
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStepwise dissolution and composition determination of samples of multiple crystals using a dissolution medium containing aqueous alcohol and fluorocarbon phases†
Humphrey A. Moynihan * and
Declan Armstrong
* and
Declan Armstrong
School of Chemistry, Analytical and Biological Chemistry Research Facility, Synthesis and Solid-State Pharmaceutical Centre, University College Cork, College Road, Cork, T12 K8AF, Ireland. E-mail: h.moynihan@ucc.ie
First published on 9th July 2019
Abstract
Crystallisations are widely used in pharmaceutical and fine chemical manufacturing to control impurity levels, however crystallisations do not always reduce impurities to acceptable levels. Information on the location and distribution of impurities in crystallised materials would be helpful in such cases. A two phase dissolution medium featuring a fluorocarbon non-solvent vehicle and a aqueous ethanol solvent phase has been used to determine the composition of multi-particle crystalline samples through a partial dissolution approach combined with particle sizing and HPLC analysis. 4-Chloro-2-nitroacetanilide (1) was chosen as the host compound for this study, with 4-methyl-2-nitroacetanilide (2) and 4-tert-butyl-2-nitroacetanilide (3) chosen as the guest impurities that were added to supersaturated toluene solutions of 1 at levels up to 5 mol%. The crystals that formed were subjected to a series of partial dissolution steps carried out using the biphasic dissolution medium composed of a 50% aqueous ethanol solvent phase and a perfluorohexane continuous phase. To inhibit particle agglomeration, the mixture also contained 13,13,14,14,15,15,16,16,17,17,18,18-dodecafluoro-2,5,8,11-tetraoxaoctadecane (4) as a non-ionic surfactant. The partial dissolution steps showed a relatively even dissolution with each sequential step as determined from particle sizing. Analysis of the solutions by HPLC from each partial dissolution step allowed the level of impurity to be determined, and when combined with the particle sizing data this allowed an impurity distribution to be generated. Impurity 2 was found to be relatively evenly distributed while impurity 3 was localised on or near the surfaces of crystals.
Introduction
The presence of impurities in crystalline solids is a common phenomenon in the manufacturing of many molecular fine chemicals and pharmaceuticals.1 Sources of impurities include reaction by-products, unreacted starting materials, stereoisomers, reagents, solvents and reaction intermediates.2,3 Crystallisations are often the most important unit processes in the manufacturing scale management of impurities, with the relative solubility of the components of the system and their lattice compatibilities as key factors in determining outcome in terms of purity.4,5 The effectiveness of washing during solid product isolation can also be a factor.6,7 The presence of impurities can have very significant impact on other crystallisation outcomes such as crystal form and morphology.8–14 Crystallisations do not always decrease the level of specific impurities to an acceptable level.15–23 In such cases, knowledge of the location or locations of impurities within batches of crystalline material can be important in designing a strategy for the management of specific impurities. The distribution of a specific impurity within a multi-particle batch of crystalline solid may not necessarily be uniform or localised, and in general it would be best to assume that the concentration of impurities can vary both within individual particles and from particle to particle.There have been investigations of the distributions of impurities within crystals using various approaches.24–27 Careful sequential dissolution and analysis studies have been used to determine the distribution of amino acids in L-asparagine monohydrate crystals,28 and of 4-hydroxyacetanilide and related substances in phenacetin crystals.1,29 Previously, we extended this approach to the determination of impurities in samples of small numbers of multiple crystals through the use of a simple medium consisting of a weak solvent to provide a degree of dissolution and a simple non-ionic surfactant to inhibit particle agglomeration.30 For this approach to begin to be of value for significantly-sized samples of fine chemicals and pharmaceuticals, more sophisticated dissolution media are required, which can provide effective particle aggregation inhibition of larger numbers of particles and a more precisely controlled degree of dissolution.
The stabilisation of suspensions of dispersed particles in liquid continuous phases has been extensively studied for systems of smaller particles in the nanometer to low micrometer size range, i.e. colloidal systems, in particular by electrostatic stabilisation or steric stabilisation.31,32 Electrostatic stabilisation is especially significant for particles suspended in water or any solvent with a high dielectric constant.33 Steric stabilisation is provided by surface layers of polymer chains which interact favourably with the continuous phase, such that particle agglomeration is disfavoured.34,35 As the present work is concerned with achieving stabilisation and controlled partial dissolution of particles of organic compounds in typical organic solvents of low dielectric constant, comparable to those used in ‘slurrying’ of suspensions in process chemistry,36 steric stabilisation would be the more appropriate approach. The dissolution of particles has been studied, especially in the context of the dissolution of drug product particles in the gastrointestinal tract, the fundamental theory being provided by the Noyes–Whitney/Nernst–Brunner model, in which the dissolution of a solid is viewed as a process by which molecules migrate from the solid into a diffusion layer surrounding the particle in which the concentration is determined by the solubility of the compound.37 Molecules then transfer from the diffusion layer into the bulk solution with the difference in concentrations between the diffusion layer and that in the bulk solution as the driver of the dissolution process.38
The aim of the present work is to provide a medium which inhibits particle agglomeration and which also allows for a defined degree of dissolution of individual particles, and to apply this medium to determining the distribution of impurities in samples of crystals of a molecular compound. In particular, it is intended to allow for controlled dissolution through selecting the amount of solvent such that the volume used is sufficient to dissolve only a specific proportion of the crystal mass. To provide for a manageable suspension in which the crystal particle can the stabilised, the solvent will be supplemented by an additional non-solvent phase, with an appropriate surfactant added to stabilise the particles against agglomeration. The dissolution medium would therefore consist of two liquid phases, one capable of dissolving the particles to the required extent and one acting as a non-solvent continuous phase. The anti-agglomeration measures would need to be compatible with this medium and provide further stabilisation. A largely organic or non-aqueous medium was preferred to facilitate isolation and drying of particles and of dissolved components. For these reasons, it was decided to investigate the possibility of a fluorous continuous phase with an organic dissolution layer. Fluorous compounds can often be immiscible with organic solvents under standard conditions, allowing the generation of fluorous/organic biphasic systems.39,40 Amphiphiles bearing fluorocarbon domains play a key role in such assemblies.41–43
In this work, perfluorohexane is used as the continuous, i.e. non-solvent, phase. Perfluorohexane is a very non-polar solvent with a dielectric constant of 1.69 at 25 °C, whereas the equivalent value for moderate organic solvents such as benzene is 2.27 and for a solvent such as THF is 7.58.44,45 However, perfluorinated solvents such as perfluorohexane are also immiscible with many organic solvents under standard conditions, for example, the Hildebrand solubility parameter for perfluoroheptane is 11.9 MPa at 25 °C, whereas the equivalent values for benzene and THF are 18.8 MPa and 18.6 MPa respectively; while for solvents to be miscible requires that their Hildebrand solubility parameters differ by no more than 3.0 MPa.45 The chosen compound for crystallisation was 4-chloro-2-nitroacetanilide (1) (Fig. 1). 50% aqueous ethanol was selected as the solvent phase, as this is a known solvent for compound 1;26 hence the degree of dissolution would be determined by the volume of 50% aq. ethanol added. 4-Methyl-2-nitroacetanilide (2) is known to act as an isosteric crystal lattice replacement for molecules of compound 1, and hence can be added as an impurity which can be incorporated into crystals of compound 1 to a controllable extent and in a relatively evenly distributed manner.26 2-Nitro-4-tert-butylacetanilide (3), an analogue of compound 2 in which the arene methyl group is replaced by the more sterically demanding tert-butyl group, has also been examined as an impurity which can provide contrasting, less predictable behaviour as the increase in size of the tert-butyl group compared to the methyl group will result in very different lattice compatibilities for the two compounds.
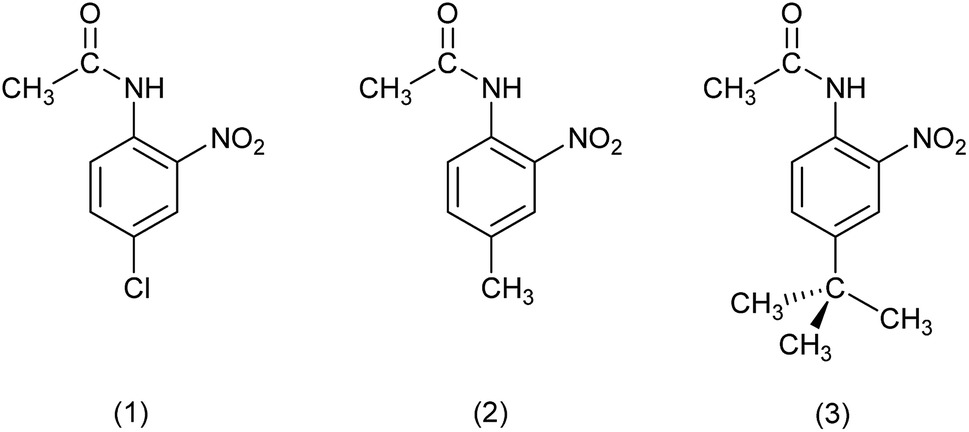 | ||
| Fig. 1 Molecular structures of 4-chloro-2-nitroacetanilide (1), 4-methyl-2-nitroacetanilide (2) and 2-nitro-4-tert-butylacetanilide (3). | ||
Materials and methods
Materials
HPLC grade solvents such as acetonitrile and deionised water were purchased from the Honeywell CHROMASOLV Plus range. All other reagents and solvents were obtained from commercial suppliers and used without further purification. The synthesis of 2-nitro-4-tert-butylacetanilide (3) is described in the ESI.† 4-Chloro-2-nitroacetanilide (1) and 4-methyl-2-nitroacetanilide (2) were prepared according to previously reported methods.26,46,47Synthesis of 13,13,14,14,15,15,16,16,17,17,18,18-dodecafluoro-2,5,8,11-tetraoxaoctadecane (4)
A surfactant with both fluorocarbon and organic (ethyleneoxy) domains was synthesized (Scheme 1). 2,2,3,3,4,4,5,5,6,6,7,7-Dodecafluoroheptan-1-ol (50.6 g, 152.4 mmol, 3 eq.) and sodium hydride (60% in mineral oil, (6.26 g, 156.5 mmol, 3.08 eq.)) were stirred in 100 mL of THF in an ice bath for 20 minutes, and then for a further 1 hour as the reaction mixture warmed to room temperature, after which time a hazy orange solution had formed. 10-p-Toluenesulfonyloxy-2,5,8-trioxadecane48 (16.18 g, 50.819 mmol, 1 eq.) was taken up in 20 mL of dioxane and added to the reaction mixture. A further rinse of 10 mL of dioxane was applied to the addition neck. The reaction mixture was then heated to reflux (∼95 °C) under N2 and stirred for 20 hours. After 1 hour, a white precipitate was observed to have formed. After 20 hours, the reaction mixture was cooled to room temperature and analysed by TLC. The reaction was diluted with water (100 mL) and extracted three times with 100 mL portion of dichloromethane. The extractions were combined and the solvent was removed under reduced pressure to produce an orange oil. The oil was bound to silica gel (∼200 g) using dichloromethane to form a slurry and subsequent removal of the solvent under reduced pressure. The silica gel was then dry loaded to a column containing a previously loaded layer of silica gel-in-hexane. Column chromatography was performed using a mobile phase of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hexane:ethyl acetate, and once impurities were removed from the column, the residual product was removed with a mobile phase of 100% ethyl acetate. The fractions were combined and the solvent was removed under reduced pressure to produce a light yellow oil which was further dried under high vacuum. Yield: 24.00 g, 98.75%. 1H NMR (300 MHz, CDCl3) δ 6.03 (tt, 2JHF = 51.9, 3JHF = 5.2 Hz, 1H, –CF2H), 4.02 (t, 3JHF = 14.0 Hz, 2H, –OCH2CF2–), 3.79–3.73 (m, 2H, –OCH2CH2O–), 3.68–3.57 (m, 8H, –OCH2CH2O–), 3.54–3.46 (m, 2H, –OCH2CH2O–), 3.35 (s, 3H, –CH3). 13C{1H} NMR (151 MHz, CDCl3): δ 115.65 (tt, 1JCF = 257.8 Hz, 2JCF = 30.6 Hz, –CH2CF2–), 113.55–107.55 (m, 3x CF2), 107.65 (tt, 1JCF = 254.5 Hz, 2JCF = 31.3 Hz, –CF2H), 72.23 (s, CH2), 71.86 (s, CH2), 70.71–70.34 (m, 4x CH2), 68.21 (t, 2JCF = 25.2 Hz, –CH2CF2–), 58.78 (s, CH3). 19F NMR (282 MHz, CDCl3); All signals displayed a large multiplicity, but approximate assignments were made with an exponential apodization of 2.5 Hz: δ −119.93 (p, 3JFF = 13.6 Hz, 2F), −122.24 to −122.51 (m, 2F), −123.64 (s, 4F), −129.70 (s, 2F), −137.26 (d, 2JHF = 51.9 Hz, 2F, –CHF2). HR-ESI-TOF MS (positive mode): m/z calculated for C14H18F12O4Na+ [M + Na]+ 501.0911 found 501.0884. Elemental analysis: found (calculated) for C14H18F12O4 (478.10 g mol−1): C, 34.86 (35.16); H, 3.73 (3.79).
