
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of healing phenotype-inducing cytokine formulations within soft hydrogels on encapsulated monocytes and incoming immune cells
Ivana Ščigalkováa,
Julie Bystroňováa,
Lenka Kovářováag,
Martin Pravdaa,
Vladimír Velebnýa,
Vladimir Riabovbc,
Harald Klüterbd,
Julia Kzhyshkowskabcd and
Nihal Engin Vrana *ef
*ef
aContipro a.s., Dolni Dobrouc 401, 561 02 Dolni Dobrouc, Czech Republic
bInstitute for Transfusion Medicine and Immunology, Medical Faculty Mannheim, University of Heidelberg, Theodor-Kutzer Ufer 1-3, 68167 Mannheim, Germany
cLaboratory for Translational Cellular and Molecular Biomedicine, Tomsk State University, 36 Lenin Prospekt, Tomsk 634050, Russia
dRed Cross Blood Service Baden-Württemberg–Hessen, Friedrich-Ebert Str. 107, D-68167 Mannheim, Germany
eProtip Medical, 8 Place de l'Hôpital, 67000 Strasbourg, France. E-mail: e.vrana@protipmedical.com
fInserm UMR 1121, Biomaterials and Bioengineering, 11 rue Humann, 67085 Strasbourg, France
gInstitute of Physical Chemistry, Faculty of Chemistry, Brno University of Technology, Purkynova 464/118, 612 00 Brno, Czech Republic
First published on 10th July 2019
Abstract
The adverse immune responses to implantable biomedical devices is a general problem with important consequences for the functionality of implants. Immunomodulatory soft hydrogel-based interfaces between the implant and the host can attenuate these reactions. Moreover, encapsulation of the patient's own immune cells into these interfaces can lead to the personalisation of implants from the immune reaction point of view. Herein, we described a co-crosslinkable composite hydrogel (composed of gelatin and hyaluronic acid), which could be used for the encapsulation of macrophages in the presence of an anti-inflammatory phenotype-fixing cytokine cocktail. To mimick the incoming immune cells on the coating surface in vivo, peripheral blood mononuclear cells were seeded on the hydrogels. The encapsulation of monocytic cells into the composite hydrogels in the presence of cytokine cocktails at 5× or 10× concentrations led to the spreading of the encapsulated cells instead of the formation of clusters. Moreover, the secretion of the anti-inflammatory cytokines IL-1RA and CCL-18 was significantly increased. The attachment of PBMC to the surface of the hydrogel is dependent on the hydrogel composition and also significantly increased in the presence of the cytokine cocktail together with the number of CD68+ cells on the hydrogel surface. Our study demonstrates that the delivery of a polarisation cocktail with biocompatible hydrogels can control the initial response by the incoming immune cells. This effect can be improved by the encapsulation of autologous monocytes that are also polarised by the cytokine cocktail and secrete additional anti-inflammatory cytokines. This interface can fine tune the initial immune response to an implanted biomaterial in a personalised manner.
Introduction
Implantable medical devices have established themselves as a robust solution to myriad medical conditions in a variety of forms. From pacemakers to dental implants to engineered artificial tissues, these systems can take over the original function of the lost organ or tissue to a certain extent, which provide the patients with an improved quality of life. However, although these systems are generally biocompatible, the presence of foreign materials generate immune responses,1 which start due to the presence of immune cells related to innate immunity. The extent and severity of the response depends on the location of the intervention, the physicochemical properties (surface roughness, hydrophilicity, topography, etc.) of the implanted material2,3 and the nature (natural, synthetic, degradable, non-degradable, etc.) of the material. Moreover, the patient's unique immunoprofile plays an important part in the outcome of these implantations. Currently, the personalization of implants using additive manufacturing techniques for better anatomical and biomechanical fits has made significant advances.4,5 Another potential aspect of personalization that has been advancing slowly but is equally important is the personalization of the implant/host tissue interface for controlling patient-specific immune reactions.Macrophages are innate immune cells that act as phagocytes and antigen-presenting cells. Tissue-resident macrophages either originate from the yolk sac and the fetal liver or they are recruited from the peripheral blood mononuclear cells, namely, the peripheral blood monocytes. The recruitment and differentiation of monocytes into macrophages are triggered by various stimuli, mostly related to infection, tissue damage and inflammation. Depending on the local microenvironment and the present cytokines, macrophages can polarize to M1 or M2 subtypes.6–8 The M1-type macrophages are associated with inflammation during the early stages of wound healing. They produce pro-inflammatory cytokines and clear the tissue of microorganisms, damaged cells and matrix debris. Later, the M1-type macrophages switch to the M2-type macrophages, which contribute to tissue repair by releasing various growth factors and cytokines that promote angiogenesis and cell proliferation.9,10
Macrophage differentiation and polarization in vitro can be promoted by the signals observed in vivo: the monocytes are activated by M-CSF (for M2) or GM-CSF (for M1)11–13 and then polarized by the cytokines. Usually, IFN-γ, TNF-α and bacterial lipopolysaccharide (LPS) are used for M1 polarization, whereas IL-4 and IL-13 or IL-10 are used for M2 polarization.6,9,14 More recently, we have developed a mixture of M-CSF, IL-4, IL-10 and TGF-β1 and demonstrated that it is efficient in macrophage polarization and stabilization of the M2 phenotype.15,16 M-CSF is a hematopoietic growth factor present in blood. It is used for the in vitro activation of monocytes prior to their M2 polarization.15 IL-4 was the first cytokine recognized to have an immunomodulatory effect.17 It was originally recognized as an inhibitor of classical macrophage activation; however, now, it is considered to be an inducer of M2 alternative activation.6 TGF-β1 is a growth factor with a wide range of actions in various tissues: it is involved in the development and homeostasis of bone, cartilage and skin, T-cell and B-cell maturation and regulation of the immune response.18 IL-10 is an anti-inflammatory cytokine that is produced similarly as IL-4 by Th2 lymphocytes; however, macrophages are also a major source of IL-10 during infection. The production of IL-10 leads to the inhibition of inflammation and enhanced survival of cells.19
However, the M1 or M2 phenotype is not permanent. Macrophage differentiation is a dynamic process influenced by both exogenous and endogenous stimuli. In response to them, the macrophages can rapidly switch from one phenotype to the other.6,20 Therefore, a smooth transition from the inflammatory stage to the healing stage (M1 to M2 switch) and stabilization of the M2 phenotype are essential for the successful integration of the implanted system with the host tissue; significant attention is being paid towards controlling the immune response around the implants, and various immunomodulatory strategies have been evolved.
