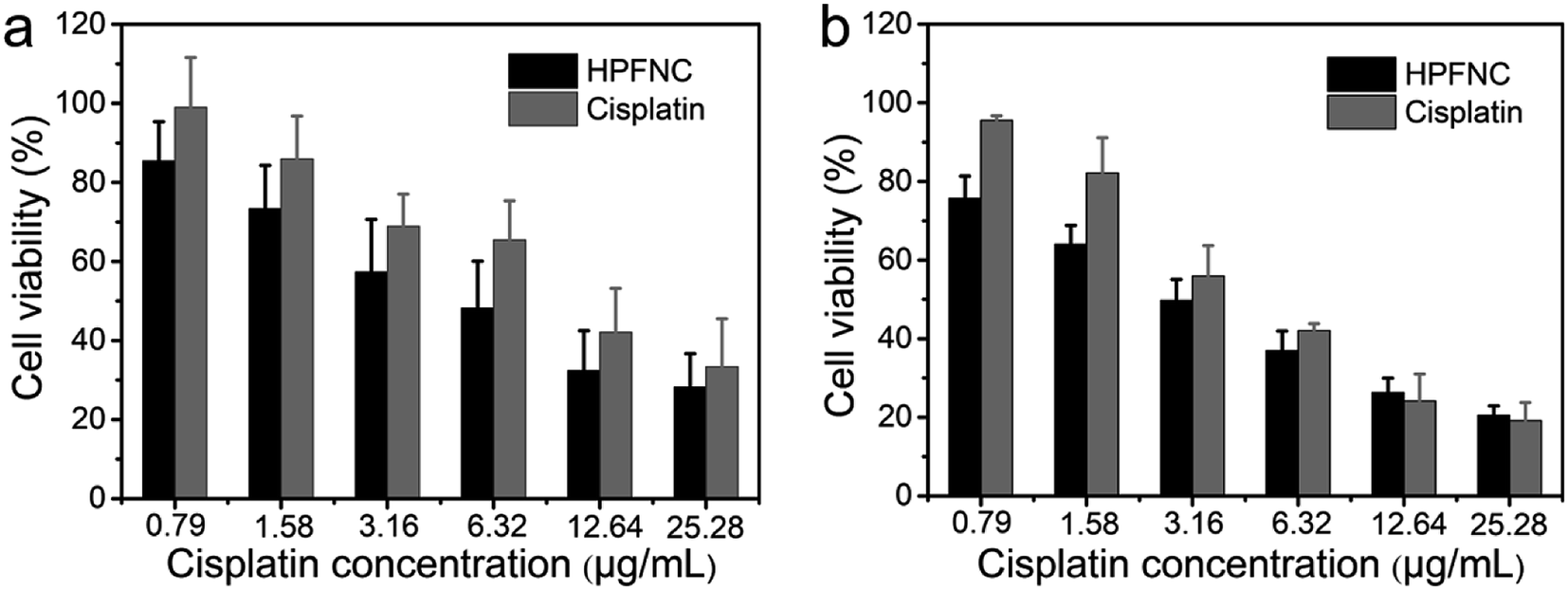 | ||
| Fig. 7 Viability of HeLa cells treated with free cisplatin and HPFN-cisplatin with increasing cisplatin concentrations for (a) 24 h and 48 h (b). | ||
4. Conclusions
In this study, we successfully developed a hollow and porous ferrogadolinium oxide nanonetwork (HPFN) using a simple solvothermal method. This nanoparticle with high T1 and T2 relaxivity (r1 = 6.63 s−1 mM−1, r2 = 244.07 s−1 mM−1) could be simultaneously used as T1 and T2 dual-modal MRI CA. The model anticancer drug cisplatin was efficiently loaded into HPFN with a large specific surface area (175.3 m2 g−1), and the loading amount of cisplatin was up to 7.9%. The HPFN displayed satisfactory dual-modal MRI contrast ability. In the in vitro experiments, the HPFNC can efficiently kill cancer cells in a dose/time-dependent manner. Therefore, HPFN with dual-modal MRI and drug loading ability could be used as a potential nanomedicine for simultaneous cancer diagnosis and chemotherapy.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (81670966), Priority Academic Program Development of Jiangsu Higher Education Institutions.References
- P. Mi, D. Kokuryo, H. Cabral, H. L. Wu, Y. Terada, T. Saga, I. Aoki, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2016, 11, 724–730 CrossRef CAS PubMed.
