DOI:
10.1039/C8RA10525A
(Paper)
RSC Adv., 2019,
9, 6770-6778
tert-Butylphenylthiazoles with an oxadiazole linker: a novel orally bioavailable class of antibiotics exhibiting antibiofilm activity†
Received
23rd December 2018
, Accepted 16th February 2019
First published on 26th February 2019
Abstract
The structure–activity and structure–kinetic relationships of a new tert-butylphenylthiazole series with oxadiazole linkers were conducted with the objective of obtaining a new orally available antibacterial compounds. Twenty-two new compounds were prepared, purified and identified. Their activity against methicillin-resistant Staphylococcus aureus were examined. Compound 20 with 3-hydroxyazetidine as a nitrogenous side chain showed promising activity against twenty-four clinical isolates, including vancomycin-resistant staphylococcal and enterococcal species with MIC values ranging from 4–8 μg mL−1. Additional advantages of this compound include an ability to eradicate staphylococcal biofilm mass in a dose-dependent manner as well as high metabolic stability after an oral dose of 25 mg kg−1 with a biological half-life that exceeds 5 hours and a plasma concentration (Cmax) that exceeds the MIC values.
1. Introduction
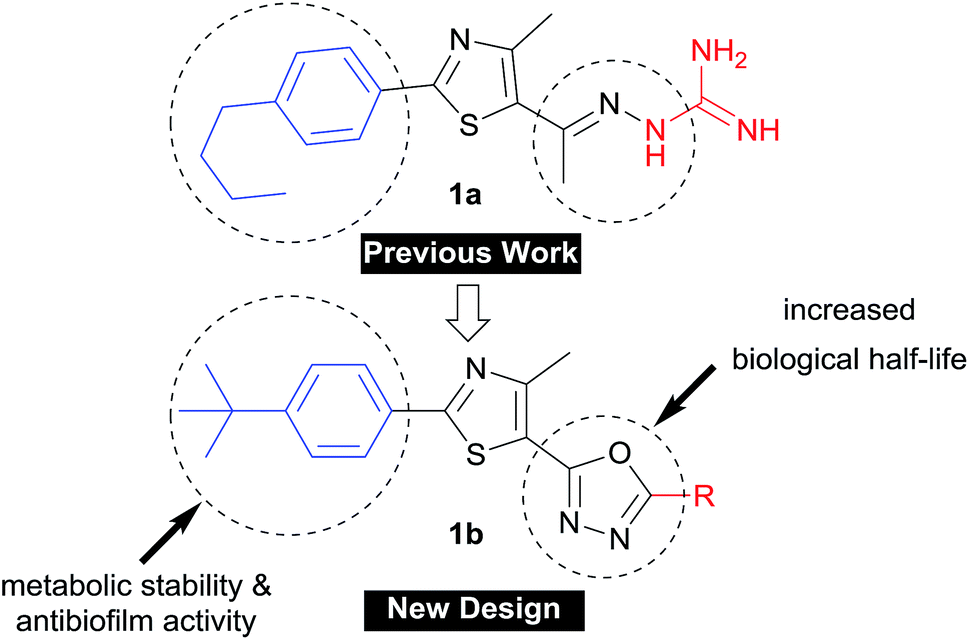
The rapid development of antimicrobial resistance is a major global health concern due to the exponentially decreased utility of antibiotics available for clinicians. The World Health Organization (WHO) recently reported a set of innovation criteria for new antibiotic developments in order to maintain long-term clinical efficacy.1 These criteria include introducing novel chemical scaffolds, broad spectrum activity versus multidrug-resistant strains, and targeting novel pathways.1 These criteria are met by our newly-developed arylthiazole 1a, as an inhibitor of two novel proteins (undecaprenyl pyrophosphatase and undecaprenyl pyrophosphate synthase) involved in the peptidoglycan synthesis.2–5 However, the main drawback of 1a (Fig. 1) was the extremely short plasma half-life, for which the t1/2 was only 29 minutes.5 The benzylic methylene carbon was identified as a metabolic soft spot.6 The replacement of the n-alkyl moiety with branched tert-butyl or alkoxy analogues provided more metabolically-stable derivatives with enhanced antibiofilm activity.6,7 We did additional structural modification focused on the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N linker, where it has been replaced with different heterocyclic rings.1 Of the tested heterocyclic linkers,4,5,8 only the oxadiazole-containing derivatives were orally bioavailable.1
N linker, where it has been replaced with different heterocyclic rings.1 Of the tested heterocyclic linkers,4,5,8 only the oxadiazole-containing derivatives were orally bioavailable.1
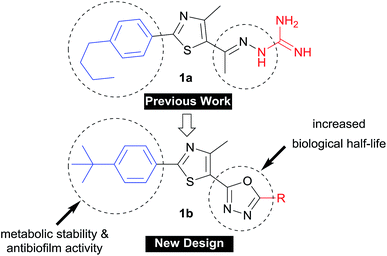 |
| | Fig. 1 Progress in the development of phenylthiazole antibiotics. | |
In this study, we address the structure–activity and structure–kinetic relationships of the oxadiazole based hybrid scaffold 1b at position-5 (Fig. 1), aiming to prepare new derivatives with enhanced antibacterial activity against multidrug-resistant staphylococcal strains, improved oral bioavailability and antibiofilm activity.
2. Results and discussion
2.1. Chemistry
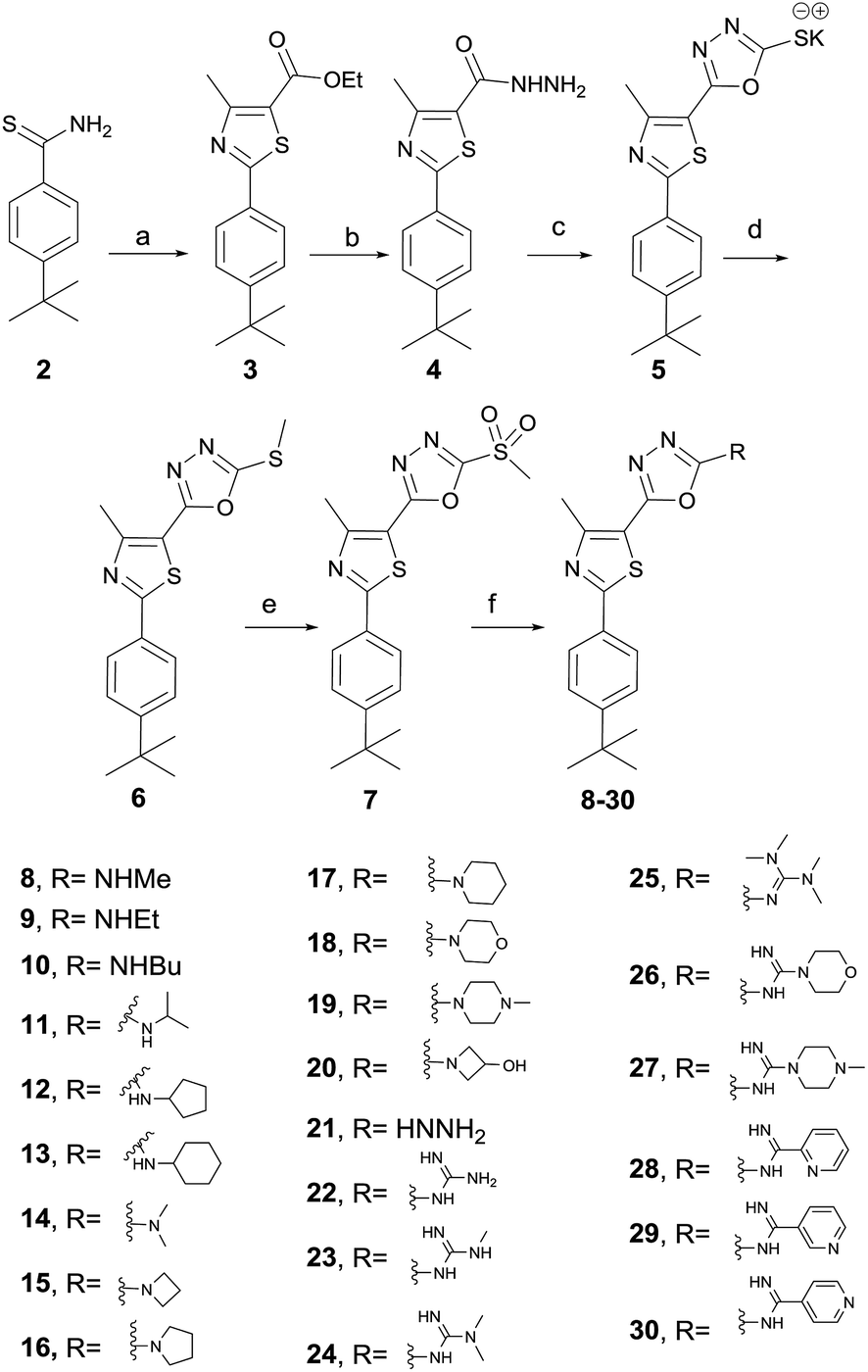
The synthesis of the target compounds started with the preparation of phenylthiazole ethyl ester 3 from commercially available thioamide 2. The treatment of ethyl ester 3 with hydrazine hydrate provided the corresponding acid hydrazide 4, which was converted into methylmercapto-1,3,4-oxadiazole upon reaction with carbon disulfide followed by the methylation of the free mercaptyl group with dimethyl sulphate (Scheme 1). The oxidation of the methylmercaptyl moiety with chloroperbenzoic acid furnished the key intermediate 7, which was then utilized to prepare the entire series of oxadiazolylphenylthiazole final products 8–30.