In this regard, one of the immunomodulatory strategies is the employment of biomaterials as a coating for the implant. The coating would affect the differentiation and polarization of the patient's incoming monocytes/macrophages.10 It is advantageous to use materials derived from or mimicking the extracellular matrix for this purpose. Hyaluronic acid (HA) and gelatin (GTN) fulfill these requirements. Hyaluronan is one of the main components of the extracellular matrix, and it is involved in the regulation of wound healing, angiogenesis and immunomodulation.11,21–24 Due to its polyanionic nature, it has been widely used in coatings.25 Gelatin is a natural polymer prepared by the partial hydrolysis of collagen, the chief component of connective tissues. The hyaluronic acid-tyramine conjugate (HA-TA) and gelatin modified by hydroxyphenyl propionic acid (GNT-HPA) have been recently described as biopolymer derivatives for the preparation of covalently crosslinked hydrogels suitable for application in the field of tissue engineering and regenerative medicine.26 Due to the presence of hydroxyphenyl moieties, both modified biopolymers undergo a horseradish peroxidase (HRP)-mediated co-crosslinking reaction, which can be performed under physiological conditions in the presence of living cells.27
Although GTN/HA hydrogels have been widely used in cell encapsulation applications, mostly for cartilage tissue engineering,28 they have not been studied with combined anti-inflammatory cytokine cocktail secretion. HA and collagen hydrogels have been used separately for immunomodulation but not in a co-crosslinkable configuration.29,30 Both GTN and HA have pendant groups that enable the development of their derivatives. The advantages of a blend of GTN and HA are the simultaneous use of the cell adhesive sequences in gelatin for the viability and the movement of the encapsulated cells, the ability of HA to retain the growth factors and high volume of water and the intrinsic interactions between GTN and HA that further reinforce the structures. Moreover, the immunomodulatory effect of the coating can be intensified when the material is combined with appropriate cytokines, and the use of multiple cytokines/hormones/growth factors can have distinct synergistic effects. These cytokines are released after implantation and modulate the local microenvironment in the wound. Both GTN and HA-TA have the ability to bind and release small molecules such as cytokines, growth factors or some drugs.16,25,31,32 Thin coatings based on HA were used as reservoirs for the anticancer drug paclitaxel, which was released via diffusion. The anti-inflammatory cytokine IL-4 was loaded onto the HA-aldehyde-based films with subsequent attenuation of the secretion of pro-inflammatory cytokines by primary macrophages. Transglutaminase-crosslinked gelatin structures can be loaded and can release growth factors and cytokines. The loading efficacy of the cytokines on these coatings was estimated to be 10%. The films based on transglutaminase-crosslinked GTN and HA-TA were prepared by spin coating and used as a release system for the cytokine mixture. The coating can be loaded with a single cytokine25 or with a mixture of two or more cytokines. A release system with sequential delivery of IFN-γ and IL-4 was developed to facilitate the M1 to M2 switch.33,34 The employment of a mixture of anti-inflammatory cytokines, i.e. IL-4, IL-10 and TGF-β1, resulted in the stabilization of the M2 phenotype in the monocytes seeded on the HA-TA + GTN-based coatings.16
In addition to the employment of biomimetic coatings and the delivery of cytokines, another strategy is the isolation of the patient's immune cells followed by their encapsulation in the coating-material and in vitro polarization into the M2-type macrophages for personalization of the implant/host interface. After implantation, the encapsulated M2 cells would produce desired cytokines and create a local healing supporting microenvironment that would lead to the M1-to-M2 polarization of the incoming monocytes/macrophages. Previously, we have shown the beneficial effects of the encapsulation of cells in degradable natural polymer-based hydrogels on the development of anti-inflammatory phenotypes using the model monocyte cell line THP-1.35,36 However, how the presence of an M2 phenotype-inducing cytokine mixture would affect the polarization of encapsulated monocytes/macrophages in a 3D system has not been investigated to date. Moreover, the potential effects of the abovementioned cytokine cocktail on other immune cells (cells present in the peripheral mononuclear blood – PBMCs) have not been analyzed.
