
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly crystalline mesoporous SSZ-13 zeolite obtained via controlled post-synthetic treatment†
Maryani K. Wardania,
Grandprix T. M. Kadja *a,
Adroit T. N. Fajara,
Subagjob,
I. G. B. N. Makertiharthab,
Melia L. Gunawanb,
Veinardi Suendoac and
Rino R. Muktiac
*a,
Adroit T. N. Fajara,
Subagjob,
I. G. B. N. Makertiharthab,
Melia L. Gunawanb,
Veinardi Suendoac and
Rino R. Muktiac
aDivision of Inorganic and Physical Chemistry, Institut Teknologi Bandung, Jl. Ganesha No. 10, Bandung, 40132, Indonesia. E-mail: kadja@chem.itb.ac.id; Fax: +62-22-250-4154; Tel: +62-22-250-2103
bDepartment of Chemical Engineering, Institut Teknologi Bandung, Jl. Ganesha No. 10, Bandung, 40132, Indonesia
cResearch Center for Nanosciences and Nanotechnology, Institut Teknologi Bandung, Jl. Ganesha No. 10, Bandung, 40132, Indonesia
First published on 20th December 2018
Abstract
The generation of mesoporosity in SSZ-13 zeolite by means of desilication via post alkaline treatment normally results in severe damage to the microporous framework hence giving an undesirable decline in catalytic performance. Herein, we propose a post-synthetic desilication treatment that is controllable with an aim to preserve the high crystallinity of SSZ-13 zeolite during the formation of mesopores. The extent of desilication in alkaline media is controlled by deliberately leaving the organics within SSZ-13 frameworks as they can effectively hinder the attack of hydroxyl ions on siloxane bonds. The resulting SSZ-13 exhibits substantial development of mesoporosity with preserved high crystallinity and microporosity that can then be used to relieve the mass transport issues and lead to an increased activity of LDPE pyrolysis.
1. Introduction
Small pore zeolites with eight-membered ring (8-MR) aperture have attracted significant attention due to their usefulness in a wide range of applications, such as adsorption, separation and catalysis.1–6 One of the most attractive small pore zeolites is SSZ-13 zeolite (CHA topology) with a typical pore architecture of a large ellipsoidal cage (cha) and double six-membered ring (d6r) as composite building units. This material has shown remarkable catalytic activity in MTO (methanol-to-olefin) owing to the structure of the cha cage which provides steric hindrance to the bulky and branched molecules, while small and linear molecules can easily diffuse out of the small, 8-MR pore.6,7 Furthermore, Cu2+-exchanged SSZ-13 has been an excellent, hydrothermally-stable and commercial catalyst for the selective reduction of nitrogen oxides (NOx) by ammonia (NH3-SCR) for mitigating the emission levels of NOx from stationary as well as mobile sources. Nevertheless, the presence of micropores means that SSZ-13 can undergo faster deactivation and/or be not fully utilized owing to diffusion constraints6,8–10 and this is considered as a major issue in using SSZ-13 as a catalyst.The synthesis of nanosized zeolites which have increased external surface area and shortened diffusion path length is often offered as a solution to address the diffusion issue. Previous reports have shown nanosized SSZ-13 to exhibit comparable, even higher activity, and longer lifetime in its application as a catalyst.11–14 Nevertheless, the synthesis conditions must be tailored very delicately to prevent the growth to bulk, micron-sized crystals. Moreover, the synthesis of nanosized zeolites are often limited by very low yield and difficulty in separation. Recently, Wakihara and co-workers developed milling-recrystallization techniques11 and two-stage crystallization14 and to prepare nanosized SSZ-13 which shows a promising yield and separability.
There have been strong pieces of evidence that the diffusion constraints in zeolite can be suppressed by the addition of extra-large pores i.e. mesopores (2–50 nm). The presence of mesopore results in enhanced mass transport into and out of the zeolite active sites. To date, the study on the creation of mesopores within SSZ-13 zeolite has just begun and is still at the initial phase. Wu et al.9 pioneered the synthesis of mesoporous SSZ-13 zeolite by using dual-template strategy in which N,N,N-trimethyl-1-1-adamantammonium hydroxide (TMAdaOH) as zeolite structure-directing agent (SDA) and C22H45–N+(CH3)2(CH2)4–N+(CH3)2C4H9Br2 (C22-4-4Br2) as mesopores-generating agent (mesoporogen). The mesoporous SSZ-13 shows significant stability improvement when compared to that of conventional microporous SSZ-13. Notably, the mesoporogen-assisted synthesis may sometimes result in competing phases between zeolite and mesoporous aluminosilicate. For example, the use of other mesoporogen such as C22-6-6Br2 resulted in a low crystallinity due to the presence of such competing phases.10 In the fluoride medium, this dual template strategy may give SSZ-13 with trimodal pores and the catalyst could be used in a prolonged lifetime for methanol-to-olefin.15 The use of organic mesoporogen is considered to be not environmentally benign. This is due to the increased carbon emission after it is removed by the typical calcination method. Furthermore, C22-4-4Br2 is not commercially available and, thus, should be synthesized through a series of complex organic reactions.
Alternatively, post-synthesis treatment in alkaline media has been used to introduce mesopores within zeolite structure.16–18 It has attracted wide attention since it is a straightforward, economically feasible and scalable method. The first attempt on the desilication of SSZ-13 zeolite was reported by Sommer et al.19 Even though the mesopore in the range of 2–10 nm was successfully introduced, the mesoporous SSZ-13 exhibited a shorter lifetime in MTO reaction than that of purely microporous SSZ-13 zeolite. These unexpected yet interesting results are explained by substantial loss of crystallinity as well as microporosity which also lead to the alteration of Brønsted acid sites. In agreement with Sommer et al.,19 Zhang et al.8 found that crystallinity of alkaline-treated SSZ-13 showed a dramatic loss to that of its parent. More recently, Oord et al.20 performed desilication of SSZ-13 using three different NaOH concentrations, i.e. 0.1, 0.15 and 0.2 M. Under higher concentration (0.15 and 0.2 M), SSZ-13 underwent a structural collapse. On the other hand, the crystallinity of SSZ-13 treated using NaOH 0.1 M displayed a preserved crystallinity. However, the external surface area was very low, <20 m2 g−1, showing the negligible development of mesoporosity.
It was firstly reported by Čimek et al.21 that the as-synthesized ZSM-5 zeolite which still contains OSDA undergo much slower desilication rate than that of calcined ZSM-5 zeolite since OSDA may prevent the hydroxyl ions attack to siloxane bonds. A similar result has also been reported in ZSM-12 zeolite.22 Pérez-Ramírez and his co-workers adopted and further modified these results into the so-called partial detemplation–desilication method to control mesopores formation in Beta zeolite.23 The remaining organics in as-synthesized and partially-detemplated zeolites acts to control the extent of desilication. Beta zeolites comprise low framework stability which provoke the significant loss of crystallinity during desilication of calcined samples, without the presence of remaining organics.24 The low framework stability was attributed to the high density of four-membered rings (4-MRs) in Beta frameworks. The similar phenomenon was also found in the pioneering work of SSZ-13 desilication.19
In the present study, motivated by the similar situation of Beta and SSZ-13, that is the high density of 4-MRs, we extend the applicable scope of the remaining organics controlled-desilication to SSZ-13 frameworks. The highly crystalline SSZ-13 with significant mesoporosity can be realized via post-synthetic treatment. The obtained mesoporous SSZ-13 exhibits increased activity towards the low-density polyethylene (LDPE) pyrolysis, showing that the introduced mesoporosity has functioned to relieve the diffusion constraints.
2. Experimental
2.1 Synthesis of SSZ-13 zeolites
SSZ-13 zeolites were synthesized with N,N,N-trimethyl-1-1-adamantammonium hydroxide (TMAdaOH) using initial gel with molar composition of 0.01 Al2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0 SiO2:0.09 NaOH:20 H2O:0.1 TMAdaOH. Sodium hydroxide 50 wt% (Merck) solution was mixed with deionized water, followed by the addition of TMAdaOH (25%wt) (Sachem) and NaAlO2 (Sigma-Aldrich) under stirring until the clear solution was obtained. Subsequently, to this clear solution, Ludox HS-40 (Sigma-Aldrich) was added and stirred for 24 h. The final mixture was transferred into a Teflon-lined stainless autoclave and subsequently heated in an oven at 175 °C for 36 h, under static condition. After crystallization, the zeolite products were filtered, washed with deionized water and dried at 110 °C overnight. The obtained samples were denoted as AS, corresponding to the as-synthesized sample.
:1.0 SiO2:0.09 NaOH:20 H2O:0.1 TMAdaOH. Sodium hydroxide 50 wt% (Merck) solution was mixed with deionized water, followed by the addition of TMAdaOH (25%wt) (Sachem) and NaAlO2 (Sigma-Aldrich) under stirring until the clear solution was obtained. Subsequently, to this clear solution, Ludox HS-40 (Sigma-Aldrich) was added and stirred for 24 h. The final mixture was transferred into a Teflon-lined stainless autoclave and subsequently heated in an oven at 175 °C for 36 h, under static condition. After crystallization, the zeolite products were filtered, washed with deionized water and dried at 110 °C overnight. The obtained samples were denoted as AS, corresponding to the as-synthesized sample.