:1 hexane:ethyl acetate, and once impurities were removed from the column, the residual product was removed with a mobile phase of 100% ethyl acetate. The fractions were combined and the solvent was removed under reduced pressure to produce a light yellow oil which was further dried under high vacuum. Yield: 24.00 g, 98.75%. 1H NMR (300 MHz, CDCl3) δ 6.03 (tt, 2JHF = 51.9, 3JHF = 5.2 Hz, 1H, –CF2H), 4.02 (t, 3JHF = 14.0 Hz, 2H, –OCH2CF2–), 3.79–3.73 (m, 2H, –OCH2CH2O–), 3.68–3.57 (m, 8H, –OCH2CH2O–), 3.54–3.46 (m, 2H, –OCH2CH2O–), 3.35 (s, 3H, –CH3). 13C{1H} NMR (151 MHz, CDCl3): δ 115.65 (tt, 1JCF = 257.8 Hz, 2JCF = 30.6 Hz, –CH2CF2–), 113.55–107.55 (m, 3x CF2), 107.65 (tt, 1JCF = 254.5 Hz, 2JCF = 31.3 Hz, –CF2H), 72.23 (s, CH2), 71.86 (s, CH2), 70.71–70.34 (m, 4x CH2), 68.21 (t, 2JCF = 25.2 Hz, –CH2CF2–), 58.78 (s, CH3). 19F NMR (282 MHz, CDCl3); All signals displayed a large multiplicity, but approximate assignments were made with an exponential apodization of 2.5 Hz: δ −119.93 (p, 3JFF = 13.6 Hz, 2F), −122.24 to −122.51 (m, 2F), −123.64 (s, 4F), −129.70 (s, 2F), −137.26 (d, 2JHF = 51.9 Hz, 2F, –CHF2). HR-ESI-TOF MS (positive mode): m/z calculated for C14H18F12O4Na+ [M + Na]+ 501.0911 found 501.0884. Elemental analysis: found (calculated) for C14H18F12O4 (478.10 g mol−1): C, 34.86 (35.16); H, 3.73 (3.79).
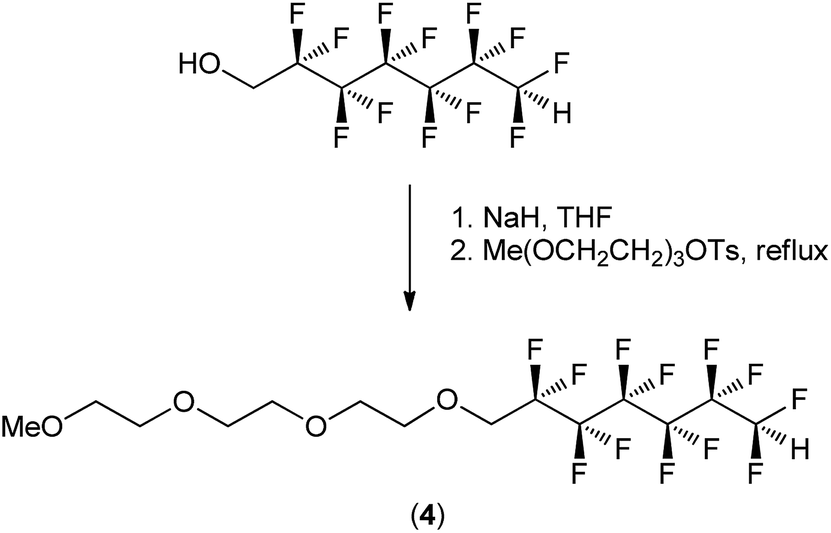 | ||
| Scheme 1 Synthetic scheme outlining the synthesis of 13,13,14,14,15,15,16,16,17,17,18,18-dodecafluoro-2,5,8,11-tetraoxaoctadecane (4). | ||
High-performance liquid chromatography
Data was obtained from an Agilent 1290 Infinity II LC System interfaced to a Dell OptiPlex 5040. A reverse-phase C-18 column (5 μm, length 150 mm, i.d. 4.6 mm, Eclipse XDB-C18-993967-902) was used to separate compounds 1, 2, and 3. The mobile phase for separations of 1 and 2 consisted of water:acetonitrile (70:30 respectively) for 0–6 minutes with a flow rate of 1.8 mL min−1. The injection volume was 10 μL and the detector was set at 234 nm. Good baseline resolutions were obtained for both compounds eluting at 3.643 and 5.115 min for 2 and 1 respectively. Calibration curves were constructed for approximate compound concentrations of 332.5, 133.0, 66.5, 33.25, 13.3, 6.65, 3.325, and 1.33 μg mL−1 for 2 and for approximate compound concentrations of 322.5, 129.0, 64.5, 32.25, 12.9, 6.45, 3.225, and 1.29 μg mL−1 for 1 (ESI†). The mobile phase for separations of 1 and 3 consisted of water:acetonitrile (55:45 respectively) for 0–5 minutes with a flow rate of 2.25 mL min−1. The injection volume was 10 μL and the detector was set at 234 nm. Good baseline resolutions were obtained for both compounds eluting at 1.694 and 3.665 min for 1 and 3 respectively. Calibration curves were constructed for approximate compound concentrations of 405.0, 162.0, 81.0, 40.5, 16.2, 8.1, 4.05, and 1.62 μg mL−1 for 1 and for approximate compound concentrations of 437.5, 175.0, 87.5, 43.75, 17.5, 8.75, 4.375, and 1.75 μg mL−1 for 3 (ESI†).