- B. R. Smith and S. S. Gambhir, Chem. Rev., 2017, 117, 901–986 CrossRef CAS PubMed.
- C. Sun, J. S. Lee and M. Q. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1252–1265 CrossRef CAS PubMed.
- Y. Chen, K. Ai, J. H. Liu, X. Y. Ren, C. H. Jiang and L. H. Lu, Biomaterials, 2016, 77, 198–206 CrossRef CAS PubMed.
- J. N. Liu, W. B. Bu and J. L. Shi, Chem. Rev., 2017, 117, 6160–6224 CrossRef CAS PubMed.
- X. Sun, R. H. Du, L. Zhang, G. L. Zhang, J. C. Qian, X. H. Tian, J. W. Zhou, J. C. He, Y. Y. Wang, Y. Q. Wu, K. Zhong, D. Q. Cai, D. H. Zou and Z. Y. Wu, ACS Nano, 2017, 11, 7049–7059 CrossRef CAS PubMed.
- Z. J. Zhou, D. T. Huang, J. F. Bao, Q. L. Chen, G. Liu, Z. Chen, X. Y. Chen and J. H. Gao, Adv. Mater., 2012, 24, 6223–6228 CrossRef CAS PubMed.
- D. Frascione, C. Diwoky, G. Almer, P. Opriessnig, C. Vonach, K. Gradauer, G. Leitinger, H. Mangge, R. Stollberger and R. Prassl, Int. J. Nanomed., 2012, 7, 2349–2359 CAS.
- A. Yadollahpour, S. A. Hosseini, S. Rashidi and F. Farhadi, Int. J. Pharm. Res. Allied Sci., 2016, 5, 251–257 CAS.
- J. Estelrich, M. J. Sanchez-Martín and M. A. Busquets, Int. J. Nanomed., 2015, 10, 1727–1741 CAS.
- A. Alipoura, Z. Soran-Erdema, M. Utkura, V. K. Sharmaa, O. Algina, E. U. Saritasa and H. V. Demir, Magn. Reson. Imaging, 2018, 49, 16–24 CrossRef PubMed.
- Z. J. Zhou, C. Q. Wu, H. Y. Liu, X. L. Zhu, Z. H. Zhao, L. R. Wang, Y. Xu, H. Ai and J. Gao, ACS Nano, 2015, 9, 3012–3022 CrossRef CAS PubMed.
- S. Santra, S. D. Jativa, C. Kaittanis, G. Normand, J. Grimm and J. M. Perez, ACS Nano, 2012, 6, 7281–7294 CrossRef CAS PubMed.
- J. Choi, J. Lee, T. Shin, H. Song, E. Kim and J. Cheon, J. Am. Chem. Soc., 2010, 132, 11015–11017 CrossRef CAS PubMed.
- K. H. Bae, Y. B. Kim, Y. Lee, J. Y. Hwang, H. W. Park and T. G. Park, Bioconjugate Chem., 2010, 21, 505–512 CrossRef CAS PubMed.
- F. F. Li, D. B. Zhi, Y. F. Luo, J. Q. Zhang, X. Nan, Y. J. Zhang, W. Zhou, B. S. Qiu, L. P. Wen and G. L. Liang, Nanoscale, 2016, 8, 12826–12833 RSC.
- H. D. Cai, X. An, S. H. Wen, J. C. Li, G. X. Zhang, X. Y. Shi and M. W. Shen, Part. Part. Syst. Charact., 2015, 32, 934–943 CrossRef CAS.