In this study, our aim was to determine the effect of a cytokine cocktail on the encapsulated monocytes in an ECM mimicking hydrogel and the potential responses of the incoming immune cells. In the first part of this study, we investigated the effect of the cytokine mixture and concentrations of cytokines on the encapsulated THP-1 cells. The second part of this study deals with the interaction of peripheral blood mononuclear cells (PBMC) with the hydrogel layers that imitates the interaction of the coated implant with the patient's incoming immune cells. We investigated the ability of PBMCs to adhere to the hydrogel coatings with different contents of HA-TA and GTN-HPA and the effect of the cytokine mixture on the PBMC attachment and phenotype of its components.
Results and discussion
Elastic modulus
The viscoelastic properties of hydrogels based on three initial types of material (HA-TA, HA-TA with GTN-HPA and GTN-HPA) were determined. Elastic (G′) modulus, viscous (G′′) modulus and loss angle (δ) were chosen as the parameters for the evaluation of the viscoelastic properties. Since material stiffness can affect the cell behavior, it is necessary to ensure that the tested materials have similar properties. The concentration of the crosslinking agents was decreased in the case of the HA-TA material (Fig. 1A) to achieve hydrogel viscoelastic properties comparable to those of the HA-TA + GTN-HPA (Fig. 1B) and GTN-HPA (Fig. 1C) materials. A comparison of the elastic moduli of hydrogels determined from the plateau region of different material types is shown in the graph in Fig. 1D. The HA-TA + GTN-HPA hydrogels had only slightly higher G′ values (∼1.2 kPa) as compared to the GTN-HPA hydrogels (∼1.0 kPa).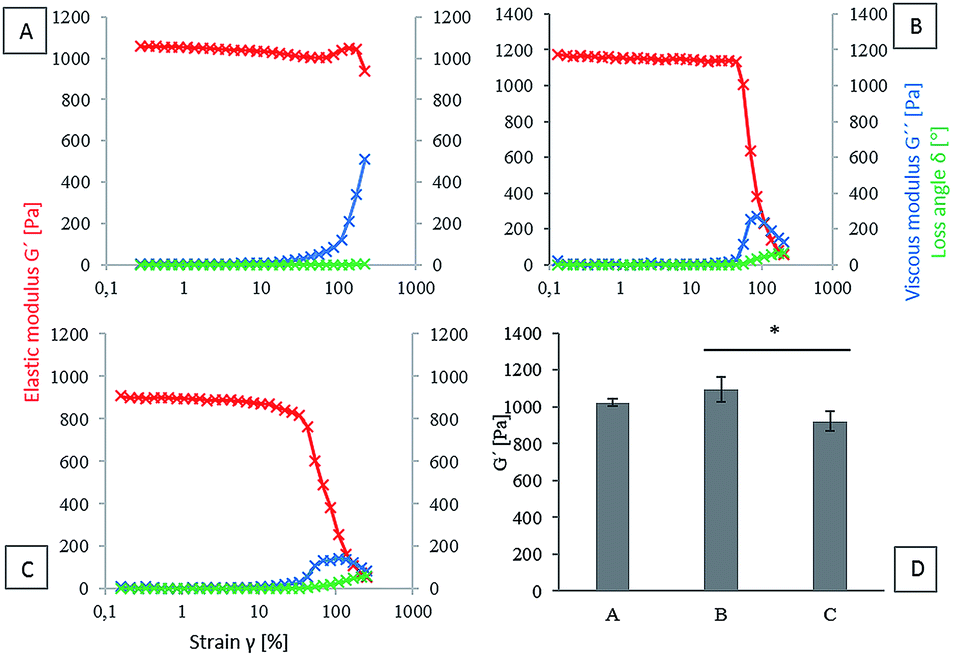 | ||
| Fig. 1 (A–C) The elastic modulus G′, viscous modulus G′′ and the loss angle δ of the hydrogels of different compositions prepared using crosslinking agents at various concentrations. (A) The hydrogel based on HA-TA, HRP 0.16 U ml−1, H2O2 0.002% (B) the hydrogel based on HA-TA with GTN-HPA, HRP 0.4 U ml−1, H2O2 0.005% (C) the hydrogel based on GTN-HPA, HRP 0.4 U ml−1, H2O2 0.005%. (D) The hydrogel elastic modulus determined from the plateau region of various types of materials. The data are presented as mean ± SD, n = 3. | ||
Effect of increased concentration of the CK cocktail on the encapsulated THP-1
In the first part of this study, the THP-1 cells were encapsulated into the HA-TA + GNT-HPA hydrogels followed by culture for 8 days in a standard culture medium (w/o CK) or in a medium containing cytokines at doses ranging from 10 ng ml−1 M-CSF, IL-10, and TGF-β1 and 3 ng ml−1 IL-4 (CK 1×) to 100 ng ml−1 M-CSF, IL-10, and TGF-β1 and 30 ng ml−1 IL-4 (CK 10×). After 8 days, the medium with the cytokines was removed, and the encapsulated cells were cultured in a culture medium without the cytokines for the next 7 days (Fig. 2A).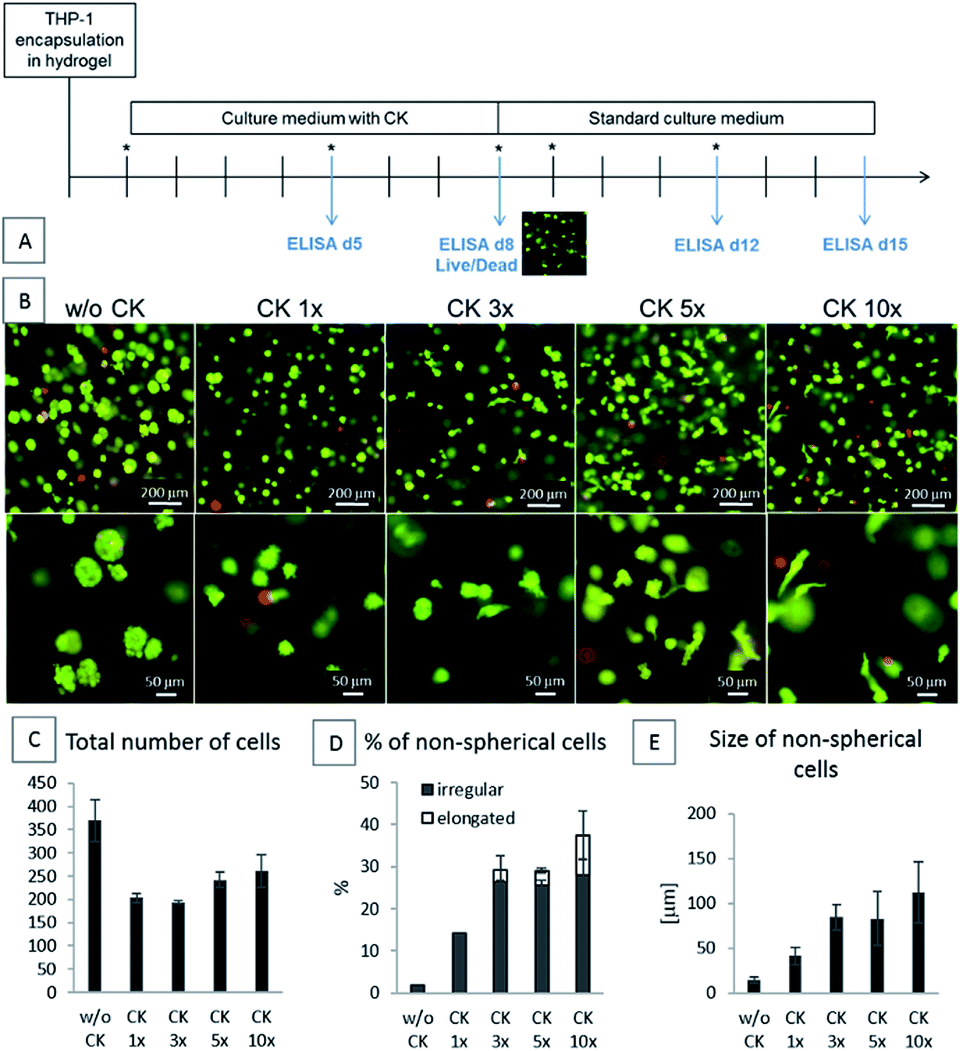 | ||
| Fig. 2 Influence of the CK concentration on the morphology of the encapsulated cells after 8 days. (A) Schematic of the experiment. Asterisks represent the change of medium, and the blue arrows represent the collection of supernatants for ELISA. (B) Live/dead staining; first row – overall view, second row – detailed view focused on the cell shape. (C) Total number of cells per image. (D) Percentage of cells in the hydrogels with irregular and elongated morphology. (E) Average size of the elongated cells in the hydrogels. The graphs (C–E) were obtained by image analysis of the live/dead images. Image analysis was performed for at least 3 images, and each image was obtained from a different piece of hydrogel. The data are presented as mean ± SD. | ||
The phenotype of the encapsulated cells was dependent on the concentration of CK in the culture medium. The cells cultured in the hydrogels in a medium without CK remained spherical and created spheroid-like clusters. The cells cultured with CK at increasing concentrations gained either irregular or elongated shapes and did not create clusters. With the increasing concentration of CK, more elongated cells were present in the hydrogel (Fig. 2).
The production of all cytokines by the encapsulated cells was proportional to the concentration of CK used for their differentiation. The release of IL-1RA, TNF-α and IL-1β was dependent on the presence of CK in the culture medium and decreased rapidly after CK was removed. The production of the M2 marker CCL18 was delayed, which manifested in the second week of cultivation and reached its maximum at day 12. The production of M2 markers in the encapsulated cells corresponded to their production in the non-encapsulated cells when CK 5× and CK 10× were applied. Under the encapsulation conditions with CK 10×, by day 12 and 15, CCL-18 was produced more significantly as compared to the case of the non-encapsulated control (p = 0.004). However, when CK at these concentrations was present in the medium (i.e. at day 5 and day 8), significantly more TNF-α was produced by the encapsulated cells than by the non-encapsulated cells (Fig. 3). However, note that even at its highest levels, the pro-inflammatory cytokine production is 2 magnitudes lower than that of the anti-inflammatory cytokines.