Solubility
Solubility measurements were obtained by first dissolving the desired compound in the experimental solvent system composed of a 1 μM solution of compound 4 in 50% (v/v) aqueous ethanol, at a set temperature of 25 °C (±1 °C) in an oil bath until further additions of the investigated compound formed a sustained suspension of material. Once a saturated solution had formed the flask contents were stirred for a further 30 minutes to allow for solvent equilibration. The flask contents were then filtered quickly through a 0.2 μm membrane filter (Agilent Technologies, PTFE). The filtrate was then stirred for 30 minutes at the previously set temperature to allow for solvent equilibration. Three 0.5 mL volumes were drawn from this solution by micropipette and transferred to sample vials and the weights of the solution and vial were determined. The solvent from these samples vials was allowed to evaporate in a fumehood with further drying of the sample vials under high vacuum for several hours. The residues in the sample vials were dissolved in acetonitrile and analysed by HPLC, making necessary dilutions until peak areas aligned with the previously established calibration curves and the solubility was calculated in mg mL−1 retrospectively. The specific volume of solvent required to dissolve a given mass was determined as follows. The average weight of the three 0.5 mL portions of 50% aq. EtOH at 25 °C was determined to be 0.4720 (0.0008) g (a density of 0.944 (0.0016) g mL−1); the average weight of solvent (50% aq. EtOH) lost from saturated solutions of compound 1 was determined to be 0.4665 (0.0012) mg, corresponding to an average of 494.174 (0.002) μL of solvent lost from the samples with average concentrations of 6.08 (0.08) mg mL−1 of compound 1 as determined by HPLC. Based on this, 1000 μL would dissolve 6.15 (0.09) mg of compound 1.Crystallisation of 2-nitro-4-chloroacetanilide (1) with additives
Samples of 1 containing additive 2 or 3 were prepared with additive levels ranging from 0.5 mol% to 5.0 mol% at 0.5 mol% intervals. Stock solutions of 1 and the additives were prepared in diethyl ether, and the appropriate volumes of the two solutions were mixed to acquire target doping levels. The diethyl ether solvent was allowed to evaporate overnight in a fumehood and the residues were further dried under high vacuum. The appropriate amount of toluene was added to the residues, the samples were heated to 80 °C with swirling of the sample vials to assist dissolution and then the samples were cooled to ambient temperature in an unassisted manner. All samples formed crystals that were then isolated by vacuum filtration and air dried.Optical microscopy and particle sizing
Optical microscopy and particle size measurement were carried out using a Nikon Eclipse 50i polarising microscope with the Nikon Digital Sight DS-Fi1 digital camera and the NIS-Elements software version BR 3.1. A perimeter was established around the visible surface area of individual crystals and the area inside this perimeter was used as the area of the crystal (μm2). The crystal lengths (μm) were determined by measuring the distance of the longest dimension of visible surface area of each individual crystal (Fig. S7†) that were selected manually to have similar sizes as determined by eye.Powder X-ray diffraction
PXRD was performed at ambient temperature using a Stoe STADI-MP diffractometer operating in transmission mode with a linear PSD detector with an anode current of 40 mA, an accelerating voltage of 40 kV and Cu Kα1 radiation (λ = 1.5406 A) scanning in steps of 0.5° for 45 seconds per step from 5 to 43° in 2θ. Samples were held between acetate foils.DSC
DSC was carried out on a TGA Q1000 calorimeter with an RCS 40 cooling system at 2 °C min−1.Partial dissolution
The solubility of 4-chloro-2-nitroacetanilide in 50% aq. EtOH as a 1 μM solution of surfactant 4 was determined to be 6.15 mg per 1 mL of applied solvent at 25 °C. Pre-weighed crystals of 4-chloro-2-nitroacetanilide (1) were suspended in 500 μL of perfluorohexane followed by the appropriate amount of surfactant-containing solvent to dissolve the desired weight-proportion of the crystalline samples. The vessel containing the mixture was submerged in a water bath set to 25 °C and the vessel was continuously inverted by rotation for approximately 1 hour using a Heidolph RZR 2020 mechanical stirrer set to a speed of 100 RPM. After this time, the liquid layers were separated from the crystals by pipette and the solvent was allowed to evaporate. The residue deposited by the solvent was further dried under high vacuum and analysed by HPLC, the residual particles were sized using optical microscopy and the partial dissolution procedure was repeated again using fresh solutions and glassware (Fig. 2). | ||
| Fig. 2 An image depicting successive partial dissolutions of one crystal of compound 1 from the series containing 4.0 mol% of compound 2. From left to right, the first crystal is the initial crystal before any dissolution media was applied, continuing progressively to the final crystal, at which point no more dissolution media was applied. | ||
Results and discussion
The aim of this study was to develop a liquid medium in which suspended crystals can be stabilised against agglomeration, while also allowing for a set degree of dissolution of each crystal. The medium would contain the required amount of solvent necessary to achieve the required degree of dissolution, while a non-solvent would provide the bulk of the liquid medium and a suitable surfactant would inhibit particle agglomeration and assist interaction of the solvent and non-solvent phases. This medium would then be used to map the distribution of impurities in the crystal sample. 4-Chloro-2-nitroacetanilide (1) was selected as the crystalline compound for study, while the methyl (2) and tert-butyl (3) analogues were selected as the impurities. Compound 2 is known to act as an isomorphic lattice replacement for compound 1,26 i.e. was selected as an impurity likely to behave in a predicable manner, while compound 3 was selected to provide likely contrasting behaviour. 