2.2 Controlled desilication of the AS samples
Prior to desilication, AS samples were partially detemplated through calcination at 250 °C and 450 °C for 1 h and labeled as C250 and C450, respectively. Full detemplation was carried out at 550 °C for 6 h and denoted as C550. Desilication through alkaline treatment (AT) with 0.2 M NaOH solution at 65 °C for 30 min were applied to AS, C250, C450 and C550 samples, respectively. After desilication, the samples were thoroughly washed with deionized water until the filtrate was neutral. Subsequently, AS, C250, C450 and C550 samples were dried and continued to calcination at 550 °C for 6 h and denoted as AS-AT, C250-AT, C450-AT, and C550-AT, respectively. Note that the used nomenclatures describe the chronological order of the samples.Ion-exchange were applied to AS-AT, C250-AT, C450-AT, and C550-AT samples using 2 M NH4Cl solution, followed by calcination at 550 °C for 6 h to obtain their proton forms. These procedures were repeated two consecutive times. All the following characterizations were applied to the proton forms for all samples.
2.3 Characterizations
Powder diffraction (XRD) patterns were measured on a Bruker D8 Advance diffractometer using Cu-Kα incident beam (λ = 1.5418 Å) equipped with nickel as filter. The crystallinity was estimated by comparing the total area of the reflection peaks in the 2θ range of 9.6–21° (ref. 25) to that of the sample with highest total area, assuming 100% crystallinity.29Si and 27Al magic angle spinning nuclear magnetic resources (MAS NMR) spectra were recorded on an Agilent DD2 500 MHz spectrometer, operated at resonance frequencies of 99.32 and 130.28 MHz, respectively. The measurements were performed at a spinning rate of 9 kHz with a pulse width of 1.0 μs and were applied with a recycle delay time of 5 s. 29Si MAS NMR spectra were referenced to tetramethylsilane (TMS), while 27Al MAS NMR spectra were reported relative to aluminium nitrate (Al(NO3)3) solution. The Si/Al ratios in the framework are calculated from 29Si MAS NMR spectra using equation below,
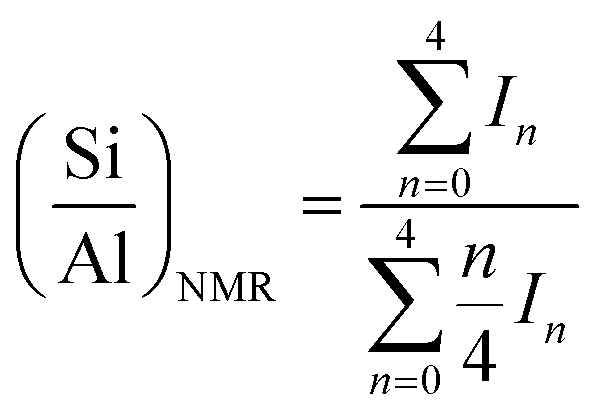 | (1) |
Ammonia-temperature programmed desorption (NH3-TPD) measurements were performed on a Quantachrome ChemBET Pulsar. A certain amount of sample was degassed at 350 °C for 1 h under He flow. Subsequently, the sample was cooled to ambient temperature, saturated with NH3 gas, and purged using He flow until a constant baseline is reached. Ammonia desorption was performed using a constant heating rate (10 °C min−1) under He flow. The calibration of TCD signal was performed using the desorption of known amounts of ammonia.
Scanning electron microscopy (SEM) images were taken using a Hitachi SU-3500 microscope at an accelerating voltage of 5 kV. Transmission electron microscopy (TEM) images were acquired on a Hitachi HT-7700 microscope equipped with a CCD camera at an accelerating voltage of 120 kV. Prior to the measurement, the samples were suspended in isopropyl alcohol and ultrasonicated for 10 min. Subsequently, a small droplet of the suspension was deposited on a Cu TEM grid.
Bruker Alpha spectrometer equipped with attenuated total reflectance (ATR) accessory was employed to collect the ATR-infrared (ATR-IR) spectra of the zeolite samples. The measurements were performed in the range of 4000–500 cm−1 using zinc selenide (ZnSe) as the ATR prism. The spectra were acquired over 256 scans and 4 cm−1 spectral resolution.
Raman spectra were measured on a Bruker Senterra spectrometer. A 532 nm laser excitation and a power output of 20 mW were applied to all measurements. All spectra were measured at room temperature with a spectral resolution of 4 cm−1.
Diffuse reflectance UV-Vis spectra (UV-Vis DRS) of powder samples were recorded on a Thermo Scientific 200 series in the range of 200 to 1000 nm. The obtained spectra were converted into the form F(R), Kubelka–Munk function, spectra.
The elemental compositions of zeolites were determined using X-ray fluorescence (XRF) analyses. The measurement was performed on a PANalytical Axios mAX spectrometer.
The weight percentage of occluded organics in SSZ-13 samples was assessed by thermogravimetric analysis (TGA) using a NETZSCH STA-449-F1 Jupiter with gas mixtures of nitrogen (80%) and oxygen (20%) as the carrier gas at a flow rate of 60 mL min−1. The temperature was ramped up to 800 °C at the rate of 20 °C min−1 with an isothermal dwell time of 10 min.
N2 physisorption isotherms were obtained on a Quantachrome Autosorb iQ-MP. Prior to the analysis, the samples were vacuumed at 300 °C for 6 h. The specific surface area (SBET) was calculated using the Brunauer–Emmet–Teller (BET) method, while t-plot method was applied to calculate the micropore volume (Vmicro). The total pore volume (Vtot) was determined at relative pressure, P/Po, ≅0.99. The non-local density functional theory (NLDFT) was employed to determine the mesopore size distribution. This method was applied to the adsorption branch of the isotherm assuming cylindrical pore model.
2.4 LDPE pyrolysis test
LDPE (Alfa Aesar) pyrolysis was performed in a Netzsch STA-449-F1 thermogravimetric (TG) analyzer. LDPE and zeolites were first mixed using LDPE:zeolites (proton form) ratio of 10, and subsequently loaded to an α-alumina crucible of TG analyzer. The temperature-programmed LDPE cracking tests were carried out from RT to 700 °C with a fixed ramping rate of 20 °C min−1 under 50 mL min−1 N2 flow.
3. Results and discussion
3.1 The effect of calcination at various temperatures
The XRD patterns of AS and the detemplated samples show the typical reflections of CHA topology with high crystallinity (Fig. 1). At calcination of 250 °C, the intensity of peaks at 2θ of 9.6 and 13.1° decreases while the peak at 2θ of 16.3° intensifies. Afterward, the intensity of the former peaks gradually rises with the increase of calcined temperatures from 250 °C to 550 °C. On the other hand, the peak at 2θ of 16.3° diminishes with the increase of calcined temperatures. These results should be originated from the existence of organics within the zeolite frameworks. The trend in the XRD patterns of SSZ-13 zeolites after calcination at various temperatures obtained in this study merits further study.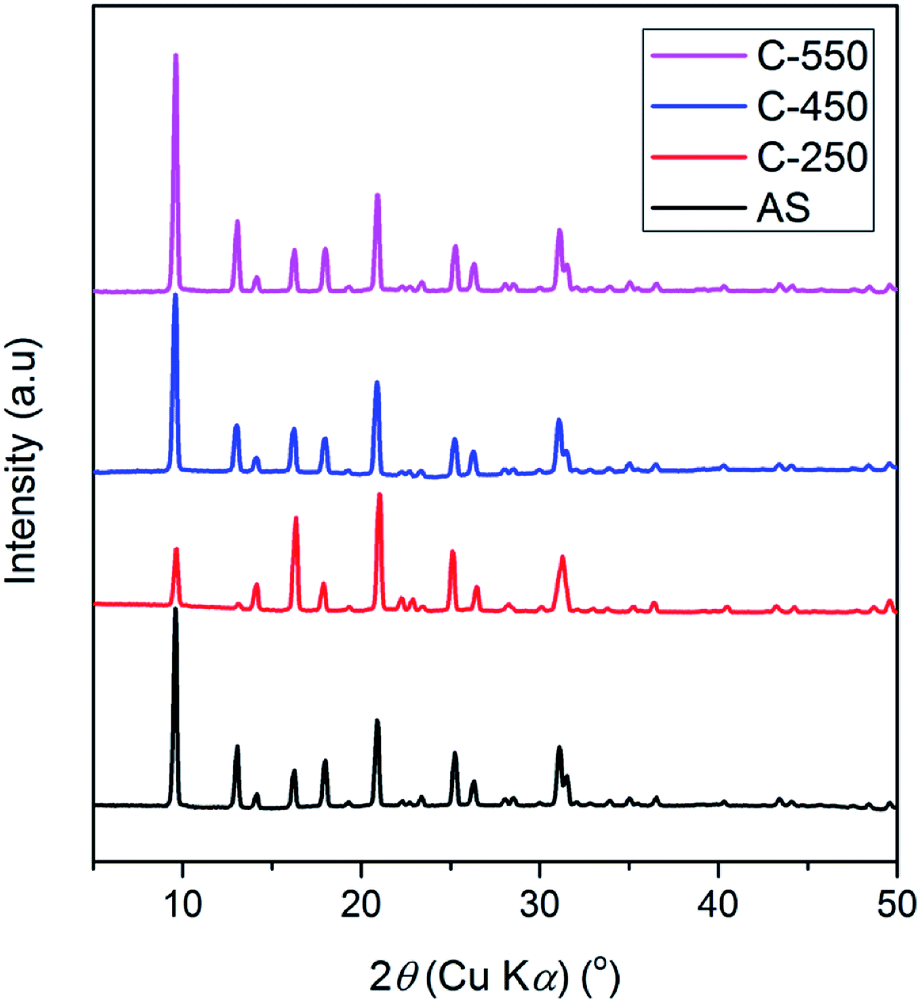 | ||
| Fig. 1 XRD patterns of as-synthesized and calcined SSZ-13. | ||
The organics within the zeolite frameworks were evaluated using thermogravimetric analyses (TGA). Fig. 2a shows the TGA curves of AS, C250, C450, and C550 samples. The weight loss at temperatures less than 250 °C is attributed to the adsorbed water molecules, while the removal of organics is observed as the weight loss at temperatures above 250 °C. As seen in Fig. 2b and c, C250 exhibits slight loss of organics, yet there is a shift toward higher temperature indicating that TMAda+ is partially converted into bulkier molecules. At high temperature, a quaternary ammonium can undergo Hofmann elimination which results in a trialkylamine and alkene. The formed alkene may be converted into (poly)aromatics through a series of polymerization and aromatization reactions during the calcination process. C450 possesses a shift toward temperature higher than AS and C250 suggesting the formation of much bulkier molecules. However, C450 has a lower amount of remained organics. Note that the calcination was performed under air atmosphere in the presence of oxygen (O2) which can convert hydrocarbons into CO2 and water. Ultimately, all of the organics were fully removed in C550 as there is no weight loss at temperatures above 250 °C.