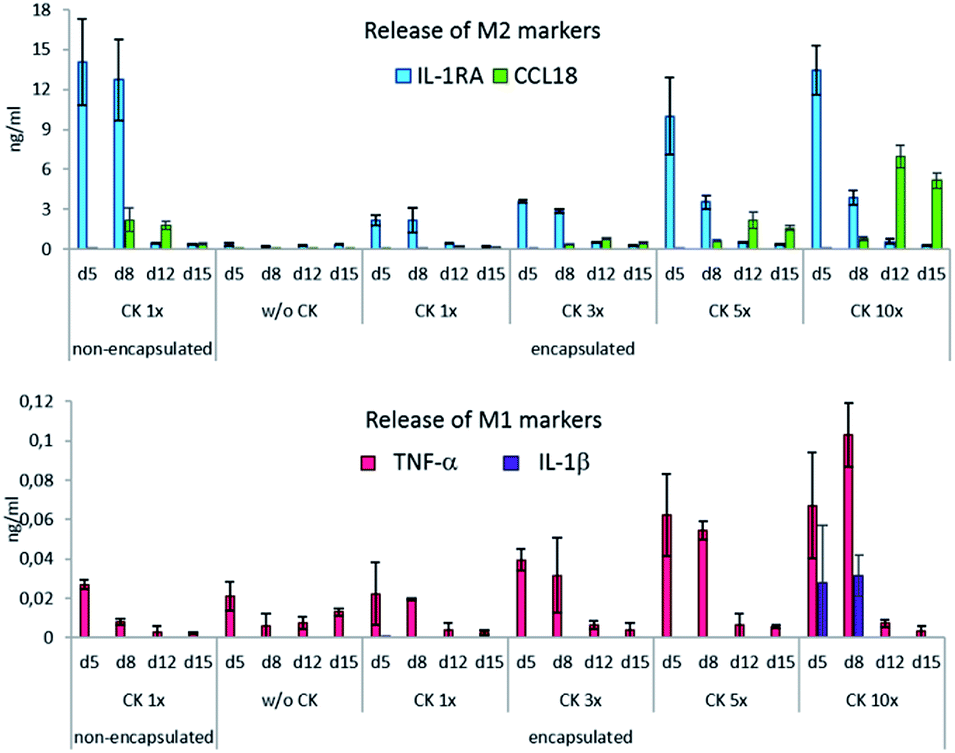 | ||
| Fig. 3 Cytokine release by the encapsulated cells. Cytokine release by the non-encapsulated and encapsulated THP-1 cells stimulated by the increasing dose of CK. ELISA. The data are presented as mean ± SD, n = 3. | ||
PBMC adhesion to coatings of different compositions
As the patient's immune cells are expected to interact with the implants, we have studied the interaction and ability of the PBMCs to attach to the coatings made from HA-TA, HA-TA + GTN-HPA and GTN-HPA. PMA was added to the culture medium to induce the adhesion of the suspended cells.The cells did not attach to the hydrogel coatings consisting of pure HA-TA but attached to the HA-TA + GNT-HPA and GTN-HPA hydrogels, where they maintained rounded morphology. When PMA was added, more cells attached to the hydrogels and tended to grow in groups (Fig. 4).
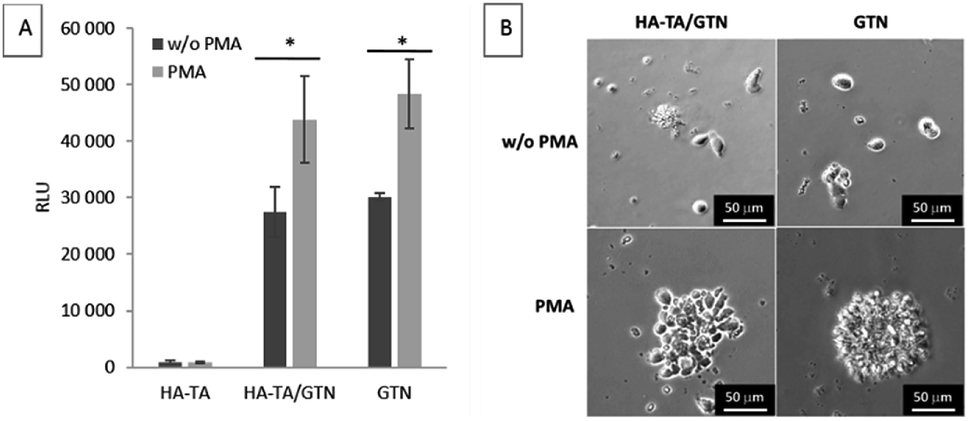 | ||
| Fig. 4 PBMC adhesion to the hydrogel coatings after 4 days. (A) Cell adhesion to the hydrogel coatings. CellTiter-Glo assay, day 4. The data are presented as mean ± SD, n = 3. (B) Morphology of the attached cells. | ||
PBMC attachment to the hydrogel coatings pretreated with CK
Since the PBMCs did not attach to the coating made from pure HA-TA, the following experiment was performed only with the coatings made from HA-TA + GTN-HPA and GTN-HPA. The coatings were allowed to absorb the culture medium with or without five- or ten-times increased concentration of the cytokine mixture (w/o CK, CK 5× or CK 10×) for three days. After this, the medium was removed, and PBMCs in a fresh medium without CK were seeded on the hydrogel coatings. The cell attachment and the phenotype were assessed after 4 days (Fig. 5A).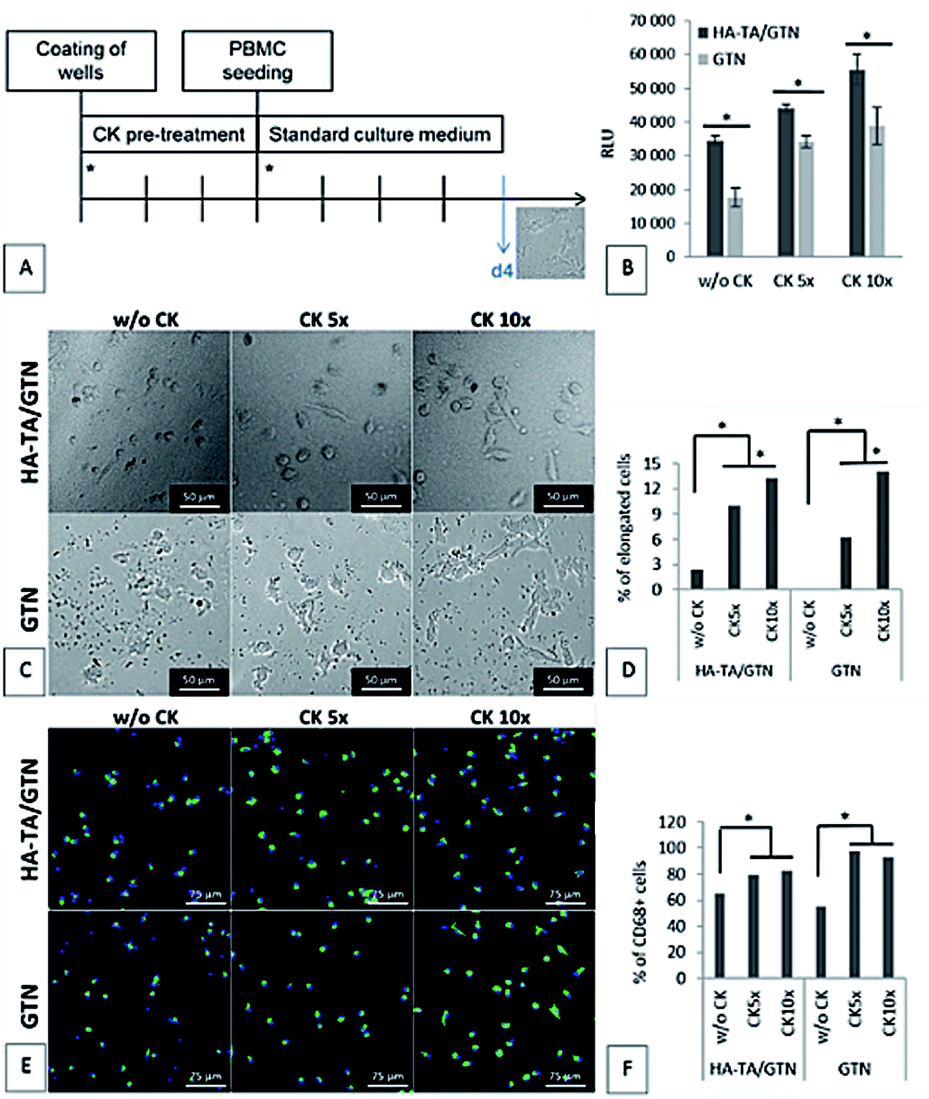 | ||
| Fig. 5 PBMC adhesion to the coatings after 4 days. (A) Schematic of the experiment. Asterisks represent change of medium. (B) Cell adhesion to the hydrogel coatings. CellTiter-Glo assay. The data are presented as mean ± SD, n = 3. (C–F) Phenotype of the cells attached to the hydrogel coatings: (C) morphology of the attached cells. (D) Percentage of the elongated cells. (E) CD68 immunostaining of the attached cells. CD68 – green, cell nuclei – blue. (F) Percentage of the CD68+ cells. The number of elongated cells and the number of CD68+ cells were obtained by image analysis of the microphotographs. | ||
The cell number and phenotype were strongly influenced by the CK pre-treatment on both types of hydrogel coatings; significantly more cells attached to the materials pretreated with CK than to the materials without CK (Fig. 5D). Although the average number of cells attached to the materials was higher for the samples pretreated with CK 10× than the case of CK 5×, the difference was not statistically significant. A similar pattern was observed for the presence of CD68+ macrophages. Moreover, the ten-times increased concentration of CK used in the pre-treatment resulted in higher portion of cells with elongated morphology than the five-times increased concentration of CK.