50% Aqueous ethanol was selected as the solvent, as this is a known solvent for compound (1).26 Perfluorohexane was selected as the non-solvent. The perfluorohexane phase is to provide a bulk liquid medium while the 50% aq. ethanol provides the dissolving capability; the volume of added 50% aq. ethanol should be the main determinant of the degree of dissolution. Given that the solvent and non-solvent phases would be 50% aq. ethanol and perfluorohexane respectively, a surfactant was required which would have domains compatible with both phases, and with the 50% aq. ethanol compatible domain also compatible with the surfaces of the 4-chloro-2-nitroacetanilide crystals. A short ethyleneoxy chain was selected as the 50% aq. ethanol and 4-chloro-2-nitroacetanilide compatible domain, and a dodecafluorohexyl chain was selected as the fluorocarbon domain. Compound 4, containing these features, was synthesised to act as the surfactant. Concentrations of 1 μM of compound 4 in the aqueous layer were found to be adequate for anti-flocculent activity.100 crystals were selected and their lengths and areas were determined by optical microscopy. The crystals were suspended in 1 mL of perfluorohexane and the required amount of 50% aqueous ethanol was added to dissolve 10% of the predetermined weight. The crystals and dissolution medium were placed into closed sample vials. These were agitated by continuous inversion using an overhead stirrer to facilitate the mixing of the crystals with the dissolution medium. The agitated sample vials were placed in a thermostated water bath. After agitation for 1 hour at 25 °C, the crystals were resized. The crystals were subjected to further such dissolution steps until the smallest of the particles would completely dissolve in further dissolution steps, at which point the final crystal measurements were taken. Fig. 3 and 4 demonstrate the use of this medium for dissolving crystals sequentially. Fig. 3 shows the area change of 100 crystals after each application of solvent and resizing of the particles, indicating the particles are getting smaller with each successive dissolution step, whereas Fig. 4 shows the same series of crystals sized by length. Table 1 lists the D10, D50, and D90 values of the particles which again demonstrate that the crystals are diminishing in size. It should also be noted that the overall spread of the size distributions was conserved as can be seen by the span values listed in Table 1. The span values for the sized particles increase in both length and area with each successive partial dissolution; length span values range from 0.4 for the initial crystals to 0.54 for the crystals sized after all the partial dissolutions had been performed; area span values follow a similar increasing trend, with values of 0.7 for the initial crystals to 0.9 for the crystals sized after the final partial dissolution.
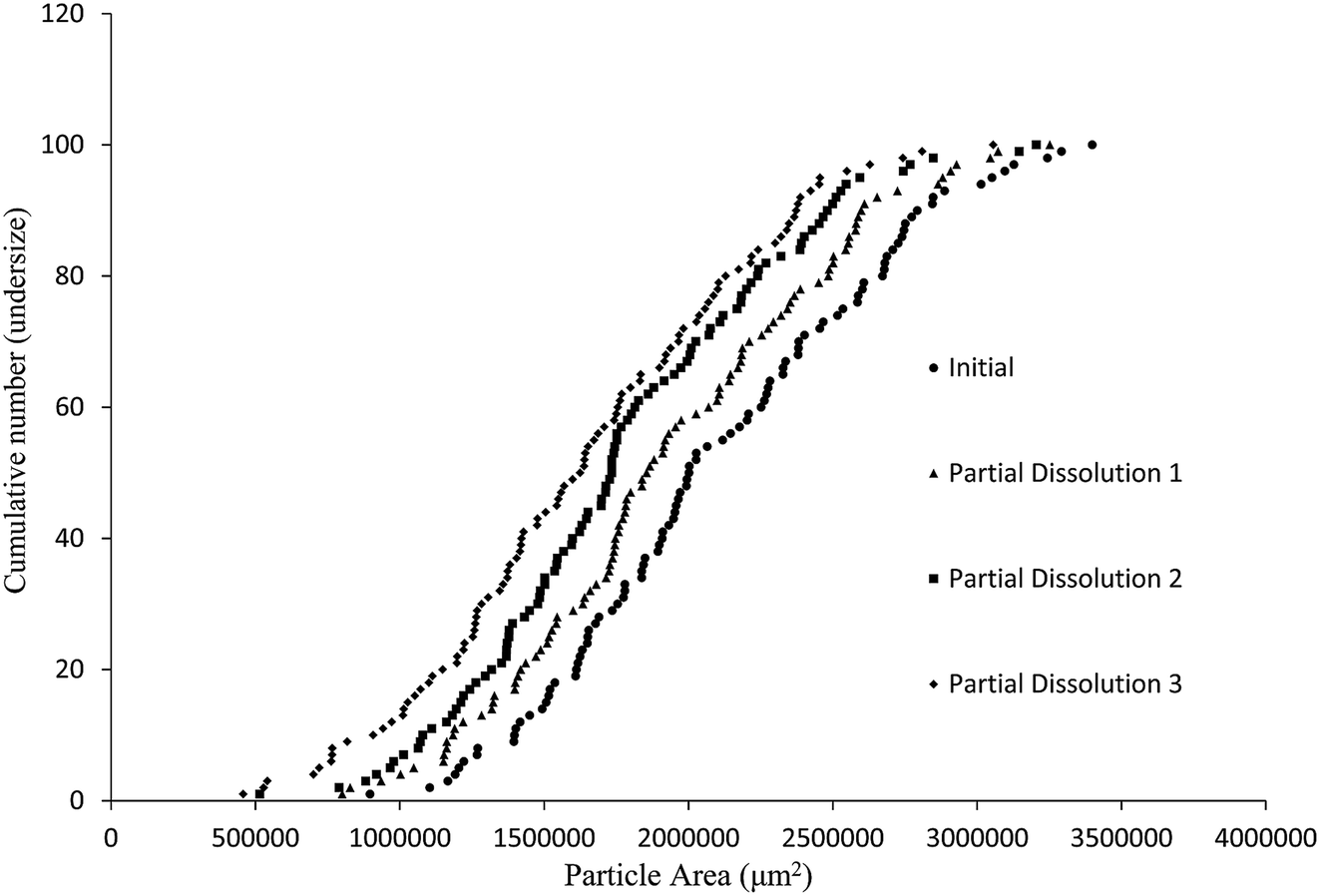 | ||
| Fig. 3 Particle size distributions (cumulative number undersize vs. area) on a sample of 100 crystals of compound 1 prior to and following three sequential dissolutions in the medium described in the text. | ||
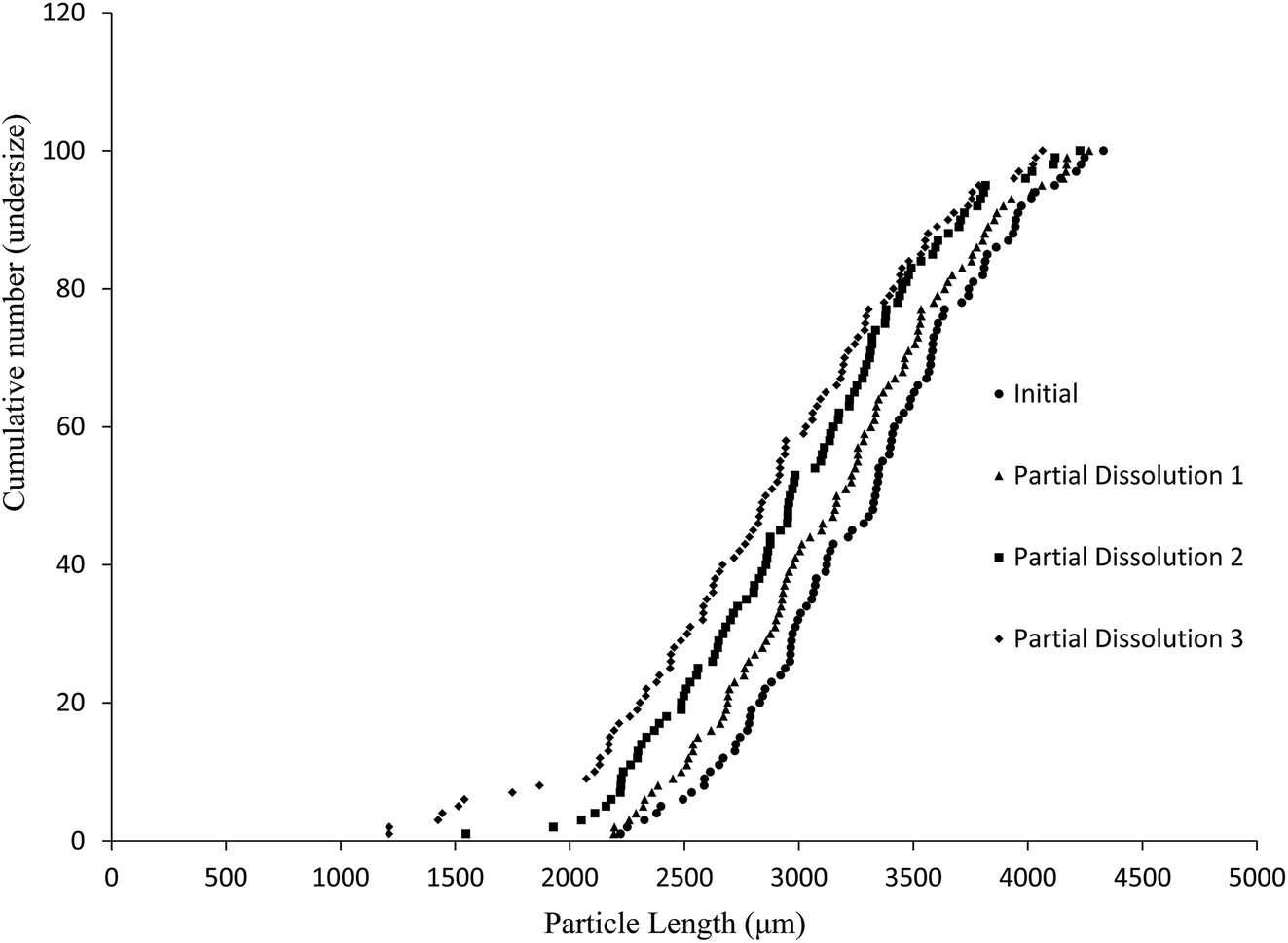 | ||
| Fig. 4 Particle size distributions (cumulative number undersize vs. length) on a sample of 100 crystals of compound 1 prior to and following three sequential dissolutions in the medium described in the text. | ||
| Measurement | Initial | 1st dissolution | 2nd dissolution | 3rd dissolution |
|---|---|---|---|---|
| Length D10 (μm) | 2613.17 | 2487.13 | 2233.77 | 2108.4 |
| Length D50 (μm) | 3333.4 | 3165.32 | 2962.1 | 2855.79 |
| Length D90 (μm) | 3947.14 | 3853.77 | 3705.49 | 3651.72 |
| Length span | 0.40 | 0.43 | 0.50 | 0.54 |
| Area D10 (μm2) | 1397093.17 |
1184415 |
1079938 |
907705.8 |
| Area D50 (μm2) | 2001728 |
1854950 |
1734432 |
1624424 |
| Area D90 (μm2) | 2792789.74 |
2598425 |
2481757 |
2373248 |
| Area span | 0.70 | 0.76 | 0.81 | 0.90 |
The spans were determined from the D values using the following formula:
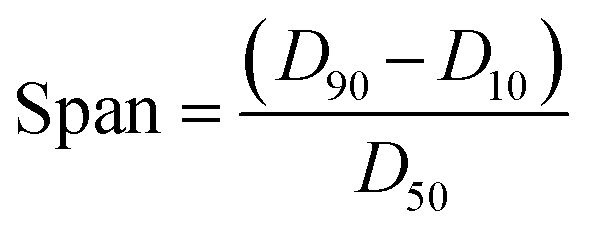
Crystals of 1 were grown from toluene at a supersaturation level of 1.5 with the levels of compound 2 in solution ranging from 0.5 to 5.0 mol%. As can be seen in Fig. 5, the relative amount of incorporation level to solution impurity level follows a consistent trend with crystal formation of 1 having an approximate uptake of 20% of the added material 2. A good comparison would be with the incorporation levels versus doping levels of compound 2 in host crystals of 4-trifluoromethyl-2-nitroacetanilide (Fig. S8†) that was reported previously;30 this further emphasises the ability of para-substituted acetanilides with similar sized functional groups to act as isomorphic additives that display an appreciable degree of mutual lattice incorporation.
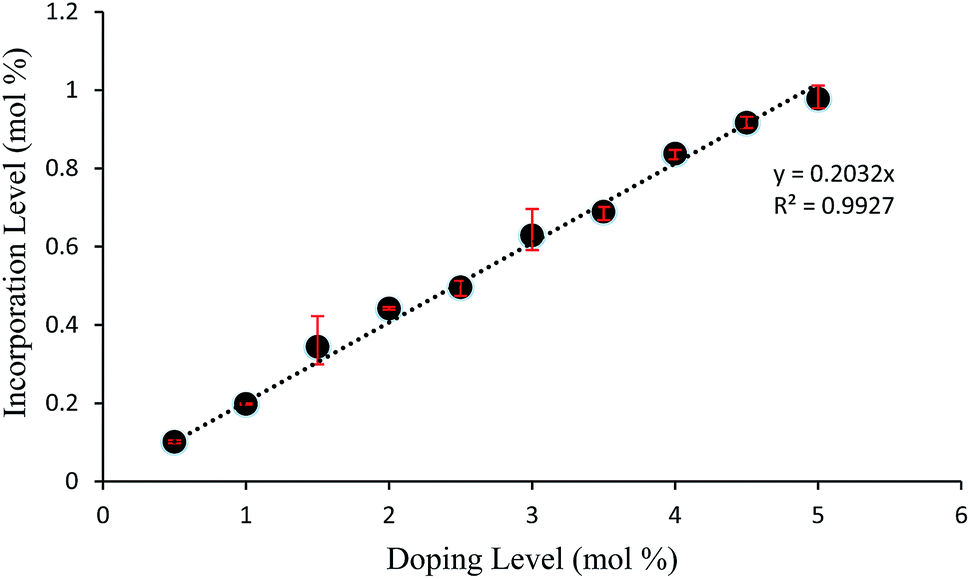 | ||
| Fig. 5 Graph displaying incorporation level versus solution impurity level of 4-methyl-2-nitroacetanilide (2) in “host” crystals of 4-chloro-2-nitroacetanilide (1). | ||
Fig. 6 displays the incorporation levels of 4-tert-butyl-2-nitroacetanilide (3) in host crystals of 4-chloro-2-nitroacetanilide (1) grown from toluene at a supersaturation level of 1.5. Compound 3 was selected for this study to provide contrasting behaviour to that of compound 2. Compound 2 differs from crystallising compound 1 in having a methyl rather than a chloro group in the aryl para position, whereas compound 3 has a sterically demanding tert-butyl group in the same position. Therefore, while compound 2 is known to behave as an isomorphic lattice replacement for compound 1,26 the space-filling requirements of compound 3 make it very unlikely to behave in such a manner, such that its distribution as an impurity is unpredictable. It is clear from Fig. 6 that, in comparison to impurity 2 (Fig. 5), there is a weaker correlation between the amount of compound 3 present in solution and the extent of incorporation of 3 into crystals of compound 1. As Fig. 6 shows, the degree of incorporation of 3 was also found to vary considerably between batches. Variation was also found even within batches, for example, three samples from the crystals obtained from solutions containing 4.5% of impurity 3 were found to show incorporation levels of 0.1867 (±0.0005) mol%, 0.1025 (±0.0002) mol%, and 0.05169 (±0.00005) mol%. The first such sample with the largest quantity of incorporated impurity was used in the partial dissolution study described below. The highly variable inclusion behaviour of compound 3 is consistent with the its lattice-incompatibility. Such an impurity is most likely to be found on the exterior of crystals from deposition from the mother liquor.
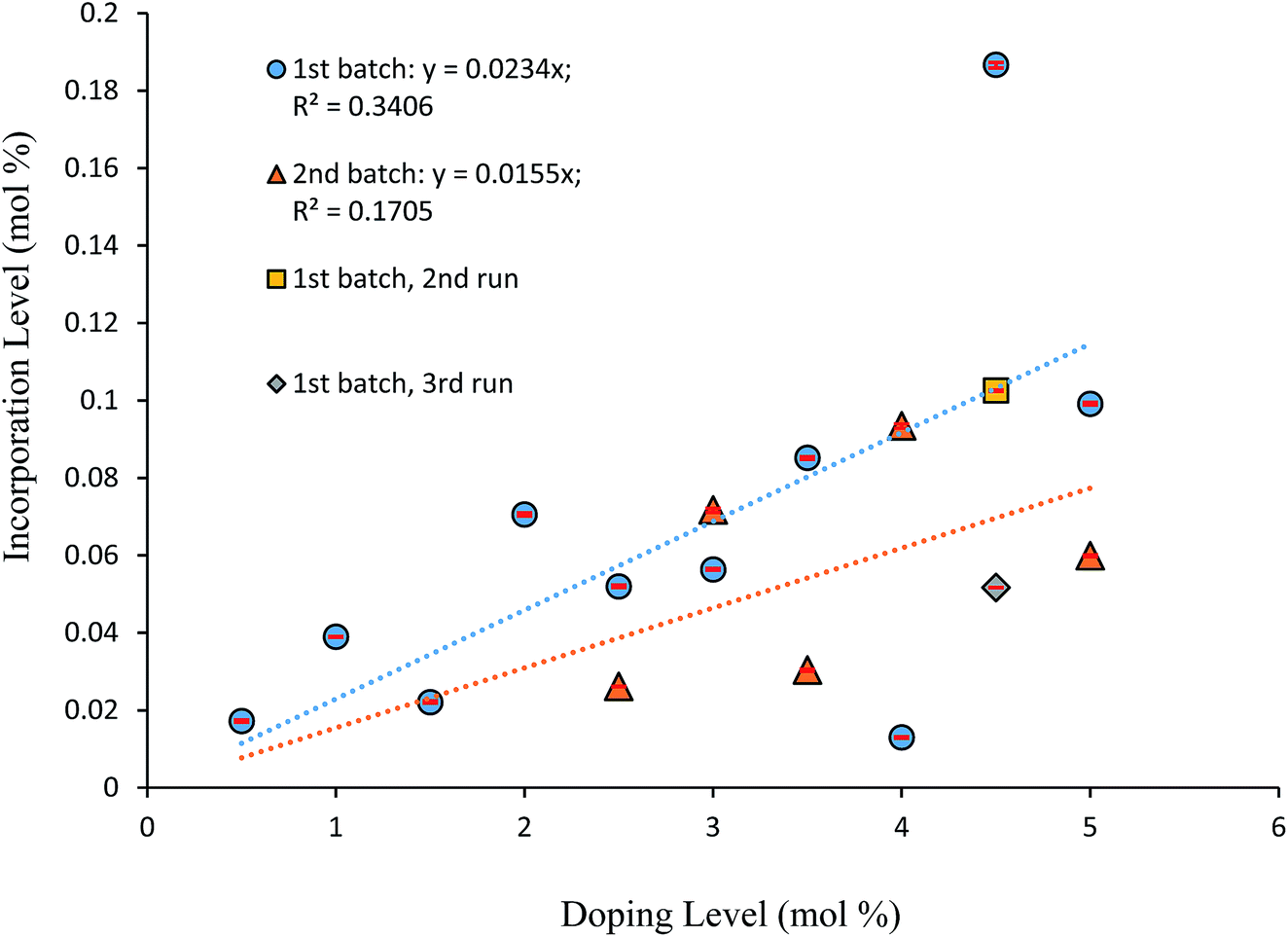 | ||
| Fig. 6 Graph displaying incorporation level of 4-tert-butyl-2-nitroacetanilide (3) in “host” crystals of 4-chloro-2-nitroacetanilide (1) versus solution impurity level. Data is shown for two crystallisation batches. | ||
PXRD and DSC data (ESI, Fig. S9–S12†) show that the addition of impurities 2 or 3 at these levels does not change the crystal form of compound 1. PXRD patterns were obtained for crystals of 1 with additive concentrations of 2 and 3 between 0.5 and 5.0 mol% grown in toluene at a supersaturation level of 1.5, however, there were no discernible formations of additional diffraction peaks when compared to spectra obtained for pure compounds of 1 (Fig. S9 and S10†). All obtained patterns displayed peaks corresponding to pure 1 with only minor differences in the intensities of some of the peaks. Consistent with the PXRD analysis, DSC analysis (Fig. S11 and S12†) of the same crystalline mixtures showed minimal difference to the melting point of compound 1, with no secondary events such as minor melting points or polymorph changes under the tested conditions.
