
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnti-phytopathogenic sesquiterpenoid-xanthone adducts from potato endophytic fungus Bipolaris eleusines†
Juan Hea,
Man-Si Yanga,
Wen-Xiang Wangb,
Zheng-Hui Lia,
Waill Ahmed Mohamed Elkhateebd,
Ting-Chi Wen*c,
Hong-Lian Ai*ab and
Tao Feng *a
*a
aSchool of Pharmaceutical Sciences, South-Central University for Nationalities, Wuhan 430074, China. E-mail: 2015050@mail.sucuec.edu.cn; tfeng@mail.scuec.edu.cn
bSchool of Agriculture and Biological Technic, Yunnan Agricultural University, Kunming 650201, China
cThe Engineering Research Center of Southwest Bio-Pharmaceutical Resources, Ministry of Education, Guizhou University, Guiyang 550025, China. E-mail: tingchiwen@yahoo.com
dDepartment of Chemistry of Microbial Natural Products, National Research Center, Dokki, Giza 12311, Egypt
First published on 21st December 2018
Abstract
A bioguided separation on the cultures of the potato endophytic fungus Bipolaris eleusines led to the isolation of two anti-phytopathogenic (Alternaria solani) sesquiterpenoid-xanthone adducts, namely bipolins I and J (1 and 2). Their structures were established via extensive spectroscopic analysis. Compounds 1 and 2 exhibit potent inhibitory activity against A. solani with MIC values of 8 and 16 μg mL−1, respectively.
Introduction
Sativene sesquiterpenoids usually feature an unusual 5/6 or 5/7 bridged ring system,1–4 which attracted great interest in total synthesis5–8 and biological studies.2,8–10 Our previous investigations on the potato endophytic fungus Bipolaris eleusines reported bipolenins A–H,11–13 and indicated that the fungus is a good resource for sativene sesquiterpenoids. Some of them show cytotoxicities to human cancer cell lines and inhibitory activity on nitric oxide production. Our recent studies found that the potato endophytic fungus B. eleusines can inhibit the growth of potato pathogen Alternaria solani (early blight) on PDA (Fig. 1), which suggested the metabolites from the fungus may possess good inhibitions on the plant pathogen. This discovery prompted us to search for anti-pathogenic agents from the endophytic fungus B. eleusines. Therefore, a bioguided isolation on the cultures of the fungus B. eleusines was carried out. As a result, two novel sativene sesquiterpenoid-xanthone adducts, namely bipolenins I and J (1 and 2, Fig. 2), were obtained from liquid fermentation of the fungus. Their structures were established via extensive spectroscopic analyses. Pure compounds 1 and 2 were evaluated for their inhibitory activities on A. solani. Herein the isolation, structural elucidation, and bioactivity of compounds 1 and 2 are reported.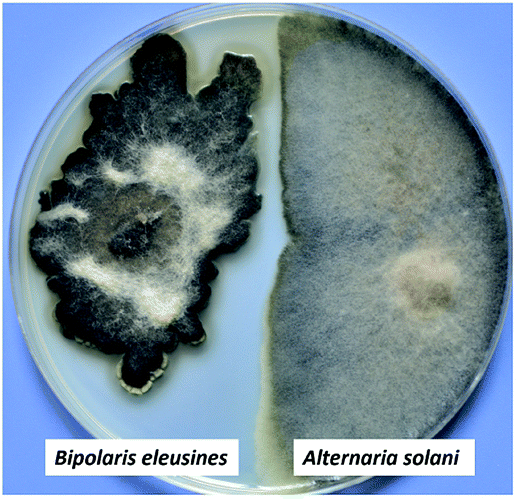 | ||
