
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEthanol dehydrogenative reactions catalyzed by copper supported on porous Al–Mg mixed oxides†
Davi D. Petrolini a,
Wellington H. Cassinellib,
Cristiane A. Pereirac,
Ernesto A. Urquieta-Gonzálezc,
Celso V. Santillia and
Leandro Martins*a
a,
Wellington H. Cassinellib,
Cristiane A. Pereirac,
Ernesto A. Urquieta-Gonzálezc,
Celso V. Santillia and
Leandro Martins*a
aInstituto de Química, UNESP – Universidade Estadual Paulista, R. Prof. Francisco Degni 55, 14800-900 Araraquara, SP, Brazil. E-mail: leandro.martins@unesp.br
bInstituto Federal de São Paulo – Campus Avaré, Av. Prof. Celso Ferreira da Silva, 1333, Jardim Europa, 18707-150, Avaré, SP, Brazil
cCentro de Pesquisas em Materiais Avançados e Energia – Universidade Federal de São Carlos, Rodovia Washington Luis, km 235, CEP 13565-905, São Carlos, SP, Brazil
First published on 24th January 2019
Abstract
Mixed aluminum and magnesium oxides (AlMgO) prepared by means of an emulsion-mediated sol–gel method was impregnated with copper nitrate solution and used in the ethanol dehydrogenative reactions to produce acetaldehyde and ethyl acetate. The emulsified system allowed to obtain a macro–mesoporous support that resulted in an outstanding dispersion of copper. The porous catalyst was about 3 times more active than the non-porous counterpart, due to the formation on the support's surface of Cu0 together with the more active Cu+ species. In fact, the simultaneous presence of Cu+ and Cu0 were advantageous for the catalytic performance, as the turnover frequencies, were 122 and 166 h−1 for the non-porous reference catalyst and for the porous one, respectively. Both catalysts deactivated due to copper particles sintering, however the porous one deactivated less, as a consequence of the better dispersion of the Cu species on the macro and mesoporous support. Acetaldehyde was the main product, however by increasing the contact time by 6.6 times, the conversion of ethanol on the non-porous catalyst reached about 90% with a selectivity to ethyl acetate of 20% by means of the coupling reaction of ethanol and acetaldehyde. The selectivity to ethyl acetate was favoured on an increased support/copper interface that is given by larger copper particles.
1. Introduction
The interest in biomass sources has mostly increased in the last decades due to the intensification of the seek for biofuels and renewable chemical products that might substitute fuels or petroleum derived products,1,2 which are currently the most important sources of the emission of greenhouse gases. Among the several processes that are being used for producing biofuels, ethanol from fermentation of biomass-derived sugars is the oldest and most important process used in the biotechnology industry. Beyond of its usage as fuel, ethanol is a versatile building block for chemical industries because it can be catalytically converted into several chemicals by means of dehydration (to give ethylene), Guerbet coupling (n-butanol), dehydrogenation (acetaldehyde and hydrogen) and by the coupling reaction of acetaldehyde with ethanol (ethyl acetate).3,4 When the last two reactions occur in a sequence, the global reaction process is called dehydrogenative coupling of ethanol, giving hydrogen and ethyl acetate.Ethanol dehydrogenation stands out by the importance of the products hydrogen and acetaldehyde, which can be used as an intermediates in the manufacture of acetic acid, acetic anhydride, ethyl acetate, butyraldehyde, crotonaldehyde and n-butanol.3,5–10 Though it has a lower hydrogen yield compared to the ethanol reforming reactions, ethanol dehydrogenation is carried out at lower temperatures. Even whether ethanol reforming is operated at mild temperatures, it still generates carbon monoxide, which causes loss of efficiency and early fuel cell poisoning.8 Moreover, the coupling reaction to further give ethyl acetate is a very attractive sustainable strategy,9,10 because it enables the production of esters directly from alcohols. Compared to the traditional esterification of ethanol with carboxylic acids, the coupling is advantageous due to the reduction of wastes and enhancement of process profit.
Some aspects are critical to guarantee high ethyl acetate yield during the catalytic ethanol coupling reaction, such as the acid–base property of the catalyst, which if not properly tuned, it can lead to unwanted by-products. The porosity of the support is another important property because it influences the particle size and the oxidation state of copper species, which are the active ones for the mentioned reactions.11 A detailed kinetic and mechanistic study on Cu/Cr2O3 showed the central role of copper in the adsorption and dehydrogenation of ethanol, which is due to its ability to preserve intact the C–C bonds and facilitate the hydrogen transfer reactions.12 In addition, it was emphasized the negative effect of the Brønsted acid sites of Cr2O3 that prevent desorption of acetaldehyde and make this a rate determinant step, which results in limited activity, without considering the unwanted capacity of the Brønsted acid sites to dehydrate ethanol.12 Therefore, the appropriate balance of acid–base sites of the support is crucial for the dehydrogenative coupling of alcohols.
The use of aluminum and magnesium mixed oxides derived from hydrotalcites as support for impregnation of copper species and forward application in the dehydrogenation of alcohols is motivating. After heat treatment, these mixed oxides have various unique properties as providing an ideal balance of acid and base sites13–17 and unique textural properties, which have impact on metals dispersion and therefore on ethanol conversion to acetaldehyde and ethyl acetate.3,5
In a previous study,18–20 an emulsion-mediated method was used to produce Al–Mg mixed oxides from hierarchically ordered hydrotalcite precursors that gave a high specific surface area. It is expected that the textural properties of these materials may reduce metal agglomeration and increase the interparticle space, and then to have an incremental effect on the catalyst performance. In fact, in situ studies have shown that the catalytic reaction rates are significantly optimized by changing the metal particle size. Moreover, the particle size in copper based catalysts affected the surface ratio of Cu0 to Cu+ and consequently the activity in reactions involving oxygenated intermediates.21–26 It is known that Cu0 supported catalysts are the most active for ethanol dehydrogenation, but further studies have shown that partially oxidized Cu+ directly influence the activity towards ethyl acetate.3,5,6,21,27
Herein, we examined how the textural properties and acid–base properties of highly porous aluminum and magnesium mixed oxides prepared from an emulsified synthesis system influence the dispersion of copper species and their oxidation state and, consequently their catalytic performance on the dehydrogenative coupling of ethanol to produce acetaldehyde and ethyl acetate with an enhanced selectivity.
2. Experimental
2.1. Synthesis of the supports and catalysts
The porous and the reference non-porous aluminum-magnesium mixed oxides, with molar fraction of Al/Mg of 0.5, were obtained by sol–gel synthesis, according to a previous method.18,19 The synthesis of the porous mixed oxides involved the use of aluminum tri-sec-butoxide (named as Al-But3), magnesium nitrate, n-dodecane as emulsification agent, a non-ionic triblock copolymer as emulsion stabilizer (Pluronic P123) and ethanol as solvent. The molar composition was: 0.01 Pluronic![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Al-But3:1 Mg(NO3)2:15 ethanol. The preparation of the sol involved the dispersion of Pluronic P123 in ethanol at room temperature, followed by addition of Al-But3 and magnesium nitrate. Subsequently, emulsification was achieved by adding 60 wt% of n-dodecane, under mechanical stirring. The addition of a solution of ammonium hydroxide drop by drop under stirring led to the sol–gel transition. The as-synthesized samples were calcined at 500 °C for 2 h under air in a conventional muffle oven. For sake of comparison, a reference support was obtained without the use of organic pore structure directing agents (PSDAs), Pluronic 123 and n-dodecane. The samples obtained without and using the PSDAs agents were named AlMgO and AlMgO–P, respectively, in which P stands for porous. The copper catalysts, with a nominal content of 10 wt% of copper, were obtained by incipient wetness impregnation of the calcined mixed oxides using an aqueous solution of copper nitrate. After impregnation, the samples were calcined at 500 °C for 3 h.