The dissolution medium (i.e. perfluorohexane/50% aqueous ethanol/compound 4) was then used to achieve the stepwise dissolution of samples of crystals containing impurities 2 and 3. The solutions obtained after each dissolution step were analysed by HPLC to provide composition data on the dissolved crystal mass. The partial dissolution data obtained for crystals of compound 1 containing quantities of impurity 2, determined by optical microscopy in terms of area (μm2) and length (μm) and grouping these values into cumulative number distributions are given in full in Fig. S13–S36.† An example of this data is also shown in Fig. 7, where it can be seen that the data representing the “Initial” series (most right-hand-side plot) displays the initial crystal areas before any partial dissolution was applied, the next plot displays the same series' areas after one partial dissolution, and each subsequent plot in a leftwards direction displays the same series after sequential partial dissolutions. The final plot is the furthest to the left of the graph gives the final recorded areas for the series and no more partial dissolution were applied after this point. It can be seen in several figures in the ESI (e.g. Fig. S28 and S31†) that occasionally distribution curves overlap with one another. This is due to crystals having stable orientations on more than one face and so being sized is different orientations for successive distributions. The small number of particles being used in these experiments allow tracking of the individual particle change from each dissolution step, which would not be possible for standard particle sizing of large batches.
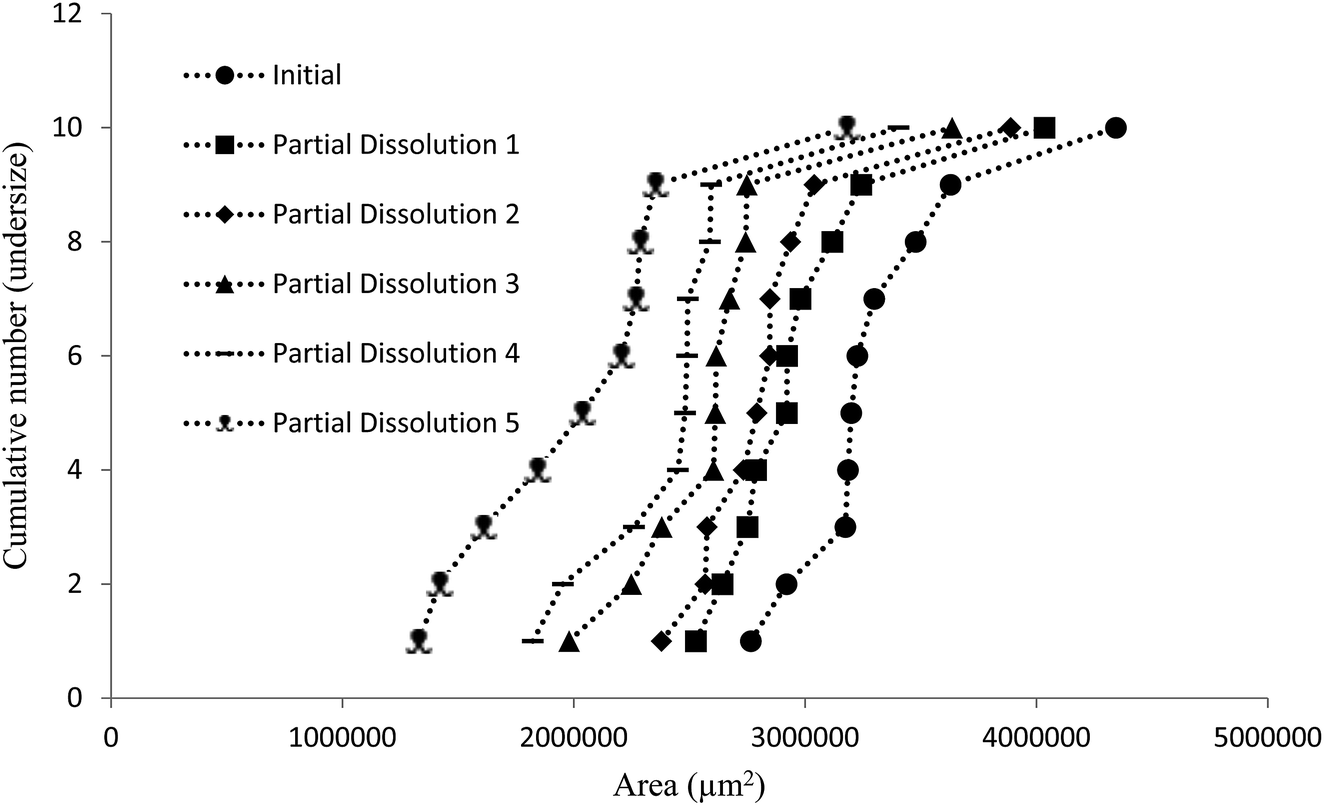 | ||
| Fig. 7 Chart comparing particle area versus the ranking of each particle in a partial dissolution series of 1 doped with 3.0 mol% of 2. | ||
The data obtained from the cumulative number vs. size charts can be combined with the HPLC data obtained to produce graphs which map the relative quantity of impurity distributed throughout crystal particles for samples of multiple crystals. These impurity percentage versus dissolution mid-point graphs are shown in Fig. 8 and S15–S36.† If the initial particles are 0% dissolved and fully dissolved particles 100% dissolved, the dissolution mid-point is the mid-point of the average percentage dissolution before and after a partial dissolution step, essentially defining the middle point of the dissolved region. The impurity percentage is the proportion of impurity-to-host as determined by HPLC from the solution obtained from a partial dissolution step. Horizontal error bars indicate the upper and lower limits of the dissolution mid-point percentages as determined from the particle sizing measurements. A pattern appears with each mid-point graph displaying a trend, albeit within a narrow range, whereby the highest levels of impurity are in the outer layer and the final dissolution layer. The highest level outer layer is attributable to the deposition of impurities from the mother liquor.
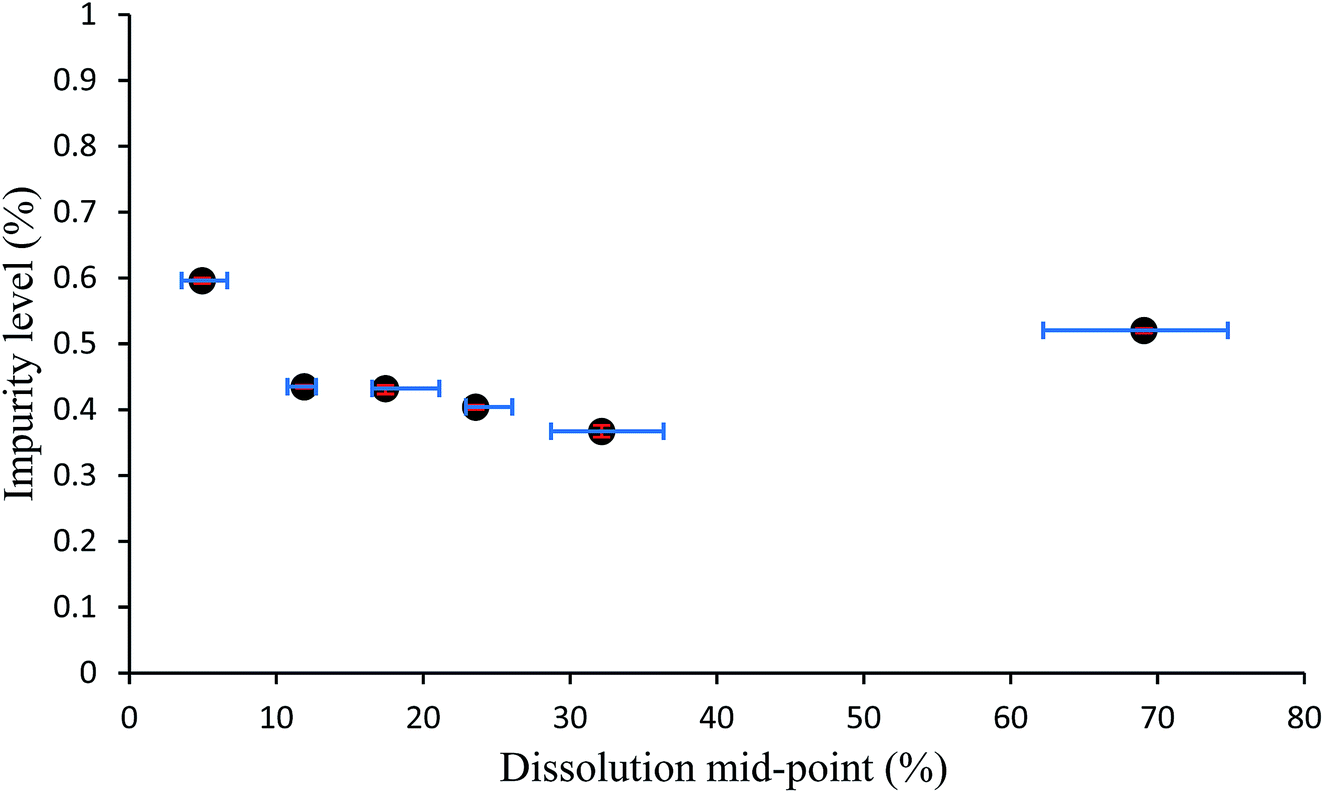 | ||
| Fig. 8 Plot of percentage by HPLC of added impurity in crystals of compound 1 vs. the dissolution mid-point for the sample of crystals grown from solutions containing 3.0 mol% of additive 2. Error bars show the upper and lower limits of experimental values from which the averages were taken to assign datapoints. | ||
One disadvantage of presenting the impurity distribution in the manner shown in Fig. 8 is that particle sizes are not shown, as the dissolution mid-point is a size independent parameter. Presentation of the data as in Fig. 9 allows the impurity distribution to be shown in relation to particle size. In Fig. 9, the impurity level is shown as an impurity density, i.e. as a percentage per unit length. The impurity density is determined by dividing the percentage impurity by the difference in average particle sizes (ΔL) between the particles before and after the dissolution which provided the solution for determination of that impurity level. The impurity density value is then assigned to a mid-point (L*) between the two size distributions, corresponding to the average size after dissolution plus ΔL/2. Fig. 9 also uses error bars to indicate the spread of the size distributions to which each impurity density is assigned, determined from the standard deviations of the before and after size distributions. This mode of presentation shows the extent of impurity incorporation with respect to size. For example, the data in Fig. 9, which is drawn for the sample grown from solutions containing 3.0 mol% of impurity 2, shows a relatively even level of incorporation during growth with a greater extent of impurity uptake at ca. 2700 μm size.