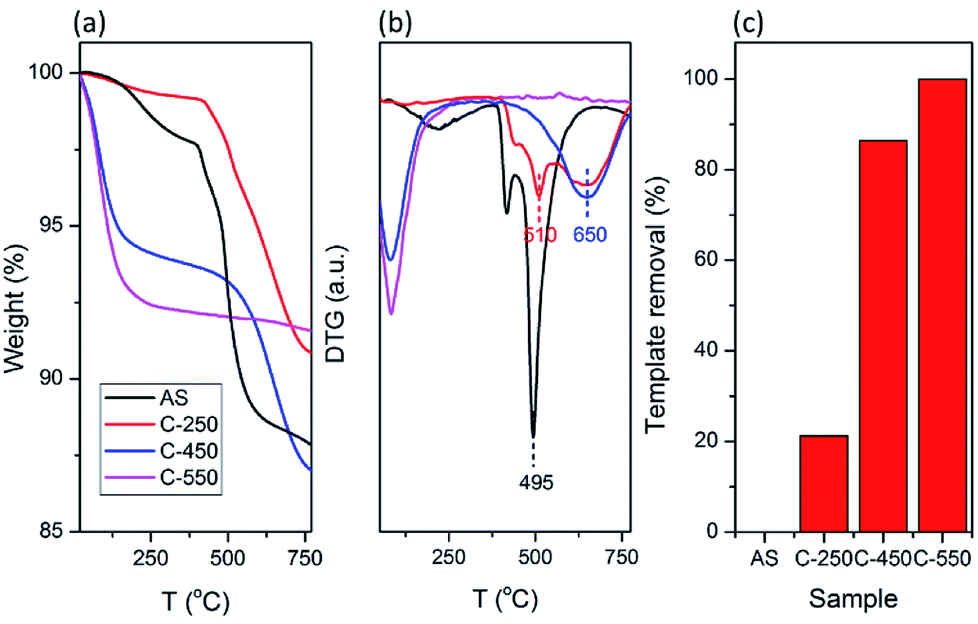 | ||
| Fig. 2 (a) TG and (b) DTG curves of as-synthesized and calcined SSZ-13. (c) Calculated template removal of the samples based on TG curve. | ||
AS and the detemplated samples are further analyzed using UV-DRS (Fig. 3). Absorption band appears in the UV range of 200–400 nm, exhibited by AS, indicates the σ–σ* and n–σ* transitions of occluded TMAda+ and zeolite frameworks. In the spectra of C250, absorption is also found in the visible range of 400–500 nm, in addition to the absorption in the UV range. This may correspond to the π–π* transitions of the formed polyaromatics as described in the previous paragraph. Moreover, C450 possesses a broad absorption band up to 1000 nm which indicate the formation of much bigger polyaromatics. When the organics have been completely removed as in C550, a narrow band is observed at 200–300 nm since the absorption is merely from zeolite frameworks.
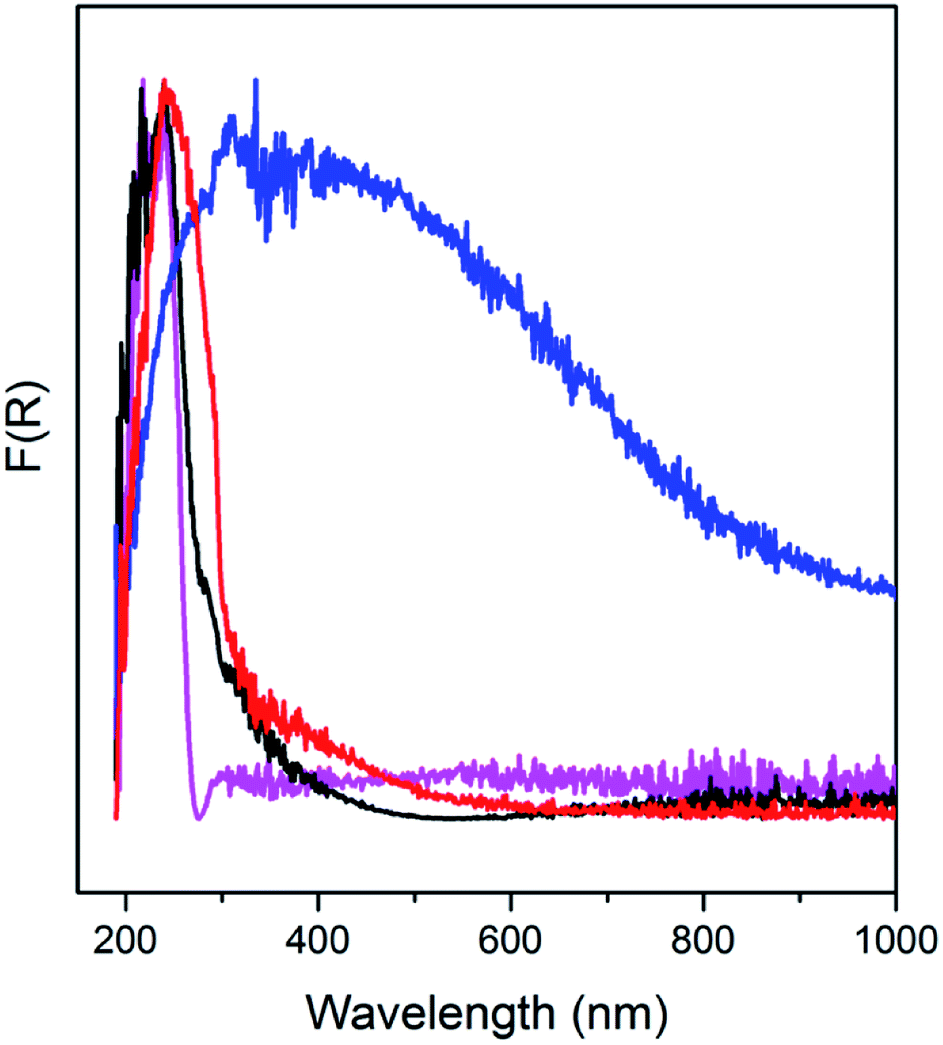 | ||
| Fig. 3 UV-Vis DRS spectra of as-synthesized and calcined SSZ-13. | ||
The ATR-IR spectra of AS, C-250, C-450 and C-550 in the C–H stretching vibration modes region (2800–3000 cm−1) are illustrated in Fig. 4. AS shows intense absorption bands in this region owing to the presence of occluded TMAda+ molecules. After calcination at 250 °C, these bands are diminished due to the formation of (poly)aromatics as described above. Nevertheless, ATR-IR spectra of C-250 still exhibit similar patterns to those of AS which indicate that TMAda+ molecules within the zeolite frameworks are partially preserved. On the other hand, C-450 and C-550 do not possess any absorption band in the C–H stretching region. The similar feature of both samples should be originated from different phenomena. As shown by TG analyses, C-450 still contains organics which are indicated to be large (poly)aromatics. These molecules are lack of C–H bonds which render their presence hard to be detected by ATR-IR spectrometer. Contrarily, C-550 does not possess any organic molecules, thus, its ATR-IR spectra show the absence of C–H stretching vibration modes.