Discussion
Elastic modulus
The aim of this study was to evaluate the influence of material composition on macrophage polarization. Since the mechanical properties of a material can affect the polarization of macrophages,39,40 it is necessary to ensure that the tested materials have similar properties. In our previous study, we have demonstrated that the HRP-mediated crosslinking reaction of HA-TA- and GTN-HPA-based polymers is a highly tunable system where the degree of crosslinking and therefore the stiffness of the hydrogel can be adjusted by the concentration of hydrogen peroxide and activity of HRP.36 To achieve comparable mechanical strength between hydrogels containing different ratios of HA-TA and GTN-HPA, decreased concentration of the crosslinking agents was used in the case of the HA-TA hydrogel. The prepared HA-TA + GTN-HPA hydrogels had only slightly higher G′ values as compared to the GTN-HPA hydrogels; the other mechanical properties were comparable among all the three materials. This ensures that the effects are due to the composition rather than the mechanical properties as it has been shown that the mechanical properties can affect the macrophage/immune cell behavior, where on the softer hydrogels, there was less pro-inflammatory cytokine secretion,40 and with polyethylene glycol (PEG) hydrogels, it was shown that the softer hydrogels (in the range of 100–800 kPa) resulted in less activation of the macrophages.Effect of increased concentration of the CK cocktail on the encapsulated THP-1
The immunomodulatory effect of a biomaterial can be achieved by the incorporation of monocytes and their in vitro polarization within the material. The monocytes are usually activated by M-CSF and then polarized with anti-inflammatory cytokines such as IL-4, IL-10, and IL-13 or their mixture.9 Polarization can also be achieved by a specific mixture of IL-4, IL-10 and TGF-β1, which seems to be more efficient in the long-term stabilization of the M2 phenotype.15,16 Although the typical concentration of cytokines for in vitro polarization ranges from 10 to 50 ng ml−1, when the cytokines are loaded onto a release system or used for the polarization of the encapsulated cells, higher concentrations are used.16 For example, in the foam configuration, up to 325 ng per scaffold of the cytokines was used to ensure biological activity.33 However, the limits for the potential adverse effects of excessive cytokine presence need to be quantified. Previously, a cocktail system was used to induce M1-like polarization using a mixture of MCP-1, IL-6 and IFN-γ,11 where the presence of sulphated HA decreased the ability to induce M1 polarization. The aim of the first part of the study was to identify how much higher concentration of CK must be present in a culture medium for the encapsulated cells to undertake a phenotype corresponding to the phenotype of the non-encapsulated cells cultured with the standard concentration of CK.We have identified that five- or ten-times increased concentration of CK must be used for the activation of the encapsulated THP-1 cells. A majority of the activated cells gained non-spherical, irregular morphology, and a smaller portion was elongated. According to some researchers, M2-type macrophages have mixed irregular and elongated morphology with predominant elongated morphology11,13 although the influence of encapsulation in a 3D scaffold has not been fully explored by them. However, deducing the polarization stage of macrophages from their morphology can be misleading since the morphology is highly variable and influenced by many factors.16,42,44 For more precise analysis of the phenotype of the encapsulated THP-1 cells, cytokine production was assessed. With five- or ten-times increased concentration of CK, the production of the M2 markers in the encapsulated cells corresponded to their production in the polarized non-encapsulated cells. However, increased production of the M2 markers was accompanied by an increased production of the M1 markers, especially TNF-α, although at much lower concentrations. Macrophage polarization is a complex process,41–43 which can be influenced by many factors including mechanical cues,40,41 growth factors from fetal bovine serum (FBS) in a culture medium,44 or even the level of glycemia.45 Previously, Riabov et al. observed the decreased production of TNF-α in the primary monocytes seeded on the surface of gelatin-based coatings containing the same cytokine cocktail.16 However, Riabov et al. studied the effect of the cytokine cocktail in a serum-free medium. In our experiment, FBS present in the culture medium could have affected the polarization of the encapsulated cells. Furthermore, the effect of a 3D microenvironment was absent in the study reported by Riabov, and Cha et al. demonstrated the effect of the properties of the hydrogel used on the macrophage polarization.35 Therefore, it is possible that the encapsulated cells are influenced by the composition of the culture medium (i.e. the presence of FBS) and the mechanical cues from the surrounding hydrogel; moreover, they are more susceptible to cytokine stimulation and also have higher potential overall for cytokine secretion, which can be advantageous within the context of personalized coatings. Another possible explanation is that the phenotype of the THP-1 cells, although widely used as an in vitro model of human macrophages, can differ from the phenotype of primary monocyte-derived macrophages.40,43 However, the distinct effects of encapsulation and the concentration of the cytokine cocktail present are transferable to the primary cells.
Although the expression of IL-1RA, TNF-α and IL-1β was directly dependent on the presence and concentration of the polarization medium and decreased shortly after it was removed, the expression of CCL18 was triggered by the CK treatment but manifested later. This phenomenon was observed on gelatin hydrogels by Dollinger, who used a commercially available polarization medium.10 According to Kodelja et al., the monocyte-to-macrophage differentiation is a prerequisite for CCL18 expression.46 In our previous studies, the production of CCL18 was detected on day 6 of the monocytes-to-macrophage differentiation;47 therefore, it could be argued that the encapsulated cells needed more time to mature and produce CCL18.
Adhesion of the cells to the coatings of different compositions
After implantation, the patient's immune cells are expected to interact with the implanted biomaterials in vivo. Therefore, in the second part of this study, we tested the interaction of PBMCs with coatings of different composition, with or without PMA in the culture medium.PMA has a stimulating effect on immune cell activation, adhesion and phagocytosis and is widely used for in vitro immune cell activation.48 However, the effect of PMA is very widespread; thus, in future studies, specific cocktails acting on different components of PBMC can be used for a more precise control over the reaction.