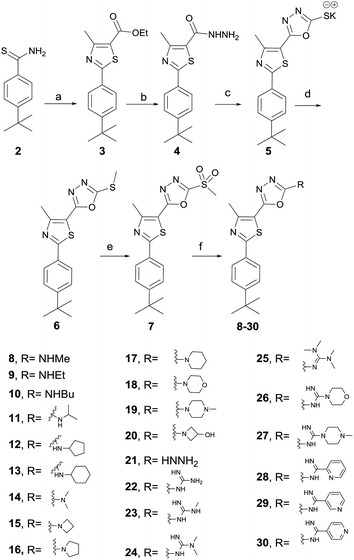 |
| | Scheme 1 Synthesis of compounds 8–30. Reagents and conditions: (a) ethyl 2-chloro-3-oxobutanoate, absolute EtOH, heat at reflux, 4 h, (b) absolute EtOH, NH2NH2·H2O, heat at reflux, 8 h; (c) CS2, KOH, EtOH, heat reflux, 12 h; (d) dimethyl sulfate, H2O, stirring at 23 °C, 2 h; (e) mCPBA, dry DCM, 23 °C, 16 h; (f) appropriate amine, hydrazine, guanidine or carboximidate; K2CO3, DMF, heat at 80 °C for 0.5–12 h. | |
The chemical structures of the newly synthesized compounds were confirmed via spectral and elemental data. For instance, the ethoxy group of compound 3 was represented in the 1H NMR spectrum by two signals at 4.33 and 1.13 ppm. These two distinguished signals were replaced with two broad singlets at δ 9.57 and 4.54 when treated with hydrazine hydrate (compound 4). Upon cyclization with carbon disulfide and methylation with dimethylsulfate, the broad signals disappeared and an additional singlet signal in the aliphatic region, appeared at δ 2.77, a characteristic of S-methyl (compound 5). Upon oxidation with mCPBA (compound 6), this particular signal shifted downfield to 3.47 ppm, and disappeared following the treatment of 6 with nitrogenous based nucleophiles. Among compounds 7–12, the characteristic methylsulfonyl moieties signal at 3.47 ppm disappeared from the 1H NMR spectra and was replaced with a broad singlet at approximately 7.83 ppm due to the NH group as well as the presence of extra aliphatic protons equivalent to the number of aliphatic side chains connected with the terminal amine. Collectively, these findings confirm the tethering of the nucleophile moiety with the oxadiazole position-5. For example, the 1H NMR spectrum of compound 10 showed, in addition to the oxadiazolylphenylthiazole nucleus protons, a broad singlet signal equivalent to one proton at δ 7.82 due to NH in addition to a multiplet signal equivalent to one proton and a doublet signal for six protons at δ 3.70 and 1.20, respectively, due to an isopropyl side chain. For the tertiary-amine side-chain-containing compounds 13–19, the most predominant features of the 1H NMR isolated products spectra were the disappearance of the methylsulfonyl singlet at 3.47 ppm and the appearance of a set of distinctive signals in the upper field region due to the corresponding protons of the alkyl nucleophile moieties. On the other hand, the hydrazinyl group of compound 20 was represented in the 1H NMR by two broad singlets at 6.94 and 4.54 ppm, due to NH and NH2 moieties, respectively.
Additionally, the 1H NMR spectrum of the guanidine-containing derivative 21 displayed two broad singlets at 7.21 and 7.06 ppm due to guanidine protons, as well as signals corresponding to the oxadiazolylphenylthiazole nucleus. The 1H NMR spectra of the carboximidate-containing nucleophiles, compounds 24–28, showed two broad singlets peaks due to NH and CNH protons, in addition to the disappearance of the methylsulfonyl singlet peak at 3.47 ppm.
2.2. Biological results and discussion
2.2.1. Antibacterial activity.
At the outset of this study, the initial antibacterial screening was performed on methicillin-resistant Staphylococcus aureus (MRSA, 2658 RCMB). The results presented in Table 1 indicated that the 2-amino-1,3,4-oxadiazole linker with aliphatic side chains (compounds 8–17) is void of anti-MRSA activity.
Table 1 Initial MIC screening of the compounds against methicillin-resistant Staphylococcus aureus (2658 RCMB)
| Compound |
MRSA (2658 RCMB) |
Compound |
MRSA (2658 RCMB) |
|
8
|
50 |
20
|
3.12 |
|
9
|
>50 |
21
|
6.25 |
|
10
|
>50 |
22
|
6.25 |
|
11
|
>50 |
23
|
12.5 |
|
12
|
>50 |
24
|
12.5 |
|
13
|
>50 |
25
|
25 |
|
14
|
>50 |
26
|
25 |
|
15
|
>50 |
27
|
25 |
|
16
|
>50 |
28
|
>50 |
|
17
|
>50 |
29
|
>50 |
|
18
|
6.25 |
30
|
>50 |
|
19
|
6.25 |
Vancomycin |
1.56 |
In contrast, the more polar side chains—morpholine, piperazine and hydroxyazetidine—provided the desired antibacterial activity, the maximum potency obtained from hydroxyazetidine-containing derivative 20, and inhibited the growth of MRSA at a minimum-inhibitory-concentration (MIC) of 3.1 μg mL−1 (one fold higher than the MIC of vancomycin against the same strain) (Table 1). Further expansion of the nitrogenous side chain provided the hydrazinyl and guanidinyl derivatives 21 and 22, both of which had a one-fold less potency than the parent compound 20. Derivatives with additional cyclic nitrogenous moieties (compounds 26–30) were found to be inactive or less active than the lead compound 20 (Table 1).
To further assess the antibacterial potency of this new class of arylthiazoles, the most promising derivative 20 was selected and its anti-staphylococcal activity was tested against twenty-four clinical isolates (Table 2).
Table 2 The minimum inhibitory concentration (MIC in μg mL−1) and minimum bactericidal concentration (MBC in μg mL−1) of compound 20 against a panel of Staphylococcus aureus clinically relevant strains
| Bacterial strains |
Compound 20 |
Vancomycin |
| MIC |
MBC |
MIC |
MBC |
| MSSA ATCC 6538 |
4 |
>64 |
1 |
2 |
| MSSA NRS 107 |
4 |
>64 |
2 |
2 |
| MRSA NRS 108 |
4 |
>64 |
1 |
2 |
| MRSA NRS 194 |
8 |
>64 |
1 |
1 |
| MRSA NRS 119 |
8 |
>64 |
1 |
1 |
| MRSA NRS 382 (USA 100) |
16 |
>64 |
2 |
2 |
| MRSA NRS 383 (USA 200) |
8 |
>64 |
1 |
1 |
| MRSA NRS384 (USA 300) |
4 |
>64 |
1 |
1 |
| MRSA NRS123 (USA 400) |
8 |
>64 |
1 |