- K. Cheng, M. Yang, R. P. Zhang, C. X. Qin, X. H. Su and Z. Cheng, ACS Nano, 2014, 8, 9884–9896 CrossRef CAS PubMed.
- X. Sun, G. L. Zhang and Z. Y. Wu, Curr. Med. Chem., 2018, 25, 3036–3057 CrossRef CAS PubMed.
- G. H. Im, S. M. Kim, D. G. Lee, W. J. Lee, J. H. Lee and I. S. Lee, Biomaterials, 2013, 34, 2069–2076 CrossRef CAS PubMed.
- P. Verwilst, S. Park, B. Yoon and J. S. Kim, Chem. Soc. Rev., 2015, 44, 1791–1806 RSC.
- X. L. Liu, C. T. Ng, P. Chandrasekharan, H. T. Yang, L. Y. Zhao, E. Peng, Y. B. Lv, W. Xiao, J. Fang, J. B. Yi, H. Zhang and K. H. Chuang, Adv. Healthcare Mater., 2016, 5, 2092–2104 CrossRef CAS PubMed.
- T. H. Shin, J. S. Choi, S. Yun, I. S. Kim, H. T. Song, Y. Kim, K. I. Park and J. Cheon, ACS Nano, 2014, 8, 3393–3401 CrossRef CAS PubMed.
- G. L. Zhang, R. H. Du, L. L. Zhang, D. Q. Cai, X. Sun, Y. Zhou, J. Zhou, J. C. Qian, K. Zhong, K. Zheng, D. Kaigler, W. Q. Liu, X. Zhang, D. H. Zou and Z. Y. Wu, Adv. Funct. Mater., 2015, 25, 6101–6111 CrossRef CAS.
- F. Ries and J. Klastersky, Am. J. Kidney Dis., 1986, 8, 368–379 CrossRef CAS PubMed.
- A. Hosseini, S. Fanoudi, H. Mollazadeh, A. Aghaei and M. Boroushaki, J. Pharm. BioAllied Sci., 2018, 10, 66–71 CrossRef PubMed.
- J. Gao, X. Ran, C. Shi, H. Cheng, T. Cheng and Y. Su, Nanoscale, 2013, 5, 7026–7033 RSC.
- X. Sun, C. Cai and Q. Wang, et al., Phys. Chem. Chem. Phys., 2016, 18, 7820–7828 RSC.
- W. Cheng, K. Tang, Y. Qi, J. Sheng and Z. Liu, J. Mater. Chem., 2010, 20, 1799–1805 RSC.
- P. Du, J. H. Lim, S. H. Kim and J. S. Yu, Opt. Mater. Express, 2016, 6, 1896–1904 CrossRef CAS.
- I. Sadiq, S. Naseem, M. Naeem Ashiq, M. Asif Iqbal, I. Ali, M. A. Khan, S. Niaz and M. U. Rana, J. Magn. Magn. Mater., 2015, 395, 159–165 CrossRef CAS.
- H. Chen, G. D. Wang, W. Tang, T. Todd, Z. P. Zhen, C. Tsang, K. Hekmatyar, T. Cowger, R. B. Hubbard, W. Z. Zhang, J. Stickney, B. Z. Shen and J. Xie, Adv. Mater., 2014, 26, 6761–6766 CrossRef CAS PubMed.
- H. Wei, O. T. Brunsa, M. G. Kaulb, E. C. Hansen, M. Barch, A. Wiśniowska, O. Chen, Y. Chen, N. Li, S. Okada, J. M. Cordero, M. Heine, C. T. Farrar, D. M. Montana, G. Adam, H. Ittrich, A. Jasanoff, P. Nielsen and M. G. Bawendi, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2325–2330 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09102a |
| ‡ These authors contributed equally to this work and should be considered as co-first authors. |
| This journal is © The Royal Society of Chemistry 2019 |