:1 Al-But3:1 Mg(NO3)2:15 ethanol. The preparation of the sol involved the dispersion of Pluronic P123 in ethanol at room temperature, followed by addition of Al-But3 and magnesium nitrate. Subsequently, emulsification was achieved by adding 60 wt% of n-dodecane, under mechanical stirring. The addition of a solution of ammonium hydroxide drop by drop under stirring led to the sol–gel transition. The as-synthesized samples were calcined at 500 °C for 2 h under air in a conventional muffle oven. For sake of comparison, a reference support was obtained without the use of organic pore structure directing agents (PSDAs), Pluronic 123 and n-dodecane. The samples obtained without and using the PSDAs agents were named AlMgO and AlMgO–P, respectively, in which P stands for porous. The copper catalysts, with a nominal content of 10 wt% of copper, were obtained by incipient wetness impregnation of the calcined mixed oxides using an aqueous solution of copper nitrate. After impregnation, the samples were calcined at 500 °C for 3 h.
2.2. Characterization of the supports and catalysts
The porosity and macro and mesopore size distributions of the calcined mixed AlMgO oxides and copper catalysts were assessed by mercury intrusion porosimetry in an AutoPore III instrument from Micromeritics. The samples were degassed at a pressure below 50 μPa before starting the measurements. The Washburn equation28 was used to calculate the pore size by using mercury surface tension of 0.489 N m−1 and contact angle of 135°.The porosity of the samples was further characterized by nitrogen adsorption–desorption isotherms at −196 °C on an ASAP 2010 instrument from Micromeritics with relative pressure ranging between 0.001 and 0.998. Prior to the analysis, the samples were vacuum-degassed at 200 °C for 12 h under a pressure below to 10 μPa. Pore volumes, mesopore size distributions (BJH method) and BET surface area for P/P0 up to 0.3 were determined from the isotherms.29
The microstructure of mixed oxides was analyzed by scanning electron microscopy (SEM), using a Philips XL 30 equipment. The samples were deposited on an aluminum sample holder and sputtered with gold.
X-ray diffraction profiles of the copper catalysts were recorded using a Siemens D5000 diffractometer equipped with a CuKα radiation (selected by a curved graphite monochromator). The protocol of the scanning steps was 2θ range from 3° to 80°, with step size of 0.02°, and counting time of 1 s.
Temperature programmed desorption of ammonia (NH3-TPD) and of carbon dioxide (CO2-TPD) were performed separately to assess the acid and base sites of the catalysts, respectively. A very similar protocol was used for both analyses. Firstly, 150 mg of catalyst was pre-treated by heating up to 500 °C and kept at this temperature for 1 h under a flow of 60 mL min−1 of He. After that, the samples were cooled to 120 °C for NH3 adsorption or to 50 °C for CO2 adsorption. Then, a flow of 30 mL min−1 of either NH3 (1% in He, v/v) during 0.5 h or pure CO2 for 2 h was fed into the reactor. The physically adsorbed NH3 or CO2 were purged for 1 h at the same temperature in which the adsorption process occurred. Upon heating from the respective adsorption temperature to 600 °C, with a heating rate of 10 °C min−1 and under a flow of 30 mL min−1 of He, NH3 or CO2 desorption was monitored by a thermal conductivity detector of the Micromeritics Pulse Chemisorb 2705 equipment.
Profiles of temperature programmed reduction with hydrogen (H2-TPR) of the calcined copper catalysts were performed using a Micromeritics Autochem 2920 equipment. A flow of 30 mL min−1 of H2 (10% in He, v/v) was fed into the reactor and the temperature was raised from room temperature to 500 °C using a heating rate of 10 °C min−1. The hydrogen consumption was monitored using a thermal conductivity detector. Using the same equipment, N2O chemisorption experiments were performed to estimate the metallic surface area of the dispersed Cu0 species. The sample was previously reduced at 250 °C for 30 min, and then cooled to 30 °C and exposed to a flow of 30 mL min−1 of N2O (1% in He, v/v) for 10 min. It is expected that the AlMgO support do not exhibit a significant interaction with N2O at this temperature. A second H2-TPR was performed by increasing the temperature up to 400 °C. The hydrogen consumption was used to calculate the amount of surface oxidized copper after N2O chemisorption. No bulk oxidation was observed for samples. The copper surface area (SCu) and particle isometric size (dCu) were estimated by using eqn (1) and (2), respectively, and a correlation of 1.46 × 1019 Cu atoms per m2 and a stoichiometry of reduction of 2 Cu/H2 were found.
| SCu = 6.4955 × 10−2 × C × D | (1) |
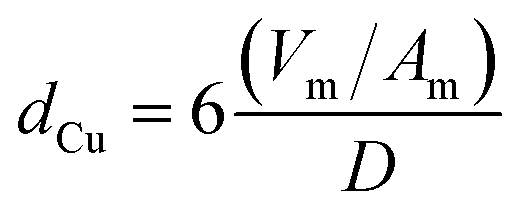 | (2) |
In eqn (1) and (2), C is the copper content in wt%, D is the copper dispersion in %, Vm is the mean atomic density of copper in g mL−1 and Am is the atomic surface area in nm2 per atom.30
In situ XANES measurements at the Cu K-edge (8979 eV) was carried out at the D06A-DXAS beamline of the Brazilian Synchrotron Light Laboratory (LNLS) at Campinas, Brazil. The D06A-DXAS beamline was equipped with a focusing curved Si (111) monochromator, operating in the Bragg mode for the selection of the desired range of X-ray wavelengths (8900–9400 eV). The samples were prepared as self-supported pellets containing 25 mg of catalyst mixed with boron nitride and placed into a tubular quartz furnace (d = 20 mm and X-ray path length = 440 mm) sealed with Kapton refrigerated windows for the transmission measurements. Temperature-resolved XANES spectra at the Cu K-edge were acquired during TPR experiments in which it was used a flow of 30 mL min−1 of H2 (5% in He, v/v) and a heating rate of 10 °C min−1 from room temperature up to 250 °C. The calibration energy and normalization of XANES spectra were performed using the Athena graphical interface program. The information about the proportion of copper species (Cu2+, Cu+ and Cu0) during the H2-TPR experiments for Cu/AlMgO samples was achieved by the linear combination fitting (LCF) of known CuO, Cu2O and metallic Cu species.
Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) of CO adsorption on copper catalysts were recorded using a Thermo Nicolet iS50 FTIR spectrophotometer equipped with a MCT detector and a Harrick diffuse reflectance infrared Fourier transform spectroscopy cell with a CaF2 window. The spectra were collected on a basis of 40 scans and a resolution of 4 cm−1 using 150 mg of sample. Initially, the samples were pre-treated at 500 °C for 30 min (10 °C min−1) under a flow of He of 100 mL min−1, and then, cooled down and reduced at 250 °C (10 °C min−1) under a flow of 100 mL min−1 of H2 (5% in He, v/v). The samples were cooled down to 25 °C under a He flow. The reduced catalysts were exposed to a flow of 100 mL min−1 of CO 2% in He, v/v during 30 min until saturation, and afterwards purged with a flow of 100 mL min−1 of He before the DRIFTS spectra were collected. The temperature was then increased to 500 °C at a rate of 10 °C min−1 in He (100 mL min−1). The CO adsorption spectra had two main signals centered at 2099 cm−1 (corresponding to an assemblage of Cu0 + Cu+ species) and 2105–2107 cm−1 (corresponding to Cu+ species).21,31
2.3. Catalytic activity
A microactivity reference system (PID Eng&Tech), operating in continuous flow mode at atmospheric pressure, was used to perform the dehydrogenative coupling of ethanol. The reactor and main valves were inside a hot box kept heated at 180 °C to avoid condensation of liquid reactants and products. The catalyst was reduced in situ at 250 °C under a flow of 30 mL min−1 of H2 (5% in N2, v/v). A fixed bed reactor was used to perform the reactions at 300 °C for 6 h using 150 mg of powdered catalyst. The reactor was fed with ethanol (99 wt%) at a flow rate of 0.1 mL min−1, through a HPLC pump (Model 307, Gilson) and a flow of 30 mL min−1 of N2, as the carrier gas. These conditions resulted in a weight hourly space velocity (WHSV, stands for weight hourly space velocity and calculated as feed room temperature mass flow/catalyst mass) of 31.1 h−1. The experiments at lower space velocity of 4.7 h−1 were achieved by using 240 mg of catalyst. The outlet products were analyzed on line using a gas chromatograph (Model 2014, Shimadzu) equipped with a flame ionization detector and a capillary Rtx-1 column of 30 m × 0.32 mm ID and a film thickness of 1 μm.3. Results and discussion
3.1. Textural characteristics of the porous supports and chemical speciation of the dispersed copper sites
The effect of PSDAs in the textural properties of the supports and, consequently, on those of the copper catalysts was studied by means of mercury intrusion porosimetry, nitrogen physisorption and scanning electron microscopy (Fig. 1 and Table 1). The mixed oxide, AlMgO–P, synthesized in the presence of PSDAs presented considerably higher pore volume and BET area than the reference non-porous support as a consequence of the voids left after removal of the emulsifier agent (n-dodecane). A previous study elucidated that the porosity is created by the stacking of layers of the hydrotalcites precursors randomly positioned around n-dodecane droplets.18,19 The macropores arose from the droplets of n-dodecane, while the mesopores were due to the slit-like arrangement of layers of the precursors. Although the hydrotalcite structure no longer exist after calcination of the samples, the porous characteristic is preserved and it can be useful for the dispersion of active metal sites and for improved accessibility of chemicals. After copper nitrate impregnation and calcination, Cu/AlMgO–P sample showed a great decrease in the pore volume and surface area. It can be related to the partial blockage of pores, as well as to a collapse of the pore walls during the process of impregnation and drying. Table 1 shows the results of a drying experiment of sample AlMgO–P that was put in contact with water and rotaevaporated in order to follow the impact of sample drying. The pores shrank from 3.7 to 1.7 cm3 g−1 only due to capillary action of evaporating water. Considering that for the sample containing copper Cu/AlMgO–P the pore volume was much smaller, i.e. 0.8 cm3 g−1, both pore blockage and capillary forces were possibly responsible for that decrease. The same behavior was observed by Cassinelli et al. that studied porous Cu/Al2O3.6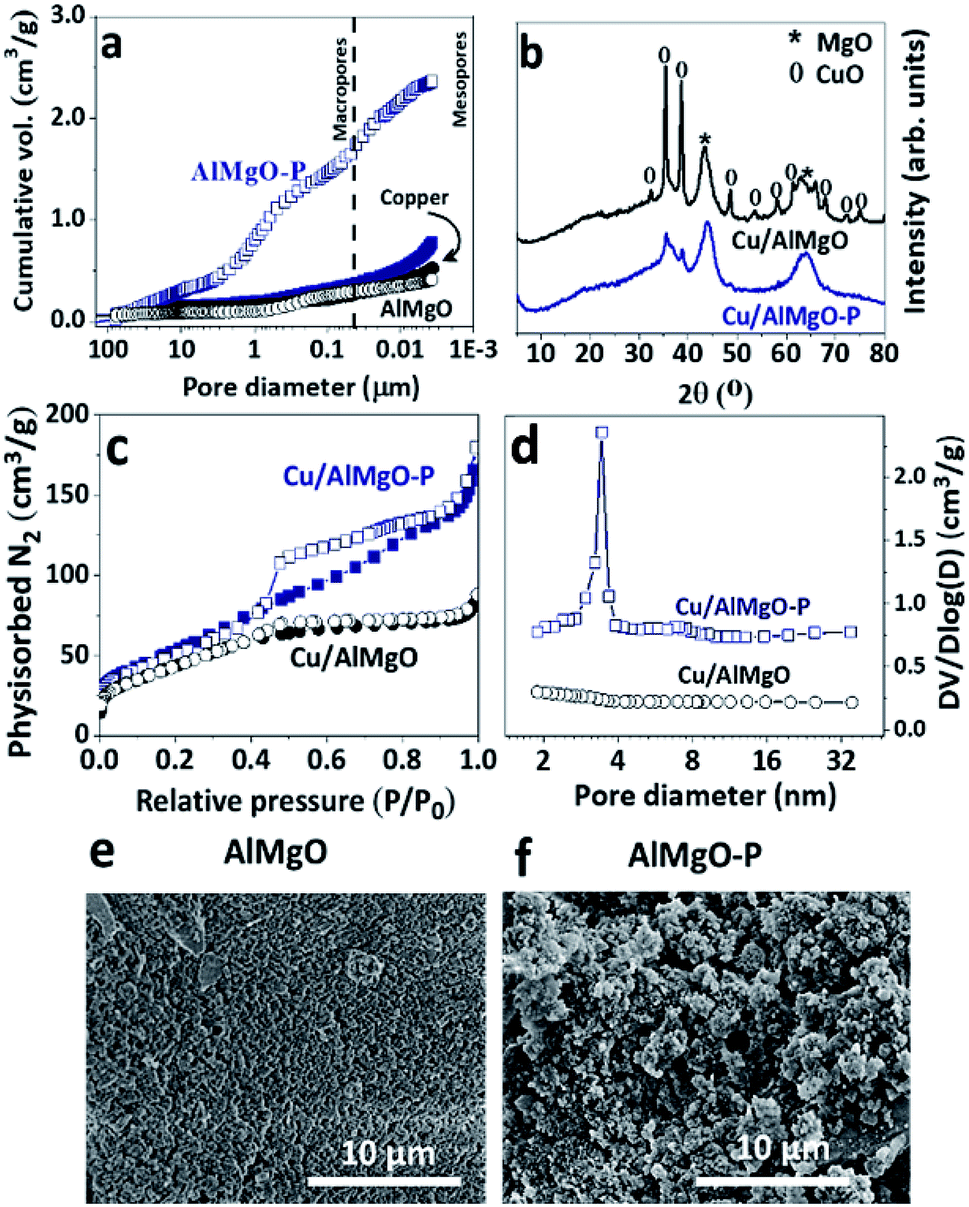 | ||
| Fig. 1 (a) Mercury intrusion porosimetry (opened and filled symbols represent the supports and the copper containing catalysts, respectively); (b) X-ray diffraction; (c) nitrogen physisorption (opened and filled symbols represent the desorption and adsorption branches, respectively); (d) BJH pore size distribution of the desorption branches for Cu/AlMgO samples; and scanning electron microscopy images of: (e) AlMgO and (f) AlMgO–P supports. | ||
| Sample | Pore volume (cm3 g−1) | BET area (m2 g−1) | Acid sites | Base sites | ||
|---|---|---|---|---|---|---|
| Meso | Macro | Total | Total NH3 (mmol g−1) | Total CO2 (mmol g−1) | ||
| a AlMgO–P sample put in water and evaporated, simulating copper wet impregnation. | ||||||
| AlMgO | 0.1 | 0.3 | 0.4 | 166 | 0.78 | 0.45 |
| AlMgO–P | 1.8 | 1.9 | 3.7 | 260 | 0.43 | 0.43 |