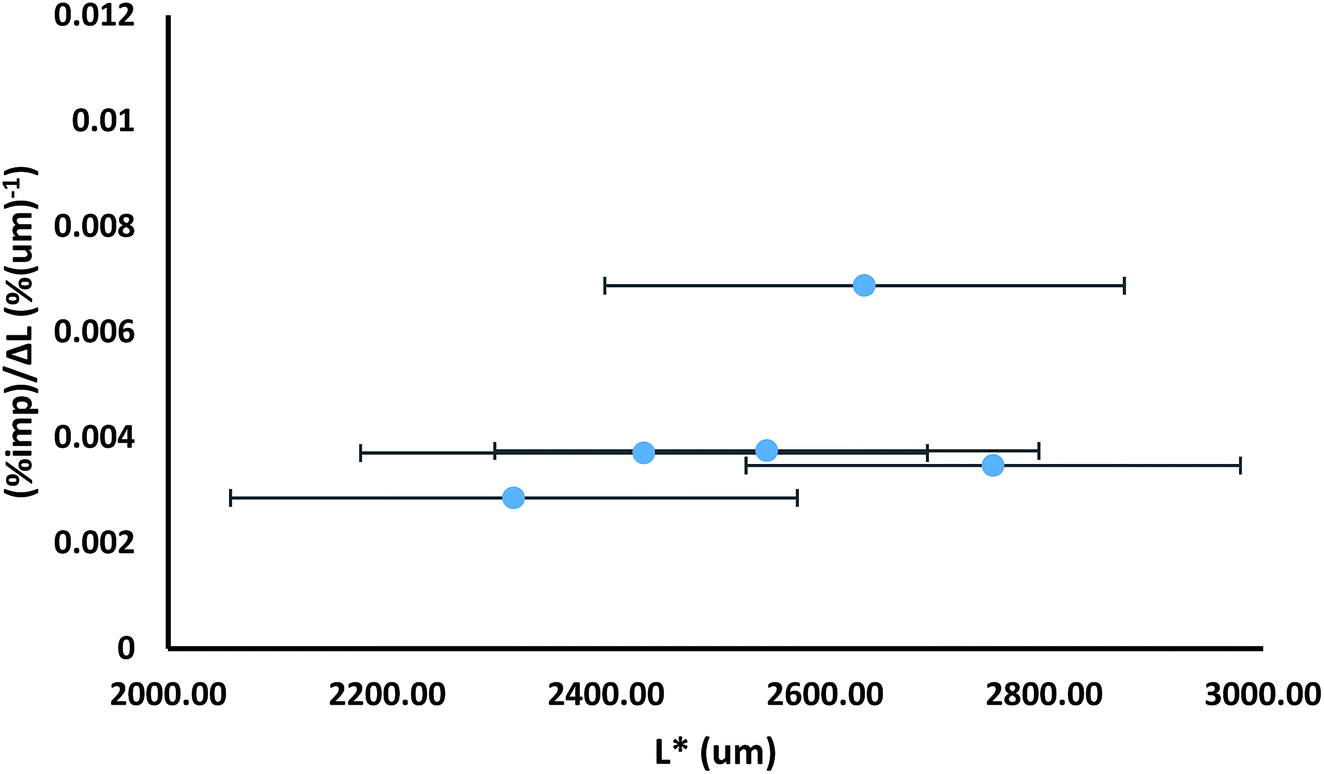 | ||
| Fig. 9 Plot of impurity density, i.e. percentage impurity per unit length [(%imp)/ΔL in which ΔL is the difference in average sizes of samples before and after a dissolution step], versus length L*, in which L* is the average length after a dissolution step plus ΔL/2; for the sample of crystals grown from solutions containing 3.0 mol% of additive 2. The error bars indicate the spread of the size distributions, as determined by the square root of the sum of the squares of the standard deviations. | ||
It is worthwhile to compare the degree of incorporation of impurities as found from the partial dissolution studies with that found for the samples as a whole (i.e., the full batch of harvested crystals prior to selection for crystals for dissolution). When a weighted average of the proportion of impurity-to-host compound is calculated from the measured mole amounts of each compound from each dissolution step and compared to the values of the whole samples, it can be seen that there is usually a decrease in the proportion of impurity-to-host (Table S2†). The smallest deviation from a parent sample is +0.025 (0.016) mol% for the 3.5 mol% 2-doped 1 sample. The largest deviation from a parent sample is +0.24 (0.03) mol% for the 5 mol% 2-doped 1. One possibility is that the larger crystals selected from the parent sample for the dissolution studies generally contained lesser quantities of the impurities. Fig. S37† displays a graphical plot of incorporation level versus doping level for the parent samples as well as for the weighted averages calculated for the partial dissolution series, showing a very similar increase in incorporation with an increase in doping level to that of the parent samples.
It is useful to try to quantify how well the dissolution medium achieved a controlled degree of dissolution. This was done using the data for samples containing impurity 2 grown from solutions containing 4.0 mol% of the impurity. Classifying a three dimensional object by a two-dimensional unit (e.g. area (μm2)) has limitations, even more so in one dimension (e.g. length (μm)), although these limitations are shared by almost all methods of particle sizing. To gauge the relative uniformity of the degree of dissolution of each particle, it would be preferable to compare the dissolution mid-points based on the microscope area determinations with the values that would be obtained based on the volumes of spheres of equivalent area (‘theoretical’ areas). It can be seen in Fig. 10 that both the length- and area-derived data series are relatively close to the data based on volumes of equivalent spheres. Both the observed length and area mid-point values show a slight increase in dissolution over the ‘theoretical’ values, which indicates that slightly more than the target 10% dissolution amount is being dissolved. To estimate a value for the volume change between partial dissolution steps and correlate it to mass change between steps, Table 2 shows the calculated percent dissolution based on the volumes (μm3) of equivalent spheres. Despite % dissolved values ranging from 7.01% to 17.83% for individual partial dissolution steps, the average % dissolved value is 11.61 (3.91) %, very close to the target value of 10% per dissolution step. This suggests that the dissolution medium is exerting a reasonable degree of control over the extent of dissolution, with the volume added of the 50% aq. ethanol solvent phase being the main determinant of the degree of dissolution.
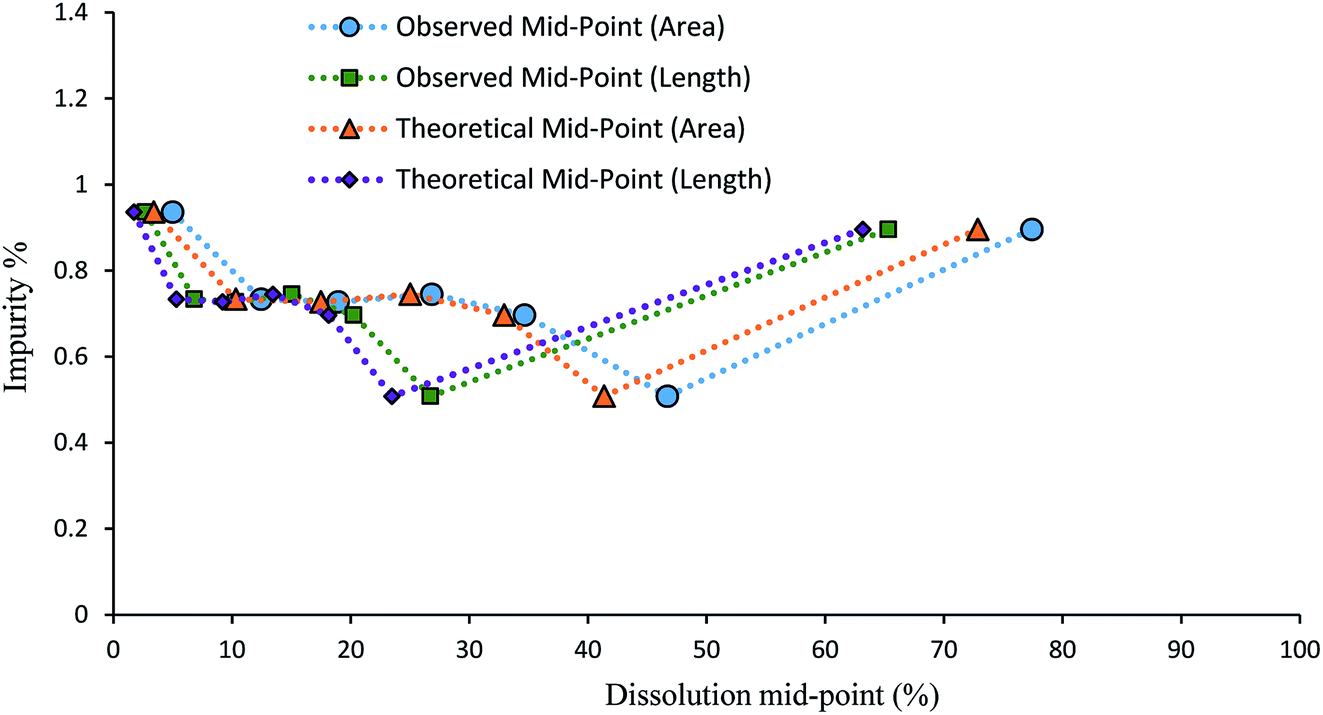 | ||
| Fig. 10 Graph comparing the dissolution mid-points for 4.0 mol% 2-doped 1. The ‘theoretical’ mid-points were calculated using the microscope image analysis data to determine the volumes of equivalent spheres, reducing these by 10% for each dissolution step, and then re-calculating the lengths and areas. | ||
| Sample | Obs. area (μm2) | Calc. volume eq. sphere (μm3) | % dissolved |
|---|---|---|---|
| Initial | 1833294 |
1867291503 |
|
| PD1 | 1650782 |
1595505713 |
14.56 |
| PD2 | 1559270 |
1464690868 |
7.01 |
| PD3 | 1413132 |
1263682314 |
10.77 |
| PD4 | 1269519 |
1076025008 |
10.05 |
| PD5 | 1126801 |
899776935 |
9.44 |
| PD6 | 828151.4 |
566928692 |
17.83 |
Additive compound 3, containing a sterically demanding tert-butyl group, was specifically selected so as to provide less predictable impurity behaviour which would contrast with that of compound 2. It was found that the total degree of incorporation of compound 3 was highly variable. Partial dissolutions were performed on crystals isolated from a batch of solutions with 4.5 mol% of compound 3, as these crystals displayed the highest proportion of impurity-to-host compound. The results for the partial dissolutions in terms of cumulative number versus area and length can be seen in Fig. 11 and 12, and display good size reductions between sets in terms of consistency. However, when the area mid-points are combined with the incorporation level percentages determined by HPLC for each dissolution step, as can be seen in Fig. 13, it becomes apparent that compound 3 does not incorporate well into crystals of compound 1. This is not surprising given the presence of a sterically demanding tert-butyl group on the structure of compound 3. The largest proportion of 3 was detected in the outer layer of the crystals, posited to be mostly from deposition of the impurities from the mother liquor. The second partial dissolution also detected compound 3. The final dissolution of the remainder of the crystals also allowed compound 3 to be detected at a level of 0.00937 (0.00005) mol%. The significant feature of this approach is that it allows determination of the very differing impurity distribution behaviour of compounds 2 and 3. The increased steric demands of compound 3, relative to those of compounds 1 or 2, resulted in it being much less effectively incorporated into growing crystals of compound 1, and was found as an impurity mostly near the surfaces of the crystals.