The cells were unable to attach firmly to the HA-TA coatings, and they attached only to GTN-HPA-containing coatings (i.e. HA-TA + GTN-HPA and GTN-HPA) via integrin-mediated adhesion to the adhesive sequences present in gelatin. When PMA is added, more cells adhere to both GTN-containing materials, which is natural since PMA stimulates β integrin-dependent adhesion. The enhanced adhesion of PMA-activated cells indicates that under inflammatory conditions in a wound, the activated immune cells will react more rapidly with the implant.49,50
PBMC attachment to the hydrogel coatings pretreated with CK
Both gelatin and hyaluronan can bind different drugs, growth factors, and serum components.16,25,28,29 Thus, an HA-TA + GTN-HPA-based coating would serve not only as a delivery system for in vitro polarized cells, but also as a reservoir and a release system for the CK mixture after implantation, which can control the phenotype of the incoming cells. In the first part of this study, we identified that CK 5× and CK 10× should be used for the in vitro polarization of the encapsulated cells. In this part, we studied the interaction of PBMC with the hydrogel coatings loaded with cytokines at these concentrations.The phenotype of the attached cells was more influenced by the concentration of loaded CK than by the composition of the material. Riabov et al. observed a similar pattern where a CK pretreatment had a generally stronger effect on the phenotype of the primary monocytes than the presence or absence of the GTN/HA coatings. On the other hand, the coatings alone inhibited TNF-α production in the monocytes regardless of the CK treatment.16
Our results suggest that the loading by CK 5× is sufficient for the activation of the incoming monocytes and their differentiation into the CD68+ macrophages; however, a further increase in the CK concentration (CK 10×) leads to a higher portion of cells with elongated morphology, indicating M2 polarization.
Conclusions
To have a better control over the response to implanted materials, the use of a hydrogel-based immunomodulatory surface coating is a potential way of interacting with the immune cells of the patients. For rendering these coatings more potent and personalised, autologous, phenotype-controlled macrophages can be used in the hydrogels as natural hydrogels are suitable for cell encapsulation. Herein, we demonstrated the potential effects of an M2 phenotype-inducing cytokine cocktail on both the encapsulated monocytes and the PBMC cells for their potential use as immunomodulatory coatings. The presence of the cytokine cocktail changed the cellular morphology in a concentration-dependent manner and also induced higher secretion of several cytokines. The presence of the cytokine cocktail also influenced the number and morphology of the cells attached to the coatings. The incorporation of specific phenotype inducing cytokine cocktails into degradable hydrogel structures can create a dynamic interface between the implanted materials and the host tissue and can aid in the resolution of inflammation and the integration of the implanted systems.Experimental
Hyaluronan polyaldehyde was produced by Contipro a.s. 3-(4-Hydroxyphenyl)propionic (HPA), 2-(N-morpholino)ethanesulfonic acid hydrate (MES), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), N-hydroxysuccinimide (HOSu), gelatin from porcine skin gel (Bloom degree 300; Type A), dimethylformamide, tetrahydrofuran, ethyl acetate, methanol, trifluoroacetic acid and horseradish peroxidase – type I (HRP) were purchased from Sigma Aldrich. Hydrogen peroxide 30% was purchased from Penta s.r.o.Synthesis of hydroxyphenyl-modified gelatin (GTN-HPA)
GTN-HPA was synthesized using a method previously described by Wang et al.:37 3-(4-hydroxyphenyl)propionic acid (3.4 g; 20.5 mmol) was dissolved in 50 ml of DMF. MES (10 g), HOSu (3.5 g; 30 mmol) and EDC (3.9 g; 20.5 mmol) were dissolved in 100 ml of demineralized water. Both solutions were mixed together and stirred for 60 minutes at room temperature. Moreover, GTN (10 g) was dissolved in demineralized water (500 ml) by heating to 40 °C. All the components were finally mixed and stirred overnight at room temperature. Conjugated GTN-HPA was purified by ultrafiltration, and it was obtained in the solid form by freeze-drying.Hyaluronan-tyramine conjugate (HA-TA) preparation
Hydrogel/coatings preparation
Hydrogel preparation was described in our previous study.36 Briefly, the HA-TA derivative was dissolved in a mixture of 0.9% NaCl + RPMI (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at RT overnight, and the GTN-HPA conjugate was dissolved in the HA-TA solution at 37 °C for next 2 h. The two-compound system was used for hydrogel/coating preparation. The solution (HA-TA or HA-TA + GTN-HPA (1:10 w/w) or GTN-HPA) was equally divided into two parts. An adequate amount of HRP stock solution (8 U ml−1) was added to one part and H2O2 stock solution (0.1%) was added to the other part. Afterwards, equipment with a static mixer was used for mixing both the parts and dosing the material into Teflon moulds with a diameter of 11 mm or into coating wells in 24-well culture plates. The gels were formed within seconds; the samples were allowed to set for 15 minutes at room temperature.
:1) at RT overnight, and the GTN-HPA conjugate was dissolved in the HA-TA solution at 37 °C for next 2 h. The two-compound system was used for hydrogel/coating preparation. The solution (HA-TA or HA-TA + GTN-HPA (1:10 w/w) or GTN-HPA) was equally divided into two parts. An adequate amount of HRP stock solution (8 U ml−1) was added to one part and H2O2 stock solution (0.1%) was added to the other part. Afterwards, equipment with a static mixer was used for mixing both the parts and dosing the material into Teflon moulds with a diameter of 11 mm or into coating wells in 24-well culture plates. The gels were formed within seconds; the samples were allowed to set for 15 minutes at room temperature.
Evaluation of the viscoelastic properties
Hydrogel samples were prepared from HA-TA or HA-TA + GTN-HPA or GTN-HPA 4% (w/v) solutions. HA-TA with Mw 292 kDa and DS 2% was used in combination with GTN-HPA with HPA substitution (15 mg g−1). Different HRP activity and H2O2 concentrations have been used to achieve comparable elastic moduli of these materials: HRP activity used for HA-TA + GTN-HPA and GTN-HPA samples was 0.4 U ml−1 and H2O2 concentration was established at 0.005%. For HA-TA hydrogel without GTN-HPA, the HRP activity and H2O2 concentration were decreased to 0.16 U ml−1 and 0.001%, respectively.The viscoelastic properties of the hydrogels were measured by an AR-G2 Rheometer (TA Instruments). A cross-hatched surface geometry was used to avoid slippage of the hydrogel. A strain sweep test with a displacement range of 10−3 to 2 radians and a frequency of 1 Hz was performed. The elastic modulus was used for the evaluation of hydrogel viscoelastic properties (n ≥ 3).
Cell culture
THP-1 cells were bought from ATCC (TIB-202™). The culture medium consisted of high glucose RPMI-1640 medium supplemented with 10% FBS, 1% penicillin–streptomycin solution, 2 mM L-glutamine, and 1 mM sodium pyruvate (all from Sigma Aldrich).PBMCs were isolated from the fresh samples of human peripheral blood from 2–3 donors using density gradient centrifugation with Ficoll® Paque Plus (GE Healthcare). Fresh PBMC were isolated for each experiment. The culture medium was the same as that used for the THP-1 cells.
Effect of increased concentration of the cytokine mixture on the encapsulated THP-1
Herein, 1.5 × 106 THP-1 cells were encapsulated per ml of the HA-TA:GTN-HPA hydrogel. The material solution was sterilized by filtration. The cell suspension was added into the solution aliquot with HRP. The solution was mildly stirred to homogenize the cells and the hydrogel samples were prepared and left 15 minutes to set. Afterwards, hydrogels were transferred in a 24-well culture plate and submerged in 1 ml of culture medium each.