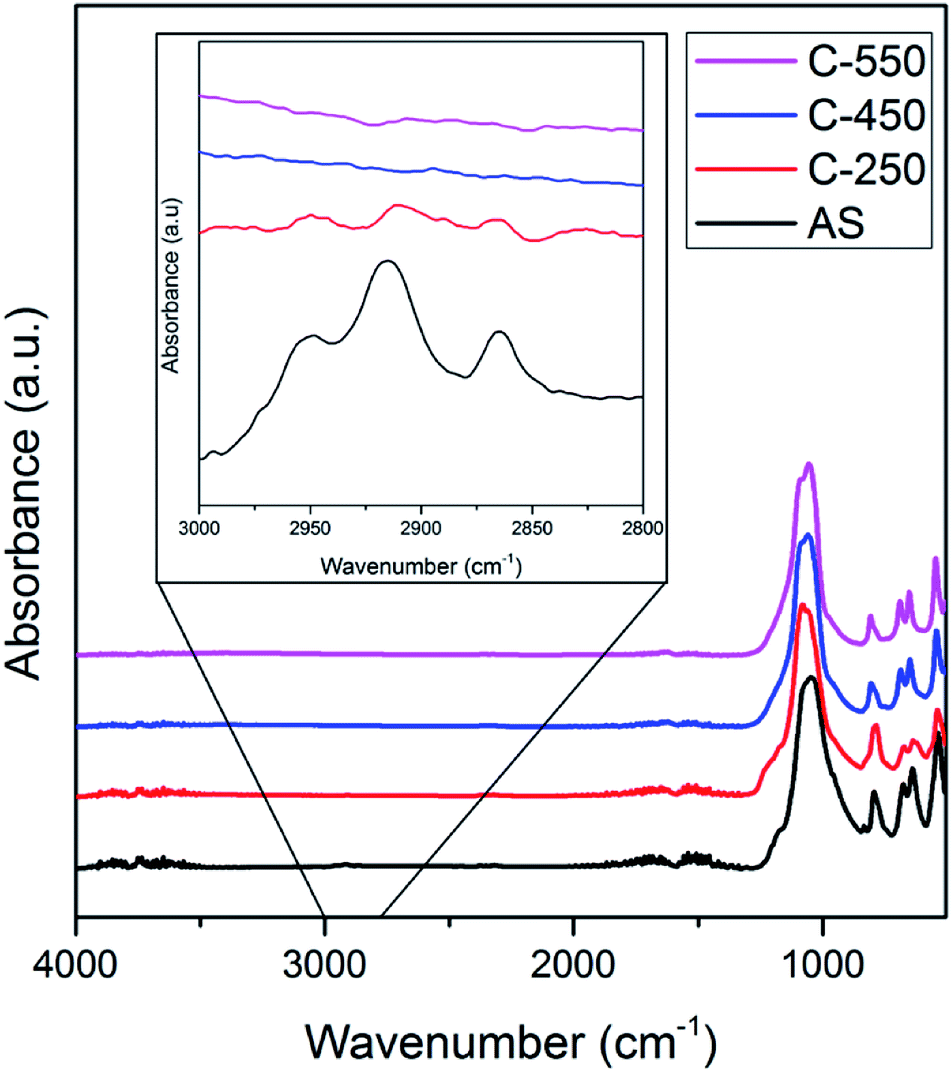 | ||
| Fig. 4 ATR-IR spectra of as-synthesized and calcined SSZ-13. | ||
3.2 The organics-controlled desilication
The samples with different amount of organics (AS, C250, C450, and C550) were subjected to the alkaline treatment followed by final calcination. XRD patterns of C550, AS-AT, C250-AT, C450-AT, and C550-AT are provided in Fig. 5. The first three samples exhibit high intense, characteristic peaks of CHA topology, while the two later samples are amorphous as the crystalline peaks have disappeared. The amorphization is due to the vulnerability of Si–O–Si bonds to the attack of hydroxyl ions. AS-AT and C250-AT can retain their crystallinity because the remained organics may protect the zeolite frameworks from the attack of hydroxyl ions. The calculated crystallinity of C550, AS-AT and C250-AT are 100, 96 and 87%, respectively.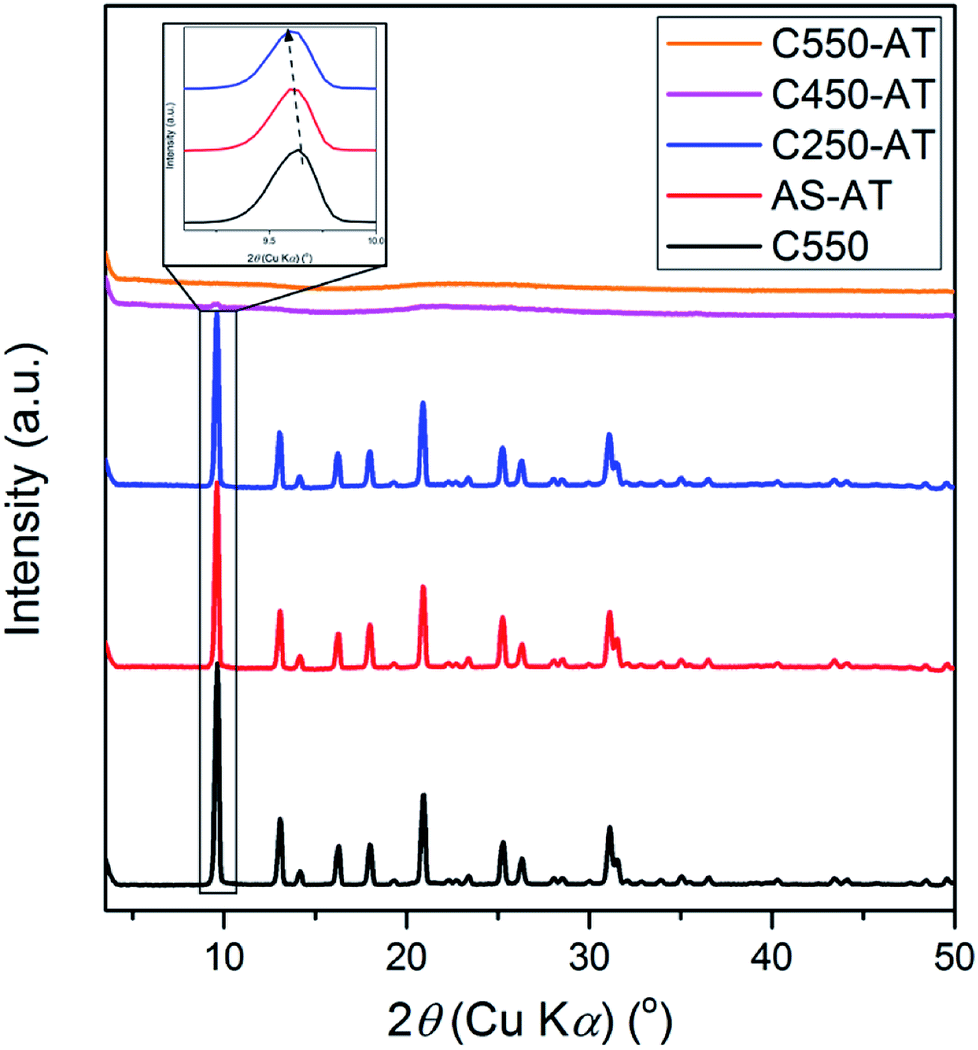 | ||
| Fig. 5 XRD samples of the desilicated samples. | ||
Due to desilication, the shift of XRD peaks towards lower 2θ (Fig. 5) indicating the expansion of the unit cell. Extraction of Si atoms from zeolite frameworks will increase the Al density within zeolite frameworks. Since Si–O possesses shorter bond length (1.61 Å) compared to that of Al–O (1.75 Å), desilication shall result in the elongation of lattice parameters, and thus expansion of the unit cell.26 Whole pattern fitting through Le-Bail method has been applied to obtain the quantified information of lattice parameters in C550, AS-AT, and C250-AT samples, as provided in Table 1 and Fig. S1.† AS-AT appears to be more protected than C250-AT based on the less-expanded unit cell. This is expected since the former possesses more organics than the latter does. Furthermore, a- and b-axes appears to be more affected by desilication compared to c-axis. This indicates that the hydroxyl ions preferentially attack (h00) and (0k0) planes over a (00l) plane. However, further investigation is strictly necessary to verify this argument.
| Samples | Cell parameters (Å) | Rp (%) | Rwp (%) | ||
|---|---|---|---|---|---|
| a | b | c | |||
| C550 | 13.558(2) | 13.558(2) | 14.767(0) | 7.56 | 9.14 |
| AS-AT | 13.572(5) | 13.572(5) | 14.773(5) | 7.96 | 9.23 |
| C250-AT | 13.582(0) | 13.582(0) | 14.776(2) | 7.06 | 8.43 |
The vibrational spectroscopy, i.e. ATR-IR and Raman, are utilized to study the local structure of the obtained SSZ-13 zeolites. ATR-IR spectra of the desilicated samples are provided in Fig. 6a. Prominent bands in the range of 1200–1000 cm−1 correspond to T–O–T (T = Si and/or Al) asymmetric stretching vibrations, while T–O–T symmetric stretching vibrations are shown by the band at 808 cm−1.27,28 In addition, there are bands located around 400–800 cm−1 (pseudo lattice vibrations) which are assigned to the T–O–T ring bending vibrations within zeolite frameworks.26 Two bands at 690 and 650 cm−1 are attributed to the presence of 4-MRs, whereas the band at 545 cm−1 shows double 6-MRs (d6r), which are typical composite building units (cbu), in addition to chabazite cage (cha).29–32 These characteristic bands correspond to 4-MRs and double 6-MRs are found in AS-AT and C250-AT samples. On the other hand, C450-AT and C550-AT do not exhibit any band in the pseudo lattice region, showing the amorphization of CHA topology. The fact that the T–O–T (T = Si or Al) asymmetric stretching vibration bands in C450-AT and C550-AT are broadened, strongly indicate the formation of less-ordered structure, i.e. the amorphous structure. The results from ATR-IR spectroscopy are in agreement with those obtained from XRD.