 | ||
| Fig. 11 Chart comparing particle area versus the ranking of each particle in a partial dissolution series of 1 doped with 4.5 mol% of 3. | ||
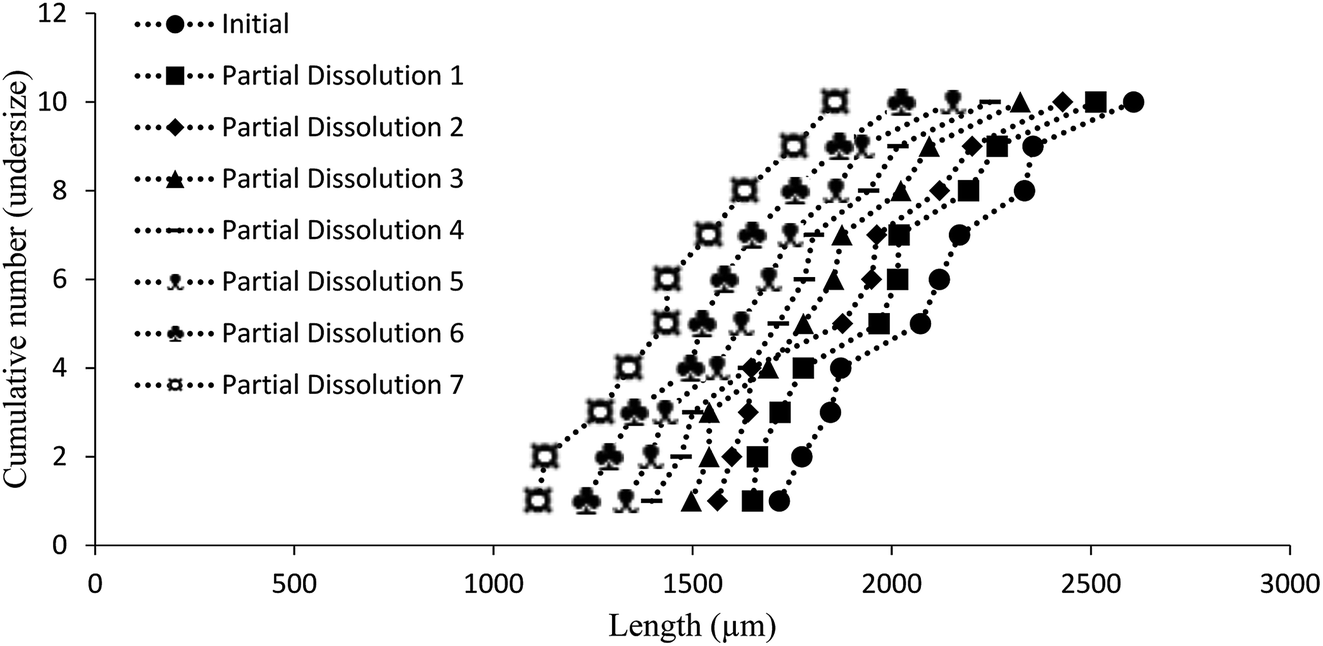 | ||
| Fig. 12 Chart comparing particle length versus the ranking of each particle in a partial dissolution series of 1 doped with 4.5 mol% of 3. | ||
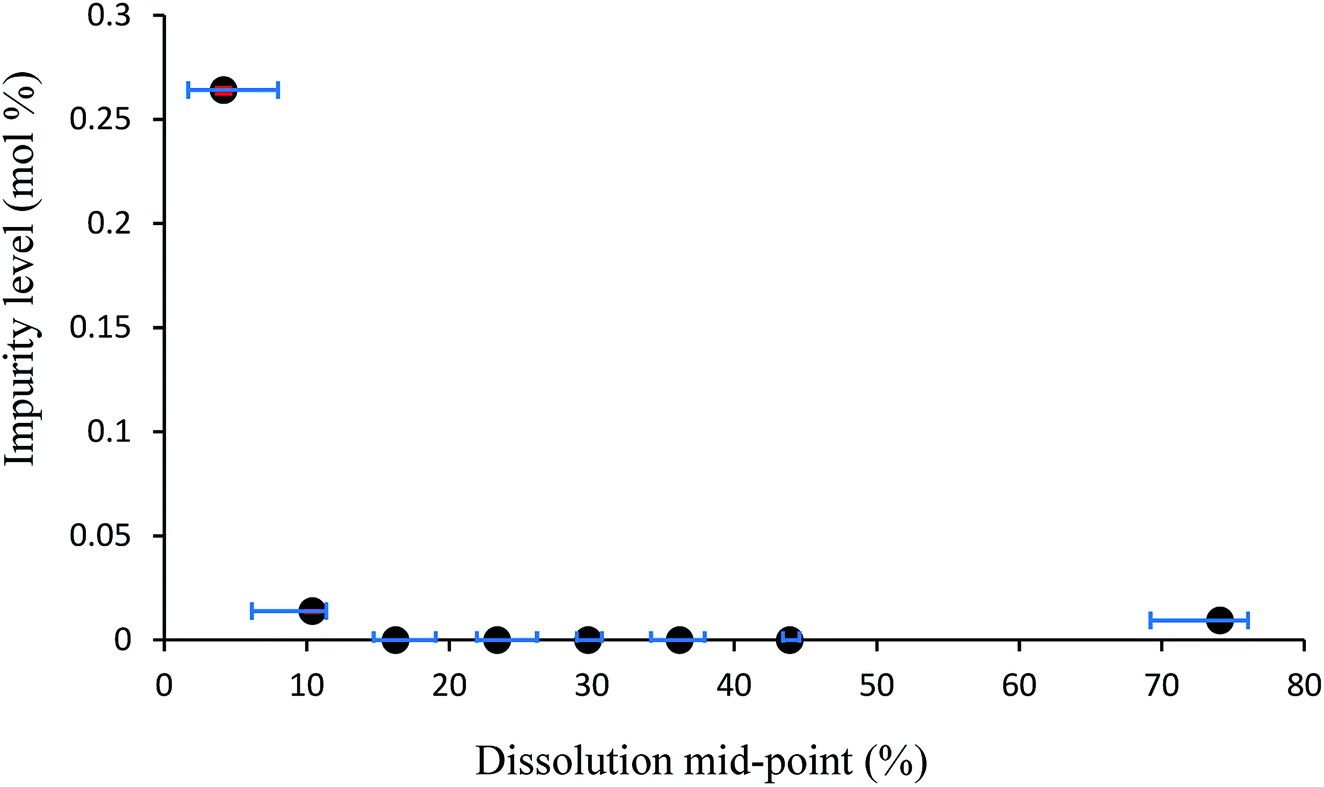 | ||
| Fig. 13 Plot of percentage by HPLC of added impurity in crystals of compound 1 vs. the dissolution mid-point for the sample of crystals grown from solutions containing 4.5 mol% of additive 3. Error bars show the upper and lower limits of experimental values from which the averages were taken to assign datapoints. | ||
Conclusions
Crystallisation is a method of purification commonly employed in the pharmaceutical and chemical manufacturing industries. The conditions under which crystallisations are carried out have varying effects on the material being precipitated such as level of included impurity, polymorphic form, particle size and morphology. During process optimisation, it is important to know how impurity incorporation progresses to ensure levels are below specification limits, which is why a method that can determine the location and proportion of impurities within batch samples would be advantageous in guiding purification procedures. Such a method would be particularly useful with low-level molecular impurities, especially when the crystal samples analysed by PXRD and DSC display no physically distinct phases as a result of impurity incorporation that could be determined by either analytical method.In previous work, a method of dissolving samples in sequential layers by agitating them in a liquid with a low solvation ability and analysing the resulting solution and partially dissolved solid sample was developed.30 For this previous method to be effective, it required the liquid phase to act as both the solvent and continuous phase. To develop partial dissolution media that could be used for larger numbers of particles, approaching the size of a sample from a process batch, it is preferable that the dissolution and suspension functions be separated. The method reported herein utilised a bi-phasic system, in which one phase (50% aq. ethanol) carried out the dissolution, while the second phase (perfluorohexane) acted as a liquid vehicle. The dissolution medium also consisted of a non-ionic surfactant (4) containing both organic and fluorocarbon segments to prevent agglomeration of particles. The samples being analysed consisted of multiple crystalline particles of host compound 4-chloro-2-nitro-acetanilide (1), with varying quantities of guest impurities 4-methyl-2-nitro-acetanilide (2) or 4-tert-butyl-2-nitro-acetanilide (3). A key feature is that the degree of dissolution would be determined by the amount of the solvent phase added, rather than, as previously, the time allowed for dissolution to proceed.