For the first 8 days, the culture medium with different concentration of the cytokine mixture (CK) was used:
(A) Standard culture medium without cytokines (w/o CK)
(B) Medium with 10 ng ml−1 M-CSF, 10 ng ml−1 IL-10, 10 ng ml−1 TGF-β1, 3 ng ml−1 IL-4 (CK 1×)
(C) Medium with 30 ng ml−1 M-CSF, 30 ng ml−1 IL-10, 30 ng ml−1 TGF-β1, 9 ng ml−1 IL-4 (CK 3×)
(D) Medium with 50 ng ml−1 M-CSF, 50 ng ml−1 IL-10, 50 ng ml−1 TGF-β1, 15 ng ml−1 IL-4 (CK 5×)
(E) Medium with 100 ng ml−1 M-CSF, 100 ng ml−1 IL-10, 100 ng ml−1 TGF-β1, 30 ng ml−1 IL-4 (CK 10×)
M-CSF and TGF-β1 were obtained from Promokine, and IL-10 and IL-4 were obtained from Sigma Aldrich.
After 8 days, the medium with the cytokines was removed, and the hydrogels were cultured in a standard culture for the next 7 days. The non-encapsulated THP-1 cells cultured in the medium with CK 1× served as the control. The cell morphology was determined from the live/dead images. At appropriate time points, cytokine release was analyzed by ELISA.
PBMC adhesion to the coatings of different compositions
The culture wells in 24-well plates were coated with HA-TA, GTN-HPA or HA-TA:GTN-HPA. Then, 3 × 105 PBMC were seeded per well in a standard culture medium or in a medium containing 100 nM PMA. After 96 h, the non-adherent cells were removed, and the number of adherent cells was evaluated via the CellTiter Glo assay.
PBMC adhesion to the coatings pretreated with cytokines
The culture wells in 24-well plates were coated with a 2 mm thick layer of HA-TA:GTN-HPA and 1 ml of medium with CK 5× or CK 10× was added. The hydrogels layers were allowed to absorb the cytokines for 72 h at 37 °C. After that, the medium with CK was removed, the coatings were rinsed with RPMI-1640, and freshly isolated PBMCs were seeded on them in a standard culture medium. After 4 days, the number of adherent cells, their morphology, and the number of CD68+ cells were evaluated.
Cell growth and phenotype assessment
Live/dead staining was used for the evaluation of cell morphology and viability. The culture medium was replaced with PBS. Calcein-AM, and propidium iodide staining solutions (both from Sigma Aldrich) were added at a final concentration of 1 ng ml−1. Everything was incubated for 15 min, protected from direct light. The images were obtained using the fluorescent microscope Nikon Eclipse-Ti.Cell number evaluation: the coatings with the cells were gently rinsed, and 200 μl PBS and 40 μl of CellTiter-Glo reagent (Promega) was added. The plate was incubated for 15 min using an orbital shaker at 200 rpm, protected from light. After this, the plate was removed from the shaker and incubated for another 10 min. The luminescent signal (RLU) was read using the fluorescent reader TECAN Infinite 200.
Immunofluorescent staining: the coatings with the cells were gently rinsed with PBS, fixed in 4% paraformaldehyde for 10 min, permeabilized with 0.2% Triton X-100 for 5 min, and washed several times with PBS. The cells were incubated with anti-CD68-Alexa488 (SantaCruz) diluted 1:100 in 0.1% BSA/PBS for 2 h. The cell nuclei were counterstained with Hoechst.
ELISA: the supernatants from the encapsulated and control THP-1 cells cultured in the media with different concentrations of the cytokine mixture were collected at 72 h interval and stored at −80 °C. ELISA was performed in Protobios LLC (Tallinn, Estonia) as per the instructions of the providers. The release of the M2 markers IL-1RA and CCL18 and the M1 markers TNF-α and IL-1β was assessed.
Statistical analysis
All the quantitative results are presented as mean ± standard deviation (SD). The Student's t test for unpaired observations was applied for statistical analysis. The differences between the samples with p-value < 0.05 were accepted as statistically significant.Conflicts of interest
Vladimír Velebný has a financial interest in the company Contipro, Dolní Dobrouč, Czech Republic, the commercial producer of Hylauronan. Ivana Ščigalková, Julie Bystroňová, Lenka Kovářová, and Martin Pravda are employees of Contipro.Acknowledgements
This project has received funding from the European Union's Seventh Framework Programme and Horizon 2020 Programme for research, technological development and demonstration under grant agreement no. 602694 (IMMODGEL) and no. 760921 (PANBioRA). We would like to thank Toomas Neuman, Kaia Palm, and Urmas Liivas (Protobios) for ELISA measurements.Notes and references
- S. Franz, S. Rammelt, D. Scharnweber and J. C. Simon JC, Biomaterials, 2011, 32, 6692–6709 CrossRef CAS.
- M. Pokorny, J. Klemes, O. Zidek, C. Dollinger, G. Ozcebe, S. Singh, V. Velebny, A. M. Ghaemmaghami, L. Wolfova and N. E. Vrana, Biomed. Phys. Eng. Express., 2017, 3, 015002 CrossRef.
- H. M. Rostam, S. Singh, N. E. Vrana, M. R. Alexander and A. M. Ghaemmaghami, Biomater. Sci., 2015, 3, 424–441 RSC.
- N. Nagarajan, A. Dupret-Bories, E. Karabulut, P. Zorlutuna and N. E. Vrana, Biotechnol. Adv., 2018, 36, 521–533 CrossRef CAS PubMed.
- L. Keller, P. Schwinté, E. Gomez-Barrena, M. Arruebo and N. Benkirane-Jessel, Trends Biotechnol., 2017, 35, 8–11 CrossRef CAS.
- S. Gordon and F. O. Martinez, Immunity, 2010, 32, 593–604 CrossRef CAS PubMed.
- A. Sica and A. Mantovani, J. Clin. Invest., 2012, 122, 787–795 CrossRef CAS.
- S. J. Van Dyken and R. M. Locksley, Annu. Rev. Immunol., 2013, 31, 317–343 CrossRef CAS.
- A. Mantovani, A. Sica, S. Sozzani, P. Allavena, A. Vecchi and M. Locati, Trends Immunol., 2004, 25, 677–686 CrossRef CAS.
- C. Dollinger, S. Ciftci, H. Knopf-Marques, R. Guner, A. M. Ghaemmaghami, C. Debry, J. Barthes and N. E. Vrana, J. Tissue Eng. Regener. Med., 2018, 12, 330–340 CrossRef CAS.
- J. Kajahn, S. Franz, E. Rueckert, I. Forstreuter, V. Hintze, S. Moeller and J. C. Simon, Biomatter, 2012, 2, 226–236 CrossRef.
- D. Y. S. Vogel, J. E. Glim, A. W. D. Stavenuiter, M. Breur, P. Heijnen, S. Amor and R. H. J. Beelen, Immunobiology, 2014, 219, 695–703 CrossRef CAS.
- W.-Q. Zhong, G. Chen, W. Zhang, X.-P. Xiong, Y. Zhao, B. Liu and Y.-F. Zhao, Sci. Rep., 2015, 5, 1–11 Search PubMed.
- M. Genin, F. Clement, A. Fattaccioli, M. Raes and C. Michiels, BMC Cancer, 2015, 15, 577–591 CrossRef.
