
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation, characterization and application in cobalt ion adsorption using nanoparticle films of hybrid copper–nickel hexacyanoferrate†
Xinxin Longa,
Rongzhi Chen *a,
Shengjiong Yangb,
Jixiang Wanga,
Tijun Huanga,
Qin Leia and
Jihua Tan*a
*a,
Shengjiong Yangb,
Jixiang Wanga,
Tijun Huanga,
Qin Leia and
Jihua Tan*a
aCollege of Resources and Environment, University of Chinese Academy of Sciences, Yuquan Road 19A, Beijing 100049, China. E-mail: crz0718@ucas.ac.cn; tanjh@ucas.ac.cn
bKey Laboratory of Environmental Engineering, Xi'an University of Architecture and Technology, No. 13, Yanta Road, Xi'an, Shaanxi 710055, China
First published on 6th March 2019
Abstract
Different mole ratios (nCu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :nNi = x:y) of hybrid copper–nickel metal hexacyanoferrates (CuxNiyHCFs) were prepared to explore their morphologies, structure, electrochemical properties and the feasibility of electrochemical adsorption of cobalt ions. Cyclic voltammetry (CV), field emission scanning electron microscopy (FE-SEM), Fourier transform infrared spectroscopy (FTIR) and X-ray diffraction (XRD) indicated that the x:y ratio of CuxNiyHCF nanoparticles can be easily controlled as designed using a wet chemical coprecipitation method. The crystallite size and formal potential of CuxNiyHCF films showed an insignificant change when 0 ≤ x:y < 0.3. Given the shape of the CV curves, this might be due to Cu2+ ions being inserted into the NiHCF framework as countercations to maintain the electrical neutrality of the structure. On the other hand, crystallite size depended linearly on the x:y ratio when x:y > 0.3. This is because Cu tended to replace Ni sites in the lattice structure at higher molar ratios of x:y. CuxNiyHCF films inherited good electrochemical reversibility from the CuHCFs, in view of the cyclic voltammograms; in particular, Cu1Ni2HCF exhibited long-term cycling stability and high surface coverage. The adsorption of Co2+ fitted the Langmuir isotherm model well, and the kinetic data can be well described by a pseudo-second order model, which may imply that Co2+ adsorption is controlled by chemical adsorption. The diffusion process was dominated by both intraparticle diffusion and surface diffusion.
:nNi = x:y) of hybrid copper–nickel metal hexacyanoferrates (CuxNiyHCFs) were prepared to explore their morphologies, structure, electrochemical properties and the feasibility of electrochemical adsorption of cobalt ions. Cyclic voltammetry (CV), field emission scanning electron microscopy (FE-SEM), Fourier transform infrared spectroscopy (FTIR) and X-ray diffraction (XRD) indicated that the x:y ratio of CuxNiyHCF nanoparticles can be easily controlled as designed using a wet chemical coprecipitation method. The crystallite size and formal potential of CuxNiyHCF films showed an insignificant change when 0 ≤ x:y < 0.3. Given the shape of the CV curves, this might be due to Cu2+ ions being inserted into the NiHCF framework as countercations to maintain the electrical neutrality of the structure. On the other hand, crystallite size depended linearly on the x:y ratio when x:y > 0.3. This is because Cu tended to replace Ni sites in the lattice structure at higher molar ratios of x:y. CuxNiyHCF films inherited good electrochemical reversibility from the CuHCFs, in view of the cyclic voltammograms; in particular, Cu1Ni2HCF exhibited long-term cycling stability and high surface coverage. The adsorption of Co2+ fitted the Langmuir isotherm model well, and the kinetic data can be well described by a pseudo-second order model, which may imply that Co2+ adsorption is controlled by chemical adsorption. The diffusion process was dominated by both intraparticle diffusion and surface diffusion.
1 Introduction
Iron hexacyanoferrate (PB, Prussian blue) and its analogues (PBAs) are collectively referred to as transition metal hexacyanoferrates (MHCFs, M = Fe, Cu, Co, Ni, etc.) Due to the electrochromism, magnetism, redox activity, and electrochemical stability associated with their unique cubic structure, MHCFs have been applied in a number of fields, including catalysis,1 electrochromics,2 electrocatalysis,3,4 detection,5 chemical sensors,6 capacitors,7 secondary batteries,8 photocatalysis,9 photochromics,10 biosensors,11,12 and metal ion sieving or capture,13,14 making great contributions to environmental technology and new-generation energy development.Recently, MHCFs have received extensive attention due to their unique electrochemical properties as modified electrode materials, such as PB (FeHCF),15 copper hexacyanoferrate (CuHCF),16 cobalt hexacyanoferrate (CoHCF),17 nickel hexacyanoferrate (NiHCF),18 and zinc hexacyanoferrate (ZnHCF).19 Each MHCF has its own unique electrochemical and spectral properties; however, materials with diversified traits are preferred. Research into hybrid-metal hexacyanoferrates (h-MHCFs) is becoming a trend due to their multiple characteristics and properties. Previous researchers reported that the prepared h-MHCFs existed as a new phase rather than a simple mixture of corresponding single metal hexacyanoferrates.20,21 Ghasemi et al.22 found that NiCoHCF has a higher capacitance than NiHCF or CoHCF; Li et al.23 revealed that CuNiHCF has a higher capacity retention than CuHCF as a cathode for sodium-ion batteries; it was also reported that the mixed MHCF showed an enhanced catalytic efficiency to H2O2 as compared to that of FeHCF.24 The surface coverage of CoNiHCF was proved to be higher than those of CoHCF and NiHCF.25
In an aqueous electrolyte containing alkali salts, MHCFs exhibit reversible redox reactions (such as FeIII/FeII) accompanied by reversible intercalation and deintercalation of hydrated alkali cations, low cost, and low toxicity.26 Within the family of MHCFs, an NiHCF modified electrode showed an excellent cation exchange capacity in redox reactions due to its open structural framework,27–29 with uniform, compact and stable films. The network structures with macropores26 of CuHCF films were characterized by nearly ideal current–potential curves.30 We also found that a CuNiHCF film modified electrode showed higher electrochemical stability in comparison to CuHCF and NiHCF in phosphate buffer solutions (PBS).25 To the best of our knowledge, few papers on different proportions of CuxNiyHCF nanoparticle films as modified electrode materials have been published, which inspired us to synthesize CuxNiyHCF films that may inherit the virtues of CuHCF and NiHCF.
Generally, MHCF film modified electrodes are prepared by electrodeposition, and are difficult to produce on a large scale,25 and fall off easily. Moreover, it is hard to control the proportion of metal phases (M1:M2) in the products (h-MHCFs), which is why few researchers have focused on the characteristic differences among varied proportions of h-MHCFs, such as morphology, structure, or electrochemical properties. Chen et al.31 prepared exact ratios of CuHCF nanoparticle ink by a wet chemical coprecipitation method, and printed them onto the electrode surface to remove cesium electrochemically; the cesium removal was comparable in magnitude to that by an electrodeposited film. Wang et al.25 prepared h-MHCF films by the same method; the surface coverage of redox-active sites of the CuCoHCF films was 35 nmol cm−2, which was much higher than the 9.02 nmol cm−2 of electrodeposited films;32 and the Co/Ni atomic ratio in CoNiHCF compounds had been proved to be controlled by the reactant ratio in the reaction mixture.
Herein, we prepared a series of CuxNiyHCF nanoparticle films with different molar ratios (nCu:nNi = x:y) by using a wet chemical coprecipitation method, and explored the characteristics of each ratio and the variations among them by multiple characterization methods. Recently, the wide application of lithium-ion batteries (LIBs) has resulted in a rapid increase in the amount of spent LIBs, containing valuable metals such as cobalt, lithium, nickel, and copper. Therefore, we also attempt to recover cobalt ions (Co2+) from solutions by using CuxNiyHCF nanoparticle films. The feasibility of electrochemical adsorption of cobalt ions onto hybrid copper–nickel hexacyanoferrate films is discussed, as well as the adsorption isotherms and kinetics properties.