| Cu/AlMgO | 0.2 | 0.3 | 0.5 | 120 | 0.29 | 0.40 |
| Cu/AlMgO–P | 0.4 | 0.4 | 0.8 | 206 | 0.36 | 0.37 |
| AlMgO–P (H2O)a | 0.6 | 0.9 | 1.7 | — | — | — |
Fig. 1b shows the X-ray diffraction patterns of the copper catalysts. The reference sample exhibited mainly diffraction peaks at 43.4° (200), 64.3° (220) and at 32.5° (110), 35.4° (002), 38.6° (111), 48.5° (202), 53.6° (021) corresponding to MgO and CuO structures, respectively. On the other hand, the porous support exhibited only diffraction peaks corresponding to MgO, as an indication of higher dispersion of copper oxides. Possibly, due to capillarity effects during drying of impregnated samples, copper solution flows into the pore voids of the porous support, allowing a high dispersion of copper species. According to earlier studies performed with copper supported on γ-Al2O3,32,33 the formation of a monolayer coverage and, consequently, saturation of the surface occurs at a loading in-between 4 and 5 wt% of copper per 100 m2 g−1 of the support. The specific area of the reference sample Cu/AlMgO is just above this limit (10 wt% of copper for 166 m2 g−1) and surface bulk copper oxide particles are prone to be formed after the saturation of the surface by a copper monolayer. Therefore, most probably, the reference sample contains heterogenous distribution of copper species, constituted by well-dispersed monolayered particles and segregated ones.
Further evidence of the improved textural properties of the porous support was provided by nitrogen physisorption measurements (Fig. 1c) and BJH pore size distribution curves (Fig. 1d). The hysteresis between the adsorption and desorption branches for Cu/AlMgO–P catalyst is typical of the existence of mesopores. The smooth slopes of the isotherms, together with the non-parallel behavior of the adsorption and desorption curves, indicated a distinct H3 hysteresis cycle for pores with a broad distribution resulting from the aggregation of plate-like particles, particularly for the catalyst Cu/AlMgO–P with an average mesopore diameter of 3.3 nm. The preparation method provided a catalyst with a BET area greater than the reference (Table 1).
The metallic copper dispersion was assessed by nitrous oxide chemisorption followed by temperature programed reduction with hydrogen (Fig. 1S†). Firstly, the catalysts were reduced under a hydrogen stream at 250 °C for complete reduction of copper, followed by exposure to dilute nitrous oxide flow that caused the oxidation of the surface copper species forming an oxygen layer around the metallic particles. The stoichiometry of the reduction of the surface copper oxides allowed to estimate the relative contribution of surface copper species. Table 2 illustrates the dispersion degrees, Cu surface area and Cu average particle size. As expected, the Cu/AlMgO–P catalyst had a superior dispersion of copper of 85% in comparison to the 50% of the reference sample, thus confirming the positive effect of the emulsion mediated synthesis in producing a porous support for dispersing copper. The dispersion degree and the average Cu particle size follow the same pattern as Cu surface area, i.e. for Cu/AlMgO–P high dispersion leads to a low particle size (1.2 nm). It is worthy to note that compared with appropriate literature data obtained over copper-based mesoporous materials, Cu/AlMgO–P catalyst shows very high dispersion and surface area for copper. For example, at similar copper loading of 10 wt%, copper supported on all-silica mesoporous SBA-15 material had only 37% of copper dispersion.34
| Sample | Cu dispersion (%) | Metallic area (m2 g−1) | Particle size (nm) | |
|---|---|---|---|---|
| B | Cu/AlMgO | 50 | 29 | 2.1 |
| Cu/AlMgO–P | 85 | 55 | 1.2 | |
| A | Cu/AlMgO | 42 | 27 | 2.5 |
| Cu/AlMgO–P | 50 | 34 | 2.1 |
Additional information about copper species distribution on supports was provided by XANES spectra (Fig. 2S†) during reduction of copper by heating from 25 to 400 °C under a H2/He atmosphere. For the reference catalyst, the CuO reduction started at approximately 25 min after heating (plateau of 220 °C in Fig. 2) promoting only a minor change in the spectra of Fig. 2S,† related to the direct transformation of Cu2+ to Cu0, without the clear identification of a shoulder in the ascending margin around 9002 eV,5,6,21,27 a fingerprint of the presence of Cu+ intermediate during the reduction process. On the other hand, the porous samples submitted to the reduction treatment exhibited a noticeable shoulder in the ascending margin of the spectrum. The LCF of the XANES spectra in Fig. 2 shows that in fact the copper on the catalyst Cu/AlMgO reduces from Cu2+ to Cu0 with a minor formation of the Cu+ intermediate. For the Cu/AlMgO–P catalyst, although the reduction of Cu2+ to Cu+ started earlier, at approximately 20 min under H2 stream, Cu+ reached a portion close to 25% that can be associated to a kinetically controlled reduction of Cu+ to Cu0, possibly due to the monolayer dispersion of copper species on the AlMgO–P support. Since, Cu+ was previously considered the most active species to promote the activation of ethanol,5,6 its formation is highly desirable for the ethanol dehydrogenation reactions.
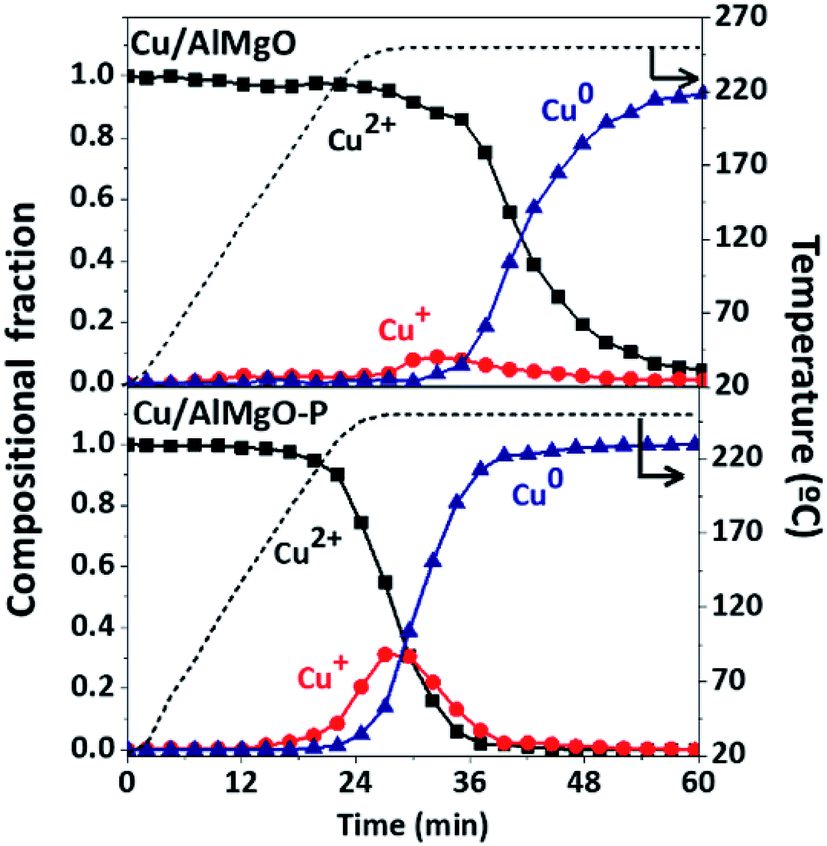 | ||
| Fig. 2 Copper species along reduction with H2 up to 250 °C for the Cu/AlMgO and Cu/AlMgO–P catalysts, obtained from XANES measurements at Cu K-edge (spectra are in Fig. 2S†). | ||
FTIR spectra of CO adsorbed on reduced copper catalysts in the high frequency (HF) region of 2160–2020 cm−1 and in the low frequency (LF) region of 1850–1150 cm−1 are shown in Fig. 3 and 3S,† respectively. It has been reported35 that CO adsorption on copper surfaces show IR bands in the HF spectral region associated with linear or bridged CO species interacting with copper species, such as, Cu2O or Cu0 sites. However, herein, based on the XANES results of copper supported samples, CuO specie is highly improbable, and the three strong overlapping bands with maximum at 2107, 2106 and 2103 cm−1 of Cu/AlMgO–P catalyst are probably related to chemisorption of CO over Cu+ and/or Cu0 species (Fig. 3S†). According to Hadjiivanov et al.,36 linear Cu0–CO carbonyls may be adsorbed at the same frequency, as Cu+–CO, when copper is highly dispersed on the surface. These copper species are better distinguished via thermal stability, once the Cu0–CO surface carbonyls are easily wrecked by increasing temperature, whereas the Cu+ cations produce more stable surface carbonyls, as a result of the strongest σ-bond.31,35 Fig. 3c shows the relative intensity of chemisorbed CO bands in the spectra of Fig. 3a and b, normalized by their maximum intensity. Carbonyl species adsorbed on Cu/AlMgO catalyst are totally absent at 150 °C, while for the Cu/AlMgO–P catalyst complete desorption of CO occurred at 255 °C, that may be related to the higher amount of surface Cu+. The LF region in Fig. 4S† presented bands at 1635, 1547, 1375, 1343 and 1220 cm−1, referring to carbonate species interacting with basic surface sites as OH groups and O2− centers or even acid–base pairs (due Mg2+–O2−–Al3+), which are very similar for both samples, as expected by the TPD-CO2, which showed minor differences for samples Cu/AlMgO and Cu/AlMgO–P (Table 1 and Fig. 5S†). The bands located at 1635 and 1220 cm−1 are attributed to bridged bidentate carbonates (b-HCO3−), whereas the bands 1547 and 1375 cm−1 and 1343 cm−1 are assigned to carbonate monodentate (m-CO32−) and bidentate (b-CO32−), respectively.13 Typically, monodentate carbonate formation requires low-coordination oxygen anions (strong base sites), provided by MgO.13
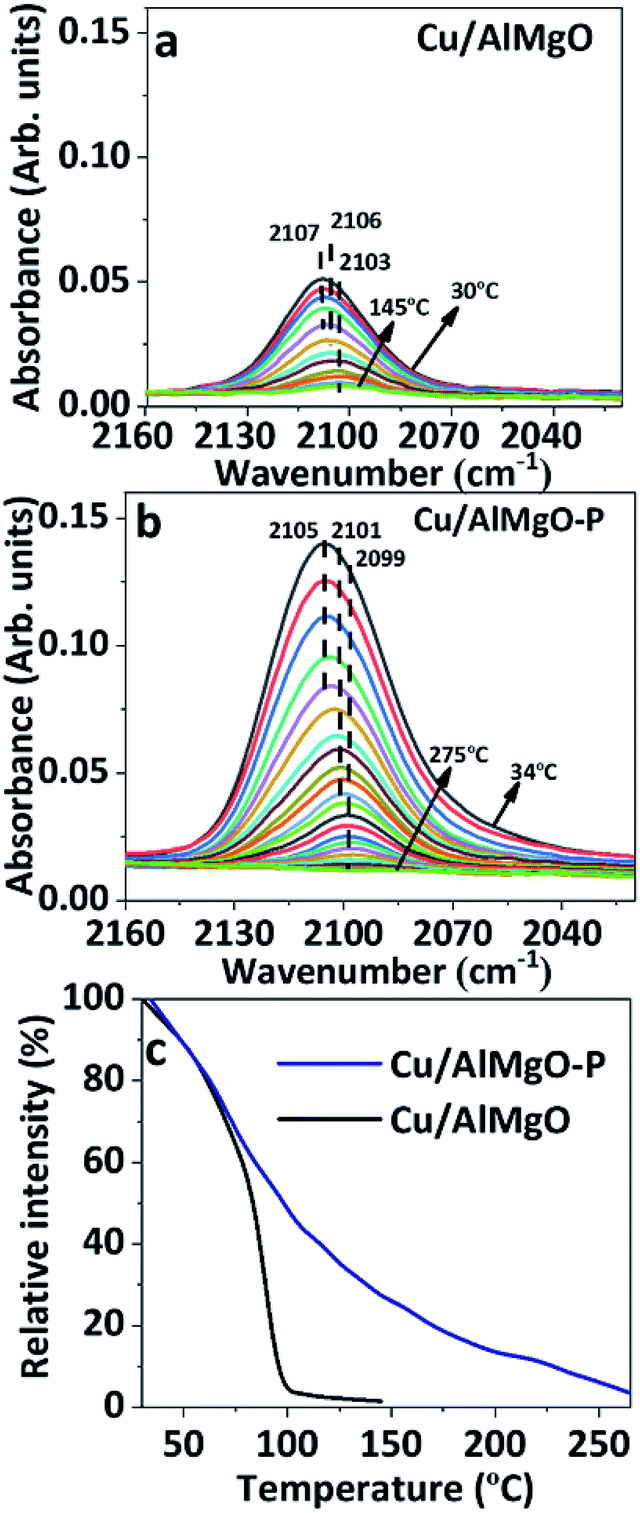 | ||
| Fig. 3 FTIR spectra of CO desorption on reduced: (a) Cu/AlMgO; (b) Cu/AlMgO–P catalysts in the region of 2160–2020 cm−1; (c) relative intensity of bands displayed in (a) and (b) vs. desorption temperature of CO. | ||
The acid and base properties of the copper catalysts were determined from the temperature programmed desorption (TPD) of NH3 or CO2, respectively. Fig. 5S† shows the TPD profiles of NH3 and CO2 and the integration of the peaks are given in Table 1. The deconvolution by means of a Gaussian function was used to discriminate the desorption peaks for weak, medium and strong sites (Fig. 5S†), which reflect the different binding acid–base sites available on the surface. The porous support (AlMgO–P) presented lower acidity than the reference (AlMgO), as reported in the previous study.19 The explanation was based on the reduction of hydroxyl groups in the surface of the AlMgO–P sample due to the interaction with the hydrophobic emulsion during the synthesis that leads to a support with a considerably low acidity and basicity. In comparison to other supports found in the literature (see data collection in Table 1S†), hydrotalcites derived oxides are among those with lower acidity/basicity. By supporting copper, the catalyst had a pronounced decrease in acidity due to the selective combination of the Lewis alkaline sites from copper oxides with acidic sites on AlMgO–P. A milder decrease in basicity was also observed, which may be related to binding of Cu to certain base sites as well. However, these events were fairly beneficial to avoid the formation of undesired by products, specially ethene and diethyl ether that can be formed on those sites.