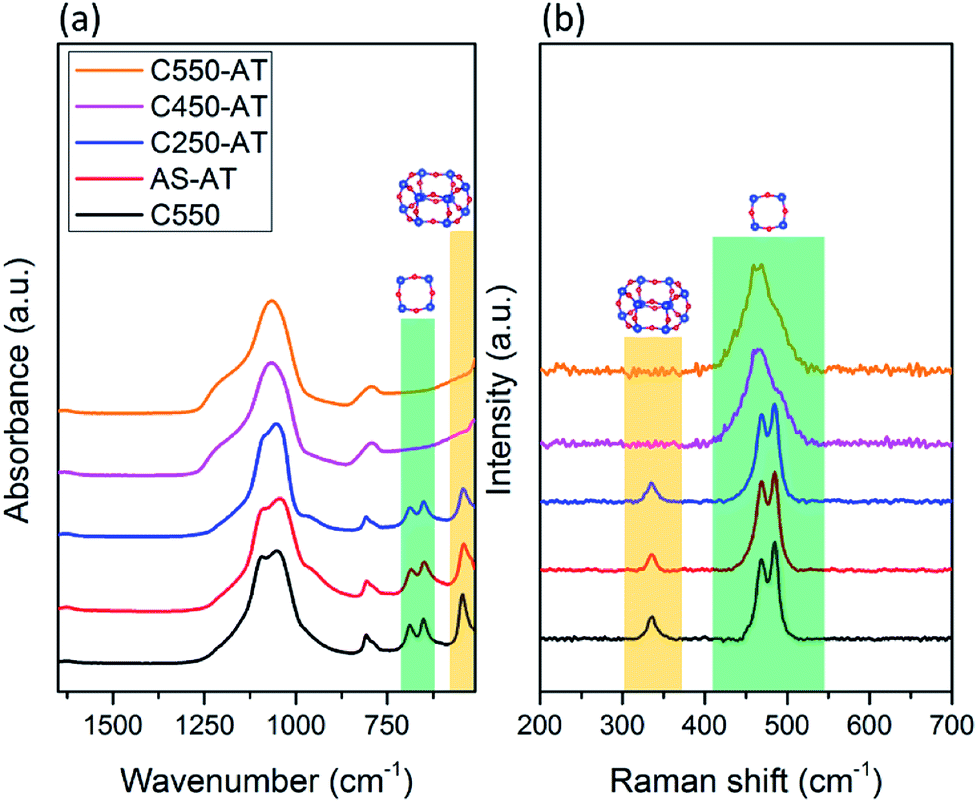 | ||
| Fig. 6 (a) ATR-IR and (b) Raman spectra of the desilicated samples. | ||
Raman spectroscopy in the region of 250–600 cm−1 is specific for the bending vibration modes of T–O–T rings. As seen in Fig. 6b, C-550, AS-AT, and C250-AT possess 4-MR and 6-MR shown by the bands at 450–520 and 335 cm−1, respectively, which are characteristic spectra of CHA topology.33 Meanwhile, C450-AT and C550-AT merely exhibit 4-MR band due to the occurring amorphization. In addition, the 4-MR band of C450-AT and C550-AT undergo a broadening because of the less-ordered structure within the amorphous phase. These results support the insights obtained from XRD and ATR-IR spectroscopy.
Fig. 7 depicts the 29Si MAS NMR spectra of the products. For C550, AS-AT and C250-AT, three distinguished bands can be observed, which are Q4(0Al) ((SiO)4Si), Q4(1Al) ((SiO)3(AlO)Si) and Q3 ((SiO)3(OH)Si) silicon species located at around −113, −106, and −103 ppm, respectively. However, C450-AT and C550-AT exhibit a broad band, owing to the formation of silicon species in the non-uniform environment, that is, amorphous phase (Fig. S2†). These results are in agreement with the previous characterizations. The fraction of each silicon species and Si/Al ratio calculated based on eqn (1) are provided in Table 2. The desilicated samples show decreased Si/Al ratio which appears to be controlled by the organics within zeolite frameworks. The calculated Si/Al ratios from 29Si MAS NMR spectra are in agreement with those calculated from XRF. Interestingly, the decrease in Si/Al ratio is accompanied by the increase of Q3 fraction. In zeolite frameworks, Q3 represents the silanol groups which are originated from the external surface and crystal defects. Thus, the increase of Q3 fraction suggests the enhancement of the external surface area.
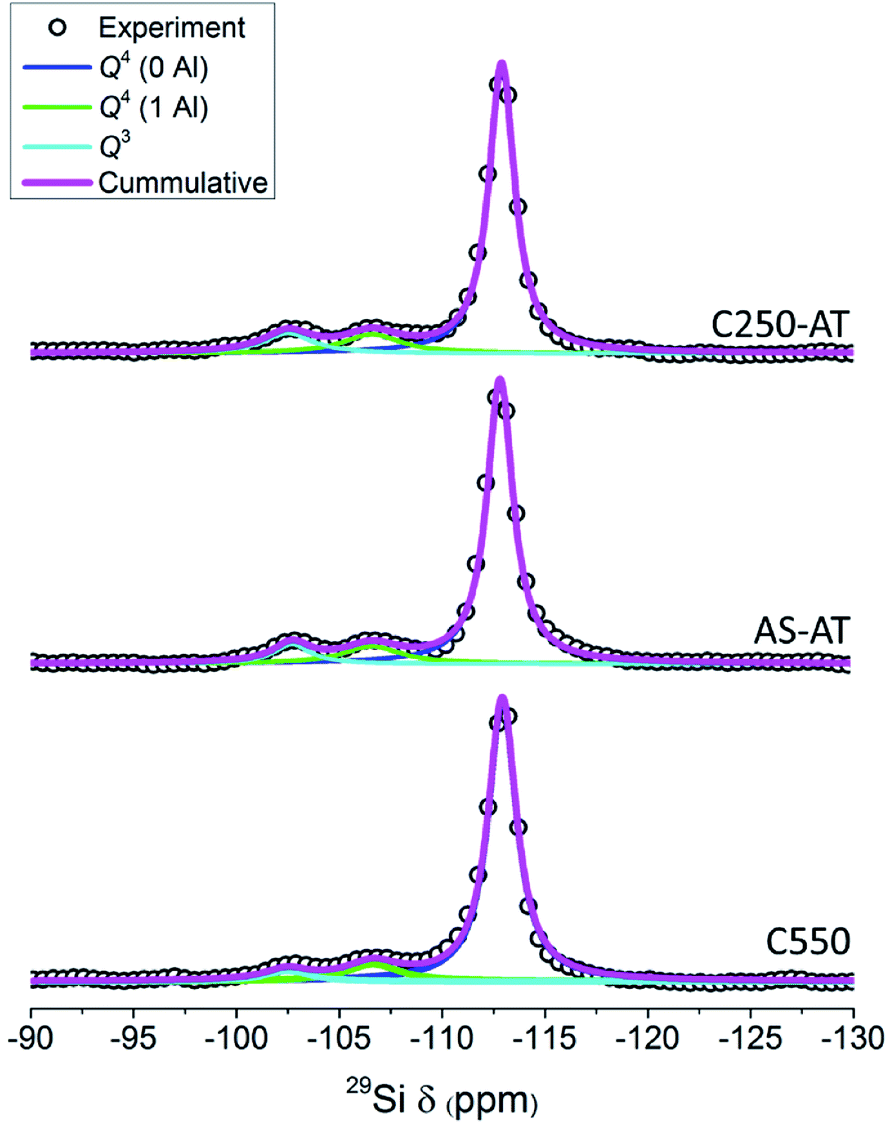 | ||
| Fig. 7 29Si MAS NMR spectra of the desilicated samples. | ||
| Samples | Q3 fraction | Si/Al frameworka | Si/Al bulkb | Total acid sites concentrationc (μmol g−1) |
|---|---|---|---|---|
| a Determined using eqn (1).b Determined using XRF.c Determined using NH3-TPD.d n.d. means not determined. | ||||
| C550 | 0.046 | 40 | 45 | 198 |
| AS-AT | 0.079 | 35 | 40 | 222 |
| C250-AT | 0.105 | 34 | 38 | 239 |
| C450-AT | n.d.d | n.d. | 28 | n.d. |
| C550-AT | n.d. | n.d. | 20 | n.d. |
27Al MAS NMR spectra of the products are depicted in Fig. 8. A prominent peak at around 62 ppm shows the presence of Al tetrahedral species, while the presence of Al octahedral (extraframework) species at around 0 ppm is negligible. Due to the alkaline treatment, the Al tetrahedral peaks undergo a broadening indicating the less homogeneous Al environment.
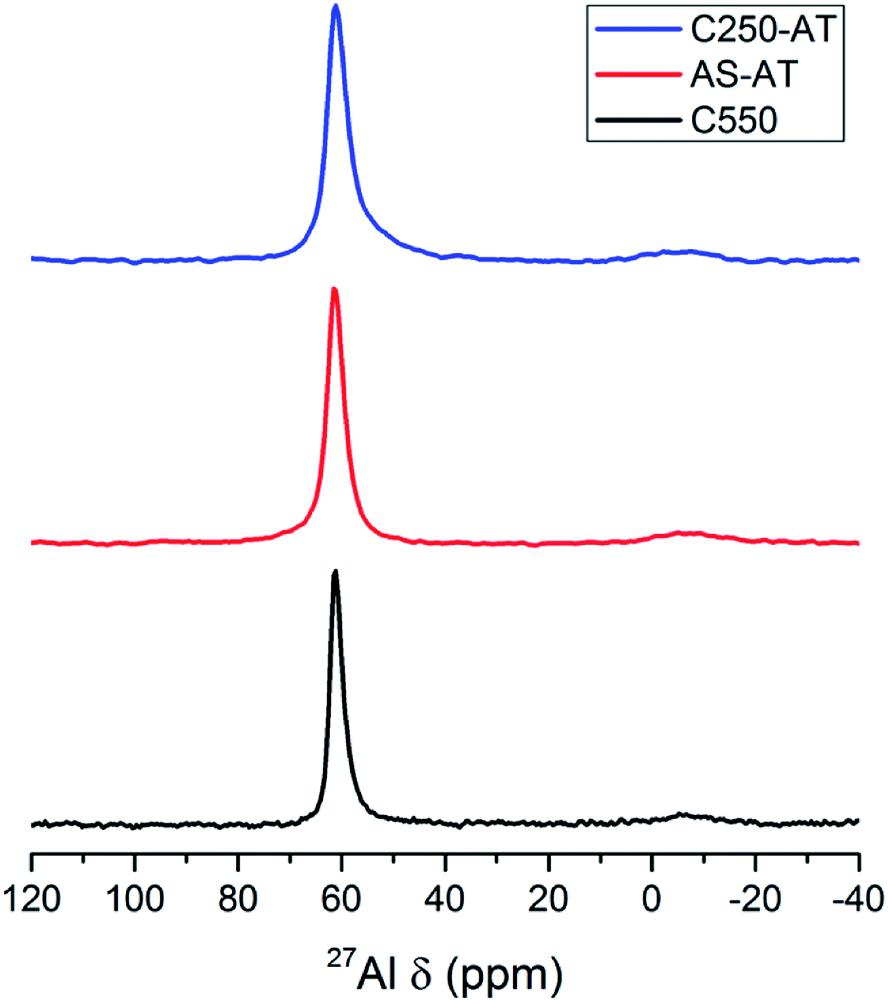 | ||
| Fig. 8 27Al MAS NMR spectra of the desilicated samples. | ||
The concentration of acid sites within the samples are determined using NH3-TPD (Fig. S3†). In each NH3-TPD curve, two peaks at around 180 °C and 450 °C associated with weak and strong acid sites, respectively. After desilication, the higher temperature peak appears to be slightly shifted to a lower temperature which may be caused by the alteration of Al environment as indicated by 27Al MAS NMR spectroscopy. By integrating the area under the curve, the concentration of acid sites within the samples can be determined by assuming one acid site adsorbs one ammonia molecule. The data are provided in Table 2. It is seen that AS-AT and C-250 have increased total acid sites concentration due the decrease in Si/Al ratio or more Al within the frameworks.
The textural properties of crystalline C-550, AS-AT and C250-AT are analyzed using N2 physisorption technique. Fig. 9 shows N2 physisorption isotherm of the three crystalline products. C-550 exhibits type I(a) isotherms, a typical type given by microporous solids having notably narrow micropores (<1 nm).34 On the other hand, AS-AT and C250-AT show a type H4 hysteresis loop with adsorption branch of isotherms resembling a hybrid of type I and II, which is often found in mesoporous zeolites.34 The specific surface area (SBET), external surface area (Sext), micropore volume (Vmicro) and total pore volume (Vtot) calculated from N2 physisorption isotherms are listed in Table 3. After post-synthetic treatment, there is no substantial loss of micropore volume, from 0.28 cm3 g−1 (C550) to 0.26 (AS-AT) and 0.23 cm3 g−1 (C250-AT). These inconsiderably lower micropore volumes within post-treated samples are due to the attack of hydroxyl ions, which is common for the preparation of mesoporous zeolites using desilication method. As seen, C250-AT is more affected by the alkali-treatment since it has fewer organics than AS-AT has.
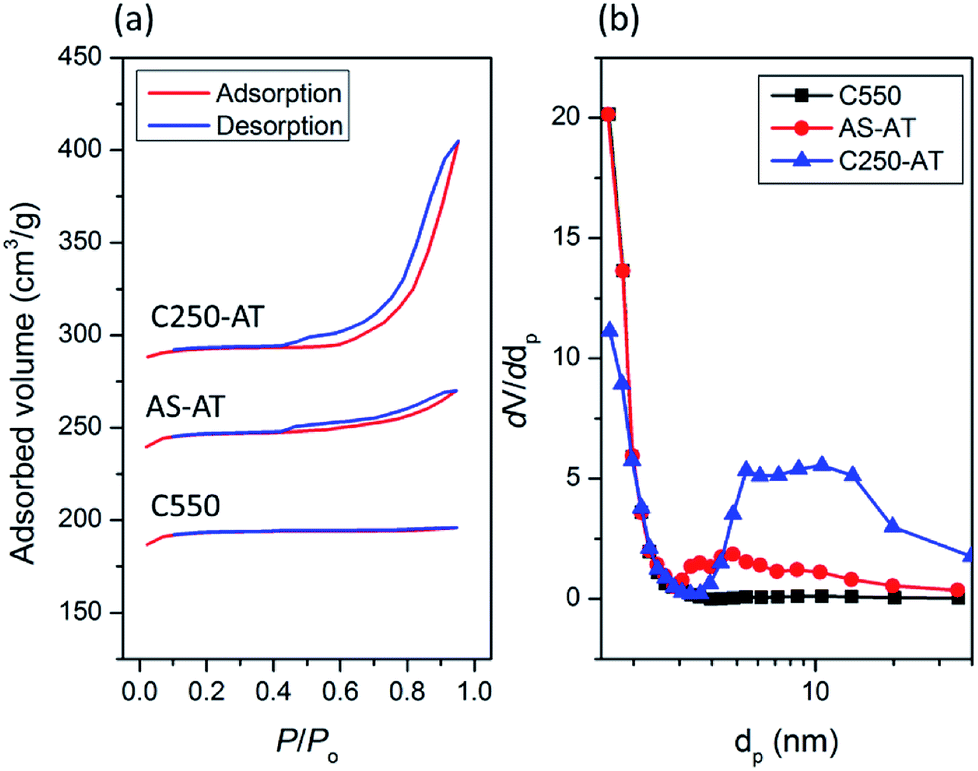 | ||
| Fig. 9 (a) N2 adsorption–desorption isotherms and (b) BJH pore size distribution of the desilicated samples. | ||
Interestingly, there is a simultaneous, significant increase of external surface area from 32 m2 g−1 (C550) to 80 (AS-AT) and 110 m2 g−1 (C250-AT). Ultimately, the pore size distribution based on BJH calculation shows the development of mesopores. C500 does not indicate the presence of mesopores as distribution found in the range of 2–50 nm. Meanwhile, AS-AT and C250-AT display mesopores in the range of 2–10 nm and 5–20 nm, respectively. These results clearly show the creation of mesoporosity with preserved microporosity. In addition, the N2 adsorption–desorption isotherms of C450-AT and C550-AT are shown in Fig. S4.† The calculated textural properties of both samples are also tabulated in Table 3. It is seen that the micropore volumes have extremely dropped showing the destruction of zeolite frameworks as also pointed out by other characterizations.
The creation of mesopores within zeolite should not excessively sacrifice the micropores. Thus, to asses the porosity, one needs to take the developed mesopores and the maintained intrinsic microporosity into account. To this extent, a normalized descriptor, so-called Indexed Hierarchy Factor (IHF), is used to compare the hierarchy (micro- and mesopores character) of a series of zeolite samples.13 It is calculated as follow, IHF = (Vmicro/Vmicro,max) × (Sext/Sext,max). Higher IHF value indicates more developed mesopores with preserved micropores, while lower IHF corresponds to either overly microporous or mesoporous. IHF of C500, AS-AT, C250-AT, C450-AT, and C550-AT are determined to be 0.06, 0.15, 0.18, 0.03 and 0, respectively. The first three samples show an increasingly IHF while C450-AT and C550-AT exhibit lower IHF than that of C550. C550-AT even has IHF of zero indicating the absence of microporous character. These results show the role of the remaining organics in controlling the extent of desilication. The more organics removed, the higher desilication extent. The role of organics in controlling the extent of desilication is also reflected in the different rate of silica dissolution (Fig. S5†). Samples with less remaining organics show faster dissolution rate. This correlation may also be influenced by another factor, presumably, the nature of the organics. Note that in AS, the organic is TMAda+ while polyaromatics are present in C250. The investigation on the interplay of the amount and nature of the organics is ongoing.
Fig. 10 displays representative SEM and TEM images of the products. All samples possess micron-sized cubic particles with rounded edges, which are typical morphology for SSZ-13. C550 shows a clean and smooth surface, while AS-AT exhibits rough surface due to desilication. A rougher surface is observed for C250-AT. The increase of surface roughness is due to the less protected frameworks by less amount of remaining organics within zeolite frameworks. Interestingly, C450-AT and C550-AT shows a hollow morphology, which is, possibly, because of the Al zoning on the periphery of the crystals. Since the core is more siliceous, the hydroxyl ion shall preferentially attack the core part of the crystals resulting in the formation of hollow morphology. A similar phenomenon has been previously reported for ZSM-5 crystals.35
 | ||
| Fig. 10 SEM and TEM images, and SAED patterns of (a) C550, (b) AS-AT, (c) C250-AT, (d) C450-AT and (e) C550-AT. | ||
Selected area electron diffraction (SAED) patterns of the products, depicted in Fig. 10, support the results of previous characterizations. C550, AS-AT, and C250-AT show distinctive patterns due to the presence of highly crystalline nature whereas C450-AT exhibits faint patterns because of the significantly less-crystalline structure. C550-AT does not show any pattern, only a halo since it is amorphous.