2 Experimental
2.1 Materials and apparatus
All reagents, such as Cu(NO3)2·xH2O, Ni(NO3)2·6H2O, Co(NO3)2·6H2O, LiNO3, K3[Fe(CN)6], K3[Fe(CN)6]·3H2O and KNO3, used in the operations were of analytical grade or better without further purification. Ultrapure water (18 MΩ cm) was used throughout all the operations. All the experiments were carried out at room temperature.Ion concentrations were determined by inductively coupled plasma mass spectrometry (ICP-MS, Thermo Fisher Scientific iCAP Q, America). The surface morphologies of CuxNiyHCF films were observed by a field-emission scanning electron microscope (FE-SEM, Hitachi SU-8010, Japan). Their structures and compositions were characterized by X-ray powder diffractometry (XRD, Rigaku SmartLab, Japan) and Fourier transform infrared spectroscopy (FTIR, Bruker Vertex 70, Germany). Cyclic voltammetry of CuxNiyHCF electrode films was performed by using an electrochemical analyzer (Chen Hua CHI660e, China).
2.2 Preparation of modified electrodes
As described in our previous studies,25 h-MHCF nanoparticle inks with different metal molar ratios were prepared by wet chemical coprecipitation. Firstly, mixed solutions with different molar ratios (x:y = 1:4, 1:2, 1:1, 2:1, and 4:1) were obtained from Cu(NO3)2·xH2O and Ni(NO3)2·6H2O, to which a solution of K3[Fe(CN)6] was added; the reactant mole ratio of total metal nitrates to K3[Fe(CN)6] was kept at 3:2. Secondly, one group of parallel samples were dried in an oven at 60 °C to obtain powder samples for subsequent chemical analysis and characterizations. The remaining samples were surface-treated by K4[Fe(CN)6]·3H2O to dissolve well in water. Thirdly, the 5% (ratio of mass to volume) ink-like solutions of nanoparticles were prepared and shaken for one week (25 °C, 150 rpm). Indium tin oxide (ITO) was washed with acetone as a support substance, and CuxNiyHCF nanoparticle inks were coated on it using a KW-4A spin-coater instrument (2000 rpm for 10 s followed by 2500 rpm for another 10 s). Finally, the modified electrodes were dried in an oven at 120 °C for 2 h.
2.3 Characterization
In order to investigate the composition ratios, the prepared CuxNiyHCF powders were decomposed in aqua regia by using microwave digestion. After microwave digestion, dilution and filtration, the elements were determined by ICP-MS. The obtained x:y ratio is linearly fitted to the theoretical values in the synthesis process. The fitness (R2) is given by eqn (1):
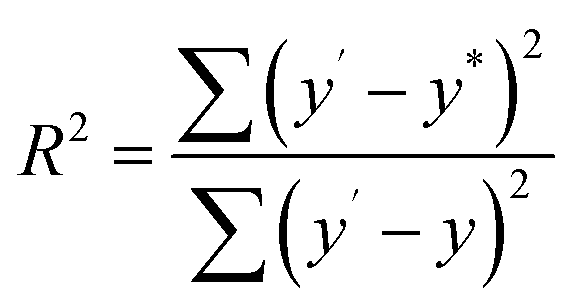 | (1) |
:y is converted to the percentage of Cu (WCu) in the nanoparticle films, with the following expression:
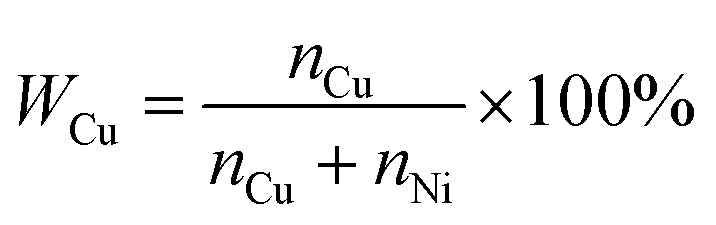 | (2) |
A three-electrode system was used with a saturated calomel electrode (SCE, Hg/Hg2Cl2/KCl) as the reference electrode, a Pt wire as the counter electrode and the ITO plate loaded with nanoparticle films as the working electrode. The cyclic voltammograms (CVs) of CuxNiyHCF films in the different cation solutions were obtained, using a CHI660e electrochemical analyzer. The electrochemical properties of CuxNiyHCF nanoparticle films were characterized by CV, formal potential (Ef), accumulated quantity of electric charge (Q) and surface coverage of redox-active sites (Γ). Ef is calculated as half of the sum of potentials of the anodic (Epa) and cathodic (Epc) current peak. Q is the charge obtained by integrating the cathodic peak after subtraction of the background. The calculation equation of Γ is as follows:
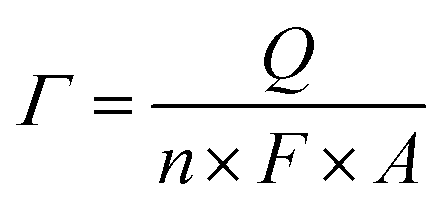 | (3) |
The stability of the CuxNiyHCF films was investigated by long-term potential cycling (500 cycles), expressed by the rate of decrease (Rd) of the integrated Faraday charge Q with the increase in the number of cycles.
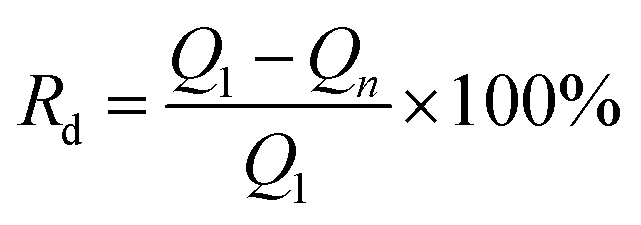 | (4) |
2.4 Electrochemical adsorption of Co2+
| K4[FeII(CN)6] → 4K+ + [FeIII(CN)6]3− + e− | (5) |
Afterwards, the films were washed with ultrapure water and transferred to 1 mg L−1 Co2+ solution to adsorb Co2+ by applying a reduction potential of −0.4 V (vs. SCE).
| M3[FeIII(CN)6]2 + 2e− + Co2+ → CoM3[FeII(CN)6]2 | (6) |
Contrast tests were conducted at the same time without applying potentials.
The results of electrochemical adsorption were given as removal efficiency (R), which was calculated according to eqn (7):
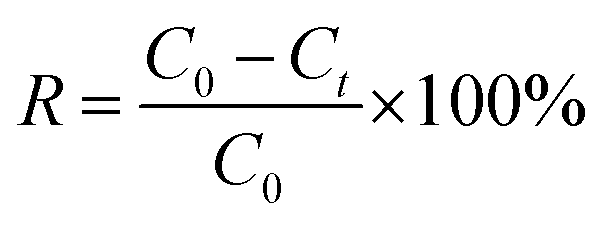 | (7) |
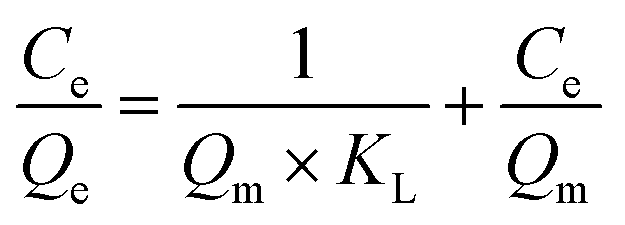 | (8) |
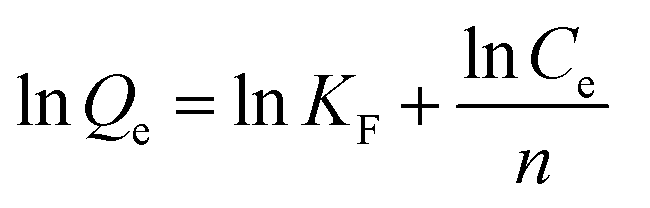 | (9) |
Here, Ce (mg L−1) is the equilibrium concentration; Qe (mg cm−2) and Qm (mg cm−2) are the saturation adsorption capacity and the maximum adsorption capacity at the pseudo-equilibrium, respectively. KL, KF and n (dimensionless) are the adsorption isotherm constants for the Langmuir isotherm and the Freundlich isotherm.
The intraparticle diffusion model, pseudo-first-order and pseudo-second-order adsorption kinetic models were used to analyze the dynamic adsorption process, as represented by eqn (10)–(12), respectively.
| Qt = kp,i × t0.5 + Ci | (10) |
|
ln(Qe − Qt) = lnQe − k1 × t
| (11) |
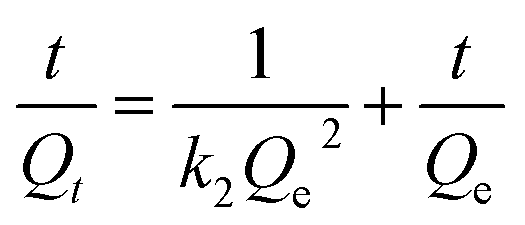 | (12) |
3 Results and discussion
3.1 Characterizations of CuxNiyHCF nanoparticle films
| x:y |
4:1 |
2:1 |
1:1 |
1:2 |
1:4 |
|---|---|---|---|---|---|
| Cu | 0.6282 | 0.5263 | 0.3860 | 0.2562 | 0.0792 |
| Ni | 0.1704 | 0.2839 | 0.4130 | 0.6022 | 0.3726 |
The fitness of the theoretical and actual values is 0.9840, showing that the two sets of values are very close. In other words, unlike electrodeposition,33,34 the x:y ratios in the CuxNiyHCF nanoparticle films can be controlled by the preparation method. This is because hexacyanoferrate (II) ions are generated firstly in the vicinity of the electrode surface and then react primarily with one kind of metal ion in the electrolyte27 when prepared by electrodeposition. For coprecipitation, metal ions and hexacyanoferrate (III) ions have been mixed and reacted evenly during the preparation stage of the ink. The results mean that CuxNiyHCF films with appropriate metal ratios can be synthesized as required.
 | ||
| Fig. 1 FE-SEM images of CuxNiyHCF nanoparticle films with different metal ratios (M1:M2). | ||
:y ratio on the properties of CuxNiyHCFs, CuxNiyHCF powders were characterized by XRD and FTIR. As shown in Fig. 2, all CuxNiyHCFs have four predominant 2θ peaks in the range of 17–40° corresponding to the (2 0 0), (2 2 0), (4 0 0) and (4 2 0) diffraction planes of a face-centered-cubic lattice structure, which are very consistent with the ref. 18, 36 and 37. 2θ values obtained for h-MHCFs fall between the NiHCF and CuHCF peak positions. With an increasing content of copper, the diffraction peak position of CuxNiyHCFs shows a slight shift from NiHCF to CuHCF.
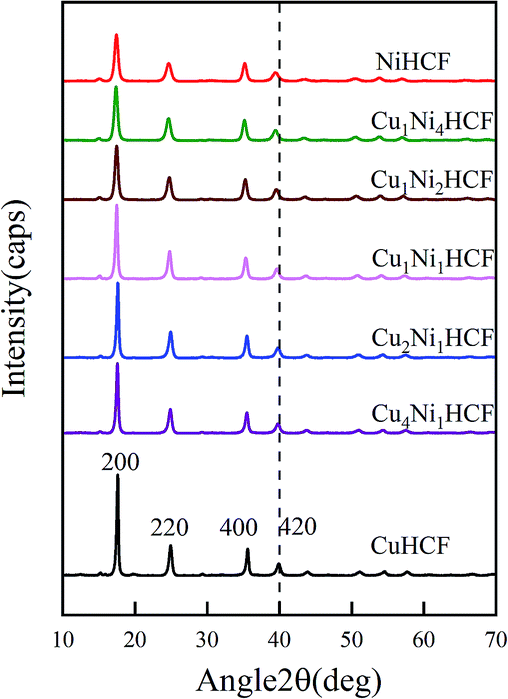 | ||
| Fig. 2 XRD patterns of CuxNiyHCF powders. | ||
The crystallite size (D) can be deduced from the diffraction peak according to Sherrer's formula:
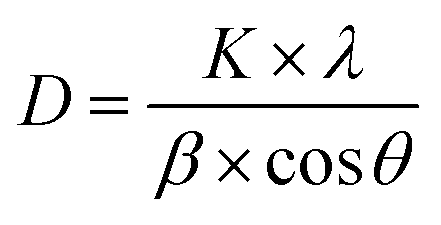 | (13) |
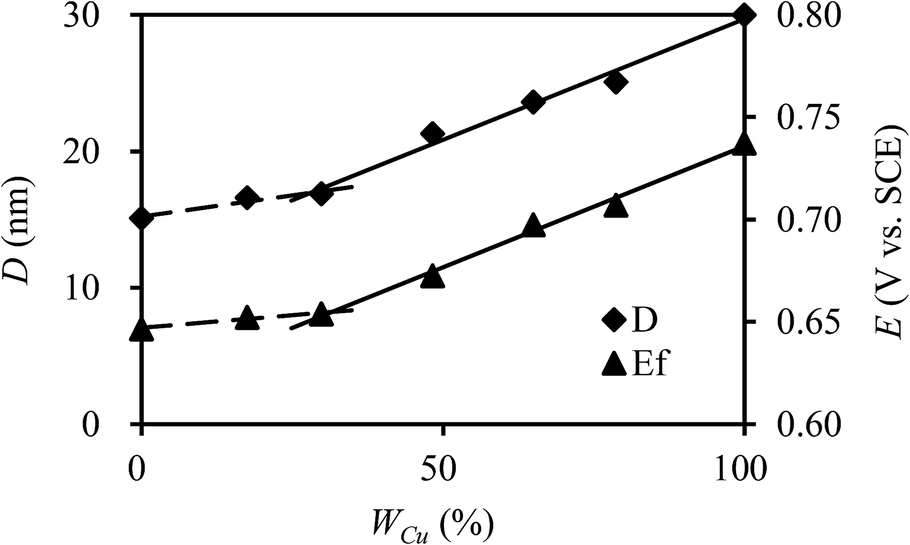 | ||
| Fig. 3 Crystallite size (D) and formal potential (Ef) of CuxNiyHCFs versus molar content of Cu (WCu). | ||
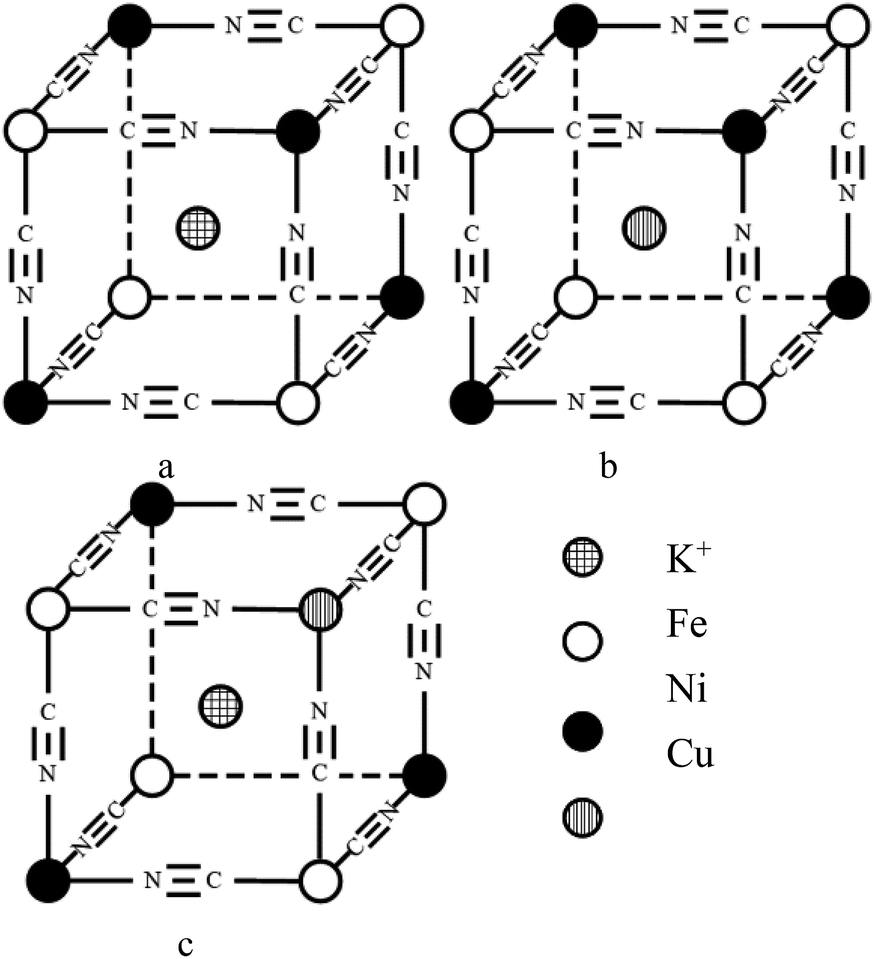 | ||
| Fig. 4 Schematic of 1/8 cell structure of MHCF (M: Cu, Ni). | ||
Fig. 5 illustrates the FTIR spectra of CuxNiyHCF powders. In particular, there is a small shoulder at 2165.21 cm−1 of NiHCF, while the other samples show only one major peak in the vicinity of 2096 cm−1. The other bands of CuxNiyHCFs are very close to each other, owing to the similar absorption peaks of NiHCF and CuHCF. The broad band at around 3330 cm−1 refers to the O–H stretching vibrations.38 The vibrational band near 2096 cm−1 corresponds to the stretching vibration absorption band of the –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N group.39,40 The bands in the region of 1400–1680 cm−1 are related to the asymmetric and symmetric stretching vibrations of COO− groups.41,42 The stretching vibration absorption band of the free –CN is at about 2060 cm−1 in aqueous solution;43 the blue shift of the absorption band observed together with the two smaller peaks at about 595 cm−1 and 491 cm−1 are attributed to the formation of Fe–CN–M.39,44
N group.39,40 The bands in the region of 1400–1680 cm−1 are related to the asymmetric and symmetric stretching vibrations of COO− groups.41,42 The stretching vibration absorption band of the free –CN is at about 2060 cm−1 in aqueous solution;43 the blue shift of the absorption band observed together with the two smaller peaks at about 595 cm−1 and 491 cm−1 are attributed to the formation of Fe–CN–M.39,44
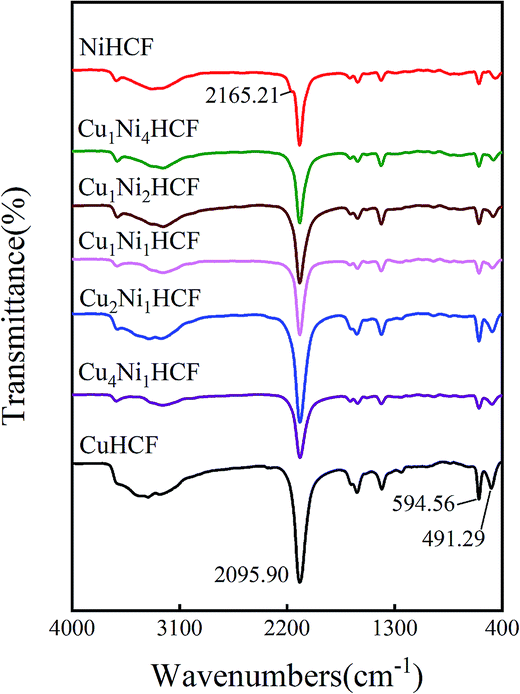 | ||
| Fig. 5 FITR spectrum of CuxNiyHCF powders. | ||
:y, which is consistent with the change in structure from XRD and FTIR results. As indicated in a previous study,47 CuxNiyHCF is a new phase rather than a simple mixture of CuHCF and NiHCF. The typical CV response of each CuxNiyHCF is attributed to its respective unique structure. In particular, two sets of redox peaks of Cu1Ni4HCF and Cu1Ni2HCF are similar to NiHCF, and, the redox peaks at 0.45 V are severely weakened, which might be because of their special structures, as illustrated in Fig. 4b. The Ef values of CuxNiyHCF films were plotted as a function of WCu, as approximated in Fig. 3. At a low molar fractions of Cu, the Ef value hardly changes with the addition of Cu, then depends linearly on the content of copper when WCu rises to about 33%. These observations confirm our previous hypothesis that Cu exists in two forms: as countercations substituted for K+, under which condition the Ef is close to that of NiHCF,48 or occupying lattice positions instead of Ni. This is similar to the electrodeposited films studied by Kulesza et al.27 It can be found from Fig. 6a that the redox peaks at more negative potential disappear when WCu > 30%, which also demonstrates that Cu started to occupy positions of the lattice.
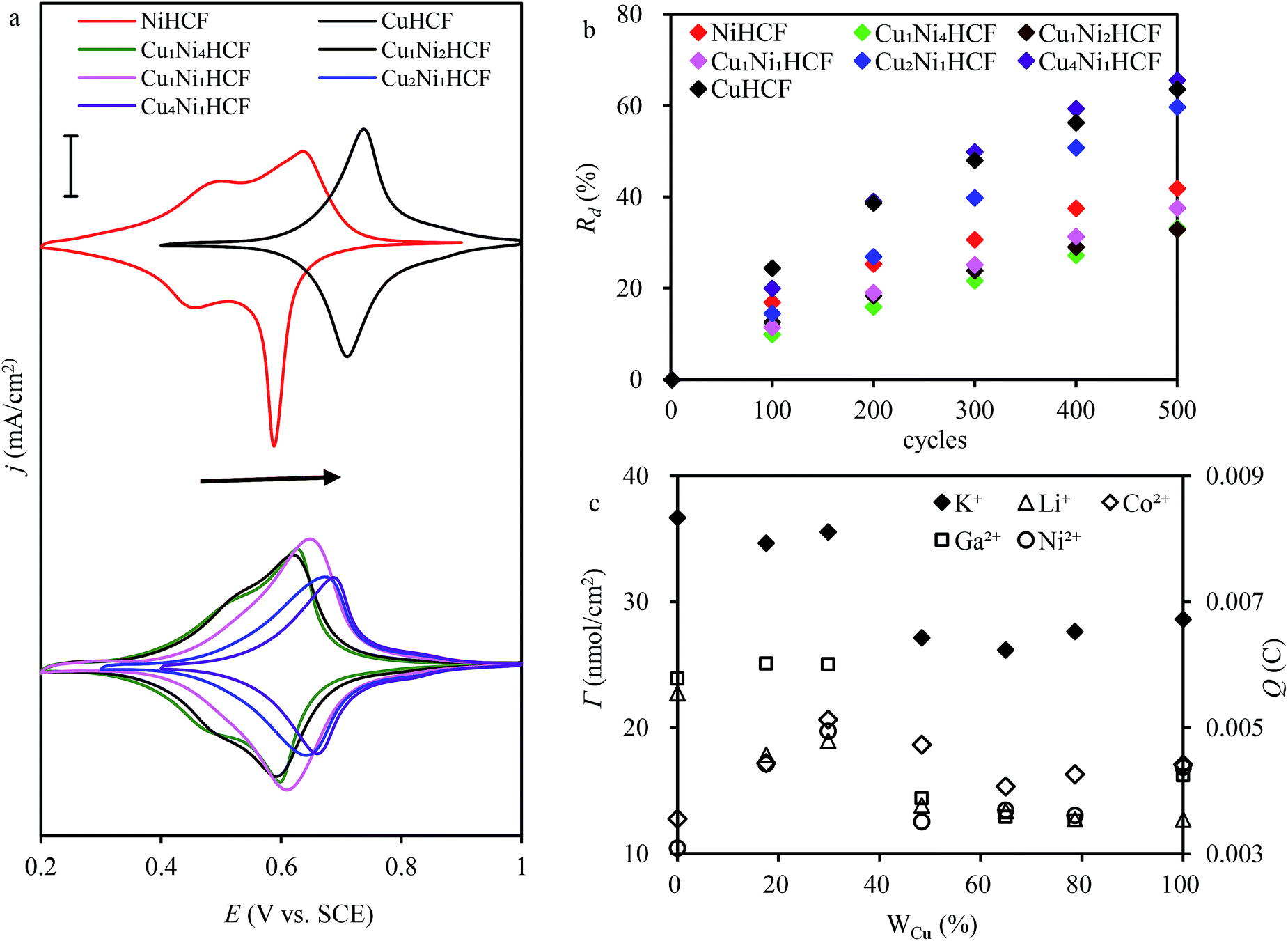 | ||
| Fig. 6 (a) Cyclic voltammograms of CuxNiyHCF modified electrodes (scan rate: 0.005 V s−1; electrode area: 15 mm × 12.5 mm); (b) rate of decrease (Rd) of the cathodic integrated charge vs. the number of sweeping cycles. (Scan rate: 0.05 V s−1; electrode area: 15 mm × 12.5 mm); (c) surface coverage and integrated Faraday charge in different electrolytes (c(K+) = 0.5 M, c(Li+) = 0.5 M, c(Co2+) = 0.5 M, c(Ga2+) = 0.1 M, c(Ni2+) = 0.5 M. Scan rate: 0.05 V s−1; electrode area: 15 mm × 12.5 mm). | ||
The ratio value of cathodic current to anodic peak (ipc/ipa) approaches one, implying the reversibility of the electrochemical redox reaction. The existence of Cu greatly improved the property of NiHCF films in this respect. The long-term CV of CuxNiyHCF films was carried out in 0.5 M K+ at a scan rate of 0.05 V s−1 to study the stability of CuxNiyHCFs. Rd values were plotted against the cycle numbers, as depicted in Fig. 6b. NiHCF films show comparatively high stability compared with CuHCF films, which is in accord with the materials synthesized previously from chlorides.25 The stability of CuxNiyHCF films increases with a decrease in x:y values in the order: Cu4Ni1HCF < Cu2Ni1HCF < Cu1Ni1HCF < Cu1Ni2HCF < Cu1Ni4HCF. It is noteworthy that the cycling stabilities of Cu1Ni4HCF, Cu1Ni2HCF and Cu1Ni1HCF films are notably better than that of NiHCF films, indicating that the stability of CuxNiyHCF films will be enhanced on the basis of NiHCF films when the copper content of copper is lower than or even equal to the nickel content. In contrast, the cycling stability will get worse when the proportion of copper is higher.
The results of the surface coverage Γ calculated from the integrated charge in 0.5 M K+ solution are shown in Fig. 6c. The Γ value of NiHCF films is apparently higher than that of CuHCF films. The Γ values of Cu1Ni4HCF films and Cu1Ni2HCF films are 1.2–1.4 times as much as the Γ values of other CuxNiyHCFs.
3.2 Electrochemical adsorption of cations
The fine CV responses imply the possibility of electrochemical adsorption of metal cations. In the electrochemical adsorption experiment, Co2+ were effectively removed by CuxNiyHCFs. That is why Co2+ was chosen to carry out the experiment on the electrochemical adsorption behavior of CuxNiyHCF films. The Cu1Ni2HCF films were used for comparison with CuHCF films and NiHCF films since they have excellent electrochemical performances. In order to study information about the mechanism, CV and electrochemical impedance spectroscopy (EIS) experiments were conducted. The CVs of CuxNiyHCF modified electrodes at various sweep rates from 0.005 to 0.15 V s−1 in 0.5 M Co2+ solutions are shown in Fig. 7a–c. The logarithmic cathodic peak current increases linearly with an increase in the logarithmic scan rate (shown as insets). The slopes of the CuxNiyHCF films are 0.75–0.88 (listed in Table S1†), indicating that the reaction at the thin films is controlled by an ion diffusion process and a surface electron transfer process, with a tendency towards the latter.49 The ΔE gradually increases with the variation in the scan rate, indicating a limitation arising from the charge-transfer kinetics50 which weakened the reversibility.51
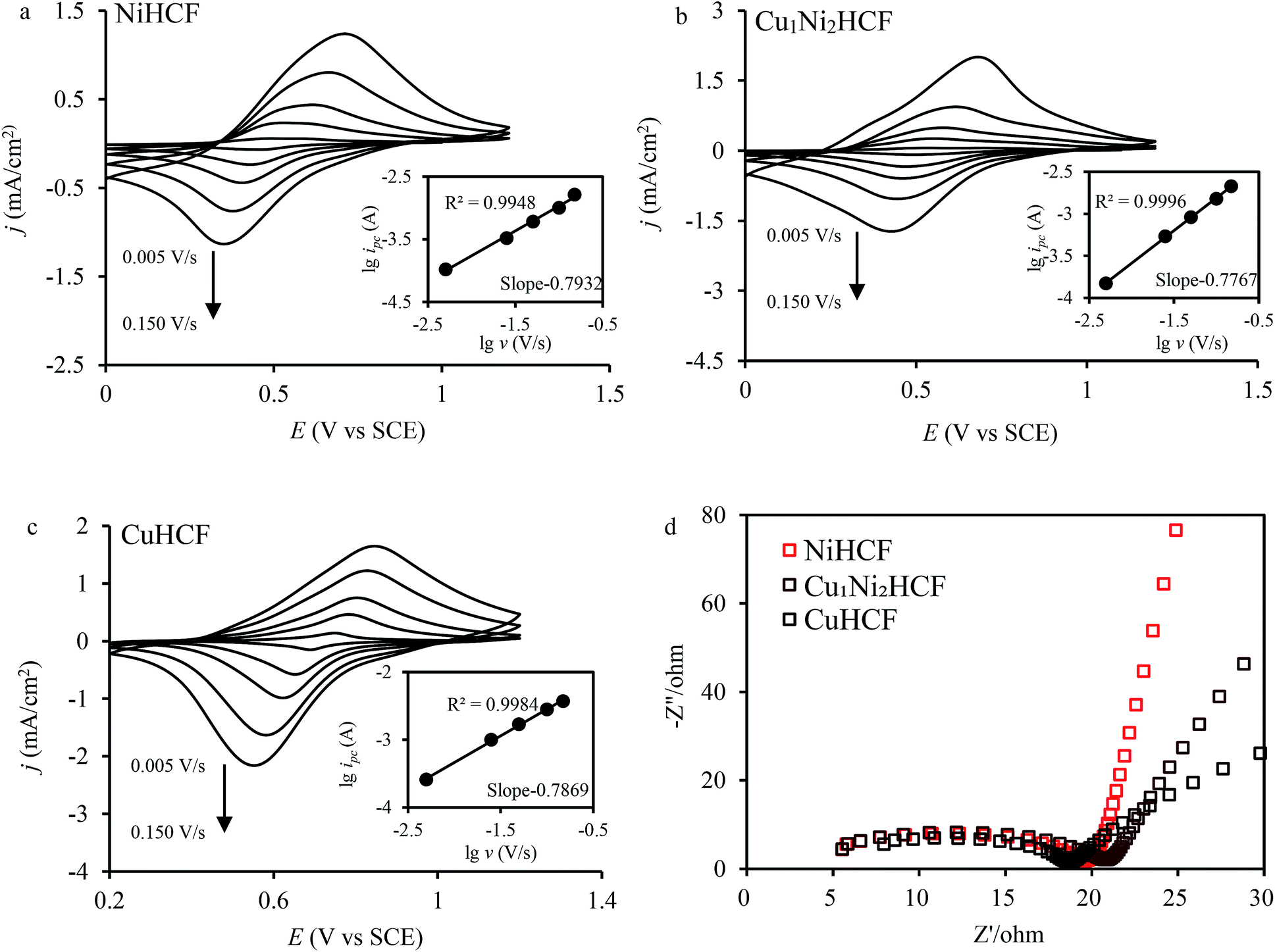 | ||
| Fig. 7 (a, b and c) CV of CuxNiyHCF films in 0.5 M Co2+ solutions at different scan rates (v = 0.005, 0.025, 0.05, 0.1, 0.15 V s−1); insets show the logarithmic values of reduction peak current (ipc) as a function of the logarithmic values of scan rate; (d) Nyquist plots of CuxNiyHCF films from 100 Hz to 1000 kHz. | ||
The Nyquist plots (Fig. 7d) are composed of a distorted semicircle in the high-frequency region assigned to the charge-transfer reaction of the electrode and a linear part at the low-frequency end due to a diffusion-controlled process.29,52 The CuHCF film is characterized by the lowest diameter of the semicircle, standing for the quickest electron transfer rate.53 The charge transfer resistance decreases in the order: Cu1Ni2HCF > NiHCF > CuHCF.
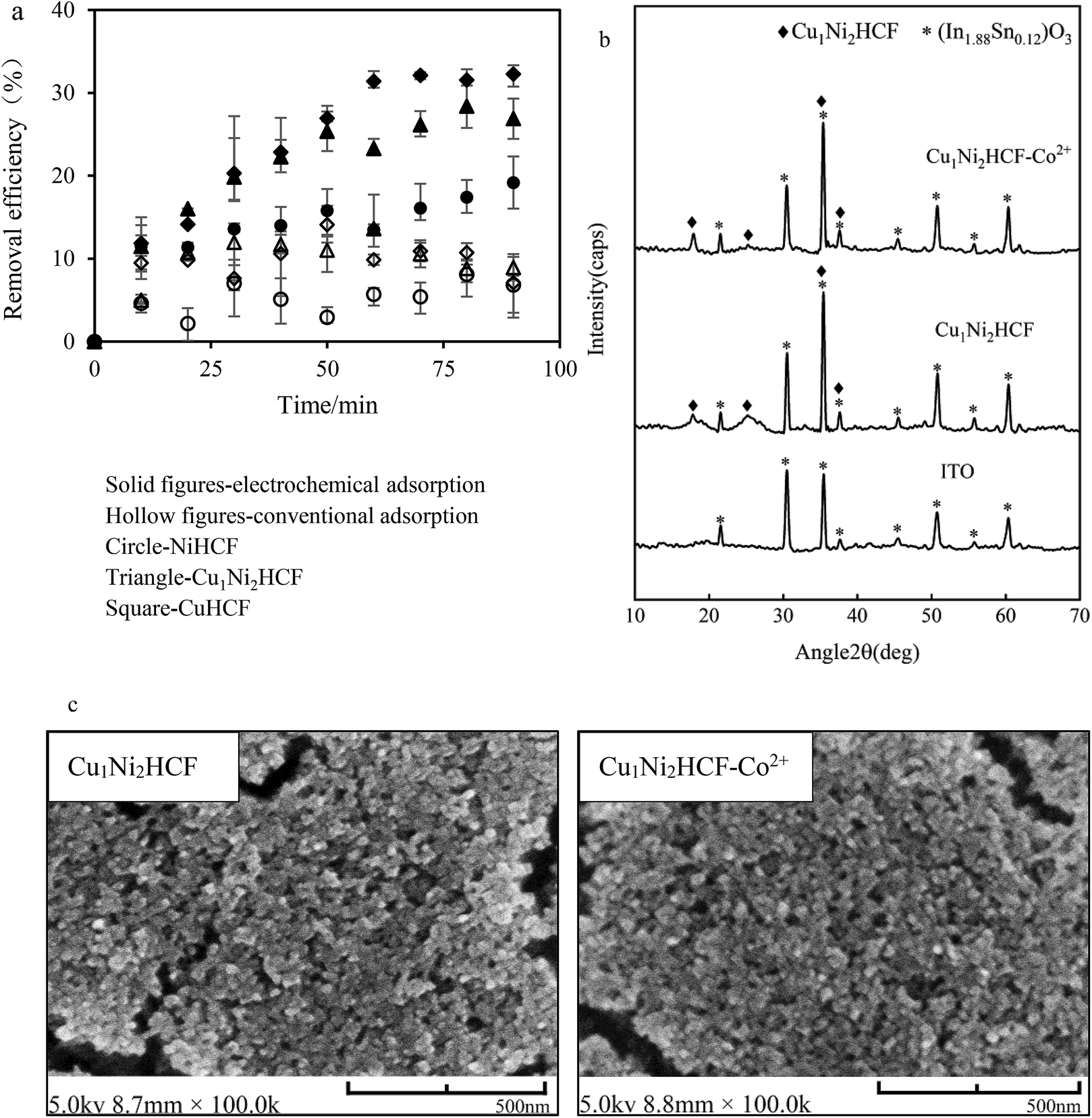 | ||
| Fig. 8 (a) Removal efficiency of Co2+ onto CuxNiyHCF films vs. contact time (Co2+ concentration: 1 mg L−1, electrode area: 15 mm × 25 mm); (b) XRD patterns and (c) SEM images of Cu1Ni2HCF film before and after adsorption (Cu1Ni2HCF-Co2+). | ||
The electrochemical adsorption capacity of CuHCF films is much higher than that of NiHCF films, verifying the results of integrated charge and EIS. The high adsorption capacity can be interpreted by the large lattice size of CuHCF. This is consistent with the fact that the sorption capacity of CuHCF is larger than that of NiHCF, as mentioned by Thierry et al.54 It is remarkable that the equilibrium removal efficiency of Cu1Ni2HCF films is higher than that of NiHCF films, and slightly lower than that of CuHCF films, which may due to the well-ordered porous structures of CuHCF films. The redox peaks of CuHCF film in Co2+ solutions are sharper than those of Cu1Ni2HCF film, indicating a faster dynamic process. The cycling stability of Cu1Ni2HCF film is obviously better than that of CuHCF film, when the equilibrium removal efficiency of Cu1Ni2HCF film is close to that of CuHCF film. That is why we chose Cu1Ni2HCF for the next step.
The crystal structure and surface morphology of Cu1Ni2HCF film before and after removing Co2+ were investigated by XRD and SEM; the results are depicted in Fig. 8b and c. Compared with the peaks of (In1.88Sn0.12)O3 (PDF # 89-4598) coated on substrates, the new peaks generated at around 17.95° and 25.35° and the sharper peaks at 35.48° and 37.65° are caused by Cu1Ni2HCF film. There is no significant change in the results of SEM and XRD after Co2+-loading, indicating that the structure of the nanocrystals had not been changed by the adsorption process. The concentrations of Cu2+ and Ni2+ before and after pretreating the films were determined by ICP-MS (Fig. S2†). The results show that there is no significant release of Cu2+ or Ni2+ from the films, indicating that the prepared nanoparticle films were stable as well. These phenomena are consistent with the expected cation insertion process, during which the essential feature of crystal structure is preserved. This may also due to the dilute solution and trace amount of Co2+ adsorbed on Cu1Ni2HCF. The same conclusions can be drawn from NiHCF and CuHCF films (as shown in Fig. S1†).
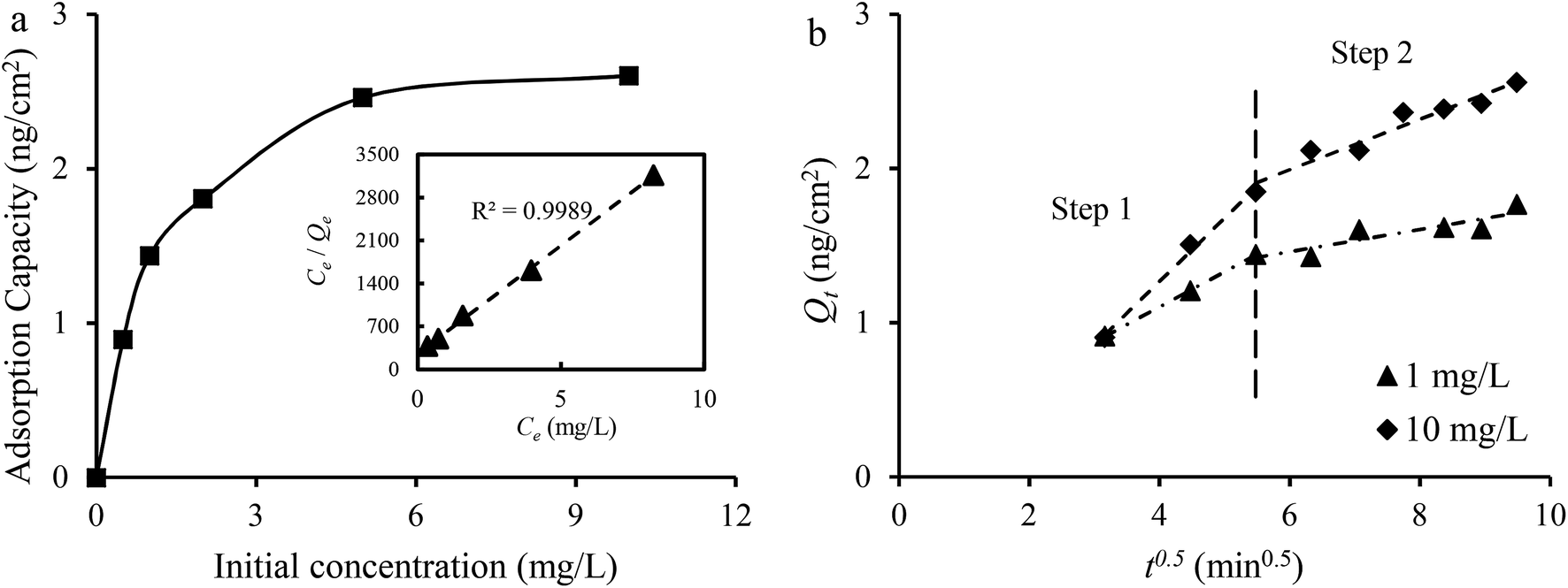 | ||
| Fig. 9 (a) The effect of initial concentration on the adsorption capacity. The inset is the fitted result with the Langmuir isotherm model; (b) adsorption kinetic data in 1 mg L−1 and 10 1 mg L−1 Co2+ solutions fitted with the intraparticle diffusion model. | ||
The Langmuir isotherm model and the Freundlich isotherm model were used to fit the experimental data. The Langmuir isotherm model assumes that only one monolayer of adsorbate molecule can be adsorbed on homogeneous sites of the adsorbent and the adsorbates do not react with each other. The Freundlich isotherm model is based on multilayer adsorption on a heterogeneous surface. The calculation results of the Qe, constants and correlation coefficient (R2) are listed in Table 2. The linear correlation coefficient (R2) from the Langmuir isotherm model is 0.9989, which is higher than that calculated from the Freundlich isotherm model, and the value of Qm from the Langmuir isotherm model is close to the practical Qe obtained from experiment. It can be concluded that the Langmuir isotherm model can describe the adsorption process better than the Freundlich isotherm model, which is in agreement with the redox reaction. The results from the data in 1 mg L−1 Co2+ solutions fitted to the intraparticle diffusion, pseudo-first-order and pseudo-second-order kinetic models are given in Table 2. The sorption process involves bulk diffusion, film diffusion and intraparticle diffusion (or pore diffusion).35 Fig. 8b shows that two linear regions exist in the intraparticle diffusion plot. The adsorption system was stirred at a speed of 500 rpm. Thus, the two curves may indicate that the adsorption of Co2+ onto Cu1Ni2HCF film occurred via two phases:57 film diffusion in the first linear part followed by intraparticle diffusion during the second linear portion. The linear portions of the two stages do not pass through the origin, and, kp,1 is much greater than kp,2, which suggests that both film and intraparticle diffusion processes are the probable rate-controlling steps.58 The constant C1 increased with an increase in ion concentration, indicating the growth of the thickness of the boundary layer. The probability of external mass transfer was reduced, and the probability of internal mass transfer increased conversely.59
| Isotherm models | Parameters | Kinetic models | Constants | Initial concentration (mg L−1) | ||
|---|---|---|---|---|---|---|
| 1 | 10 | |||||
| Langmuir | Qm (mg cm−2) | 0.0029 | Pseudo-first-order | Qe (mg cm−2) | 0.0017 | 0.0062 |
| KL | 1.3263 | k1 | 0.0659 | 0.0044 | ||
| R2 | 0.9989 | R2 | 0.9705 | 0.9083 | ||
| Freundlich | n | 3.0386 | Pseudo-second-order | Qe (mg cm−2) | 0.0019 | 0.0079 |
| KF | 0.0015 | k2 | 45.0086 | 4.8711 | ||
| R2 | 0.9370 | R2 | 0.9762 | 0.9730 | ||
| Intraparticle diffusion | kp,1 | 0.0002 | 0.0004 | |||
| C1 | 0.0002 | −0.0004 | ||||
| R2 | 0.9998 | 0.9932 | ||||
| kp,2 | 7 × 10−5 | 0.0002 | ||||
| C2 | 0.0010 | 0.0010 | ||||
| R2 | 0.8084 | 0.9398 | ||||
Both the pseudo-first-order kinetic model and the pseudo-second-order kinetic model show a good fit to the experimental data with well-matching correlation coefficients at a low initial concentration. At a higher initial concentration, the correlation coefficient (R2) obtained from the pseudo-second-order kinetic model is higher than that of the pseudo-first-order kinetic model, suggesting that the pseudo-second-order model is more suitable for describing the adsorption process. This may predict that electrochemical adsorption involved a complicated process, and chemical adsorption might be the rate-limiting step.60
4 Conclusions
In summary, the proportion of metals in CuxNiyHCF nanoparticle films can be controlled by the reactant ratio during the preparation process. With an increase in the Cu ratio, the crystallite size and Ef of CuxNiyHCF films varied from NiHCF films to CuHCF films. The results of SEM and XRD, together with the shape of the CV curves supported Cu2+ being inserted into the NiHCF lattice as countercations to maintain the electrical neutrality of the structure when the proportion of copper is less than 33%; at higher molar fractions of Cu, Ni was partially replaced by Cu. Cu1Ni2HCF showed superior electrochemical properties of stability and current responses. The electrochemical adsorption behavior of Co2+ followed the Langmuir isotherm model and the pseudo-second order model. Our study reveals the potential of CuxNiyHCF nanoparticle films as a promising technology for the recovery of Co2+ from spent LIBs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge the research staffs in University of Chinese Academy of Sciences to provide technical assistance on FE-SEM, XRD and FTIR test and analysis. This work was supported by National Natural Science Foundation of China (Contract No. 21806166), Beijing Natural Science Foundation (No. 2184128), and Key Program of Chinese Academy of Sciences (No. KFZD-SW-315).References
- X. Yao, R. Zhao, L. Chen, J. Du, C. Tao, F. Yang and L. Dong, Appl. Catal., B, 2017, 208, 82–93 CrossRef CAS.
- A. Paolella, C. Faure, V. Timoshevskii, S. Marras, G. Bertoni, A. Guerfi, A. Vijh, M. Armand and K. Zaghib, J. Mater. Chem. A, 2017, 5, 18919–18932 RSC.
- S. Zhang, P. Kang, S. Ubnoske, M. K. Brennaman, N. Song, R. L. House, J. T. Glass and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 7845–7848 CrossRef CAS PubMed.
- F. Yu, F. Li, B. Zhang, H. Li and L. Sun, ACS Catal., 2015, 5, 627–630 CrossRef CAS.
- A. Farhangfar, IEEE Sens. J., 2018, 18, 925–932 CAS.
- M. L. M. Firmino, S. Morais, A. N. Correia, P. de Lima-Neto, F. A. O. Carvalho, S. S. L. Castro and T. M. B. F. Oliveira, Chem. Eng. J., 2017, 323, 47–55 CrossRef CAS.
- X. Zhang, J. Jiang, Y. Chen, K. Cheng, F. Yang, J. Yan, K. Zhu, K. Ye, G. Wang, L. Zhou and D. Cao, Chem. Eng. J., 2018, 335, 321–329 CrossRef CAS.
- X. Bie, K. Kubota, T. Hosaka, K. Chihara and S. Komaba, J. Power Sources, 2018, 378, 322–330 CrossRef CAS.
- U. Shanker, V. Jassal and M. Rani, J. Environ. Chem. Eng., 2017, 5, 4108–4120 CrossRef CAS.
- L. Z. Cai, Q. S. Chen, C. J. Zhang, P. X. Li, M. S. Wang and G. C. Guo, J. Am. Chem. Soc., 2015, 137, 10882–10885 CrossRef CAS PubMed.
- V. Svitkova, J. Blaskovicova, M. Tekelova, B. M. Kallai, T. Ignat, V. Horackova, P. Skladal, P. Kopel, V. Adam, D. Farkasova and J. Labuda, Sens. Actuators, B, 2017, 243, 435–444 CrossRef CAS.
- M. Baghayeri, R. Ansari, M. Nodehi, I. Razavipanah and H. Veisi, Microchim. Acta, 2018, 185 Search PubMed.
- R. Z. Chen, H. Tanaka, T. Kawamoto, M. Asai, C. Fukushima, M. Watanabe, M. Kurihara, M. Arisaka and T. Nankawa, Procedia Eng., 2012, 44, 1728–1730 CrossRef.
- Z. Jia, X. Cheng, Y. Guo and L. Tu, Chem. Eng. J., 2017, 325, 513–520 CrossRef CAS.
- I. Zunke, D. Kloss, A. Heft, J. Schmidt and B. Gruenler, Surf. Coat. Technol., 2016, 289, 186–193 CrossRef CAS.
- P. Hosseini, G. Wittstock and I. Brand, J. Electroanal. Chem., 2018, 812, 199–206 CrossRef CAS.
- W.-R. Cai, G.-Y. Zhang, T. Song, X.-J. Zhang and D. Shan, Electrochim. Acta, 2016, 198, 32–39 CrossRef CAS.
- A. Senthil Kumar, P. Barathi and K. Chandrasekara Pillai, J. Electroanal. Chem., 2011, 654, 85–95 CrossRef CAS.
- K. M. Rachna, M. Rani and U. Shanker, Chem. Eng. J., 2018, 348, 754–764 CrossRef.
- A. V. N. Kumar and J. Joseph, J. Phys. Chem. C, 2015, 119, 296–304 CrossRef CAS.
- P. Xu, G. Wang, H. Wang, Y. Li, C. Miao, J. Qu, Y. Zhang, F. Ren, K. Cheng, K. Ye, K. Zhu, D. Cao and X. Zhang, Chem. Eng. J., 2017, 328, 834–843 CrossRef CAS.
- S. Ghasemi, R. Ojani and S. Ausi, Int. J. Hydrogen Energy, 2014, 39, 14918–14926 CrossRef CAS.
- W. F. Li, F. Zhang, X. D. Xiang and X. C. Zhang, ChemElectroChem, 2018, 5, 350–354 CrossRef CAS.
- P. C. Pandey and A. K. Pandey, Analyst, 2013, 138, 2295–2301 RSC.
- J. Wang, R. Chen, X. Long and Z. Li, J. Electroanal. Chem., 2018, 810, 191–198 CrossRef CAS.
- M.-S. Wu, L.-J. Lyu and J.-H. Syu, J. Power Sources, 2015, 297, 75–82 CrossRef CAS.
- P. J. Kulesza, M. A. Malik, R. Schmidt, A. Smolinska, K. Miecznikowski, S. Zamponi, A. Czerwinski, M. Berrettoni and R. Marassi, J. Electroanal. Chem., 2000, 487, 57–65 CrossRef CAS.
- S. Zamponi, M. Berrettoni, P. J. Kulesza, K. Miecznikowski, M. A. Malik, O. Makowski and R. Marassi, Electrochim. Acta, 2003, 48, 4261–4269 CrossRef CAS.
- W. Li, F. Zhang, X. Xiang and X. Zhang, ChemElectroChem, 2017, 4, 2870–2876 CrossRef CAS.
- M. Giorgetti, L. Guadagnini, D. Tonelli, M. Minicucci and G. Aquilanti, Phys. Chem. Chem. Phys., 2012, 14, 5527–5537 RSC.
- R. Chen, H. Tanaka, T. Kawamoto, M. Asai, C. Fukushima, M. Kurihara, M. Watanabe, M. Arisaka and T. Nankawa, Electrochem. Commun., 2012, 25, 23–25 CrossRef CAS.
- A. Abbaspour and M. A. Kamyabi, J. Electroanal. Chem., 2005, 576, 73–83 CrossRef CAS.
- X. Cui, L. Hong and X. Lin, J. Electroanal. Chem., 2002, 526, 115–124 CrossRef CAS.
- L. Guadagnini, M. Giorgetti, F. Tarterini and D. Tonelli, Electroanalysis, 2010, 22, 1695–1701 CrossRef CAS.
- R. Chen, H. Tanaka, T. Kawamoto, M. Asai, C. Fukushima, H. Na, M. Kurihara, M. Watanabe, M. Arisaka and T. Nankawa, Electrochim. Acta, 2013, 87, 119–125 CrossRef CAS.
- S. Q. Chang, L. Chang, W. Han, Z. Li, Y. D. Dai and H. Q. Zhang, J. Radioanal. Nucl. Chem., 2018, 316, 209–219 CrossRef CAS.
- J. Y. Liao, Q. Hu, B. K. Zou, J. X. Xiang and C. H. Chen, Electrochim. Acta, 2016, 220, 114–121 CrossRef CAS.
- S. J. Gerber and E. Erasmus, Mater. Chem. Phys., 2018, 203, 73–81 CrossRef CAS.
- Y. Zong, Y. Zhang, X. Lin, D. Ye, X. Luo and J. Wang, J. Radioanal. Nucl. Chem., 2017, 311, 1577–1591 CrossRef CAS.
- S. Manakasettharn, A. Takahashi, T. Kawamoto, K. Noda, Y. Sugiyama and T. Nakamura, Anal. Chem., 2018, 90, 4856–4862 CrossRef CAS PubMed.
- S.-C. Jang, S.-M. Kang, Y. Haldorai, K. Giribabu, G.-W. Lee, Y.-C. Lee, M. S. Hyun, Y.-K. Han, C. Roh and Y. S. Huh, Sci. Rep., 2016, 6, 7845–7848 Search PubMed.
- S. Naeimi and H. Faghihian, Sep. Purif. Technol., 2017, 175, 255–265 CrossRef CAS.
- C. W. Ng, J. Ding, Y. Shi and L. M. Gan, J. Phys. Chem. Solids, 2001, 62, 767–775 CrossRef CAS.
- M. Hu, S. Furukawa, R. Ohtani, H. Sukegawa, Y. Nemoto, J. Reboul, S. Kitagawa and Y. Yamauchi, Angew. Chem., Int. Ed., 2012, 51, 984–988 CrossRef CAS PubMed.
- G. Kalaiyarasan, K. Aswathi and J. Joseph, Int. J. Hydrogen Energy, 2017, 42, 22866–22876 CrossRef CAS.
- M. Ciabocco, M. Berrettoni, S. Zamponi, R. Spinosi and P. Conti, Int. J. Electrochem. Sci., 2018, 13, 5535–5551 CrossRef CAS.
- J. X. Wang, R. Z. Chen, X. X. Long and Z. J. Li, J. Electroanal. Chem., 2018, 810, 191–198 CrossRef CAS.
- J. Kukulka-Walkiewicz, J. Stroka, M. A. Malik, P. J. Kulesza and Z. Galus, Electrochim. Acta, 2001, 46, 4057–4063 CrossRef CAS.
- J. C. Kemmegne-Mbouguen, L. Angnes, E. Mouafo-Tchinda and E. Ngameni, Electroanalysis, 2015, 27, 2387–2398 CrossRef CAS.
- F. Jalali and S. Ranjbar, Russ. J. Electrochem., 2014, 50, 482–489 CrossRef CAS.
- Y. Zhang, Y. Liu, Z. Chu, L. Shi and W. Jin, Sens. Actuators, B, 2013, 176, 978–984 CrossRef CAS.
- Y. Wang, Y. Yang, X. Zhang, C. Liu and X. Hao, J. Solid State Electrochem., 2015, 19, 3157–3168 CrossRef CAS.
- H.-C. Zhao, P. Zhang, S.-H. Li and H.-X. Luo, Chin. J. Anal. Chem., 2017, 45, 830–836 CAS.
- T. Vincent, C. Vincent and E. Guibal, Molecules, 2015, 20, 20582–20613 CrossRef CAS PubMed.
- Y. Zhu, W. Wang, H. Zhang, X. Ye, Z. Wub and A. Wang, Chem. Eng. J., 2017, 327, 982–991 CrossRef CAS.
- Y. Wan, X. Liu, P. Liu, L. Zhao and W. Zou, Sci. Total Environ., 2018, 639, 428–437 CrossRef CAS PubMed.
- J. Q. Deng, X. D. Li, Y. G. Liu, G. M. Zeng, J. Liang, B. Song and X. Wei, Ecotoxicol. Environ. Saf., 2018, 165, 211–218 CrossRef CAS PubMed.
- L. Lv, N. Chen, C. Feng, J. Zhang and M. Li, RSC Adv., 2017, 7, 27992–28000 RSC.
- C. Theivarasu and S. Mylsamy, Int. J. Eng. Sci. Technol., 2010, 2, 6284–6292 Search PubMed.
- S. M. El-Bahy, D. A. Fadel, Z. M. El-Bahy and A. M. Metwally, J. Environ. Chem. Eng., 2018, 6, 1875–1885 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00596j |
| This journal is © The Royal Society of Chemistry 2019 |