| Fig. 1 The growth inhibition of potato endophytic fungus B. eleusines against potato pathogen Alternaria solani on PDA. | ||
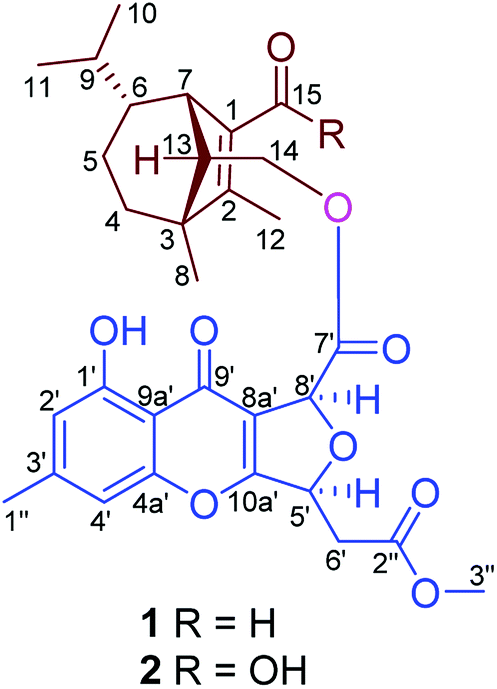 | ||
| Fig. 2 Structures of bipolenins I and J (1 and 2) from cultures of the fungus Bipolaris eleusines. | ||
Results and discussion
Bipolenin I (1) had a molecular formula C31H36O9 as determined on the basis of the positive HRESIMS, which showed a molecular ion peak at m/z 575.2250 [M + Na]+ (calcd for C31H36O9Na, 575.2252), corresponding to seven degrees of unsaturation. The UV data at 329 nm indicated a highly conjugated system, while the IR absorption bands at 3440 and 1745 cm−1 suggested the existence of hydroxy and carbonyl, respectively. The 1H and 13C NMR spectra, in association with the HSQC spectrum, revealed six methyl (one methoxy), four methene, nine methine, and twelve quaternary carbons (Table 1). Preliminary analysis on the NMR data, a possible moiety of sativene sesquiterpenoid was detected by comparison the data with those our reported previously.1,2,4,11–13 As shown in unit A of Fig. 3, a long spin system was obtained by analysis of the 1H–1H COSY spectrum. A methyl substituted 1,2-double bond-15-aldehyde moiety was established by the 13C NMR data at δC 137.2 (s, C-1), 164.9 (s, C-2), 187.9 (d, C-15), and 10.6 (q, C-12), and supported by HMBC correlations from δH 10.00 (s, H-15) and 2.01 (3H, s, H-12) to C-1 and C-2. This moiety was connected by bonds of C-1/C-7 and C-2/C-3 as deduced from HMBC correlations from δH 3.11 (1H, br s, H-7) to C-2, and from H-12 to an sp3 quaternary carbon at δC 50.9 (s, C-3). In addition, a singlet at δH 1.05 (3H, s, H-8) for a methyl group showed HMBC correlations to C-3, C-2, and to other two carbon resonances at δC 33.8 (d, C-4) and 57.5 (d, C-13). These data, as well as other HMBC correlations (Fig. 3), suggested that unit A was a sativene sesquiterpenoid related to helminthosporol.1,4 In unit B (Fig. 3), a four-substituted benzene ring was identified by two aromatic protons at δH 6.68 (1H, s, H-2′) and 6.74 (1H, s, H-4′). A methyl and a hydroxy as two substituents in the benzene ring were