3.3 Catalytic activity in LDPE pyrolysis
In this section, we evaluate the catalytic activity of C550, AS-AT, and C250-AT by performing the temperature-programmed catalytic test of LDPE pyrolysis. A blank test, in the absence of catalyst, is also carried out for comparison. Acid sites, Brønsted and Lewis, of zeolites catalyze the pyrolysis of LDPE, by the proton addition to a C–C bond and by hydride abstraction, respectively. Both routes are followed by the β-scission to form small fragments of hydrocarbon. This allows the reaction to proceed at a lower temperature compared to that of thermal pyrolysis. However, these active sites must be accessible by the bulky reactants, including LDPE, calling out the necessity of mesopores to relieve the diffusion limitations.Fig. 11 shows the conversion of LDPE versus temperature with and without the presence of catalyst. The conversion, α, is calculated as follows, α = (mo – mt)/(mo – mf), where mo and mf refer to the initial and final weight of samples, respectively, while mt is the mass of the samples at a given time, t. The relative activity of the catalyst is measured as T50, the temperature at α of 0.5 or 50% LDPE conversion. The lower the T50, the more active a catalyst. As shown in Fig. 11, pure LDPE (blank test) decomposes at the highest temperature, T50 = 476 °C, whereas, in the presence of catalyst, T50 could be reduced. The T50 is gradually shifted to lower values in the order of C550 (468 °C) > AS-AT (463 °C) > C250-AT (460 °C). This sequence is in accordance with IHF (Fig. 12) showing that the presence of mesopores along with preserved microporosity clearly alleviate the diffusion limitations.
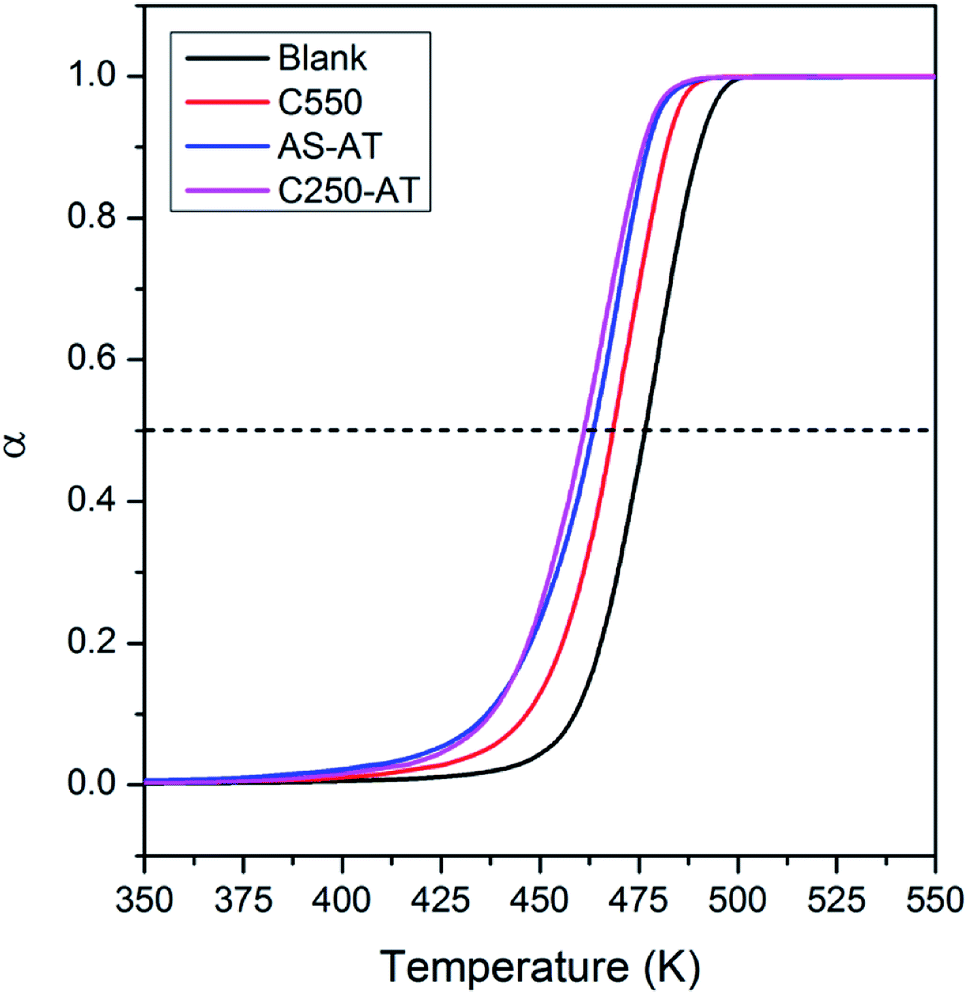 | ||
| Fig. 11 Thermogram of LDPE without and with the presence of SSZ-13 catalysts. | ||
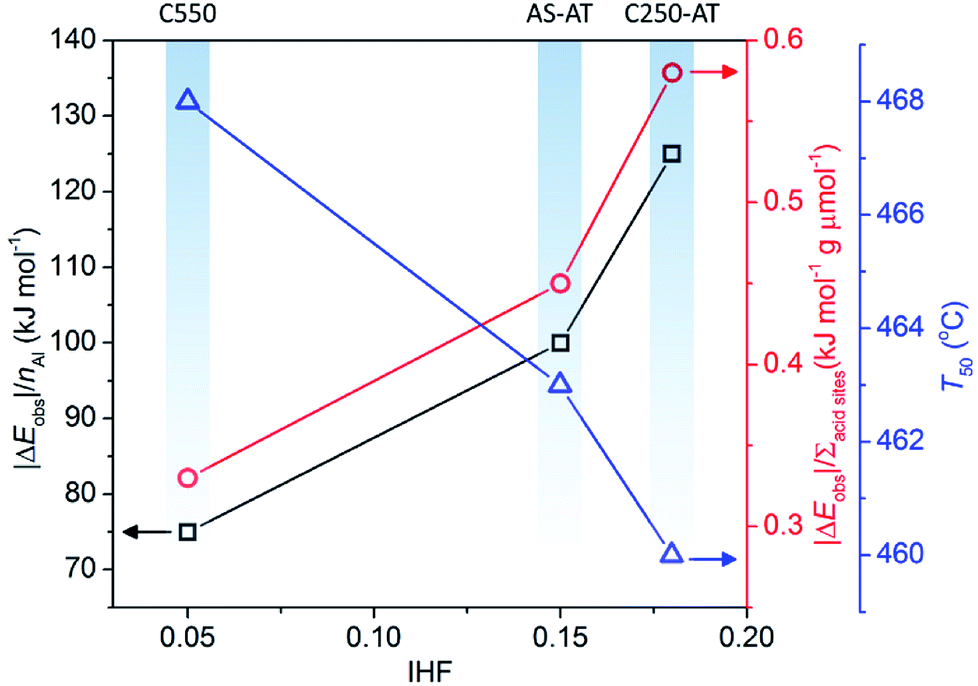 | ||
| Fig. 12 The effect of IHF on |ΔEobs|/nAl, |ΔEobs|/Σacid sites and T50. | ||
From TG data, the observed activation energy (Eobs) can be calculated using Coats-Redfern method (eqn (2)) since the LDPE pyrolysis is a first-order reaction.36,37
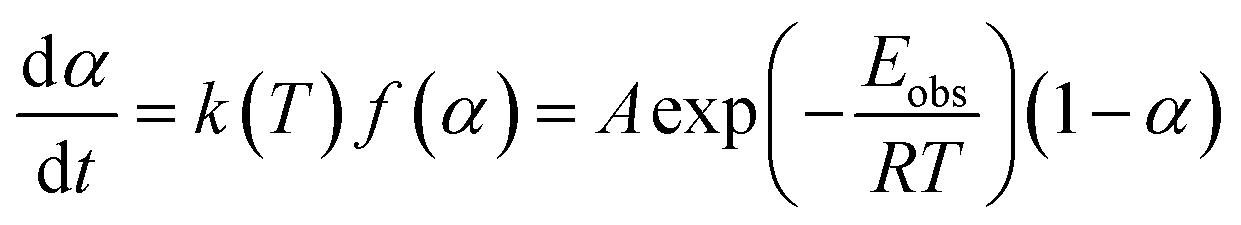 | (2) |
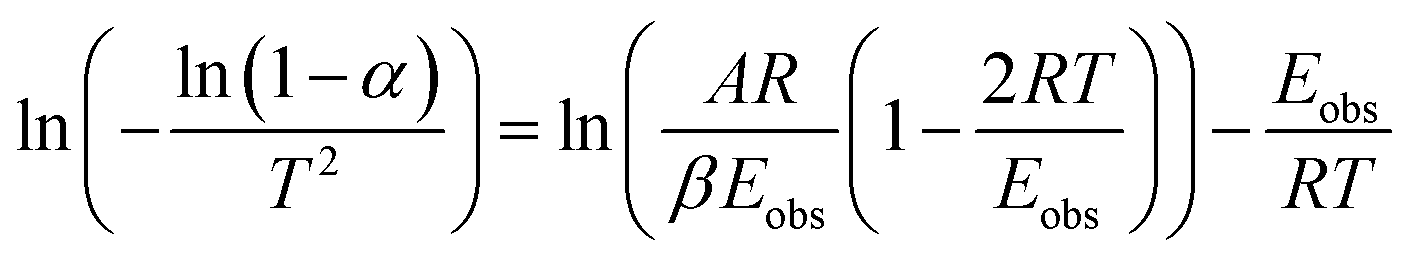 | (3) |
Plotting 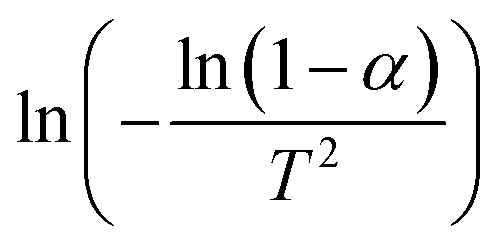 versus
versus 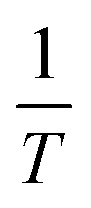 will result in a straight line with a slope of
will result in a straight line with a slope of 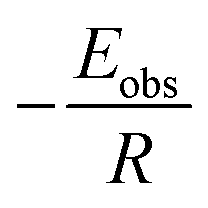 . The plots are provided in Fig. S6.† The Eobs in the absence of catalyst is 440 kJ mol−1 while the Eobs in the presence of catalysts undergoes a decrement based on the following order, C550 (374 kJ mol−1) < AS-AT (340 kJ mol−1) < C250-AT (302 kJ mol−1).
. The plots are provided in Fig. S6.† The Eobs in the absence of catalyst is 440 kJ mol−1 while the Eobs in the presence of catalysts undergoes a decrement based on the following order, C550 (374 kJ mol−1) < AS-AT (340 kJ mol−1) < C250-AT (302 kJ mol−1).
It is clear that the introduction of zeolites does lower the activation energy, functioning as catalysts. However, we realize that the enhanced activity in the hierarchically porous catalysts may also be due to decreased Si/Al ratio or increased total acid sites concentration. Therefore, we normalize |ΔEobs| (|Eobs,catalyst − Eobs,blank|) to the number of Al in a unit cell of each catalyst calculated from Si/Al framework obtained using eqn (1) and the total acid sites concentration denoted as (|ΔEobs|/nAl) and (|ΔEobs|/Σacid sites). The results are as follows, C250-AT (134 kJ mol−1) > AS-AT (100 kJ mol−1) > C550 (75 kJ mol−1) for |ΔEobs|/nAl. The similar order is also observed in the plot of |ΔEobs|/Σacid sites. Higher values of |ΔEobs|/nAl and |ΔEobs|/Σacid sites of the post-treated samples indicate the more accessible active sites to the reactant molecules. |ΔEobs|/nAl, |ΔEobs|/Σacid sites and T50 exhibit a proportional correlation with IHF (Fig. 12). Again, it is shown that mesopores play a significant role in the application of zeolites as catalysts.
4. Conclusions
In summary, the presence of organics could be used to tailor the formation of mesopores within SSZ-13 zeolites. The ability of the remaining organics to hinder the attack of hydroxyl ions to the frameworks could soften the extent of desilication, thus, inducing the formation of mesopores yet still retaining the crystallinity as well as microporosity. This fact is demonstrated by a series of detailed characterizations, including diffraction (XRD), spectroscopic (ATR-IR, Raman, 27Al and 29Si MAS NMR), NH3-TPD, electron microscopic and N2 physisorption studies. Catalytic tests on LDPE pyrolysis justify the effect of mesopores which smoothen the mass-transport, hence, increasing the catalytic performance. This method is suitable for zeolites with low framework stability, e.g. SSZ-13, which will undergo severe destruction when applied to a conventional desilication, without the protection of organics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is funded by Hibah Program Penelitian, Pengabdian kepada Masyarakat dan Inovasi (P3MI) ITB 2018. The authors thank Mr Ramadhan Dipta Maula for providing us the LDPE samples. We are grateful for OSDAs provided by SACHEM, Inc.References
- O. Cheung and N. Hedin, RSC Adv., 2014, 4, 14480–14494 RSC.
- T. D. Pham and R. F. Lobo, Microporous Mesoporous Mater., 2016, 236, 100–108 CrossRef CAS.
- M. Miyamoto, Y. Fujioka and K. Yogo, J. Mater. Chem., 2012, 22, 20186–20189 RSC.
- J. Zhang, R. Singh and P. A. Webley, Microporous Mesoporous Mater., 2008, 111, 478–487 CrossRef CAS.
- A. Shishkin, H. Kannisto, P.-E. Carlsson, J. Härelinda and M. Skoglundh, Catal. Sci. Technol., 2014, 4, 3917–3926 RSC.
- J. Yi, M. A. Deimund, Y. Bhawe and M. E. Davis, ACS Catal., 2015, 5, 4456–4465 RSC.
- J. F. Haw and D. M. Marcus, Top. Catal., 2015, 34, 41–48 CrossRef.
- T. Zhang, F. Qiu and J. Li, Appl. Catal., B, 2016, 15, 48–58 CrossRef.
- L. Wu, V. Degirmenci, P. C. M. M. Magusin, B. M. Szyja and E. J. M. Hensen, Chem. Commun., 2012, 48, 9492–9494 RSC.
- L. Wu, V. Degirmenci, P. C. M. M. Magusin, N. J. H. G. M. Lousberg and E. J. M. Hensen, J. Catal., 2013, 298, 27–40 CrossRef CAS.
- Z. Liu, N. Nomura, D. Nishioka, Y. Hotta, T. Matsuo, K. Oshima, Y. Yanaba, T. Yoshikawa, K. Ohara, S. Kohara, T. Takewaki and T. Okubo, Chem. Commun., 2015, 51, 12567–12570 RSC.
- M. Kumar, H. Luo, Y. Román-Leshkov and J. D. Rimer, J. Am. Chem. Soc., 2015, 137, 13007–13017 CrossRef CAS PubMed.
- Z. Li, M. T. Navarro, J. Martínez-Triguero, J. Yu and A. Corma, Catal. Sci. Technol., 2016, 6, 5856–5863 RSC.
- C. Peng, Z. Liu, A. Horimoto, C. Anand, H. Yamada, K. Ohara, S. Sukenaga, H. Shibata, T. Takewaki, R. R. Mukti, T. Okubo and T. Wakihara, Microporous Mesoporous Mater., 2018, 192–199 CrossRef CAS.
- X. Zhu, J. P. Hofmann, B. Mezari, N. Kosinov, L. Wu, Q. Qian, B. Weckhuysen, S. Asahina, J. Ruiz-Martínez and E. J. M. Hensen, ACS Catal., 2014, 6, 2163–2177 CrossRef.
- D. Verboekend and J. Péréz-Ramírez, Catal. Sci. Technol., 2011, 1, 879–890 RSC.
- D. Verboekend, S. Mitchell, M. Milina, J. C. Groen and J. Péréz-Ramírez, J. Phys. Chem. C, 2011, 115, 14193–14203 CrossRef CAS.
- D. P. Serrano, J. M. Escola and P. Pizarro, Chem. Soc. Rev., 2013, 42, 4004–4035 RSC.
- L. Sommer, D. Mores, S. Svelle, M. Stöcker, B. M. Weckhuysen and U. Olsbye, Microporous Mesoporous Mater., 2010, 132, 384–394 CrossRef CAS.
- R. Oord, I. C. ten Have, J. M. Arends, F. C. Hendriks, J. Schmidt, I. Lezcano-Gonzalez and B. M. Weckhuysen, Catal. Sci. Technol., 2017, 7, 3851–3862 RSC.
- A. Čimek, B. Subotić, I. Smit, A. Tonejc, R. Aiello, F. Crea and A. Nastro, Microporous Mater., 1997, 8, 159–169 CrossRef.
- X. Wei and P. G. Smirniotis, Microporous Mesoporous Mater., 2006, 97, 97–106 CrossRef CAS.
- J. Péréz-Ramírez, S. Abelló, A. Bonilla and J. C. Groen, Adv. Funct. Mater., 2009, 19, 164–172 CrossRef.
- J. C. Groen, S. Abelló, L. A. Villaescusa and J. Péréz-Ramírez, Microporous Mesoporous Mater., 2008, 114, 93–102 CrossRef CAS.
- C. Anand, T. Kaneda, S. Inagaki, S. Okamura, H. Sakurai, K. Sodeyama, T. Matsumoto, Y. Kubota, T. Okubo and T. Wakihara, New J. Chem., 2016, 40, 492–496 RSC.
- Z. Ristanovic, J. P. Hofmann, U. Deka, T. U. Schulli, M. Rohnke, A. M. Beale and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2013, 52, 13382–13386 CrossRef CAS PubMed.
- R. Al-Oweini and H. El-Rassy, J. Mol. Struct., 2009, 919, 140–145 CrossRef CAS.
- G. T. M. Kadja, V. A. Fabiani, M. H. Aziz, A. T. N. Fajar, A. Prasetyo, V. Suendo, E.-P. Ng and R. R. Mukti, Adv. Powder Technol., 2017, 28, 443–452 CrossRef CAS.
- W. Mozgawa, J. Mol. Struct., 2001, 596, 129–137 CrossRef CAS.
- M. Sitarz, W. Mozgawa and M. Handke, J. Mol. Struct., 1999, 511–512, 281–285 CrossRef CAS.
- W. Mozgawa and M. Sitarz, J. Mol. Struct., 2002, 614, 273–279 CrossRef CAS.
- A. Fernández-Jiménez and A. Palomo, Microporous Mesoporous Mater., 2005, 86, 207–214 CrossRef.
- Y. Yu, G. Xiong, C. Li and F.-S. Xiao, Microporous Mesoporous Mater., 2001, 46, 23–34 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- J. C. Groen, T. Bach, U. Ziese, A. N. M. P. Donk, K. P. de Jong, J. A. Moulijn and J. Péréz-Ramírez, J. Am. Chem. Soc., 2005, 127, 10792–10793 CrossRef CAS PubMed.
- S. Kim and Y. C. Kim, J. Anal. Appl. Pyrolysis, 2005, 73, 117–121 CrossRef CAS.
- G. Yan, X. Jing, H. Wen and S. Xiang, Energy Fuels, 2015, 29, 2289–2298 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08979e |
| This journal is © The Royal Society of Chemistry 2019 |