3.2. Dehydrogenative reactions of ethanol to acetaldehyde and ethyl acetate
The copper impregnated catalysts were investigated in the dehydrogenation of ethanol (Scheme 1) at 300 °C under space velocities of 31.1 h−1 (Fig. 4) and 4.7 h−1 (Fig. 5), and under a period of 6 h to evaluate the activity and deactivation. Catalytic tests were also performed using the Cu-free supports (Fig. 6S†) and the ethanol conversion was negligible, being below 1%. On the other hand, the copper catalysts, had a high activity in selectively converting ethanol into acetaldehyde (Fig. 4), reaching conversion values of 28 and 61% for Cu/AlMgO and Cu/AlMgO–P samples, respectively. Ethyl acetate, ethene, diethyl ether, n-butanal, crotonaldehyde, methyl ethyl ketone, acetone and butanol were minor products summing less than 2%. Thus, the porous catalyst was about 3 times more active, because of the higher dispersion of copper and creation of a singular environment that led to the formation of Cu+ species on the catalyst surface. In fact, the presence of partially reduced copper Cu+ and Cu0 are advantageous for the catalytic performance, as the initial turnover frequencies, considering the surface copper, were 122 and 166 h−1 for the reference and for the porous catalyst, respectively.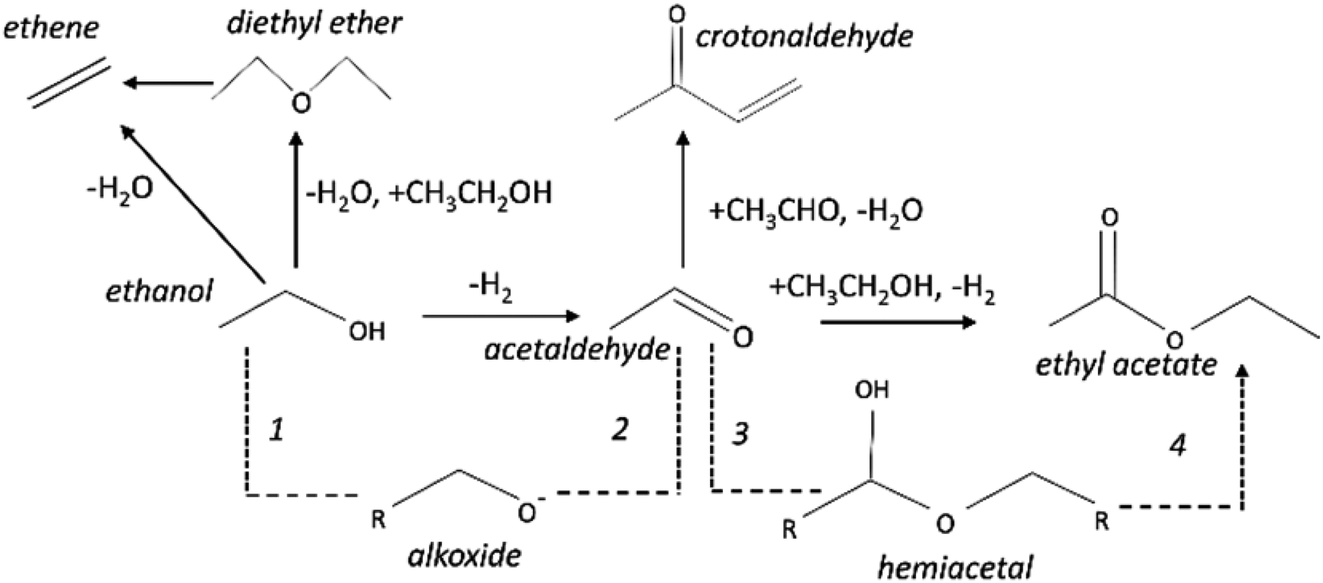 | ||
| Scheme 1 Dehydrogenative reactions of ethanol to give acetaldehyde and ethyl acetate as main products. The dashed path corresponds to the Tishchenko mechanism that might take place on the support/copper interface. | ||
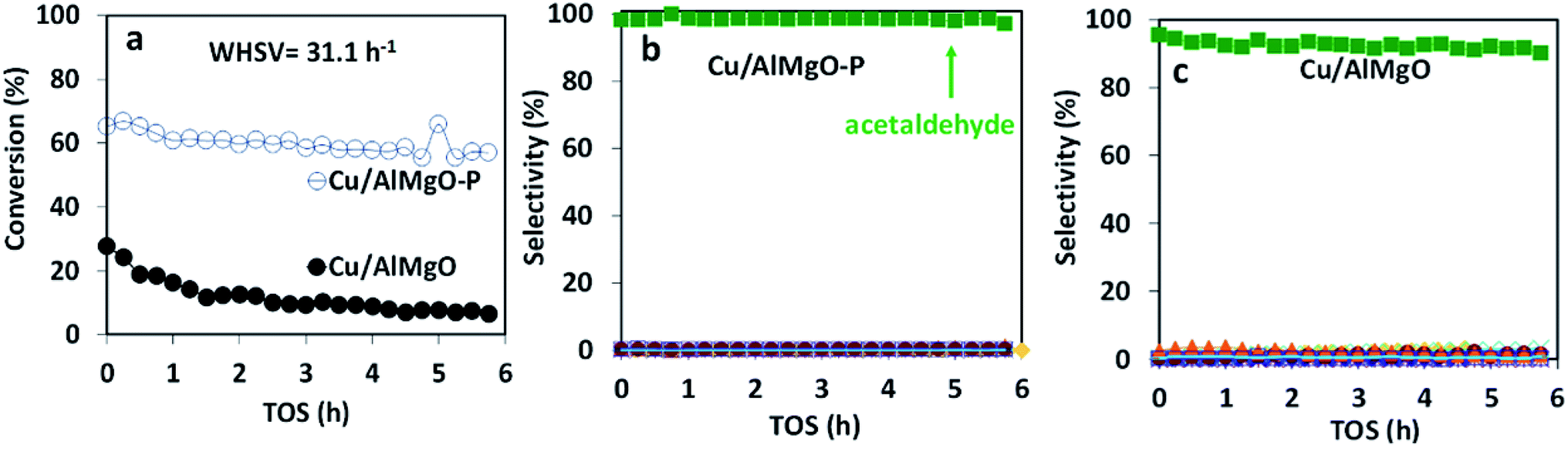 | ||
| Fig. 4 (a) Conversion of ethanol at 300 °C and WHSV of 31.1 h−1; (b) selectivity over Cu/AlMgO–P; (c) Cu/AlMgO catalysts. Ethyl acetate, ethene, diethyl ether, butanal, crotonaldehyde, methyl ethyl ketone, acetone and butanol were less than 2%. | ||
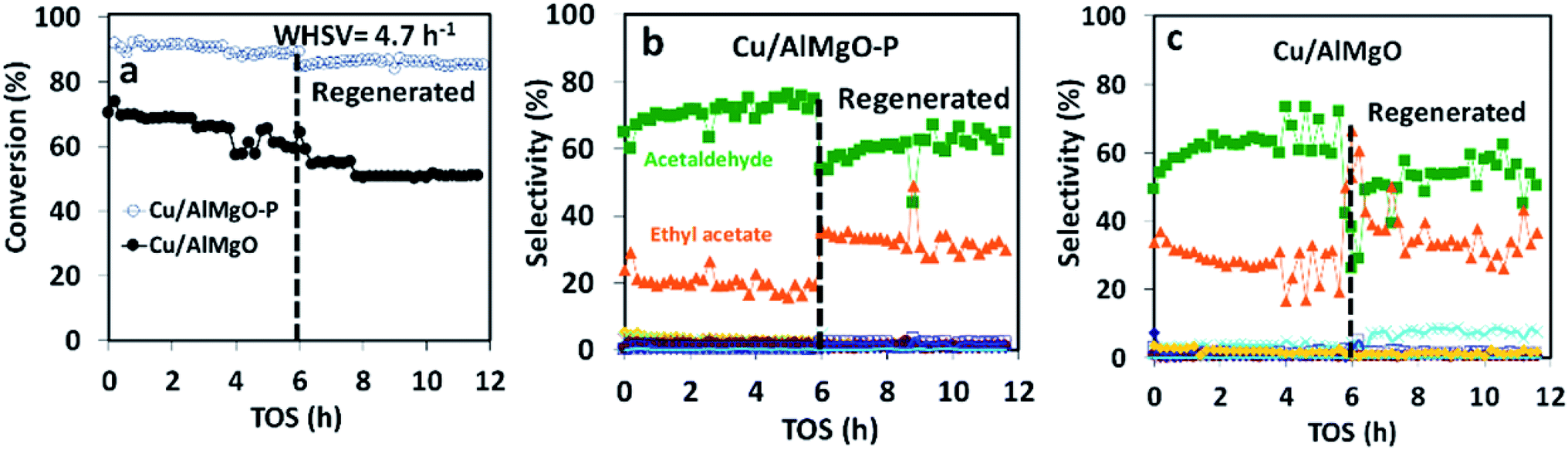 | ||
| Fig. 5 (a) Conversion of ethanol at 300 °C and WHSV of 4.7 h−1; (b) selectivity over Cu/AlMgO–P; (c) Cu/AlMgO catalysts, before and after regeneration by calcination at 500 °C and reduction at 250 °C. Diethyl ether, butanal, crotonaldehyde, methyl ethyl ketone, acetone and butanol were less than 2%. | ||
The catalysts were not completely steady for a time-on-stream (TOS) of 6 h. The notable deactivation of the Cu/AlMgO reference sample was related to the harsh copper dispersion and to the sintering of copper particles that were loosely supported on the non-porous sample. The porous catalyst, in contrast, deactivated less, because of the physical barrier that well dispersed copper particles placed on the surface of macro and mesopores of the support had to sinter. Both catalysts suffered some extent of deactivation along 6 h at 300 °C (21.1% for Cu/AlMgO against 8.2% for Cu/AlMgO–P) as a consequence of a slow copper sintering, as revealed by chemisorption experiments depicted in Table 2, however, its effect was more pronounced on the non-porous reference catalyst.
Ethanol conversion was also examined at a WHSV of 4.7 h−1 (Fig. 5), i.e. by decreasing the space velocity and consequently increasing the contact time in 6.6 times. Ethanol conversion and selectivity to all products were noticeably improved: the conversion increased by a factor of 3.5 and 1.4, for the reference and for the porous catalyst, respectively. The main product, ethyl acetate, was mostly produced on the reference non-porous catalyst (35 against 20% on the porous one). That behavior reveals that the coupling reaction of ethanol and acetaldehyde is favored on larger copper particles.37 The differences in selectivity to ethyl acetate could rely on the support/copper interface, which differs on copper particles of different sizes (Table 2S†). The result can be explained based on the Tishchenko mechanism,7,37–40 which consists of four main steps depicted in Scheme 1 and that involve the participation of the support/copper interface. Firstly, it occurs the formation an alkoxide (CH3CH2O−) adsorbed on AlMgO base sites (step 1) that reacts with the aldehyde formed on the copper phase (step 2). At this step, the presence of Cu+ in combination with Cu0 aids to promote the formation of acetaldehyde (which is more evident in the porous catalyst). Then, the alkoxide reacts with the aldehyde to form a hemiacetal (step 3). Finally, the hemiacetal dehydrogenates on copper phase (step 4). According to the results of Scotti et al. that studied Cu/ZrO2 catalysts,40 we can predict herein that the steps 1 and 4 are promoted by AlMgO/Cu interface and that improved hydrogen spillover takes place, thus allowing the interface to work fittingly. It seems that the interface plays a major role in the overall catalyst performance, because the superior capacity of the porous catalyst to dehydrogenate ethanol did not allow to obtain higher selectivity to ethyl acetate. Lastly, the cooperative effect of acid and base sites of suitable strength of the Al–Mg oxides was important to give high selectivity to ethyl acetate, as reported elsewhere.7,40
In order to confirm that the catalyst deactivation was due to copper sintering and not to coke deposition, after running ethanol dehydrogenation for 6 h the catalysts were calcined at 500 °C followed by reduction at 250 °C in H2. After regeneration, the catalytic activity was not restored at all and the copper particle size increased (Fig. 5 and Table 2). The increase in particle size between 2–2.5 nm appears to favor to increase the selectivity of ethyl acetate to about 40% and decrease acetaldehyde to about 55%. Also, the regeneration process leads to an increase of sintering of the copper particles of the catalysts which increased ethyl acetate selectivity up to about 8%. The sintering process is more evident in the non-porous Cu/AlMgO (Fig. 5c). Finally, after the thermal treatment at 500 °C and sintering of copper, the surface of the support led to the occurrence of the dehydration on ethanol to ethene, with a selectivity of about 5%.
4. Conclusions
Hydrotalcite derived mixed oxides of Al and Mg were successfully prepared by an emulsion mediated method and impregnated with copper nitrate. The combination of texture characterization techniques, such as mercury porosimetry and nitrogen physisorption, disclosed the production of a more porous material with a higher BET area. The acidity and basicity of the catalysts decreased after impregnation with copper, due to metal coating on these sites. The porous catalyst exhibited more available metallic sites and greater dispersion on the surface of the material, as demonstrated by H2-TPR together with N2O chemisorption.From the LCF of XANES spectra and FTIR-CO measurements, the presence of a larger amount of Cu+ intermediate was observed, which was advantageous for the conversion of ethanol to acetaldehyde, as the turnover frequencies, were 122 and 166 h−1 for the non-porous reference and for the porous catalyst, respectively. Both catalysts deactivated due to copper sintering, however the porous catalyst deactivated less, because of the difficulties that the physical barrier related with a higher surface area generated by the presence of macro and mesopores, which allowed to well disperse the copper particles and consequently minimizing their sintering. By increasing the contact time by 6.6 times, the formation of ethyl acetate was mostly favored on the non-porous catalyst, because its formation is favored on the support/copper interface given by larger copper particles.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support provided by the São Paulo State Research Foundation (FAPESP, grant numbers #2013/01328-0 and #2015/05321-5) and CNPq (grant number #306326/2017-5). The authors also acknowledge the XAFS1 beamline of the Brazilian Synchrotron Light Laboratory (LNLS) for the XAS measurements, LCE-UFSCar for SEM images and Prof. J. M. C. Bueno for the CO/DRIFTS adsorption experiments.References
- E. S. Shuba and D. Kifle, Renewable Sustainable Energy Rev., 2018, 81, 743–755 CrossRef CAS.
- H. H. Mardhiah, H. C. Ong, H. H. Masjuki, S. Lim and H. V. Lee, Renewable Sustainable Energy Rev., 2017, 67, 1225–1236 CrossRef CAS.
- A. G. Sato, D. P. Volanti, D. M. Meira, S. Damyanova, E. Longo and J. M. C. Bueno, J. Catal., 2013, 307, 1–17 CrossRef CAS.
- A. G. Sato, D. P. Volanti, I. C. De Freitas, E. Longo, J. Maria and C. Bueno, Catal. Commun., 2012, 26, 122–126 CrossRef CAS.
- W. H. Cassinelli, L. Martins, M. Magnani, S. H. Pulcinelli, V. Briois and C. V. Santilli, RSC Adv., 2016, 6, 20453–20457 RSC.
- W. H. Cassinelli, L. Martins, A. R. Passos, S. H. Pulcinelli, A. Rochet, V. Briois and C. V. Santilli, ChemCatChem, 2015, 7, 1668–1677 CrossRef CAS.
- F. Zaccheria, N. Scotti and N. Ravasio, ChemCatChem, 2018, 10, 1–11 CrossRef.
- D. A. Morgenstern and J. P. Fornango, Energy Fuels, 2005, 19, 1708–1716 CrossRef CAS.
- S. K. Rout, S. Guin, K. K. Ghara, A. Banerjee and B. K. Patel, Org. Lett., 2012, 14, 3982–3985 CrossRef CAS PubMed.
- J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2005, 127, 10840–10841 CrossRef CAS PubMed.
- H. Miura, K. Nakahara, T. Kitajima and T. Shishido, ACS Omega, 2017, 2, 6167–6173 CrossRef CAS.
- S. W. Colley, J. Tabatabaei, K. C. Waugh and M. A. Wood, J. Catal., 2005, 236, 21–33 CrossRef CAS.
- V. K. Diez, C. R. Apesteguia and J. I. Di Cosimo, J. Catal., 2003, 215, 220–233 CrossRef CAS.
- P. J. Luggren, C. R. Apesteguía and J. I. Di Cosimo, Fuel, 2016, 177, 28–38 CrossRef CAS.
- P. J. Luggren, C. R. Apesteguía and J. I. Di Cosimo, Top. Catal., 2016, 59, 196–206 CrossRef CAS.
- J. J. Bravo-Suárez, B. Subramaniam and R. V. Chaudhari, Appl. Catal., A, 2013, 455, 234–246 CrossRef.
- J. T. Kozlowski and R. J. Davis, ACS Catal., 2013, 3, 1588–1600 CrossRef CAS.
- D. D. Petrolini, E. A. Urquieta-González, S. H. Pulcinelli, C. V. Santilli and Martins, Microporous Mesoporous Mater., 2017, 240, 149–158 CrossRef CAS.
- D. D. Petrolini, A. V. da Silva Neto, E. A. Urquieta-González, S. H. Pulcinelli, C. V. Santilli and L. Martins, RSC Adv., 2018, 8, 6039–6046 RSC.
- D. D. Petrolini, S. H. Pulcinelli, C. V. Santilli and L. Martins, J. Sol-Gel Sci. Technol., 2014, 71, 9–15 CrossRef CAS.
- P. C. P. Caldas, J. M. R. Gallo, A. Lopez-Castillo, D. Zanchet and J. M. C. Bueno, ACS Catal., 2017, 7, 1–28 CrossRef.
- R. A. Hoyt, M. M. Montemore, E. C. H. Sykes and E. Kaxiras, J. Phys. Chem. C, 2018, 122, 21952–21962 CrossRef CAS.
- J. Quesada, L. Faba, E. Díaz and S. Ordonez, ChemCatChem, 2018, 10, 3583–3592 CrossRef CAS.
- M. Ohira, H. Liu, D. He, Y. Hirata, M. Sano, T. Suzuki and T. Miyake, J. Jpn. Pet. Inst., 2018, 61, 205–212 CrossRef CAS.
- Z. Lu, D. Gao, H. Yin, A. Wang and S. Liu, Ind. Eng. Chem. Res., 2015, 31, 301–308 CrossRef CAS.
- E. V. Shelepova, L. Y. Ilina and A. A. Vedyagin, React. Kinet., Mech. Catal., 2017, 122, 385–398 CrossRef CAS.
- I. Ro, Y. Liu, M. R. Ball, D. H. K. Jackson, J. P. Chada, C. Sener, T. F. Kuech, R. J. Madon, G. W. Huber and J. A. Dumesic, ACS Catal., 2016, 6, 7040–7050 CrossRef CAS.
- E. W. Washburn, Proc. Natl. Acad. Sci. U. S. A., 1921, 7, 115–116 CrossRef CAS.
- S. J. Gregg, K. S. W. Sing and H. W. Salzberg, J. Electrochem. Soc., 1967, 114, 279C CrossRef.
- H. Masuda, M. Suzuki, K. Ehara, M. Fuji, T. Fukui, H. Suzuki, J. Tatami, K. Hayashi and K. Toda, Nanoparticle Technology Handbook, 2008, pp. 3–48 Search PubMed.
- W. W. Wang, W. Z. Yu, P. P. Du, H. Xu, Z. Jin, R. Si, C. Ma, S. Shi, C. J. Jia and C. H. Yan, ACS Catal., 2017, 7, 1313–1329 CrossRef CAS.
- B. R. Strohmeier, D. E. Leyden, R. S. Field and D. M. Hercules, J. Catal., 1985, 94, 514–530 CrossRef CAS.
- R. Friedman, J. Catal., 1978, 55, 10–28 CrossRef CAS.
- C. S. Chen, C. C. Chen, C. T. Chen and H. M. Kao, Chem. Commun., 2011, 47, 2288–2290 RSC.
- M. Neurock, Z. Tao, A. Chemburkar, D. D. Hibbitts and E. Iglesia, Faraday Discuss., 2017, 197, 59–86 RSC.
- K. I. Hadjiivanov and G. N. Vayssilov, Adv. Catal., 2002, 47, 307–511 CAS.
- J. Sun and Y. Wang, ACS Catal., 2014, 4, 1078–1090 CrossRef CAS.
- M. Nielsen, H. Junge, A. Kammer and M. Beller, Angew. Chem., Int. Ed., 2012, 51, 5711–5713 CrossRef CAS PubMed.
- P. C. Zonetti, J. Celnik, S. Letichevsky, A. B. Gaspar and L. G. Appel, J. Mol. Catal. A: Chem., 2011, 334, 29–34 CrossRef CAS.
- N. Scotti, F. Zaccheria, C. Evangelisti, R. Psaro and N. Ravasio, Catal. Sci. Technol., 2017, 7, 1386–1393 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra10076d |
| This journal is © The Royal Society of Chemistry 2019 |