identified by the HMBC correlations as shown in Fig. 3. Analysis of UV data and the chemical shifts at δC 178.0 (s, C-9′), 114.5 (s, C-8a′), 167.5 (s, C-10a′) suggested the existence of a chromone core of unit B. Due to the existence of an intramolecular hydrogen bond, the proton of the hydroxy group appeared as a singlet at δH 12.09 (s, OH-1′), which further confirmed the existence of the chromone core. The 1H–1H COSY spectrum revealed a short spin system of CH(5′)–CH2(6′), which connected to a methyl ester group as established by the HMBC correlations from δH 2.97 (1H, dd, J = 16.0, 4.3 Hz, H-6′a), 2.82 (1H, dd, J = 16.0, 4.3 Hz, H-6′b) and 3.73 (3H, s, OMe) to δC 169.3 (s, C-2′′). The spin system also connected to C-10a′ based on the HMBC correlation from δH 5.69 (1H, ddd, J = 6.3, 4.3, 4.0 Hz, H-5′) to C-10a′. So far, only two carbons including a methine (CH-8′) and a carbonyl carbon at δC 169.4 (s, C-7′) were left. Further analysis of HMBC spectrum, the correlations from δH 5.88 (1H, d, J = 4.0 Hz, H-8′) to C-7′, C-8a′, and C-5′ suggested that C-8′ was connected to C-8a′ and C-7′, respectively, and an ether bond was established between C-5′ and C-8′. The unit B was, therefore, identified as a seco-xanthone related to a synthetic product in the literature.14 The connection by an ester bond between units A and B was revealed by an HMBC correlation from H-14 to C-7′, as well as analysis of the MS data. An ROESY experiment was carried out to establish the relative configuration of 1 (Fig. 3). In unit A, a cross peak between H-6 and H-13 suggested that H-6 and H-13 were in the same side. Another cross peak between H-12 and H-14 confirmed the relative configuration of C-13 as shown. In unit B, a key cross peak between H-5′ and H-8′ indicated that H-5′ and H-8′ were in the same side, while the cross peaks of H-1′′/H-4′ and H-1′′/H-2′ helped with the position identification of substituents in the benzene ring. The structure of 1 was, finally, established as depicted.| No. | 1 | 2 | ||
|---|---|---|---|---|
| δC | δH | δC | δH | |
| 1 | 137.2, s | 125.5, s | ||
| 2 | 164.9, s | 161.3, s | ||
| 3 | 50.9, s | 50.5, s | ||
| 4 | 33.8, t | 1.38, m | 33.6, t | 1.34, m |
| 5a | 25.0, t | 1.72, m | 24.9, t | 1.71, m |
| 5b | 0.84, m | 0.97, m | ||
| 6 | 44.6, d | 0.98, m | 44.9, d | 0.96, m |
| 7 | 41.7, d | 3.11, br s | 43.6, d | 3.10, br s |
| 8 | 18.4, q | 1.05, s | 18.9, q | 1.00, s |
| 9 | 31.5, t | 0.97, m | 31.5, d | 1.17, m |
| 10 | 20.6, q | 0.71, s | 20.8, q | 0.74, d (6.5) |
| 11 | 21.3, q | 0.95, s | 21.4, q | 0.93, d (6.5) |
| 12 | 10.6, q | 2.01, s | 12.5, q | 2.00, s |
| 13 | 57.5, d | 1.81, dd (8.6, 5.6) | 58.0, d | 1.79, dd (8.3, 5.5) |
| 14a | 65.8, t | 4.21, dd (11.1, 5.6) | 66.0, t | 4.26, dd (11.2, 5.5) |
| 14b | 3.89, dd (11.1, 5.6) | 3.94, dd (11.2, 8.3) | ||
| 15 | 187.9, d | 10.00, s | 168.6, s | |
| 1′ | 160.9, s | 160.9, s | ||
| 2′ | 113.5, d | 6.68, s | 113.5, d | 6.66, br s |
| 3′ | 147.4, s | 147.4, s | ||
| 4′ | 107.9, d | 6.74, s | 107.9, d | 6.73, br s |
| 4a′ | 157.2, s | 157.2, s | ||
| 5′a | 78.0, d | 5.69, ddd (6.3, 4.3, 4.0) | 78.0, d | 5.70, ddd (6.4, 4.3, 4.0) |
| 5′b | ||||
| 6′a | 37.6, t | 2.97, dd (16.0, 4.3) | 37.6, t | 2.97, dd (16.1, 4.3) |
| 6′b | 2.82, dd (16.0, 6.3) | 2.82, dd (16.1, 6.4) | ||
| 7′ | 169.4, s | 169.5, s | ||
| 8′ | 79.4, d | 5.58, d (4.0) | 79.4, d | 5.60, d (4.0) |
| 8a′ | 114.5, s | 114.6, s | ||
| 9′ | 178.0, s | 177.9, s | ||
| 9a′ | 108.9, s | 108.9, s | ||
| 10a′ | 167.5, s | 167.5, s | ||
| 1′′ | 22.3, q | 2.41, s | 22.3, q | 2.40, s |
| 2′′ | 169.3, s | 169.3, s | ||
| OMe | 52.2, q | 3.73, s | 52.2, q | 3.73, s |
| 1′-OH | 12.09, s | 12.10, s | ||
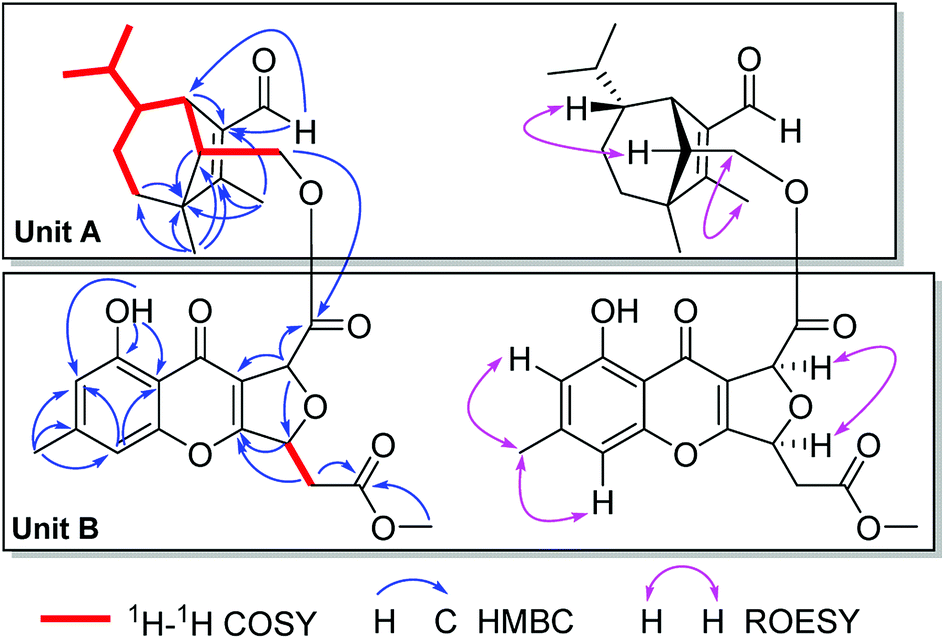 | ||
| Fig. 3 Key 2D NMR correlations of 1. | ||
Compound 2 was isolated as a light yellow gum. Its molecular formula C31H36O10 was determined by an HRESIMS ion peak at m/z 567.2240 [M − H]− (calcd for C31H35O10, 567.2236), 16 unit more than that of 1. All the spectroscopic data of 2 displayed almost the same patterns to those in 1, except that a carboxyl signal at δC 168.6 (s, C-15) in the 13C NMR of 2 replaced the aldehyde signal at δC 187.9 in that of 1. It was supported by an HMBC correlation from δH 3.10 (1H, br s, H-7) to C-15, while the other parts of 2 were elucidated as the same to those of 1 by detailed analysis of 2D NMR data. The structure of 2 was established, and named as bipolenin J.
Compounds 1 and 2 were evaluated for their anti-phytopathogenic activities against four plant pathogens including Phytophthora infestane, Alternaria solani, Rhizoctonia solani, and Fusarium oxysporum. As a result (Table 2), compounds 1 and 2 all exhibited potent inhibitory activity against A. solani with MIC values of 8 and 16 μg mL−1, respectively. While compound 1 also weakly inhibited F. oxysporum with an MIC values of 64 μg mL−1.
Conclusions
In summary, two novel sesquiterpenoid-xanthone adducts, bipolenins I and J, were obtained from potato endophytic fungus Bipolaris eleusines by a bioguided isolation. The potent inhibitory activities of B. eleusines and its metabolites on phytopathogens revealed a possible application prospect on biological pesticide for B. eleusines.Experimental section
General experimental procedures
The melting points were tested by a Putiantongchuang WRX-5A apparatus. Optical rotations were measured on a Jasco-P-1020 polarimeter. IR spectra were obtained by using a Bruker Tensor 27 FT-IR spectrometer with KBr pellets. NMR spectra were acquired with instrument of a Bruker DRX-500. HSESIMS was measured on an API QSTAR Pulsar spectrometer. Silica gel (200–300 mesh, Qingdao Marine Chemical Inc., China) and Sephadex LH-20 (Amersham Biosciences, Sweden) were used for column chromatography (CC). Fractions were monitored by TLC (Qingdao Marine Chemical Inc., China) and spots were visualized by heating silica gel plates immersed in vanillin–H2SO4 in EtOH, in combination with Agilent 1200 series HPLC system (Eclipse XDB-C18 column, 5 μm, 4.6 × 150 mm). Preparative HPLC was performed on an Agilent 1100 series with a Zorbax SB-C18 (5 μm, 9.4 × 150 mm) column.Fungal material and cultivation conditions
The fungus Bipolaris eleusines was isolated from fresh potatoes sampled from the nursery of Yunnan Agricultural University at random in July 2012. The fungus was identified by observing the morphological characteristics and analysis of the internal transcribed spacer (ITS) regions (max identity: 99%; query cover: 98%; accession: KJ909768.1). The strain is deposited at South-Central University for Nationalities, China (No. PE20120728-SCUN-201603).This fungal strain was cultured on potato dextrose agar (PDA) medium at 24 °C for 10 days. The agar plugs were inoculated in 500 mL Erlenmeyer flasks, each containing 100 mL of potato dextrose media. Flask cultures were incubated at 28 °C on a rotary shaker at 160 rpm for two days as seed culture. One hundred and twenty 500 mL Erlenmeyer flasks each containing 150 mL of potato dextrose broth (PDB) were individually inoculated with 25 mL of seed culture, and were incubated at 25 °C on a rotary shaker at 160 rpm for 20 days.
Extraction and isolation
A total of 20 L culture broth was extracted three times with EtOAc. The organic layer was evaporated to give a crude extract (15 g). The crude extract was subjected to silica gel CC (200–300 mesh) eluted with a CHCl3–MeOH solvent system (a gradient from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 to 0:1) to afford seven fractions A–G. Fraction C (1.2 g) was further separated by silica gel CC eluted with petroleum ether–Me2CO (12:1) to afford subfractions C1–C6. Fraction C2 (90 mg) was isolated by Sephadex LH-20 CC (MeOH) and by HPLC (MeCN–H2O, from 3:7 to 6:4 in 25 min, 10 mL min−1) to give 1 (4.2 mg) and 2 (3.5 mg).
ε) 203.0 (3.65), 250.5 (3.66), 329 (2.88) nm; IR (KBr) νmax: 3440, 2924, 1745, 1661, 1635, 1619, 1462, 1383, 1208 cm−1; 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) data, see Table 1, HRESIMS m/z 575.2250 [M + Na]+ (calcd for C31H36O9Na, 575.2252).ε) 203 (3.53), 246 (3.52), 329.5 (2.58), 382.5 (2.37) nm; IR (KBr) νmax 3441, 2923, 1740, 1632, 1462, 1384, 1206, 1061 cm−1; 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) data, see Table 1, HRESIMS m/z 567.2240 [M − H]− (calcd for C31H35O10, 567.2236).
:0 to 0:1) to afford seven fractions A–G. Fraction C (1.2 g) was further separated by silica gel CC eluted with petroleum ether–Me2CO (12:1) to afford subfractions C1–C6. Fraction C2 (90 mg) was isolated by Sephadex LH-20 CC (MeOH) and by HPLC (MeCN–H2O, from 3:7 to 6:4 in 25 min, 10 mL min−1) to give 1 (4.2 mg) and 2 (3.5 mg).
ε) 203.0 (3.65), 250.5 (3.66), 329 (2.88) nm; IR (KBr) νmax: 3440, 2924, 1745, 1661, 1635, 1619, 1462, 1383, 1208 cm−1; 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) data, see Table 1, HRESIMS m/z 575.2250 [M + Na]+ (calcd for C31H36O9Na, 575.2252).ε) 203 (3.53), 246 (3.52), 329.5 (2.58), 382.5 (2.37) nm; IR (KBr) νmax 3441, 2923, 1740, 1632, 1462, 1384, 1206, 1061 cm−1; 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) data, see Table 1, HRESIMS m/z 567.2240 [M − H]− (calcd for C31H35O10, 567.2236).Anti-phytopathogenic assay
Compounds 1 and 2 were subjected to minimal inhibitory concentration (MIC) tests against four phytopathogenic fungi (Phytophthora infestane, Alternaria solani, Rhizoctonia solani, Fusarium oxysporum) on the PDA medium using a twofold serial dilution in the microplate wells over the range of 4–256 μg mL−1. To this end, into each well of 96 well plate was placed 80 μL of PDA along with a 20 μL volume of an aqueous test sample solution. The test solutions held different concentrations of the sample being tested. The control well held 80 μL PDA and 20 μL of sterile water. Then, agar plugs (1 mm3) with fresh phytopathogens were inoculated into each well. Observations of the plates were made after 24 h of incubation at 26 °C in order to acquire the MICs with no growth in the well taken as that value. Three replicates were maintained to confirm the antifungal activity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (31560010, 81872762, 31870513) and the Fundamental Research Funds for the Central University, South-Central University for Nationalities (CZT18014, CZT18013). The authors thank the Analytical & Measuring Center of Kunming Institute of Botany, CAS for the spectral test.Notes and references
- S. Tamura, A. Sakurai, K. Kainuma and M. Takai, Agric. Biol. Chem., 1963, 27, 738–739 CrossRef CAS.
- P. de Mayo and R. E. Williams, J. Am. Chem. Soc., 1965, 87, 3275 CrossRef CAS.
- F. Dorn and D. Arigoni, Experientia, 1974, 30, 851–852 CrossRef CAS.
- C. Osterhage, G. M. König, U. Höller and A. D. Wright, J. Nat. Prod., 2002, 65, 306–313 CrossRef CAS.
- G. L. Hodgson, D. F. MacSweeney and T. Money, J. Chem. Soc., Perkin Trans. 1, 1973, 19, 2113–2130 RSC.
- P. Bakuzis, O. O. S. Campos and M. L. F. Bakuzis, J. Org. Chem., 1976, 41, 3261–3264 CrossRef CAS.
- S. Karimi, J. Nat. Prod., 2001, 64, 406–410 CrossRef CAS.
- A. Khanum Shaukath, S. Shashikanth, G. Sathyanarayana Syagadadu, S. Lokesh and A. Deepak Saligrama, Pest Manage. Sci., 2009, 65, 776–780 CrossRef CAS PubMed.
- H. Miyagawa, S. Nagai, T. Tsurushima, M. Sato, T. Ueno and H. Fukami, Biosci., Biotechnol., Biochem., 1994, 58, 1143–1145 CrossRef CAS.
- J. K. Park, K. Hasumi and A. Endo, J. Antibiot., 1993, 46, 1303–1305 CrossRef CAS PubMed.
- H. L. Ai, M. S. Yang, S. H. Zi and H. C. Guo, J. Asian Nat. Prod. Res., 2015, 17, 982–987 CrossRef CAS.
- M. S. Yang, X. Y. Cai, Y. Y. He, M. Y. Lu, S. Liu, W. X. Wang, Z. H. Li, H. L. Ai and T. Feng, Nat. Prod. Bioprospect., 2017, 7, 147–150 CrossRef CAS PubMed.
- Z.-H. Li, H.-L. Ai, M.-S. Yang, J. He and T. Feng, Phytochem. Lett., 2018, 27, 87–89 CrossRef CAS.
- T. Sassa, H. Kachi and M. Nukina, J. Antibiot., 1985, 38, 439–441 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09861a |
| This journal is © The Royal Society of Chemistry 2019 |