1 |
| MRSA NRS 385 (USA 500) |
4 |
>64 |
0.5 |
1 |
| MRSA NRS 386 (USA 700) |
8 |
>64 |
1 |
1 |
| MRSA NRS 387 (USA 800) |
4 |
>64 |
0.5 |
0.5 |
| MRSA NRS 483 (USA 1000) |
8 |
>64 |
1 |
1 |
| MRSA NRS 484 (USA 1100) |
8 |
>64 |
2 |
2 |
| VISA NRS 1 |
4 |
>64 |
4 |
4 |
| VISA NRS 19 |
8 |
>64 |
4 |
4 |
| VISA NRS 37 |
4 |
>64 |
4 |
8 |
| VRSA 2 |
4 |
>64 |
64 |
64 |
| VRSA 5 |
8 |
>64 |
>64 |
>64 |
| VRSA 6 |
8 |
>64 |
>64 |
>64 |
| VRSA 7 |
8 |
>64 |
>64 |
>64 |
| VRSA 9 |
8 |
>64 |
>64 |
>64 |
| VRSA 10 |
8 |
>64 |
64 |
>64 |
| VRSA 11a |
8 |
>64 |
>64 |
>64 |
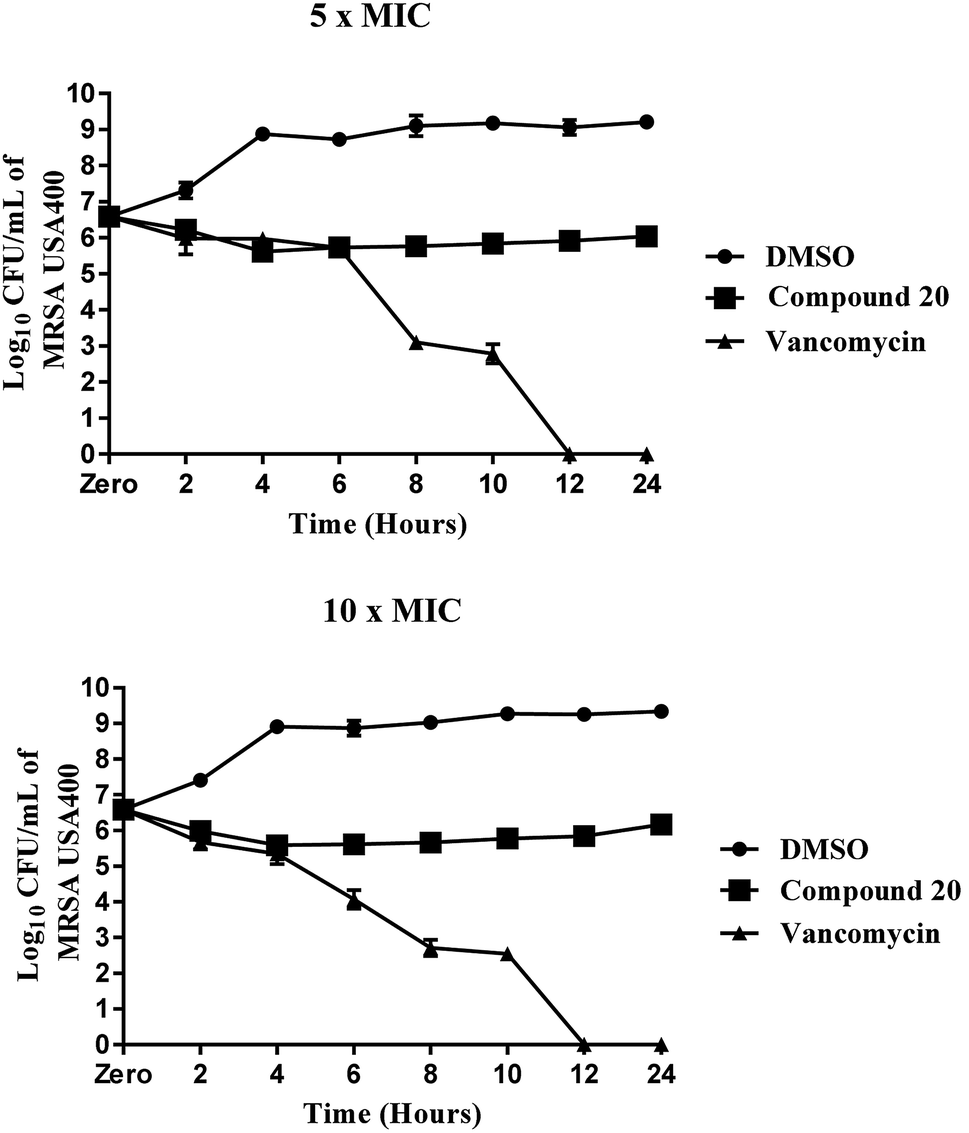
Compound 20 exhibited moderate antibacterial activity against all methicillin-sensitive S. aureus (MSSA) and tested MRSA strains at concentrations ranging from 4 to 16 μg mL−1. Its MBC values were higher than 64 μg mL−1, more than threefold higher than its MIC values against the tested strains and evidence that the compound is a bacteriostatic agent. This mode of anti-staphylococcal inhibition was further confirmed using a time-killing assay at two different concentrations (5× MIC and 10× MIC) (Fig. 2). Despite a moderate potency against MRSA clinical isolates, compound 20 maintained its antibacterial potency against all nine-tested vancomycin-intermediate and vancomycin-resistant Staphylococcus aureus strains (VISA and VRSA). These findings suggest the advantages of this compound over vancomycin, the drug of choice for the treatment of invasive staphylococcal infections.
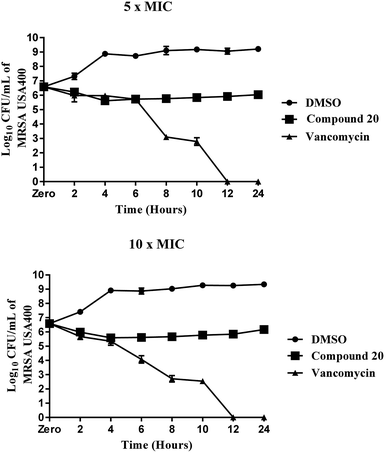 |
| | Fig. 2 Time-kill assay of compound 20 and vancomycin tested in triplicates (at 5× MIC and 10× MIC) against methicillin-resistant Staphylococcus aureus (MRSA USA400) over a 24 hour incubation period at 37 °C. DMSO (solvent for the compound) served as a negative control. The error bars represent standard deviation values. | |
Similar to the results obtained with S. aureus strains, compound 20 exhibited moderate antibacterial activity against other clinically-important Gram-positive pathogens, and inhibited the growth of the tested strains at concentrations of 4 to 16 μg mL−1 (Table 3).
Table 3 The minimum inhibitory concentration (MIC in μg mL−1) and minimum bactericidal concentration (MBC in μg mL−1) of compound 20 against a panel of Gram-positive bacterial pathogens
| Bacterial strains |
Compound 20 |
Vancomycin |
| MIC |
MBC |
MIC |
MBC |
| Methicillin-resistant S. epidermidis NRS101 |
4 |
>64 |
1 |
1 |
|
Enterococcus faecalis ATCC 51299 (VRE)1 |
16 |
>64 |
32 |
64 |
|
Enterococcus faecium ATCC 700221 (VRE)1 |
8 |
>64 |
>64 |
>64 |
|
Listeria monocytogenes ATCC 19111 |
8 |
>64 |
1 |
1 |
| Cephalosporin-resistant Streptococcus pneumoniae ATCC 51916 |
16 |
>64 |
1 |
1 |
| Methicillin-resistant Streptococcus pneumoniae ATCC 700677 |
16 |
>64 |
2 |
4 |
Most importantly, compound 20 also maintained its potency against vancomycin-resistant enterococci strains (i.e. vancomycin-resistant Enterococcus faecium and Enterococcus faecalis) (Table 3). Vancomycin-resistant enterococci (VRE) are a leading cause of healthcare-associated infections. VRE colonization can eventually lead to life-threatening endocarditis and urinary tract infections.9–11E. faecium is of high clinical interest and responsible for the vast majority of nosocomial enterococcal infections that result in patients with vancomycin-resistant enterococcal endocarditis with limited therapeutic options. Moreover, the MBC values for compound 20 against the tested strains were more than threefold higher than its MIC values, evidence that compound 20 may be bacteriostatic against these strains. In addition, the preliminary safety profile of compound 20 was evaluated against two types of cell (vero and caco-2 cells) and it showed high tolerability against both (Fig. 1S and 2S†).
2.2.2. Antibiofilm activity.
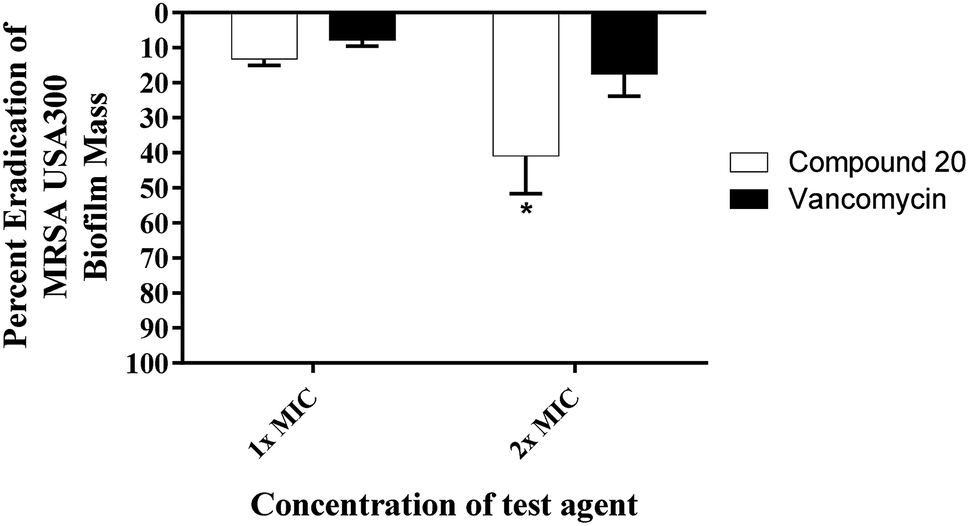
Given that approximately 65% of current human microbial infections are biofilms,12 it is predicted that biofilm will be the major source of future bacterial infections. Biofilm is a surmountable barrier to most of the antibiotic arsenal. Hence, common antibiotics such as penicillin and ciprofloxacin are losing their potency as a result of biofilm-induced infections.13 Staphylococci (in particular S. aureus and S. epidermidis) cause various biofilm-related infections.14 They tend to form harmful biofilms on implanted prosthetic devices and tissues.15 Few antibacterial agents can disrupt these biofilms (MRSA biofilms in particular). Therefore, we evaluated the capacity of compound 20 to disrupt mature MRSA biofilm. Due to its large molecular structure and polar nature, vancomycin is unable to effectively penetrate such bacterial biofilms. At 1× MIC, vancomycin disrupted only about 8% of the MRSA biofilm (Fig. 3). This value increased to around 18% eradication of adherent biofilm when the concentration was doubled. Even at a concentration of 32× MIC, vancomycin was only capable of reducing the biofilm mass by 34% (data not published). Remarkably, the hydroxyzetidine derivative 20 was superior to vancomycin in MRSA biofilm eradication and exhibited a concentration-dependent biofilm disruption (Fig. 3). At 1× MIC, compound 20 disrupted 13% of the mature MRSA300 biofilm mass. This increased to about 41% eradication of the adherent biofilm when its concentration was increased by one-fold (2× MIC) (Fig. 3).
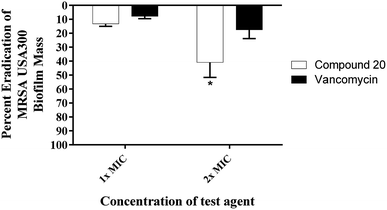 |
| | Fig. 3 Efficiency of compound 20 and vancomycin (tested in quartets) in disrupting the established MRSA biofilm. Data are presented as percent eradication of MRSA USA300 mature biofilm compared to the control (DMSO; the solvent for the tested compound). Error bars represent standard deviation values. Asterisk (*) denotes statistical significance (P < 0.05) between results for compound 20 and vancomycin analyzed via one-way ANOVA with post hoc Dennett's test for multiple comparisons. | |
2.2.3.
In vivo pharmacokinetic evaluation.
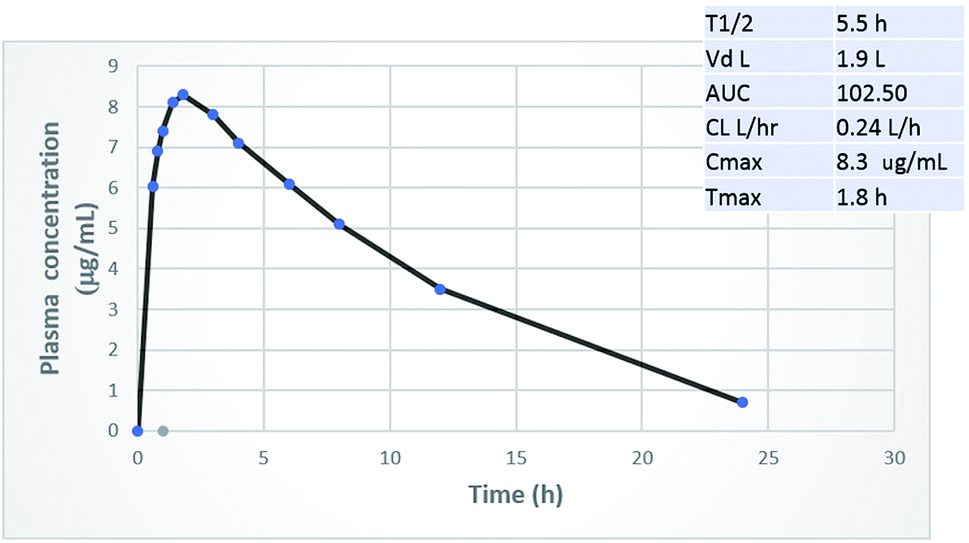
The principal challenge of designing an orally administered antibiotic is that its plasma concentration has to exceed its MIC values. So far, most of the newly-introduced antibiotics, such as ceftaroline16 and lipoglycopeptides are taken parenterally.17 When phenylthiazoles were first discovered, their main drawback was the extremely short plasma half-life of the early generation, for which the t1/2 of the lead compound 1 (Fig. 1) was only 29 minutes.2 To assess whether the chemical modifications presented in this study have the expected positive impact on the pharmacokinetic profile, a rat was administered a 25 mg kg−1 oral dosage of compound 20. Fig. 4 indicates that compound 20 possesses high metabolic stability with a biological half-life of 5.5 hours and low clearance rate. Most importantly, compound 20 reaches a plasma concentration that surpasses its MIC values against most of the tested multidrug-resistant clinical isolates.
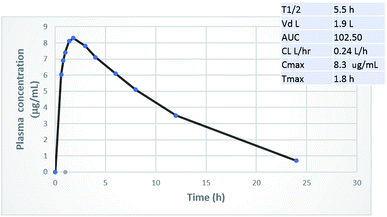 |
| | Fig. 4 Oral pharmacokinetic curve and key PK parameters in the rat after 25 mg kg−1 oral dose of compound 20. | |
3. Conclusions
Building a hybrid scaffold with the t-butylphenyl lipophilic part and oxadiazole linker followed by a study of the proper nitrogenous substituent at oxadiazole position-5 provided compound 20 with 3-hydroxyazetidine moiety as the most promising derivative. Interestingly, the unsubstituted azetidinyl analogue 15 was void of any antibacterial activity. The moderate antibacterial potency of hydroxyazetidinyl derivative 20, when tested against around thirty Gram-positive multidrug-resistant strains, was advantageous over vancomycin, the drug of choice for the treatment of invasive, life-threatening staphylococcal infections, and effective against all tested vancomycin-resistant species (i.e. VRSA and VRE). Furthermore, it was highly tolerable on vero cells and caco-2 cells. Moreover, compound 20 was more than two-fold better than vancomycin in its ability to disrupt bacterial biofilm mass. Another advantage of compound 20 over vancomycin and other phenylthiazoles is its highly acceptable pharmacokinetic profile: a biological half-life that is 11-fold more than the lead compound 1a with oral absorbability.
4. Experimental
4.1. General
1H NMR spectra were run at 400 MHz and 13C NMR spectra were determined at 100 MHz in deuterated chloroform (CDCl3), or dimethyl sulfoxide (DMSO-d6) on a Varian Mercury VX-400 NMR spectrometer. Chemical shifts are given in parts per million (ppm) on the delta (δ) scale. Chemical shifts were calibrated relative to those of the solvents. Flash chromatography was performed on 230–400 mesh silica. The progress of reactions was monitored with Merck silica gel IB2-F plates (0.25 mm thickness). The infrared spectra were recorded in potassium bromide disks on pye Unicam SP 3300 and Shimadzu FT IR 8101 PC infrared spectrophotometer. Mass spectra were recorded at 70 eV. High-resolution mass spectra for all ionization techniques were obtained from a FinniganMAT XL95. Melting points were determined using capillary tubes with a Stuart SMP30 apparatus and are uncorrected. All yields reported referring to isolated yields.
4.1.1. Ethyl 2-(4-(tert-Butyl)phenyl)-4-methylthiazole-5-carboxylate (3).
Compound 2 (3 g, 14 mmol) and ethyl 2-chloro-3-oxobutanoate (3.88 mL, 4.62 g, 28 mmol) were added to absolute ethanol (30 mL). The reaction mixture was heated at reflux for 4 h. After removal of solvent under reduced pressure, the solid residue was purified by crystallization from ethanol to provide the desired product as white crystals (4.4 g, 95%) mp = 105 °C; 1H NMR (DMSO-d6) δ: 7.79 (d, J = 8.4 Hz, 2H), 7.49 (d, J = 8.4 Hz, 2H), 4.33 (q, J = 4.8 Hz, 2H), 2.67 (s, 3H), 1.29 (s, 9H), 1.13 (t, J = 4.8 Hz, 3H); 13C NMR (DMSO-d6) δ: 169.1, 154.5, 153.8, 139.5, 130.8, 126.7, 126.1, 115.3, 61.8, 35.1, 28.8, 17.7, 14.5; MS (m/z) 303; anal. calc. for: (C17H21NO2S): C, 67.30; H, 6.98; N, 4.62%; found: C, 67.32; H, 6.99; N, 4.63%.
4.1.2. 2-(4-(tert-Butyl)phenyl)-4-methylthiazole-5-carbohydrazide (4).
To a solution of 3 (1.29 g, 4 mmol) in ethanol (15 mL), hydrazine hydrate (99%, 1 mL, 20 mmol) was added dropwise. The reaction mixture was heated at reflux for 8 h then allowed to cool down to room temperature. The formed solid was separated by filtration and crystallized from ethanol to provide the desired product as white crystals (1.1 g, 93%) mp = 204–205 °C; 1H NMR (DMSO-d6) δ: 9.57 (brs, 1H), 7.82 (d, J = 8.4 Hz, 2H), 7.49 (d, J = 8.4 Hz, 2H), 4.54 (brs, 2H), 2.65 (s, 3H), 1.30SS (s, 9H); 13C NMR (DMSO-d6) δ: 165.6, 155.2, 153.5, 142.3, 130.9, 126.7, 126.4, 115.3, 35.1, 18.7, 17.3; MS (m/z) 289; anal. calc. for: (C15H19N3OS): C, 62.26; H, 6.62; N, 14.52%; found: C, 62.27; H, 6.63; N, 14.53%.
4.1.3. 2-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)-5-(methylthio)-1,3,4-oxadiazole (6).
Potassium hydroxide (0.4 g, 10 mmol) was added to a solution of 4 (3 g, 10 mmol) in ethanol (15 mL), followed by drop-wise addition of carbon disulphide (3 mL, 110 mmol) over 0.5 h. The reaction mixture was stirred at room temperature for an additional 15 min and then heated to reflux until the evolution of hydrogen sulfide gas ceased. After completion of the reaction, as monitored by TLC, the obtained intermediate was poured on cold water (50 mL), filtered, washed with water, dried and crystallized from ethanol to provide potassium salt 5 as yellow crystals (3.4 g, 89%) mp > 300 °C; 1H NMR (DMSO-d6) δ: 7.89 (d, J = 8.4 Hz, 2H), 7.53 (d, J = 8.4 Hz, 2H), 2.64 (s, 3H), 1.29 (s, 9H); 13C NMR (DMSO-d6) δ: 167.6, 159.4, 158.1, 155.1, 154.3, 130.2, 126.6, 126.5, 111.7, 35.1, 31.3, 18.7; MS (m/z) 369. The obtained salt 5 (0.8 g, 2.1 mmol) was dissolved in water (15 mL). Then, dimethyl sulfate (0.5 mL, 4 mmol) was added dropwise with vigorous stirring. After 2 h, the formed solid was filtered and washed with copious amounts of water to yield the titled compound 6 as a yellowish white solid (0.68 g, 91%); mp = 173 °C; 1H NMR (DMSO-d6) δ: 7.93 (d, J = 8.4 Hz, 2H), 7.56 (d, J = 8.4 Hz, 2H), 2.77 (s, 3H), 2.68 (s, 3H), 1.31 (s, 9H); 13C NMR (DMSO-d6) δ: 166.5, 162.5, 159.4, 153.9, 153.4, 130.5, 126.5, 126.4, 106.5, 39.3, 35.1, 28.2, 18.6; MS (m/z) 345; anal. calc. for: (C17H19N3OS2): C, 59.10; H, 5.54; N, 12.16%; found: C, 59.12; H, 5.55; N, 12.17%.
4.1.4. 2-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)-5-(methylsulfonyl)-1,3,4-oxadiazole (7).
To a solution of 6 (0.5 g, 1.3 mmol) in dry DCM (5 mL), m-CPBA (0.514 g, 2.9 mmol) diluted with DCM (5 mL) was added portion-wise with continuous stirring. Afterward, the reaction mixture was kept at 23 °C for 16 h, additional DCM (10 mL) was added and the reaction mixture was washed with 25 mL of 5% aqueous solution of sodium metabisulfite, and 25 mL of 5% aqueous sodium carbonate. The organic layer was separated, dried and concentrated under reduced pressure to give the desired product as yellow crystals (0.5 g, 93%) mp = 144 °C; 1H NMR (DMSO-d6) δ: 7.88 (d, J = 8.4 Hz, 2H), 7.50 (d, J = 8.4 Hz, 2H), 3.47 (s, 3H), 2.76 (s, 3H), 1.29 (s, 9H); 13C NMR (DMSO-d6) δ: 165.3, 159.2, 159.0, 158.3, 157.0, 130.1, 126.7, 126.6, 118.4, 42.2, 35.1, 31.2, 18.9; MS (m/z) 377; anal. calc. for: (C17H19N3O3S2): C, 54.09; H, 5.07; N, 11.13%; found: C, 54.11; H, 5.08; N, 11.15%.
4.2. Compounds 8–30
4.2.1. General procedure.
To a solution of 7 (0.1 g, 0.25 mmol) in dry DMF (5 mL), appropriate amine, hydrazine, guanidine or carboximidate (0.4 mmol); namely: methylamine, ethylamine, butylamine, isopropylamine, cyclopentylamine, cyclohexylamine, dimethylamine, azetidine hydrochloride, pyrrolidine, piperidine, morpholine, 4-methylpiperazine, azetidin-3-ol hydrochloride, hydrazine hydrate, guanidine hydrochloride, methylguanidine hydrochloride, 1,1-dimethylguanidine hydrochloride, N,N-tetramethyl guanidine, morpholine-4-carboximidamide hydroiodide, 4-methylpiperazine-1-carboximidamide hydroiodide, picolinimidamide hydrochloride, nicotinimidamide hydrochloride, isonicotinimidamide hydrochloride, was added. The reaction mixture was heated at 80 °C for 0.5–12 h and then poured over ice water (50 mL). The formed solid was extracted with ethyl acetate (10 mL). The organic layer was evaporated under reduced pressure. The obtained crude material was then purified by crystallization or column chromatography. Physical properties and spectral analysis of isolated products are listed below:
4.2.2. 5-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)-N-methyl-1,3,4-oxadiazol-2-amine (8).
Following the general procedure 4.2.1., and using methylamine (13 μL, 0.4 mmol), compound 8 was obtained as yellow solid (0.08 g, 88%) mp = 223 °C; 1H NMR (DMSO-d6) δ: 7.85 (d, J = 8.4 Hz, 2H), 7.77 (brs, 1H), 7.51 (d, J = 8.4 Hz, 2H), 2.87 (s, 3H), 2.61 (s, 3H), 1.30 (s, 9H); 13C NMR (DMSO-d6) δ: 166.1, 164.2, 154.3, 154.1, 153.8, 130.0, 126.5, 126.5, 115.0, 35.1, 29.5, 26.8, 17.3; MS (m/z) 328; HRMS (EI) m/z 328.1358 M+, calcd for C17H20N4OS 328.1358; anal. calc. for: (C17H20N4OS): C, 62.17; H, 6.14; N, 17.06%; found: C, 62.18; H, 6.15; N, 17.07%.
4.2.3. 5-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)-N-ethyl-1,3,4-oxadiazol-2-amine (9).
Following the general procedure 4.2.1., and using ethylamine (18 μL, 0.4 mmol), compound 9 was obtained as yellow solid (0.05 g, 55%) mp = 200 °C; 1H NMR (DMSO-d6) δ: 8.26 (brs, 1H),7.90 (d, J = 8.4 Hz, 2H), 7.54 (d, J = 8.4 Hz, 2H), 3.21 (m, 2H), 2.68 (s, 3H), 1.37 (s, 9H), 1.14 (m, 3H); 13C NMR (DMSO-d6) δ: 166.0, 163.5, 154.3, 154.1, 153.7, 130.0, 126.8, 126.6, 115.0, 37.9, 34.6, 31.3, 17.4, 15.17; MS (m/z) 342; HRMS (EI) m/z 342.1514 M+, calcd for C18H22N4OS 342.1514; anal. calc. for: (C18H22N4OS): C, 63.13; H, 6.48; N, 16.36%; found: C, 63.15; H, 6.50; N, 16.37%.
4.2.4.
N-Butyl-5-(2-(4-(tert-butyl)phenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-amine (10).
Following the general procedure 4.2.1., and using ethylamine (29 μL, 0.4 mmol), compound 10 was obtained as yellow solid (0.09 g, 93%) mp = 145 °C; 1H NMR (DMSO-d6) δ: 7.88 (d, J = 8.4 Hz, 2H), 7.83 (brs, 1H), 7.53 (d, J = 8.4 Hz, 2H), 3.37 (t, J = 3.2 Hz, 2H), 2.66 (s, 3H), 1.94 (m, 2H), 1.70 (m, 2H), 1.54 (m, 3H), 1.29 (s, 9H); 13C NMR (DMSO-d6) δ: 166.4, 162.9, 154.3, 153.7, 152.7, 130.0, 126.6, 126.5, 128.6, 127.9, 127.3, 127.1, 115.4, 42.7, 31.2, 19.8, 17.4, 14.0; MS (m/z) 370; HRMS (EI) m/z 370.1827 M+, calcd for C20H26N4OS 370.1827; anal. calc. for: (C20H26N4OS): C, 64.83; H, 7.07; N, 15.12%; found: C, 64.84; H, 7.09; N, 15.13%
4.2.5. 5-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)-N-isopropyl-1,3,4-oxadiazol-2-amine (11).
Following the general procedure 4.2.1., and using ethylamine (23 μL, 0.4 mmol), compound 11 was obtained as yellow solid (0.06 g, 81%) mp = 220 °C; 1H NMR (DMSO-d6) δ: 7.88 (d, J = 8.4 Hz, 2H), 7.82 (brs, 1H), 7.53 (d, J = 8.4 Hz, 2H), 3.70 (m, 1H), 2.65 (s, 3H), 1.29 (s, 9H), 1.20 (d, J = 6.8 Hz, 6H); 13C NMR (DMSO-d6) δ: 166.5, 162.9, 154.3, 153.7, 152.8, 130.0, 126.6, 126.6, 115.0, 45.4, 35.1, 31.3, 22.7, 17.3; MS (m/z) 356; HRMS (EI) m/z 356.1671 M+, calcd for C19H24N4OS 356.1671; anal. calc. for: (C19H24N4OS): C, 64.02; H, 6.79; N, 15.72%; found: C, 64.02; H, 6.80; N, 15.73%.
4.2.6. 5-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)-N-cyclopentyl-1,3,4-oxadiazol-2-amine (12).
Following the general procedure 4.2.1., and using cyclopentylamine (34 μL, 0.4 mmol), compound 12 was obtained as yellow solid (0.06 g, 60%) mp = 195 °C; 1H NMR (DMSO-d6) δ: 7.95 (brs, 1H), 7.86 (d, J = 8.4 Hz, 2H), 7.51 (d, J = 8.4 Hz, 2H), 3.87 (m, 1H), 2.65 (s, 3H), 1.91 (m, 2H), 1.65 (m, 2H), 1.58 (m, 4H), 1.28 (s, 9H); 13C NMR (DMSO-d6) δ: 166.4, 163.2, 154.3, 153.7, 152.8, 130.0, 126.6, 126.5, 115.0, 54.8, 35.1, 32.6, 31.2, 23.6, 17.3; MS (m/z) 382; HRMS (EI) m/z 382.1827 M+, calcd for C21H26N4OS 382.1827; anal. calc. for: (C21H26N4OS): C, 65.94; H, 6.85; N, 14.65%; found: C, 65.96; H, 6.86; N, 14.67%.
4.2.7. 5-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)-N-cyclohexyl-1,3,4-oxadiazol-2-amine (13).
Following the general procedure 4.2.1., and using cyclohexylamine (39 μL, 0.4 mmol), compound 13 was obtained as light brown solid (0.08 g, 76%) mp = 186 °C; 1H NMR (DMSO-d6) δ: 7.95 (brs, 1H), 7.88 (d, J = 8.4 Hz, 2H), 7.53 (d, J = 8.4 Hz, 2H), 3.22 (m, 1H), 2.65 (s, 3H), 1.54 (m, 4H), 1.32 (m, 4H), 1.29 (s, 2H), 0.90 (m, 2H); 13C NMR (DMSO-d6) δ: 166.5, 163.6, 154.3, 153.7, 152.8, 130.0, 126.6, 126.5, 115.0, 42.7, 35.1, 31.3, 31.2, 19.8, 17.3, 14.0; MS (m/z) 396; HRMS (EI) m/z 396.1984 M+, calcd for C22H28N4OS 396.1984; anal. calc. for: (C22H28N4OS): C, 66.63; H, 7.12; N, 14.13%; found: C, 66.64; H, 7.13; N, 14.14%.
4.2.8. 5-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)-N,N-dimethyl-1,3,4-oxadiazol-2-amine (14).
Following the general procedure 4.2.1., and using dimethylamine (18 μL, 0.4 mmol), compound 14 was obtained as yellow solid (0.07 g, 80%) mp = 185 °C; 1H NMR (DMSO-d6) δ: 7.89 (d, J = 8.4 Hz, 2H), 7.53 (d, J = 8.4 Hz, 2H), 3.04 (s, 6H), 2.66 (s, 3H), 1.29 (s, 9H); 13C NMR (DMSO-d6) δ: 166.6, 164.6, 154.4, 153.9, 153.5, 130.0, 126.6, 126.6, 114.9, 38.1, 35.1, 31.3, 17.3; MS (m/z) 342; HRMS (EI) m/z 342.1514 M+, calcd for C18H22N4OS 342.1514; anal. calc. for: (C18H22N4OS): C, 63.13; H, 6.48; N, 16.36%; found: C, 63.14; H, 6.50; N, 16.37%.
4.2.9. 2-(Azetidin-1-yl)-5-(2-(4-(tert-butyl)phenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazole (15).
Following the general procedure 4.2.1., and using azetidine hydrochloride (0.04 g, 0.4 mmol), compound 15 was obtained as yellow solid (0.07 g, 70%) mp = 228 °C; 1H NMR (DMSO-d6) δ: 7.89 (d, J = 8.4 Hz, 2H), 7.52 (d, J = 8.4 Hz, 2H), 4.14 (t, J = 8 Hz, 4H), 2.87 (m, 2H), 2.69 (s, 3H), 1.28 (s, 9H); 13C NMR (DMSO-d6) δ: 166.9, 164.7, 162.7, 154.4, 154.3, 129.9, 126.6, 126.5, 114.6, 52.5, 35.1, 31.2, 17.8, 17.3; MS (m/z) 354; HRMS (EI) m/z 354.1514 M+, calcd for C19H22N4OS 354.1514; anal. calc. for: (C19H22N4OS): C, 64.38; H, 6.26; N, 15.81%; found: C, 64.40; H, 6.27; N, 15.83%.
4.2.10. 2-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)-5-(pyrrolidin-1-yl)-1,3,4-oxadiazole (16).
Following the general procedure 4.2.1., and using pyrrolidine (28 μL, 0.4 mmol), compound 16 was obtained as yellow solid (0.089 g, 91%) mp = 226 °C; 1H NMR (DMSO-d6) δ: 7.89 (d, J = 8.4 Hz, 2H), 7.53 (d, J = 8.4 Hz, 2H), 3.48 (m, 4H), 2.65 (s, 3H), 1.96 (m, 4H), 1.27 (s, 9H); 13C NMR (DMSO-d6) δ: 166.5, 162.4, 154.4, 153.8, 153.2, 130.0, 126.63, 126.61, 114.2, 48.0, 35.1, 31.3, 25.5, 17.3; MS (m/z) 368; HRMS (EI) m/z 368.1671 M+, calcd for C20H24N4OS 368.1671; anal. calc. for: (C20H24N4OS): C, 65.19; H, 6.57; N, 15.20%; found: C, 65.20; H, 6.58; N, 15.21%
4.2.11. 2-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)-5-(piperidin-1-yl)-1,3,4-oxadiazole (17).
Following the general procedure 4.2.1., and using piperidine (34 μL, 0.4 mmol), compound 17 was obtained as yellow solid (0.098 g, 97%) mp = 285 °C; 1H NMR (DMSO-d6) δ: 7.89 (d, J = 8.4 Hz, 2H), 7.54 (d, J = 8.4 Hz, 2H), 3.45 (m, 4H), 2.66 (s, 3H), 1.59 (m, 6H), 1.29 (s, 9H); 13C NMR (DMSO-d6) δ: 166.7, 164.0, 154.4, 154.0, 153.5, 130.0, 126.64, 126.63, 114.8, 47.1, 35.1, 31.3, 24.8, 23.6, 17.4; MS (m/z) 382; HRMS (EI) m/z 382.1827 M+, calcd for C21H26N4OS 382.1827; anal. calc. for: (C21H26N4OS): C, 65.94; H, 6.85; N, 14.65%; found: C, 65.95; H, 6.87; N, 14.67%.
4.2.12. 4-(5-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)morpholine (18).
Following the general procedure 4.2.1., and using morpholine (35 μL, 0.4 mmol), compound 18 was obtained as yellow solid (0.098 g, 97%) mp = 285 °C; 1H NMR (DMSO-d6) δ: 7.99 (d, J = 8.4 Hz, 2H), 7.57 (d, J = 8.4 Hz, 2H), 3.73 (m, 4H), 3.48 (m, 4H), 2.68 (s, 3H), 1.32 (s, 9H); 13C NMR (DMSO-d6) δ: 167.0, 164.0, 154.5, 154.0, 153.1, 130.0, 126.6, 126.3, 114.7, 65.6, 46.2, 35.2, 31.3, 17.4; MS (m/z) 384; HRMS (EI) m/z 384.1620 M+, calcd for C20H24N4O2S 384.1620; anal. calc. for: (C20H24N4O2S): C, 62.48; H, 6.29; N, 14.57%; found: C, 62.49; H, 6.30; N, 14.59%
4.2.13. 2-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)-5-(4-methylpiperazin-1-yl)-1,3,4-oxadiazole (19).
Following the general procedure 4.2.1., and using 4-methylpiperazine (40 μL, 0.4 mmol), compound 19 was obtained as yellow solid (0.099 g, 97%) mp = 177 °C; 1H NMR (DMSO-d6) δ: 7.84 (d, J = 8.4 Hz, 2H), 7.52 (d, J = 8.4 Hz, 2H), 3.47 (m, 4H), 2.66 (s, 3H) 2.25 (m, 4H), 2.14 (s, 3H), 1.28 (s, 9H); 13C NMR (DMSO-d6) δ: 166.8, 163.8, 161.8, 157.1, 156.4, 130.2, 126.6, 126.5, 114.7, 53.7, 46.0, 43.8, 35.1, 31.3, 17.5; MS (m/z) 397; HRMS (EI) m/z 397.1936 M+, calcd for C21H27N5OS 397.1936; anal. calc. for: (C21H27N5OS): C, 63.45; H, 6.85; N, 17.62%; found: C, 63.47; H, 6.87; N, 17.64%.
4.2.14. 1-(5-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)azetidin-3-ol (20).
Following the general procedure 4.2.1., and using azetidin-3-ol hydrochloride (0.04 g, 0.4 mmol), compound 20 was obtained as yellow solid (0.05 g, 54%) mp = 210 °C; 1H NMR (DMSO-d6) δ: 7.88 (d, J = 8.4 Hz, 2H), 7.52 (d, J = 8.4 Hz, 2H), 6.06 (brs, 1H), 4.66 (m, 1H), 4.35 (dd, J = 7.6 Hz, J = 8.4 Hz, 2H), 3.97 (dd, J = 5.2 Hz, J = 8.2 Hz, 2H), 2.65 (s, 3H), 1.29 (s, 9H); 13C NMR (DMSO-d6) δ: 166.9, 164.7, 154.49, 154.45, 154.42, 129.9, 126.6, 126.4, 114.6, 62.3, 62.1, 35.1, 31.3, 17.4; MS (m/z) 370; HRMS (EI) m/z 370.1463 M+, calcd for C19H22N4O2S 370.1463; anal. calc. for: (C19H22N4O2S): C, 61.60; H, 5.99; N, 15.12%; found: C, 61.61; H, 6.01; N, 15.13%.
4.2.15. 2-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)-5-hydrazinyl-1,3,4-oxadiazole (21).
Following the general procedure 4.2.1., and using hydrazine hydrate (5 mL), compound 21 was obtained as yellow fluffy powder (0.06 g, 75%) mp = 260 °C; 1H NMR (DMSO-d6) δ: 7.85 (d, J = 8.4 Hz, 2H), 7.50 (d, J = 8.4 Hz, 2H), 6.94 (brs, 1H), 4.54 (brs, 2H), 2.62 (s, 3H), 1.31 (s, 9H); 13C NMR (DMSO-d6) δ: 166.3, 163.2, 155.7, 155.2, 154.1, 130.3, 126.6, 126.5, 124.6, 35.1, 31.3, 17.5; MS (m/z) 329; HRMS (EI) m/z 329.1310 M+, calcd for C16H19N5OS 329.1310; anal. calc. for: (C16H19N5OS): C, 58.34; H, 5.81; N, 21.26%; found: C, 58.36; H, 5.82; N, 21.26%.
4.2.16. 1-(5-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)guanidine (22).
Following the general procedure 4.2.1., and using guanidine hydrochloride (0.05 g, 0.5 mmol), compound 22 was obtained as grayish solid (0.07 g, 76%) mp = 198 °C; 1H NMR (DMSO-d6) δ: 7.88 (d, J = 8.4 Hz, 2H), 7.53 (d, J = 8.4 Hz, 2H), 7.21 (brs, 1H), 7.06 (brs, 3H), 2.65 (s, 3H), 1.29 (s, 9H); 13C NMR (DMSO-d6) δ: 167.2, 166.5, 159.5, 154.3, 153.7, 152.6, 130.0, 126.6, 126.5, 115.7, 35.1, 31.3, 17.3; MS (m/z) 356; HRMS (EI) m/z 356.1419 M+, calcd for C17H20N6OS 356.1419; anal. calc. for: (C17H20N6OS): C, 57.28; H, 5.66; N, 23.58%; found: C, 57.30; H, 5.67; N, 23.60%.
4.2.17. 1-(5-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)-3-methylguanidine (23).
Following the general procedure 4.2.1., and using methylguanidine hydrochloride (0.06 g, 0.5 mmol), compound 23 was obtained as yellowish brown solid (0.07 g, 75%) mp = 194 °C; 1H NMR (DMSO-d6) δ: 7.89 (d, J = 8.4 Hz, 2H), 7.81 (brs, 1H), 7.54 (d, J = 8 Hz, 2H), 7.37 (brs, 1H), 2.87 (s, 3H), 2.78 (brs, 1H), 2.67 (s, 3H), 1.30 (s, 9H); 13C NMR (DMSO-d6) δ: 166.5, 164.2, 154.3, 153.8, 153.0, 130.1, 130.0, 126.6, 126.5, 115.0, 35.1, 31.3, 29.5, 17.4; MS (m/z) 370; HRMS (EI) m/z 370.1576 M+, calcd for C18H22N6OS 370.1576; anal. calc. for: (C18H22N6OS): C, 58.36; H, 5.99; N, 22.68%; found: C, 58.37; H, 5.99; N, 22.69%.
4.2.18. 3-(5-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)-1,1-dimethylguanidine (24).
Following the general procedure 4.2.1., and using 1,1-dimethylguanidine hydrochloride (0.06 g, 0.5 mmol), compound 24 was obtained as yellowish brown solid (0.08 g, 75%) mp = 194 °C; 1H NMR (DMSO-d6) δ: 7.89 (d, J = 8.4 Hz, 2H), 7.75 (brs, 2H), 7.54 (d, J = 8 Hz, 2H), 3.01 (s, 6H), 2.68 (s, 3H), 1.29 (s, 9H); 13C NMR (DMSO-d6) δ: 166.8, 166.6, 157.8, 154.3, 153.9, 152.8, 130.1, 126.63, 126.60, 115.7, 37.5, 35.1, 31.3, 17.5; MS (m/z) 384; HRMS (EI) m/z 384.1732 M+, calcd for C19H24N6OS 384.1732; anal. calc. for: (C19H24N6OS): C, 59.35; H, 6.29; N, 21.86%; found: C, 59.35; H, 6.29; N, 21.86%.
4.2.19. 2-(5-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)-1,1,3,3-tetramethylguanidine (25).
Following the general procedure 4.2.1., and using N,N-tetramethyl guanidine (50 μL, 0.4 mmol), compound 25 was obtained as yellowish brown solid (0.08 g, 75%) mp = 194 °C; 1H NMR (DMSO-d6) δ: 7.88 (d, J = 8.4 Hz, 2H), 7.52 (d, J = 8.4 Hz, 2H), 2.81 (s, 12H), 2.66 (s, 3H), 1.28 (s, 9H); 13C NMR (DMSO-d6) δ: 166.4, 164.9, 163.7, 154.2, 154.1, 153.4, 130.1, 126.6, 126.5, 115.8, 139.3, 35.1, 31.3, 17.4; MS (m/z) 412; HRMS (EI) m/z 412.2045 M+, calcd for C21H28N6OS 412.2045; anal. calc. for: (C21H28N6OS): C, 61.14; H, 6.84; N, 20.37%; found: C, 61.15; H, 6.86; N, 20.38%.
4.2.20.
N-(5-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)morpholine-4-carboximidamide (26).
Following the general procedure 4.2.1., and using morpholine-4-carboximidamide hydroiodide (0.1 g, 0.4 mmol), compound 26 was obtained as yellow solid (0.07 g, 65%) mp = 260 °C; 1H NMR (DMSO-d6) δ: 7.96 (brs, 2H), 7.90 (d, J = 8.4 Hz, 2H), 7.54 (d, J = 8.4 Hz, 2H), 3.64 (m, 4H), 3.56 (m, 4H), 2.69 (s, 3H), 1.31 (s, 9H); 13C NMR (DMSO-d6) δ: 166.9, 166.8, 157.2, 154.3, 154.1, 153.2, 130.0, 126.6, 126.5, 115.4, 66.1, 44.8, 35.1, 31.3, 17.5; MS (m/z) 426; HRMS (EI) m/z 426.1838 M+, calcd for C21H26N6O2S 426.1838; anal. calc. for: (C21H26N6O2S): C, 59.13; H, 6.14; N, 19.70%; found: C, 59.15; H, 6.15; N, 19.72%.
4.2.21.
N-(5-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)-4-methylpiperazine-1-carboximidamide (27).
Following the general procedure 4.2.1., and using 4-methylpiperazine-1-carboximidamide hydroiodide (0.11 g, 0.4 mmol), compound 27 was obtained as yellow solid (0.07 g, 63%) mp = 262 °C; 1H NMR (DMSO-d6) δ: 7.98 (brs, 2H), 7.92 (d, J = 8.4 Hz, 2H), 7.52 (d, J = 8.4 Hz, 2H), 3.58 (m, 4H), 2.68 (s, 3H), 2.35 (m, 4H), 2.21 (s, 3H), 1.30 (s, 9H); 13C NMR (DMSO-d6) δ: 166.9, 166.6, 156.9, 154.3, 154.0, 153.0, 130.0, 126.59, 126.56, 115.5, 54.5, 45.9, 44.3, 35.1, 31.3, 17.5; MS (m/z) 439; HRMS (EI) m/z 439.2154 M+, calcd for C22H29N7OS 439.2154; anal. calc. for: (C22H29N7OS): C, 60.11; H, 6.65; N, 22.31%; found: C, 60.12; H, 6.67; N, 22.33%.
4.2.22.
N-(5-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)picolinimidamide (28).
Following the general procedure 4.2.1., and using picolinimidamide hydrochloride (0.06 g, 0.4 mmol), compound 28 was obtained as yellow solid (0.1 g, 91%) mp = 225 °C; 1H NMR (DMSO-d6) δ: 9.43 (brs, 1H), 8.92 (brs, 1H), 8.76 (m, 1H), 8.34 (m, 1H), 7.94–7.82 (m, 3H), 7.57–7.49 (m, 3H), 2.68 (s, 3H), 1.30 (s, 9H); 13C NMR (DMSO-d6) δ: 167.2, 166.9, 162.8, 158.4, 158.0, 154.2, 153.9, 149.0, 138.0, 131.7, 130.5, 126.5, 126.4, 122.6, 111.4, 35.1, 31.3, 18.7; MS (m/z) 418; HRMS (EI) m/z 418.1576 M+, calcd for C22H22N6OS 418.1576; anal. calc. for: (C22H22N6OS): C, 63.14; H, 5.30; N, 20.08%; found: C, 63.15; H, 5.31; N, 20.09%.
4.2.23.
N-(5-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)nicotinimidamide (29).
Following the general procedure 4.2.1., and using nicotinimidamide hydrochloride (0.06 g, 0.4 mmol), compound 29 was obtained as yellow solid (0.1 g, 91%) mp = 289 °C; 1H NMR (DMSO-d6) δ: 9.78 (brs, 1H), 9.24 (s, 1H), 9.02 (brs, 1H), 8.75 (d, J = 6.8 Hz, 1H), 8.46 (t, J = 6.6 Hz, 1H), 7.95 (d, J = 8.4 Hz, 2H), 7.56 (m, 3H), 2.71 (s, 3H), 1.28 (s, 9H); 13C NMR (DMSO-d6) δ: 167.6, 166.4, 160.5, 155.4, 155.1, 154.5, 152.8, 149.2, 135.9, 130.6, 129.6, 126.5, 126.4, 123.9, 114.8, 35.1, 31.3, 17.6; MS (m/z) 418; HRMS (EI) m/z 418.1576 M+, calcd for C22H22N6OS 418.1576; anal. calc. for: (C22H22N6OS): C, 63.14; H, 5.30; N, 20.08%; found: C, 63.16; H, 5.32; N, 20.10%.
4.2.24.
N-(5-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)isonicotinimidamide (30).
Following the general procedure 4.2.1., and using isonicotinimidamide hydrochloride (0.06 g, 0.4 mmol), compound 30 was obtained as yellow solid (0.07 g, 64%) mp = 257 °C; 1H NMR (DMSO-d6) δ: 9.83 (brs, 1H), 9.05 (brs, 1H), 8.77 (d, J = 7.6 Hz, 2H), 7.89 (d, J = 7.6 Hz, 2H), 7.83 (d, J = 8.4 Hz, 2H), 7.52 (d, J = 8.4 Hz, 2H), 2.73 (s, 3H), 1.29 (s, 9H); 13C NMR (DMSO-d6) δ: 167.7, 166.3, 160.2, 155.5, 154.5, 153.9, 150.7, 141.1, 130.0, 126.6, 126.4, 121.9, 114.7, 35.1, 31.2, 17.6; MS (m/z) 418; HRMS (EI) m/z 418.1576 M+, calcd for C22H22N6OS 418.1576; anal. calc. for: (C22H22N6OS): C, 63.14; H, 5.30; N, 20.08%; found: C, 63.16; H, 5.31; N, 20.09%.
4.3. Microbiological assays
4.3.1. Determination of minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC).
Broth microdilution method was used18,19 against a panel of clinically important Gram positive bacterial pathogens according the previous reports.19,20
4.3.2. Time-kill assay against MRSA.
It was performed against MRSA USA400 as described previously.20,21 In brief, bacterial cells in logarithmic phase were diluted and drugs were added at 5× MIC and 10× MIC (in triplicates). After time points, cells were collected, diluted and plated on tryptic soy agar plates. Plates were incubated for 18–20 hours and the viable CFU mL−1 was determined.
4.3.3. MRSA biofilm eradication assessment.
Compound 20 was examined for its ability to eradicate established, mature staphylococcal biofilm using the microtiter plate biofilm formation assay following the procedure described before.7,22,23
4.3.4.
In vivo pharmacokinetics.
Pharmacokinetic studies were performed following the procedure described in a previous report.5
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was funded by Science & Technology Development Funds (STDF-Egypt) and the Egyptian Ministry of Higher Education and Scientific Research (MHESR). The work is derived from the Subject Data funded in whole or part by NAS and USAID, and that any opinions, findings, conclusions, or recommendations expressed in such article are those of the authors alone, and do not necessarily reflect the views of USAID or NAS. The authors would like to thank BEI Resources for providing clinical isolates of S. aureus used in this study.
Notes and references
- M. Hagras, Y. A. Hegazy, A. H. Elkabbany, H. Mohammad, A. Ghiaty, T. M. Abdelghany, M. N. Seleem and A. S. Mayhoub, Eur. J. Med. Chem., 2018, 143, 1448–1456 CrossRef CAS PubMed.
- H. Mohammad, A. S. Mayhoub, A. Ghafoor, M. Soofi, R. A. Alajlouni, M. Cushman and M. N. Seleem, J. Med. Chem., 2014, 57, 1609–1615 CrossRef CAS.
- H. Mohammad, W. Younis, L. Chen, C. E. Peters, J. Pogliano, K. Pogliano, B. Cooper, J. Zhang, A. Mayhoub, E. Oldfield, M. Cushman and M. N. Seleem, J. Med. Chem., 2017, 60, 2425–2438 CrossRef CAS.
- M. Hagras, H. Mohammad, M. S. Mandour, Y. A. Hegazy, A. Ghiaty, M. N. Seleem and A. S. Mayhoub, J. Med. Chem., 2017, 60, 4074–4085 CrossRef CAS.
- M. A. Seleem, A. M. Disouky, H. Mohammad, T. M. Abdelghany, A. S. Mancy, S. A. Bayoumi, A. Elshafeey, A. El-Morsy, M. N. Seleem and A. S. Mayhoub, J. Med. Chem., 2016, 59, 4900–4912 CrossRef CAS.
- E. Yahia, H. Mohammad, T. M. Abdelghany, E. Fayed, M. N. Seleem and A. S. Mayhoub, Eur. J. Med. Chem., 2017, 126, 604–613 CrossRef CAS.
- A. Kotb, N. S. Abutaleb, M. A. Seleem, M. Hagras, H. Mohammad, A. Bayoumi, A. Ghiaty, M. N. Seleem and A. S. Mayhoub, Eur. J. Med. Chem., 2018, 151, 110–120 CrossRef CAS.
- I. Eid, M. M. Elsebaei, H. Mohammad, M. Hagras, C. E. Peters, Y. A. Hegazy, B. Cooper, J. Pogliano, K. Pogliano, H. S. Abulkhair, M. N. Seleem and A. S. Mayhoub, Eur. J. Med. Chem., 2017, 139, 665–673 CrossRef CAS.
- H. Mohammad, A. AbdelKhalek, N. S. Abutaleb and M. N. Seleem, Int. J. Antimicrob. Agents, 2018, 51, 897–904 CrossRef CAS.
- A. AbdelKhalek, N. S. Abutaleb, K. A. Elmagarmid and M. N. Seleem, Sci. Rep., 2018, 8, 8353 CrossRef.
- A. AbdelKhalek, N. S. Abutaleb, H. Mohammad and M. N. Seleem, PLoS One, 2018, 13, e0199710 CrossRef.
- R. J. McLean, J. S. Lam and L. L. Graham, J. Bacteriol., 2012, 194, 6706–6711 CrossRef CAS.
- R. Cavaliere, J. L. Ball, L. Turnbull and C. B. Whitchurch, MicrobiologyOpen, 2014, 3, 557–567 CrossRef CAS.
- R. Wang, B. A. Khan, G. Y. Cheung, T. H. Bach, M. Jameson-Lee, K. F. Kong, S. Y. Queck and M. Otto, J. Clin. Invest., 2011, 121, 238–248 CrossRef CAS.
- D. Davies, Nat. Rev. Drug Discovery, 2003, 2, 114–122 CrossRef CAS.
- L. D. Saravolatz, G. E. Stein and L. B. Johnson, Clin. Infect. Dis., 2011, 52, 1156–1163 CrossRef CAS.
- C. R. Caulder, A. Sloan, A. Yasir and P. B. Bookstaver, Hosp. Pharm., 2014, 49, 644–646 CrossRef.
- S. Thangamani, H. Mohammad, M. F. Abushahba, T. J. Sobreira, V. E. Hedrick, L. N. Paul and M. N. Seleem, Sci. Rep., 2016, 6, 22571 CrossRef CAS.
- H. Mohammad, M. Cushman and M. N. Seleem, PLoS One, 2015, 10, e0130385 CrossRef PubMed.
- M. F. Mohamed, M. I. Hamed, A. Panitch and M. N. Seleem, Antimicrob. Agents Chemother., 2014, 58, 4113–4122 CrossRef.
- M. F. Mohamed, G. K. Hammac, L. Guptill and M. N. Seleem, PLoS One, 2014, 9, e116259 CrossRef.
- G. A. O'Toole, J. Visualized Exp., 2011, 47, 2437 Search PubMed.
- H. Mohammad, A. S. Mayhoub, M. Cushman and M. N. Seleem, J. Antibiot., 2015, 68, 259–266 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Scanned copies of all spectral data of all new reported compounds. See DOI: 10.1039/c8ra10525a |
| ‡ These two authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ae
*ae