Multi-particle samples were dissolved in several partial dissolution steps attempting to control the quantity of material dissolved in each step. The dissolution was provided by the 50% aq. ethanol phase while perfluorohexane provided the vehicle for the medium. Residual solid samples were measured using a particle sizing microscope to determine the area and length changes with each subsequent step. HPLC analysis was performed on samples containing the dissolved material and the proportions of guest impurity and host compound were determined. It can be concluded that the method was effective in controlling the relative amount of material dissolved by comparing it to theoretical values of the expected sizing measurements. HPLC analysis for particles of compound 1 containing impurity 2 showed a general trend of a slight increase in concentration of guest impurity towards the inner sections of the crystal, with the largest quantities found in the final dissolution step. HPLC analysis for particles composed of compound 1 containing impurity 3 showed a general trend of low to no incorporation of the guest impurity within the crystals. Inclusion of impurity 3 chiefly remained on the outer layer of the crystal, likely to be from solution deposition. The contrast between the behaviours of impurities 2 and 3 is not surprising, given that compound 2 is a known isomorphic lattice replacement for compound 1,26 whereas compound 3 contains a sterically demanding tert-butyl group which was unlikely to be well accommodated with the crystal lattice of compound 1. The key point of the study is that it provides an independent determination of those behaviours. The approach is compatible with conventional composition and size analytical methods and, importantly, the degree of dissolution is determined by the volume of solvent (at a particular temperature) rather than the time allowed for agitation.
The two phase dissolution medium was successful in providing a controlled degree of dissolution while providing for particle flocculation to be minimised or inhibited. The approach allowed mapping of the distribution of two contrasting impurities, one (compound 2) known to act as an isosteric lattice replacement for the crystallising compound, and hence likely to be evenly distributed; the other featuring a sterically demanding group which renders its distribution in the crystallisation host less predictable and less uniform, as was confirmed by the stepwise dissolution study. While the compounds studied were relatively simple, the principles of the method is general and could be advanced to more complex pharmaceutical and fine chemical systems.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research has been carried out with the support of Science Foundation Ireland under Grant Number 12/RC/2275.References
- H. A. Moynihan and D. E. Horgan, Org. Process Res. Dev., 2017, 21, 689–704 CrossRef CAS.
- D. A. Pierson, B. A. Olsen, D. K. Robbins, K. M. DeVries and D. L. Varie, Org. Process Res. Dev., 2009, 13, 285–291 CrossRef CAS.
- J. P. Bercu and C. M. Callis, Org. Process Res. Dev., 2009, 13, 938 CrossRef CAS.
- S. N. Black, K. Quigley and A. Parker, Org. Process Res. Dev., 2006, 10, 241–244 CrossRef CAS.
- S. Black, L. Dang, C. Liu and H. Wei, Org. Process Res. Dev., 2013, 17, 486–492 CrossRef CAS.
- C. G. Moyers, Chem. Eng. Prog., 1986, 82, 42–46 CAS.
- J. A. Hayes, K. S. Eccles, S. E. Lawrence and H. A. Moynihan, Carbohydr. Res., 2012, 349, 108–112 CrossRef CAS.
- G. J. Tanoury, R. Hett, D. W. Kessler, S. A. Wald and C. H. Sennayake, Org. Process Res. Dev., 2002, 6, 855–862 CrossRef CAS.
- N. Blagden, Powder Technol., 2001, 121, 46–52 CrossRef CAS.
- K. A. Solanko and A. D. Bond, CrystEngComm, 2011, 13, 6991–6996 RSC.
- T. Mukuta, A. Y. Lee, T. Kawakami and A. S. Myerson, Cryst. Growth Des., 2005, 5, 1429–1436 CrossRef CAS.
- M. Mirmehrabi, S. Rohani, K. S. K. Murthy and B. Radatus, Cryst. Growth Des., 2006, 6, 141–149 CrossRef CAS.
- Z. Berkovitch-Yellin, L. Addadi, M. Idelson, M. Lahav and L. Leiserowitz, Angew. Chem., Int. Ed. Engl., 1982, 1336–1345 CrossRef.
- N. Blagden, M. Song, R. J. Davey, L. Seton and C. C. Seaton, Cryst. Growth Des., 2005, 5, 467–471 CrossRef CAS.
- M. M. Hansen, D. J. Jarmer, E. Arslantas, A. C. DeBaillie, A. L. Frederick, M. Harding, D. W. Hoard, A. Hollister, D. Huber, S. P. Kolis, J. E. Kuenhe-Willmore, T. Kull, M. E. Laurila, R. J. Linder, T. J. Martin, J. R. Martinelli, M. J. McCulley, R. N. Richey, D. R. Starkey, J. A. Ward, N. Zaborenko and T. Zweifel, Org. Process Res. Dev., 2015, 19, 1214–1230 CrossRef CAS.
- N. Yoshikawa, F. Xu, J. D. Arrendondo and T. Itoh, Org. Process Res. Dev., 2011, 15, 824–830 CrossRef CAS.
- A. Isidro-Llobet, M. Alverez and F. Albericio, Chem. Rev., 2009, 109, 2455–2504 CrossRef CAS.
- S. Sankareswaran, M. Mannam, V. Chakka, S. R. Mandapati and P. Kumar, Org. Process Res. Dev., 2016, 20, 1461–1468 CrossRef CAS.
- A. Stumpf, A. McClory, H. Yajima, N. Segraves, R. Angelaud and F. Gosselin, Org. Process Res. Dev., 2016, 20, 751–759 CrossRef CAS.
- S. Duan, D. Place, H. H. Perfect, N. D. Ide, M. Maloney, K. Sutherland, K. E. Price Wiglesworth, K. Wang, M. Olivier, F. Kong, K. Leeman, J. Blunt, J. Draper, M. McAuliffe, M. O'Sullivan and D. Lynch, Org. Process Res. Dev., 2016, 20, 1191–1202 CrossRef CAS.
- Q. Wen, J. Jin, L. Zhang, Y. Luo, P. Lu and Y. Wang, Tetrahedron Lett., 2014, 55, 1271–1280 CrossRef CAS.
- Y. Deng, Q. Xie, M. G. LaPoprte, A. T. A. Chasnoff, M. A. Mortensen, D. Patra, S. A. Putrelo, R. S. Antonovich, H. Coa, Y. Yan, A. J. Cooper, S. R. Rippin, M. D. Alexander, P. T. Kumar, M. S. Hendi, Y.-H. Lee, T. Hainmowitz and S. M. Condon, Org. Process Res. Dev., 2016, 20, 242–252 CrossRef CAS.
- M. T. Maloney, B. P. Jones, M. A. Olivier, J. Magano, K. Wang, N. D. Ide, A. S. Palm, D. R. Bill, K. R. Leeman, K. Sutherland, J. Draper, A. M. Daly, J. Keane, D. Lynch, M. O'Brien and J. Touhy, Org. Process Res. Dev., 2016, 20, 1203–1216 CrossRef CAS.
- B. Y. Shekunov, S. Bristow, A. H. L. Chow, L. Cranswick, D. J. W. Grant and P. York, Cryst. Growth Des., 2003, 3, 603–610 CrossRef CAS.
- H. Kaemmerer, H. Lorenz, S. N. Black and A. Seidel-Morgenstern, Cryst. Growth Des., 2009, 9, 1851–1862 CrossRef CAS.
- X. He, J. G. Stowell, K. R. Morris, R. R. Pfeiffer, H. Lui, P. Stahly and S. R. Byrn, Cryst. Growth Des., 2001, 1, 305–312 CrossRef CAS.
- S. Ottoboni, M. Chrubasik, L. M. Bruce, T. T. H. Nguyen, M. Robertson, B. Johnson, I. D. H. Oswald, A. Florence and C. Price, Cryst. Growth Des., 2018, 18, 2750–2758 CrossRef CAS.
- L. Addadi, S. Weinstein, E. Gati, I. Weissbuch and M. Lahav, J. Am. Chem. Soc., 1982, 104, 4610–4617 CrossRef CAS.
- D. E. Horgan, L. M. Crowley, S. P. Stokes, S. E. Lawrence, H. A. Moynihan, Crystallization, ed. Y. Mastai, InTech, 2015, Rijeka, Croatia, ISBN 978-953-51-2125-1 Search PubMed.
- H. A. Moynihan and D. Armstrong, CrystEngComm, 2018, 20, 2617–2633 RSC.
- J. N. Israelachvili, Intermolecular and Surface Forces, Elsevier, Singapore, 3rd edn, 2012, ch. 14 and 16, ISBN 978-0-12-375182-9 Search PubMed.
- M. Selin, S. Nummelin, J. Deleu, J. Ropponen, T. Viitala, M. Lahtinen, J. Koiovisto, J. Hirvonen, L. Peltonen, M. A. Kostiainen and L. M. Bimbo, Biomacromolecules, 2018, 19, 3983–3993 CrossRef CAS.
- H. Oguzlu, C. Damumah and Y. Boluk, Curr. Opin. Colloid Interface Sci., 2017, 29, 46–56 CrossRef CAS.
- B. Akpinar, L. A. Fielding, V. J. Cunningham, Y. Ning, O. O. Mykhaylyk, P. W. Fowler and S. P. Armes, Macromolecules, 2016, 49, 5160–5171 CrossRef CAS.
- D. H. Napper, Ind. Eng. Chem. Prod. Res. Dev., 1970, 9, 467–477 CrossRef CAS.
- H. Batra, R. Penmasta, K. Phares, J. Staszewski, S. M. Tuladhar and D. A. Walsh, Org. Process Res. Dev., 2009, 13, 242–249 CrossRef CAS.
- A. Dokoumetzidis and P. Macheras, Int. J. Pharm., 2006, 321, 1–11 CrossRef CAS.
- A. P. Tinke, K. Vanhoutte, R. De Maesschalck, S. Verheyen and H. De Winter, J. Pharm. Biomed. Anal., 2005, 39, 900–907 CrossRef CAS.
- M. Cametti, B. Crousse, P. Metrangelo, R. Milani and G. Resnati, Chem. Soc. Rev., 2012, 41, 32–42 RSC.
- M. P. Krafft and J. G. Riess, Chem. Rev., 2009, 109, 1714–1792 CrossRef CAS.
- M. P. Krafft, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4251–4258 CrossRef CAS.
- W. Huang, C. Jin, D. K. Derzon, T. A. Huber, J. A. Last, P. P. Provencio, A. S. Gopalan, M. Dugger and D. Y. Sasaki, J. Colloid Interface Sci., 2004, 272, 457–464 CrossRef CAS.
- J. G. Riess and J. Greiner, Carbohydr. Res., 2000, 327, 147–168 CrossRef CAS.
- T. Hiyama, Organofluorine Compounds, Chemistry and Applications, Springer Science and Business Media, 2013, ISBN 9783662041642 Search PubMed.
- C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, VCH, 1988, ISBN 0-89573-684-5 Search PubMed.
- G. Berti, G. Catelani, B. Lombardi and L. Monti, Gazz. Chim. Ital., 1977, 107, 175–180 CAS.
- A. V. Zeiger and M. M. Joullié, J. Org. Chem., 1977, 42, 542–545 CrossRef CAS.
- S. Krakert, N. Ballav, M. Zharnikov and A. Terfort, Phys. Chem. Chem. Phys., 2010, 12, 507–515 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02781e |
| This journal is © The Royal Society of Chemistry 2019 |