- S. Mia, A. Warnecke, X.-M. Zhang, V. Malmström and R. A. Harris, Scand. J. Immunol., 2014, 79, 305–314 CrossRef CAS.
- V. Riabov, F. Salazar, S. S. Htwe, A. Gudima, C. Schmuttermaier, J. Barthes, H. Knopf-Marques, H. Klüter, A. M. Ghaemmaghami, N. E. Vrana and J. Kzhyshkowska, Acta Biomater., 2017, 53, 389–398 CrossRef CAS.
- W. E. Paul, Blood, 1991, 77, 1859–1870 CAS.
- L. Kubiczkova, L. Sedlarikova, R. Hajek and S. Sevcikova, J. Transl. Med., 2012, 3, 183–207 CrossRef PubMed.
- K. N. Couper, D. G. Blount and E. M. Riley, J. Immunol., 2008, 180, 5771–5777 CrossRef CAS.
- N. Wang, H. Liang and K. Zen, Front. Immunol., 2014, 5, 614–623 Search PubMed.
- D. West, I. Hampson, F. Arnold and S. Kumar, Science, 1985, 228, 1324–1326 CrossRef CAS.
- C. M. McKee, M. B. Penno, M. Cowman, M. D. Burdick, R. M. Strieter, C. Bao and P. W. Noble, J. Clin. Invest., 1996, 98, 2403–2413 CrossRef CAS.
- R. Deed, P. Rooney, P. Kumar, J. D. Norton, J. Smith, A. J. Freemont and S. Kumar, Int. J. Cancer, 1997, 71, 251–256 CrossRef CAS.
- D. Jiang, J. Liang and P. W. Noble, Physiol. Rev., 2011, 91, 221–264 CrossRef CAS.
- H. Knopf-Marques, S. Singh, S. S. Htwe, L. Wolfova, R. Buffa, J. Bacharouche, G. Francius, J. C. Voegel, P. Schaaf, A. M. Ghaemmaghami, N. E. Vrana and P. Lavalle, Biomacromolecules, 2016, 17, 2189–2198 CrossRef CAS.
- F. Lee, J. E. Chung and M. Kurisawa, J. Controlled Release, 2009, 134, 186–193 CrossRef CAS.
- L. Wolfová, M. Pravda, M. Foglarová, M. Měmcová, K. Niedoba and V. Velebny, US Pat., US9492586, National Center for Biotechnology Information, 2013.
- P. A. Levett, F. P. Melchels, K. Schrobback, D. W. Hutmacher, J. Malda and T. J. Klein, Acta Biomater., 2014, 10, 214–223 CrossRef CAS.
- T. Yuan, L. Zhang, K. Li, H. Fan, Y. Fan, J. Liang and X. Zhang, J. Biomed. Mater. Res., Part B, 2014, 102, 337–344 CrossRef.
- S. E. Hanson, S. N. King, J. Kim, X. Chen, S. L. Thibeault and P. Hematti, Tissue Eng., Part A, 2011, 17, 2463–2471 CrossRef CAS.
- C. Vodouhê, E. Le Guen, J. M. Garza, G. Francius, C. Déjugnat, J. Ogier, P. Schaaf, J. C. Voegel and P. Lavalle, Biomaterials, 2006, 27, 4149–4156 CrossRef.
- J. Barthes, N. E. Vrana, H. Özçelik, R. Gahoual, Y. N. François, J. Bacharouche, G. Francius, J. Hemmerlé, M. H. Metz-Boutigue, P. Schaaf and P. Lavalle, Biomater. Sci., 2015, 3, 1302–1311 RSC.
- K. L. Spiller, S. Nassiri, C. E. Witherel, R. R. Anfang, J. Ng, K. R. Nakazawa, T. Yu and G. Vunjak-Novakovic, Biomaterials, 2015, 37, 194–207 CrossRef CAS.
- J. Chen, M. Li, C. Yang, X. Yin, K. Duan, J. Wang and B. Feng, Colloids Surf., B, 2018, 163, 336–345 CrossRef CAS.
- B. H. Cha, S. R. Shin, J. Leijten, Y. C. Li, S. Singh, J. C. Liu, N. Annabi, R. Abdi, M. R. Dokmeci, N. E. Vrana, A. M. Ghaemmaghami and A. Khademhosseini, Adv. Healthcare Mater., 2017, 6, 1700289 CrossRef.
- J. Bystroňová, I. Ščigalková, L. Wolfová, M. Pravda, N. E. Vrana and V. Velebný, RSC Adv., 2018, 8, 7606–7614 RSC.
- L.-S. Wang, J. E. Chung, P. P.-Y. Chan and M. Kurisawa, Biomaterials, 2010, 31, 1148–1157 CrossRef CAS.
- P. Šedová, R. Buffa, S. Kettou, G. Huerta-Angeles, M. Hermannová, V. Leierová, D. Šmejkalová, M. Moravcová and V. Velebný, Carbohydr. Res., 2013, 371, 8–15 CrossRef.
- N. R. Patel, M. Bole, C. Chen, C. C. Hardin, A. T. Kho, J. Mih, L. Deng, J. Butler, D. Tschumperlin and J. J. Fredberg, PLoS One, 2012, 7, e41024 CrossRef CAS.
- A. K. Blakney, M. D. Swartzlander and S. J. Bryant, J. Biomed. Mater. Res., Part A, 2012, 100, 1375–1386 CrossRef.
- K. Garg, N. A. Pullen, C. A. Oskeritzian, J. J. Ryan and G. L. Bowlin, Biomaterials, 2013, 34, 4439–4451 CrossRef CAS PubMed.
- F. Y. McWhorter, T. Wang, P. Nguyen, T. Chung and W. F. Liu, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17253–17258 CrossRef CAS.
- C. E. Witherel, P. L. Graney, D. O. Freytes, M. S. Weingarten and K. L. Spiller, Wound Repair Regen., 2016, 24, 514–524 CrossRef.
- F. Rey-Giraud, M. Hafner and C. H. Ries, PLoS One, 2012, 7, e42656 CrossRef CAS.
- K. Moganti, F. Li, C. Schmuttermaier, S. Riemann, H. Klüter, A. Gratchev, M. C. Harmsen and J. Kzhyshkowska, Immunobiology, 2017, 222, 952–959 CrossRef CAS.
- V. Kodelja, C. Müller, O. Politz, N. Hakij, C. E. Orfanos and S. Goerdt, J. Immunol., 1998, 160, 1411–1418 CAS.
- H. Özçelik, N. E. Vrana, A. Gudima, V. Riabov, A. Gratchev, Y. Haikel, M. H. Metz-Boutigue, A. Carradò, J. Faerber, T. Roland, H. Klüter, J. Kzhyshkowska, P. Schaaf and P. Lavalle, Adv. Healthcare Mater., 2015, 4, 2026–2036 CrossRef.
- T. Starr, T. J. Bauler, P. Malik-Kale and O. Steele-Mortimer, PLoS One, 2018, 13, e0193601 CrossRef.
- L. Liu, B. R. Schwartz, J. Tupper, N. Lin, R. K. Winn and J. M. Harlan, J. Biol. Chem., 2002, 277, 40893–40900 CrossRef CAS.
- D. F. Kucik, M. L. Dustin, J. M. Miller and E. J. Brown, J. Clin. Invest., 1996, 97, 2139–2144 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |