
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptimizing electron-rich arylamine derivatives in thiophene-fused derivatives as π bridge-based hole transporting materials for perovskite solar cells†
Xiaorui Liu * and
Xing Liu
* and
Xing Liu
Key Laboratory of Luminescent and Real-Time Analytical Chemistry (Southwest University), Ministry of Education, School of Chemistry and Chemical Engineering, Southwest University, Chongqing 400715, China. E-mail: liuxiaorui@swu.edu.cn
First published on 8th August 2019
Abstract
Based on the observations of thienothiophene derivatives as π-bridged small molecule hole transporting materials (HTMs), adjusting their electron-rich arylamine derivatives is an effective approach to obtain the alternative HTMs for perovskite solar cells (PSCs). In this work, starting from a new electron-rich arylamine derivative and different π-bridged units of thienothiophene derivatives, a series of arylamine derivative-based HTMs were designed, and their properties were investigated using density functional theory combined with the Marcus charge transfer theory. Compared with the parental Z26 material, the designed H01–H04 exhibit appropriate frontier molecular orbitals, good optical properties, better solubility, good stability and higher hole mobilities. H01–H04 materials with high hole mobility (∼× 10−2) can serve as promising HTMs for improving the efficiency of PSCs. The results confirm that the design strategy of adjusting the electron-rich arylamine derivatives in thienothiophene derivatives as π-bridged HTMs is a reliable approach to obtain the promising HTMs for PSC applications.
1. Introduction
Organic–inorganic hybrid perovskite materials (e.g., CH3NH3PbX3, X = Cl, Br and I) as light absorbers have excellent optoelectronic properties such as broad optical absorption, tunable band gaps, long charge-diffusion length and ambipolar charge transporting behaviour.1–3 Up to now, the power conversion efficiency (PCE) of perovskite solar cells (PSCs) on the basis of a fluorene-terminated HTM has exceeded 23%.4 In general, the PSC device architecture of indium tin oxide (ITO)/electron-transporting layer (ETL)/perovskite (CH3NH3PbI3)/hole-transporting layer (HTL)/Au is the most commonly used.5–7 As has been demonstrated, HTMs as the HTLs play a key role in hole extraction and transportation to Au electrodes for conventional devices.5 It is reported that the organic HTMs have been considered one of the most important factors restricting the device stability.8,9 In general, spiro-OMeTAD is used as a standard HTM to be widely used in traditional PSCs. Owing to the poor conductivity (9.1 × 10−7 S cm−1) of spiro-OMeTAD itself, lithium bis-trifluoromethanesulfonyl imide (LiTFSI) and tert-butylpyridine (TBP) must be added to improve the conductivity.9,10 However, the dopants LiTFSI and TBP in HTL are the main factors that lead to the degradation of perovskites as LiTFSI tends to absorb water.9,10 To overcome the shortcomings of doped spiro-OMeTAD, the development of alternative HTMs with high hole transporting ability and good stability is important.At present, diphenylamine derivatives [e.g., bis(4-methoxyphenyl)amine] and triphenylamine derivative [e.g., 4-methoxy-N-(4-methoxyphenyl)-N-phenylbenzenamine] are widely used as electron-rich units in the construction of HTMs, such as FDT,11 Z26,12 H101,13 X59,14 TQ2 (ref. 15) and TTH101.5,16–18 In the aforementioned HTMs, adjusting the electron-deficient π-bridged units of diphenylamine or triphenylamine derivative-based HTMs is the usual method to improve their performance. For example, Sun and co-workers19,20 reported a class of star HTMs that were theoretically designed via adjusting the electron-deficient cores in order to obtain efficient HTMs with appropriate energy levels and high hole mobility for PSC applications. In fact, electron rich arylamine derivatives are also important parts that affect the properties of HTMs. For example, Jang and co-workers21 reported two HTMs (pDPA-DBTP and pTPA-DBTP), which contained the electron-deficient di(1-benzothieno)[3,2-b:2′,3′-d]pyrrole (DBTP) as the π-bridged units and electron-rich units of the diphenylamine derivative and triphenylamine derivative. The PCEs of PSCs based on HTMs of pDPA-DBTP and pTPA-DBTP are 14.10% and 15.63%, respectively.21 Moreover, based on the investigations via density-functional theory (DFT) and Marcus theory, Li and co-workers22–24 reported that the introduction of a highly symmetrical core to diphenylamine- or triphenylamine-derivative-based HTMs resulted in some organic HTMs with suitable energy levels and improved hole mobility. Therefore, modifying the electron rich arylamine derivatives of HTMs may be a reliable approach to obtain promising HTMs in PSC applications.
Herein, on the basis of the potential compounds such as thiophene, thienothiophene, dithienothiophene and terthienothiophene as electron-deficient π-bridged units, a strategy was devised to introduce the new electron-rich arylamine [N,N-bis(4-methoxyphenyl)naphthalen-2-amine] derivatives to construct HTMs (see H01–H04 in Fig. 1) in order to provide theoretical models and obtain the alternative HTMs. These electron-deficient π-bridged units with good photoelectronic properties have been proved in previous works.24–26 Moreover, Z26 materials in Fig. S1† reported by Grätzel and co-workers were chosen as a reference.12 Z26 as a HTM in PSC applications exhibits good performance such as a hole mobility of 1.34 × 10−4 cm2 V−1 s−1 and PCE of 20.1%.12 Thereupon, the properties of these designed HTMs, such as their geometric structures, frontier molecular orbitals and reorganization energies were investigated by DFT. The optical properties of absorption and emission were calculated using time-dependent DFT (TD-DFT). The electron coupling and hole mobility were predicted on the basis of the first-principle calculations combined with Marcus theory.
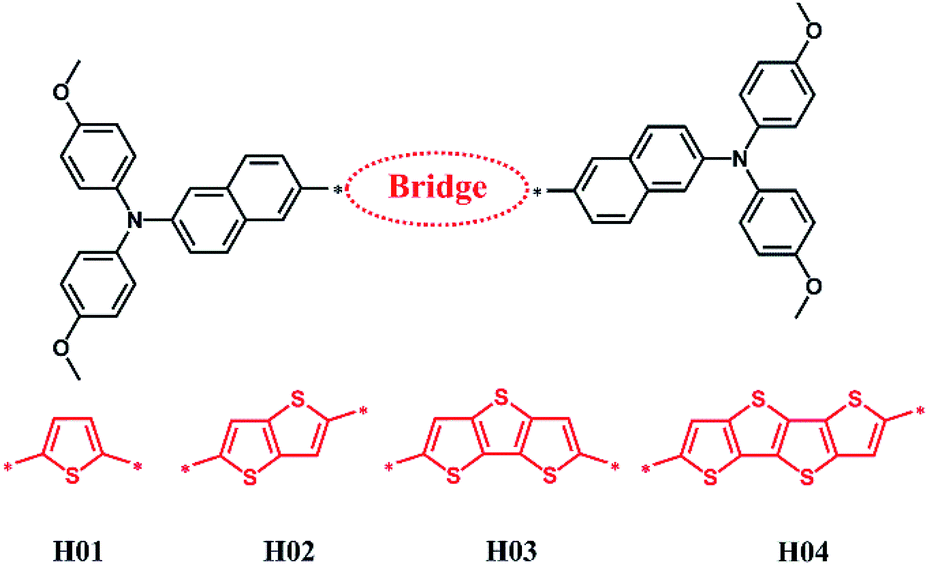 | ||
| Fig. 1 Chemical structures of designed molecules H01–H04. | ||
2. Computational details
2.1 Geometry optimization and electronic properties
Investigated systems are shown in Fig. 1 and S1.† Geometric structures and electronic properties of the investigated systems were fully optimized using B3P86 at 6-311G(d,p) and BMK/6-31G(d) in a gaseous phase or tetrahydrofuran solution.27 The energies of all of the obtained geometries are ensured to be the lowest as the optimized structures do not exhibit imaginary frequency. In fact, frontier orbital energies of an organic molecule are very sensitive to the functionals and basis sets. For the arylamine derivative-based HTMs (Z26), HOMO energy was calculated using the methods of B3LYP, B3P86 and PBE0 at 6-311G(d,p) levels. The calculated results indicate that the calculated HOMO energy (−5.05 eV) of Z26 with B3P86/6-311G(d,p) functional and basis set is close to its experimental result (−5.16 eV (ref. 12)). The HOMO energies of Z26 from the B3LYP/6-311G(d,p) and PBE0/6-311G(d,p) calculations are −4.45 and −4.61 eV, respectively. Moreover, the calculated HOMO energy (−5.09 eV) of FDT with B3P86/6-311G(d,p) functional and basis set is in line with its experimental result (−5.16 eV (ref. 28)). Therefore, the frontier orbital energies of the designed HTMs are calculated using DFT with B3P86/6-311G(d,p) functional and basis set.2.2 Calculation of optical absorption
On the basis of the ground-state geometries from the B3P86, BMK, B3LYP and PBE0 at 6-31G(d) level in tetrahydrofuran solution, the optical absorptions of Z26 were simulated by TD-DFT with B3P86, BMK, B3LYP and PBE0 at 6-31G(d) level in tetrahydrofuran solution with a polarizable continuum model (PCM).27,29 Compared with the experimental value (456 nm (ref. 12)) of Z26, using the TD-BMK/6-31G(d) method in tetrahydrofuran solution could yield an accurate absorption peak at 450 nm. Under the same conditions, the maximum absorption of Z26 obtained using the TD-B3LYP (529 nm), TD-PBE0 (530 nm) and TD-B3P86 (507 nm) methods are overestimated. Therefore, the optical absorptions of designed HTMs are calculated by TD-BMK/6-31G(d) on the basis of the same functional and basis set in tetrahydrofuran solution. The calculation of electron density difference including the overlap between the regions of density depletion and the increment and transferred charge amount can be obtained from our previous work.30 The DFT and TD-DFT calculations were carried out by the Gaussian 09 program.312.3 Energy calculations
On the basis of the adiabatic potential energy surfaces, the inner reorganization energy λh for holes of HTMs could be calculated as follows:25,32,33
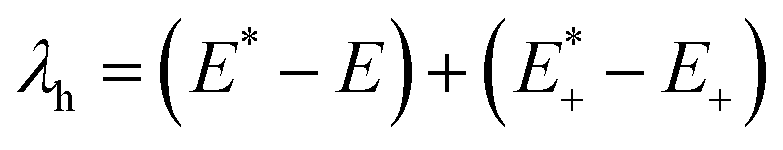 | (1) |
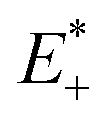 are the energies of the neutral and cationic states with the geometries of the cationic and neutral species.34 Moreover, energy calculations, including electron affinities, adiabatic ionization potential and absolute hardness of the investigated systems, were performed using the B3P86/6-311G(d,p) method. The solvation free energy for all the molecules were calculated using the TD-BMK/6-31G(d) method in tetrahydrofuran solution or in a gaseous phase. Geometry optimization and single point energy calculations were implemented using the Gaussian 09 program.
are the energies of the neutral and cationic states with the geometries of the cationic and neutral species.34 Moreover, energy calculations, including electron affinities, adiabatic ionization potential and absolute hardness of the investigated systems, were performed using the B3P86/6-311G(d,p) method. The solvation free energy for all the molecules were calculated using the TD-BMK/6-31G(d) method in tetrahydrofuran solution or in a gaseous phase. Geometry optimization and single point energy calculations were implemented using the Gaussian 09 program.
2.4 Calculations of charge mobility
The hole mobility of the designed HTMs was calculated from the Einstein relation:35,36
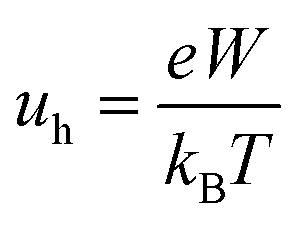 | (2) |
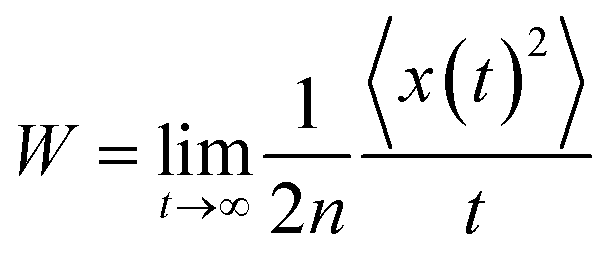 | (3) |
For a spatially isotropic system, the homogeneous diffusion constant W can be approximately evaluated by:38
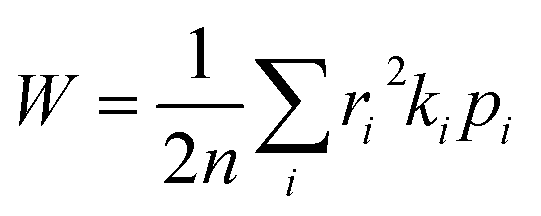 | (4) |
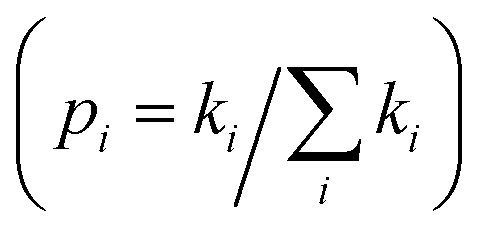 , respectively.
, respectively.
Here, the parameter of hole transfer rate (kh) for organic molecules can be calculated from the Marcus–Hush equation:36,39,40
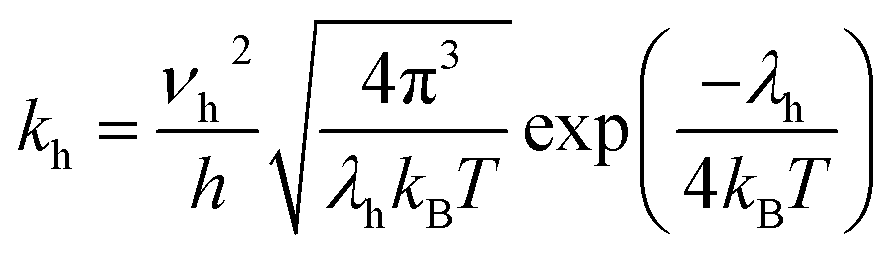 | (5) |
The parameter of hole electronic coupling (vh) could be obtained from the equation as shown below:44,45
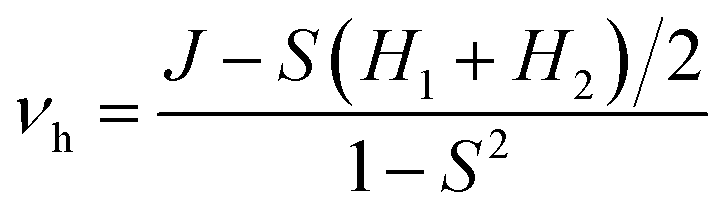 | (6) |
| J = 〈φ1HOMO|hks|φ2HOMO〉 | (7) |
2.5 Simulation of crystal structures
In order to calculate the electronic coupling of HTMs, it was necessary to obtain the dimer structure, which was defined as adjacent segments from the crystal structures of molecules. The crystal structure of the investigated HTMs can be obtained from the polymorph predictor module in the Material Studio software.50,51 The geometry of the cluster models used in the present study was taken from the B3P86/6-311G(d,p) level. The Dreiding force field was used for the prediction.52 For the investigated HTMs, the polymorph calculations are restricted to the ten most probable space groups, namely, C2, P21/c, P1, P212121, C2/c, P21, Pbca, Cc, Pna21 and Pbcn.53 To verify the rationality of the selected models herein, the FDT crystal structure was predicted using the same methods. The simulated results indicate that the predicted FDT crystal structure share the space group of P1, consistent with the experimental one.28 This validates that the calculation method is reasonable to a certain degree. Further, the crystal structures were sorted according to their total energy. On the basis of the crystal structures, we selected a molecule as the center. All of the adjacent fragments with the center are defined as the transport pathways. Thus, each transport pathway is the paired dimer between neighboring and center molecules.3. Results and discussion
3.1 Molecular design and electronic properties
In PSC devices, the conventional architecture of indium tin oxide(ITO)/electron-transporting layer (ETL)/perovskite/hole-transporting layer (HTL)/Au is the most commonly used.6,7 As has been demonstrated, HTMs as the HTL play a key role in hole extraction and transportation to the Au electrode in conventional devices.5 However, the frontier molecular orbitals are the basic parameters to estimate the properties of the HTMs in PSCs. For example, the HTMs should have an energy higher than that of the valence band for CH3NH3PbI3 (−5.43 eV (ref. 54 and 55)) to ensure the hole injection or transfer on the perovskite/HTM interface. Analogously, to prevent the electron movement from the absorber (e.g., CH3NH3PbBr3) to the metal electrodes, the HTMs should have a higher lowest unoccupied molecular orbital (LUMO) level than the conduction band of CH3NH3PbI3 (−3.93 eV (ref. 54)). To further improve the HTM performances in PSC devices, the first aim of this work is to introduce the new electron-rich arylamine derivatives to construct HTMs with a suitable frontier molecular orbital and high hole mobility. Therefore, starting from the new electron-rich arylamine derivatives and different π-bridged units of thienothiophene derivatives, a series of arylamine derivative-based HTMs were designed (as shown in Fig. 1) on the basis of the structure of the reported Z26 material (as shown in Fig. S1†). Optimized ground-state geometries of HTMs are presented in Fig. S2.†The functional and basis set of B3P86/6-311G(d,p) can reproduce accurate HOMO energies for the reported Z26 and FDT. To further ensure the accurate HOMO and LUMO energies with which we can evaluate the electronic properties of the designed HTMs in PSC devices, we calculated the values of HOMO and LUMO energies by employing the B3P86/6-311G(d,p) functional and basis set for 13 arylamine derivative-based HTMs. The detailed information on the HTMs in PSC applications that have been reported are listed in Table S1.† Based on the calculated results, important information regarding the theoretical and experimental HOMO and LUMO values for these HTMs were obtained. The data are presented in Table S1.† A correlation can be established between the theoretical HOMO/LUMO energy levels and the experimental HOMO/LUMO energy levels of these reported HTMs (in Fig. S3†). Hence, the empirical correlations between theoretical HOMO(th.)/LUMO(th.) and experimental HOMO(exp.)/LUMO(exp.) for these HTMs are expressed in the following equations:
| HOMO(exp.) = 0.66HOMO(th.) − 1.75, R = 0.79 | (8) |
| LUMO(exp.) = 0.69LUMO(th.) − 1.07, R = 0.88 | (9) |
According to eqn (8) and Table S2,† the calculated results show that the values of LUMO energy for Z26 (−5.08 eV) and FDT (−5.11 eV) are in accord with those of the experimental values (Z26: −5.16 eV (ref. 12) and FDT: −5.16 eV (ref. 28)), respectively. According to eqn (9) and Table S2,† the calculated results show that the LUMO values of Z26 (−2.69 eV) and FDT (−2.28 eV) are in accord with those of the experimental values (Z26: −2.77 eV (ref. 12) and FDT: −2.28 eV (ref. 28)), respectively. Therefore, eqn (8) and (9) can give an accurate result to predict the HOMO and LUMO energies of the designed HTMs on the basis of the B3P86/6-311G(d,p) calculation. In fact, the HOMO and LUMO energies of HTMs can be evaluated by adiabatic ionization potentials (IPs) and electron affinities (EAs), respectively.25,56 The IPs and EAs of the reported Z26 and FDT are calculated by B3P86/6-311G(d,p) functional and basis set. Table S2† shows that the calculated IPs and EAs of the reported Z26 and FDT are divorced from the HOMO and LUMO energies of these HTMs, respectively. Analogously, in order to further verify the reliability of the theoretical model that was used to predict the HOMO and LUMO energies of the designed HTMs, we calculated the IPs and EAs of the reported arylamine derivative-based HTMs at B3P86/6-311G(d,p) level in comparison with the HOMO and LUMO energies of these HTMs (see Table S1†). Hence, two empirical correlation between theoretical IP/EA and experimental HOMO/LUMO of these reported HTMs were fitted (see Fig. S3 and Table S2†). As shown in Table S2,† the calculated results reveal that the empirical correlation (Fig. S3c and d†) can yield a reliable data about the HOMO and LUMO values of Z26 and FDT, in line with the results from eqn (8) and (9). Therefore, eqn (8) and (9) could be used to predict the HOMO and LUMO energies of the designed HTMs. According to eqn (8) and (9), the calculated values of HOMO and LUMO energies for the designed HTMs (H01–H04) are presented in Fig. 2A.
 | ||
| Fig. 2 (A) Energy levels and frontier molecular orbitals of all investigated molecules at the B3P86/6-311G(d,p) levels; (B) simulated absorption spectra of molecules H01–H04 using the TD-BMK/6-31g(d) functional and basis set in tetrahydrofuran; (C) electron density difference plots of electronic transition S0 → S1 for H01–H04 (isovalue: 2 × 10−2 e au−3), w is overlap between the regions of density depletion and increment, Δq is transferred charge amount (|e−|). | ||
As shown in Fig. 2A, the values of HOMO energy are H01 (−5.25 eV), H02 (−5.25 eV), H03 (−5.26 eV) and H04 (−5.28 eV), respectively. For the H01–H04, their HOMO levels are higher than the valence band of the perovskite. Therefore, the HOMO levels of designed HTMs are in good agreement with the valence band of CH3NH3PbI3 (−5.43 eV (ref. 48 and 49)), which ensure a good hole injection from the perovskite to the HTMs. In addition, the illustration of the frontier molecular orbitals in Fig. 2A indicates that both the HOMOs and LUMOs of H01–H04 are delocalized on the entire molecular chain with π characteristics. The HOMO orbital of HTMs with extensive delocalization on the molecular chain is favorable for increasing the electron coupling between the adjacent molecules.18,31 According to the HOMO distribution, H01–H04 as HTMs in PSCs may provide large electron coupling that increases the hole mobility.
As shown in Fig. 2A, the LUMO values for H01–H04 are −2.56, −2.62, −2.68 and −2.76 eV, respectively. Obviously, the LUMO levels of H01–H04 that are higher than the conduction band of the perovskite (e.g. CH3NH3PbI3) can effectively prevent the electron from moving from the absorber (e.g., CH3NH3PbBr3) to the metal electrodes. Moreover, the energy gaps (Egs) of HTMs are in the following sequence: 2.69 eV (H01) > 2.63 eV (H02) > 2.58 eV (H03) > 2.52 eV (H04). The decreased energy gaps from H01 to H04 are mainly driven from the downshifted LUMO contributions. Fig. 2A indicates that the LUMO values of the designed H01–H04 are in the order of H01 > H02 > H03 > H04. There is no significant change in the HOMO values of the designed H01–H04. For the designed HTMs, an increased hybridization and π-conjugation degree of electron-deficiency of π-bridged units in the LUMO from H01 to H04 should have a significant impact on the decreasing LUMO level, resulting in the reduction of the band gap. Optimizing electron-deficient π-bridged units in small-molecule arylamine derivative-based HTMs is an effective strategy to adjust the electronic properties for obtaining suitable frontier molecular orbital levels.
3.2 Optical absorptions and emission
Optical properties of HTMs can provide some useful information to evaluate the performance of HTM in PSC devices. Therefore, the optical properties of all HTMs were simulated by the TD-BMK/6-31G(d) functional and basis set on the basis of the ground state structures with the same functional and basis set in tetrahydrofuran solution. Simulated absorption and photoluminescence spectra of Z26 together with the experimental spectra are presented in Fig. S4,† which indicates that the TD-BMK/6-31G(d) level in tetrahydrofuran solution can yield an accurate absorption peak in comparison with the experimental one. The optical parameters of H01–H04 in S0–S1 states, such as absorption wavelength (λabs), oscillator strength (f), major configurations, emission wavelength (λem) and Stokes shift are presented in Table 1. In the S0–S1 states of the designed HTMs, the absorption is mostly from the HOMO to LUMO transitions. As shown in Table 1 and Fig. 2B, the maximum absorption wavelengths and oscillator strength (in parentheses) of H01–H04 are 397 nm (2.19), 407 nm (2.66), 416 nm (2.86), and 438 nm (3.08), respectively.| Absorption | Emission | ||||
|---|---|---|---|---|---|
| λabs | f | Assignments | λem | Shift (nm) | |
| H01 | 397 | 2.19 | H → L (85%) | 519 | 122 |
| H02 | 407 | 2.66 | H → L (84%) | 536 | 129 |
| H03 | 416 | 2.86 | H → L (80%) | 552 | 136 |
| H04 | 438 | 3.08 | H → L (81%) | 561 | 123 |
As shown in Table 1, the λem values for H01–H04 in the S0–S1 states are 519, 536, 552 and 561 nm, respectively. Stokes shifts from the maximum absorption wavelength to the emission wavelengths of H01–H04 are 122, 129, 136 and 123 nm, respectively. It has been reported that a large Stokes shifts of organic HTMs can give rise to a large change in the structure between ground and excited states.13 This also contributes to the pore filling of the HTM in PSCs by optical post-treatment or simple annealing.13 H01–H04 molecules with large Stokes shifts may improve the performance of HTMs in PSC applications.
To further explore the charge transfer properties of new designed HTMs during the optical excitation process, the overlap between the regions of density depletion and increment (w), transferred charge amount (Δq) in (|e−|) and electron density difference (EDD) plots of H01–H04 are provided (in Fig. 2C). Fig. 2C shows that the decreased electron densities (blue) are obviously located in the entire chains. The increased electron densities (red) are limited to the electron-deficiency region of π-bridged units in the molecules. In addition, the corresponding parameters, such as Δq and w are presented in Fig. 2C. The Δq values of H01–H04 are 0.82, 0.81, 0.80 and 0.79|e−|, respectively, and the w values of H01–H04 are 0.50, 0.52, 0.45 and 0.52, respectively. The Δq and w values of Z26 are 0.78 and 0.50|e−|. For these parameters of EDD, there are no significant differences in comparison with the Z26. This indicates that the designed HTMs can ensure an efficient charge separation, which promotes HTM performance in PSC applications.
3.3 Solubility and stability of HTMs
For the fabrication of PSC devices, the HTMs are coated on ITO or perovskite films. In order to ensure that the HTMs can be coated as a uniform film on the ITO or perovskite, a good solubility of the HTMs is needed. Theoretically, solvation free energy (ΔG) can be used to estimate the solubility of organic materials.57,58 ΔG of organic materials is equal to the difference between the free energy in the solvent and that in the gaseous phase.57,58 Herein, tetrahydrofuran was selected as the solvent. Table 2 shows that the calculated ΔG values of Z26 and H01–H04 are −0.54 eV, −0.53 eV, −0.58 eV, −0.66 eV and −0.65 eV, respectively. It can be seen that the investigated H01–H04 molecules with similar ΔG in comparison with that of Z26 (in Table S3†) may exhibit good solubility.| H01 | H02 | H03 | H04 | |
|---|---|---|---|---|
| IPa | 6.03 | 6.01 | 6.00 | 6.01 |
| EAa | 1.51 | 1.58 | 1.67 | 1.80 |
| η | 2.26 | 2.21 | 2.17 | 2.10 |
| ΔG | −12.70 | −13.91 | −15.66 | −15.51 |
In order to obtain an excellent HTM, stability is a very important parameter. Absolute hardness (η) can be used to estimate the stability of an organic material to a certain extent.22 η can be described by the equation, η = (IP − EA)/2,22 where IP is the adiabatic ionization potential and EA the adiabatic electron affinity.59 As shown in Table 2, the calculated η values of H01–H04 are 2.26, 2.21, 2.17 and 2.10 eV, respectively. The values of η for the designed HTMs are close to that (2.06 eV) of Z26 (in Table S3†). It indicates that these designed HTMs may have good stability.
3.4 Hole mobility
Development of HTMs with high hole mobility is an important way to improve the performance of PSC devices. According to the Marcus theory (eqn (5)), the hole reorganization energy (λh) and hole-transfer integral (vh) are key factors affecting the charge-transfer rate. Generally speaking, a small recombination energy is in favor of improving the charge-transfer rate. According to the calculated results presented in Table 3, the λh values of HTMs are H01 (0.194 eV), H02 (0.195 eV), H03 (0.188 eV) and H04 (0.161 eV), respectively. In comparison with the λh value of Z26 (0.280 eV), the designed HTMs with smaller λh values may improve the charge-transfer rate or hole mobility. This result can be explained by introducing the electron-deficient units in arylamine derivative-based HTMs, which is an effective strategy to reduce the recombination energy.| Compounds | Pathways | λh | vh | kh | D | uh |
|---|---|---|---|---|---|---|
| H01 | 1 | 0.194 | 4.682 × 10−3 | 1.28 × 1011 | 8.190 | 1.372 × 10−2 |
| 2 | −5.695 × 10−4 | 1.89 × 109 | 15.382 | |||
| 3 | 1.868 × 10−5 | 2.03 × 106 | 15.649 | |||
| 4 | 7.789 × 10−3 | 3.53 × 1011 | 8.557 | |||
| 5 | −2.000 × 10−5 | 2.33 × 106 | 21.941 | |||
| 6 | −2.000 × 10−5 | 2.33 × 106 | 19.851 | |||
| H02 | 1 | 0.195 | 3.240 × 10−5 | 6.03 × 1012 | 4.781 | 8.886 × 10−2 |
| 2 | 1.934 × 10−5 | 2.15 × 106 | 21.664 | |||
| 3 | 1.424 × 10−3 | 1.17 × 1010 | 20.938 | |||
| 4 | −9.939 × 10−4 | 5.68 × 109 | 21.288 | |||
| 5 | 1.560 × 10−4 | 1.40 × 108 | 31.839 | |||
| 6 | 6.650 × 10−4 | 2.54 × 109 | 31.826 | |||
| 8 | 1.794 × 10−5 | 1.85 × 106 | 15.843 | |||
| 7 | 2.000 × 10−5 | 2.30 × 106 | 16.993 | |||
| 9 | 6.151 × 10−4 | 2.17 × 109 | 16.093 | |||
| H03 | 1 | 0.188 | 9.254 × 10−3 | 5.36 × 1011 | 8.922 | 2.624 × 10−2 |
| 2 | −3.391 × 10−4 | 7.20 × 108 | 17.796 | |||
| 3 | 1.736 × 10−3 | 1.89 × 1010 | 12.811 | |||
| 4 | 6.564 × 10−4 | 2.70 × 109 | 13.066 | |||
| 5 | −3.833 × 10−5 | 9.20 × 106 | 18.186 | |||
| 6 | −5.333 × 10−4 | 1.78 × 109 | 15.638 | |||
| 7 | 4.392 × 10−4 | 1.21 × 109 | 17.820 | |||
| 8 | −5.073 × 10−4 | 1.61 × 109 | 18.842 | |||
| 9 | −2.419 × 10−4 | 3.67 × 108 | 13.993 | |||
| 10 | 8.037 × 10−4 | 4.05 × 109 | 13.993 | |||
| H04 | 1 | 0.161 | −3.419 × 10−3 | 1.03 × 1011 | 9.551 | 5.197 × 10−3 |
| 2 | −6.399 × 10−5 | 3.60 × 107 | 13.104 | |||
| 3 | 3.000 × 10−5 | 7.91 × 106 | 15.375 | |||
| 5 | 2.179 × 10−4 | 4.17 × 108 | 16.407 | |||
| 4 | 1.717 × 10−3 | 2.59 × 1010 | 10.373 |
Another important parameter influencing the hole-transporting ability of HTMs is the hole-transfer integral vh. According to Marcus theory (eqn (5)), a maximized vh value is helpful in promoting the hole-transfer rate of HTM. For HTMs, it is well known that the intermolecular arrangement and frontier molecular orbitals of neighboring molecules are the key factors influencing vh.60 The intermolecular arrangement of a HTM can be obtained from the crystal structure. On the basis of the predicted crystal structure of Z26 and designed HTMs, one fragment is arbitrarily chosen as the center. All of the adjacent fragments with the center are defined as the transport pathways; that is, each transport pathway is the paired dimer between neighboring and center molecules. The main pathways of Z26, FDT and H01–H04 are presented in Fig. S5† and 3. Predicted crystal data of investigated molecules are listed in Table S4.† As shown in Table S4,† the calculated results indicate that the crystal structures of Z26 and H01–H04 with the lowest total energies belong to the space groups of C2, P1, P1, Pna21 and P212121, respectively. As shown in Fig. S5,† the calculated uh (8.050 × 10−4 cm2 V−1 s−1) of Z26 has the same order of magnitude as that of the experimental value (1.34 × 10−4 cm2 V−1 s−1).12 The calculated results indicate that the current theoretical level in this work is reliable based on the estimation of hole mobility for the designed HTMs.
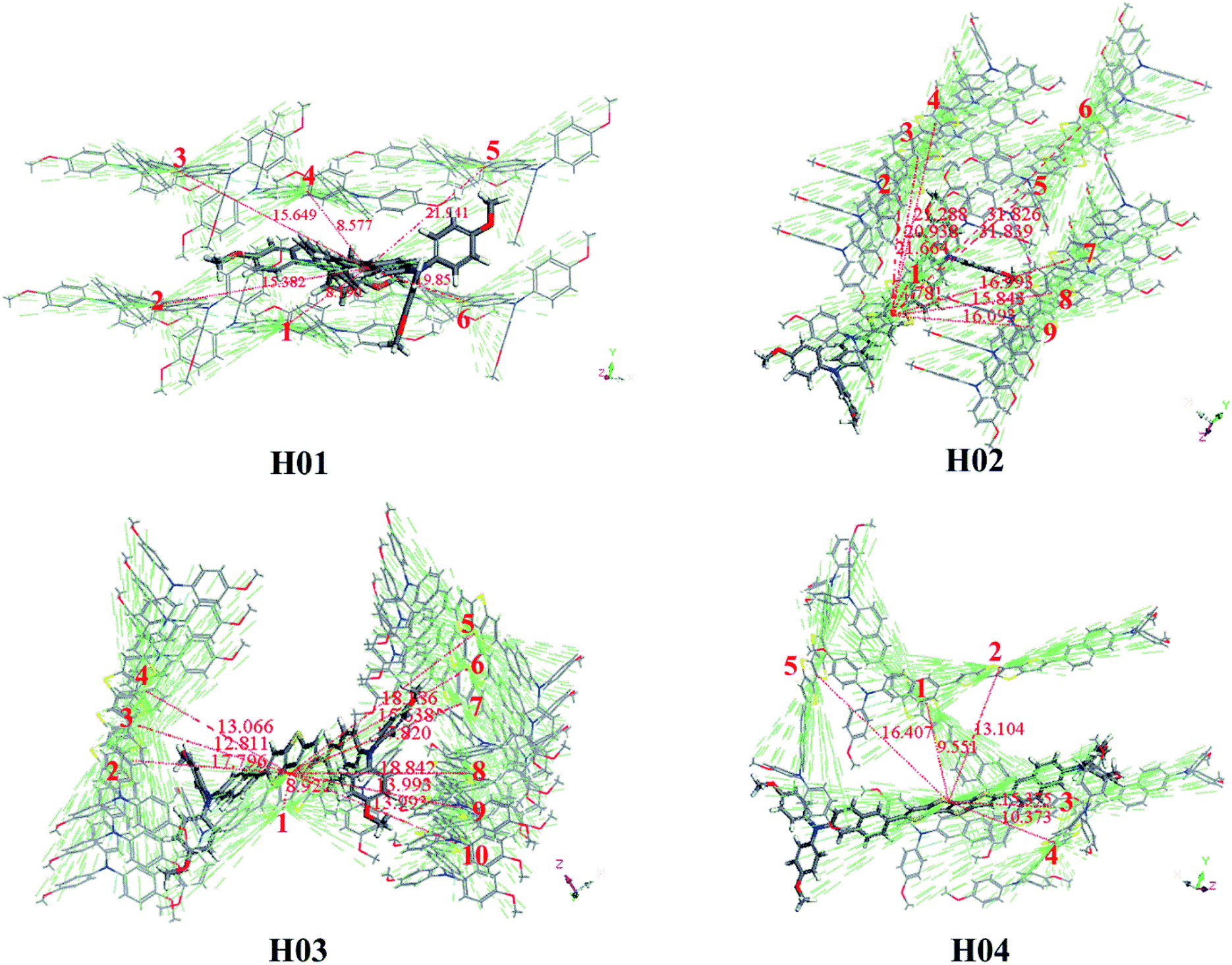 | ||
| Fig. 3 Main hole hopping pathways selected on basis of the crystal structures for all investigated molecules. | ||
The reorganization energy λh (eV), hole coupling vh (eV), hole transport rate kh (s−1), center-of-mass distance D (Å) and hole mobility uh (cm2 V−1 s−1) of main hopping pathway selected on the basis of the crystal structure for molecules H01–H04 are listed in Table 3. Table 3 shows that the uh values of H01–H04 are 1.372 × 10−2, 8.886 × 10−2, 2.624 × 10−2 and 5.197 × 10−3 cm2 V−1 s−1, respectively. In comparison with the calculated uh of Z26 (8.050 × 10−4 cm2 V−1 s−1) and FDT (2.848 × 10−3 cm2 V−1 s−1) (see Fig. S5†), the designed H01–H04 with higher hole mobility may improve the performance of PSC devices.
It can be seen that the uh values of H01–H04 are higher than those of Z26 and FDT, which can largely promote the performance of HTMs in PSC applications. H01–H04 with high uh values are attributed to a small recombination energy and large transfer integral. Therefore, in view of high uh values, the designed H01–H04 as HTMs can act as promising materials for improving the performance of PSCs.
On the basis of investigating the designed H01–H04, it can be concluded that these molecules serve as promising candidates as HTMs for PSC applications. Therefore, our proposed H01–H04 can be synthesized by the judicious design of the route, as shown in Fig. S6,† on the basis of the previously reported procedures.21
4. Conclusions
In summary, by adjusting the electron-rich arylamine derivatives in thienothiophene derivatives as π-bridged small molecule HTMs, four HTMs (H01–H04) were built on the basis of the reported excellent Z26. According to the requirement and working mechanism of HTM in the PSC devices, we investigated the electronic properties, optical absorptions, solubility, stability and hole transporting ability of these HTMs using DFT, TD-DFT and Marcus theory. The calculated results indicate that the selected methods we used herein could reproduce reliable HOMO/LUMO energy, absorption maximum and hole mobility of the reported Z26 in line with its experimental values. The frontier molecular orbital levels of molecules H01–H04 match the conduction and valence bands of CH3NH3PbI3 very well. Compared with Z26, the designed H01–H04 exhibit good optical properties, better solubility, good stability and higher hole mobilities. The designed HTMs of H01–H04 exhibit high hole mobilities of 1.372 × 10−2, 8.886 × 10−2, 2.624 × 10−2 and 5.197 × 10−3 cm2 V−1 s−1, respectively. Conclusively, the results confirmed that the design strategy of adjusting the electron-rich arylamine derivatives in thienothiophene derivatives as π-bridged HTMs is a reliable approach to obtain the promising HTMs for PSC applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant No. 21803043), Fundamental Research Funds for the Central Universities (Grant No. SWU117050), and Project Funds for Staying (Working) at Chongqing of the Outbound Postdoc.References
- G. Xing, N. Mathews, S. Sun, S. S. Lim, Y. M. Lam, M. Grätzel, S. Mhaisalkar and T. C. Sum, Science, 2013, 342, 344–347 CrossRef CAS PubMed.
- D. Shi, V. Adinolfi, R. Comin, M. Yuan, E. Alarousu, A. Buin, Y. Chen, S. Hoogland, A. Rothenberger and K. Katsiev, Science, 2015, 347, 519–522 CrossRef CAS PubMed.
- Q. Dong, Y. Fang, Y. Shao, P. Mulligan, J. Qiu, L. Cao and J. Huang, Science, 2015, 347, 967–970 CrossRef CAS PubMed.
- N. J. Jeon, H. Na, E. H. Jung, T.-Y. Yang, Y. G. Lee, G. Kim, H.-W. Shin, S. Il Seok, J. Lee and J. Seo, Nat. Energy, 2018, 3, 682–689 CrossRef CAS.
- X. Sun, D. Zhao and Z. a. Li, Chin. Chem. Lett., 2018, 29, 219–231 CrossRef CAS.
- X. Zhao and M. Wang, Materials Today Energy, 2018, 7, 208–220 CrossRef.
- P.-Y. Chen and S.-H. Yang, Opt. Mater. Express, 2016, 6, 3651 CrossRef CAS.
- I. García-Benito, I. Zimmermann, J. Urieta-Mora, J. Aragó, J. Calbo, J. Perles, A. Serrano, A. Molina-Ontoria, E. Ortí, N. Martín and M. K. Nazeeruddin, Adv. Funct. Mater., 2018, 28, 1801734 CrossRef.
- X. Liu, F. Kong, F. Guo, T. Cheng, W. Chen, T. Yu, J. Chen, Z. a. Tan and S. Dai, Dyes Pigm., 2017, 139, 129–135 CrossRef CAS.
- J. Zhang, T. Zhang, L. Jiang, U. Bach and Y.-B. Cheng, ACS Energy Lett., 2018, 3, 1677–1682 CrossRef CAS.
- M. Saliba, S. Orlandi, T. Matsui, S. Aghazada, M. Cavazzini, J.-P. Correa-Baena, P. Gao, R. Scopelliti, E. Mosconi, K.-H. Dahmen, F. De Angelis, A. Abate, A. Hagfeldt, G. Pozzi, M. Graetzel and M. K. Nazeeruddin, Nat. Energy, 2016, 1, 15017 CrossRef CAS.
- F. Zhang, Z. Wang, H. Zhu, N. Pellet, J. Luo, C. Yi, X. Liu, H. Liu, S. Wang, X. Li, Y. Xiao, S. M. Zakeeruddin, D. Bi and M. Grätzel, Nano Energy, 2017, 41, 469–475 CrossRef CAS.
- H. Li, K. Fu, A. Hagfeldt, M. Grätzel, S. G. Mhaisalkar and A. C. Grimsdale, Angew. Chem., Int. Ed., 2014, 53, 4085–4088 CrossRef CAS PubMed.
- D. Bi, B. Xu, P. Gao, L. Sun, M. Grätzel and A. Hagfeldt, Nano Energy, 2016, 23, 138–144 CrossRef CAS.
- H. Zhang, Y. Wu, W. Zhang, E. Li, C. Shen, H. Jiang, H. Tian and W. H. Zhu, Chem. Sci., 2018, 9, 5919–5928 RSC.
- P. Agarwala and D. Kabra, J. Mater. Chem. A, 2017, 5, 1348–1373 RSC.
- R. A. Belisle, P. Jain, R. Prasanna, T. Leijtens and M. D. McGehee, ACS Energy Lett., 2016, 1, 556–560 CrossRef CAS.
- A. Fakharuddin, L. Schmidt-Mende, G. Garcia-Belmonte, R. Jose and I. Mora-Sero, Adv. Energy Mater., 2017, 7, 1700623 CrossRef.
- Z.-Z. Sun, Y.-L. Xu, W.-L. Ding and W.-J. Chi, Synth. Met., 2019, 247, 157–162 CrossRef CAS.
- Y. L. Xu, W. L. Ding and Z. Z. Sun, Nanoscale, 2018, 10, 20329–20338 RSC.
- R. Azmi, S. Y. Nam, S. Sinaga, Z. A. Akbar, C.-L. Lee, S. C. Yoon, I. H. Jung and S.-Y. Jang, Nano Energy, 2018, 44, 191–198 CrossRef CAS.
- W. J. Chi, Q. S. Li and Z. S. Li, Nanoscale, 2016, 8, 6146–6154 RSC.
- W. J. Chi, P.-P. Sun and Z. S. Li, Nanoscale, 2016, 8, 17752–17756 RSC.
- W.-J. Chi, D.-Y. Zheng, X.-F. Chen and Z.-S. Li, J. Mater. Chem. C, 2017, 5, 10055–10060 RSC.
- E. G. Kim, V. Coropceanu, N. E. Gruhn, R. S. Sanchez-Carrera, R. Snoeberger, A. J. Matzger and J. L. Bredas, J. Am. Chem. Soc., 2007, 129, 13072–13081 CrossRef CAS PubMed.
- W. Hu, Z. Zhang, J. Cui, W. Shen, M. Li and R. He, Nanoscale, 2017, 9, 12916–12924 RSC.
- A. Cohen, P. Mori-Sánchez and W. Yang, Chem. Rev., 2012, 112, 289–320 CrossRef CAS PubMed.
- M. Saliba, S. Orlandi, T. Matsui, S. Aghazada, M. Cavazzini, J.-P. Correa-Baena, P. Gao, R. Scopelliti, E. Mosconi and K.-H. Dahmen, Nat. Energy, 2016, 1, 15017 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- X. Liu, M. Li, R. He and W. Shen, Phys. Chem. Chem. Phys., 2014, 16, 311–323 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, et al., Gaussian 09, Revision A.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- W. Q. Deng, L. Sun, J. D. Huang, S. Chai, S. H. Wen and K. L. Han, Nat. Protoc., 2015, 10, 632–642 CrossRef CAS.
- J. L. Bredas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971–5004 CrossRef CAS.
- B. C. Lin, C. P. Cheng and Z. P. M. Lao, J. Phys. Chem. A, 2003, 107, 5241–5251 CrossRef CAS.
- M. Y. Kuo, H. Y. Chen and I. Chao, Chem.–Eur. J., 2007, 13, 4750–4758 CrossRef CAS PubMed.
- L. Wang, G. Nan, X. Yang, Q. Peng, Q. Li and Z. Shuai, Chem. Soc. Rev., 2010, 39, 423–434 RSC.
- J. Bisquert, Phys. Chem. Chem. Phys., 2008, 10, 3175–3194 RSC.
- W. Q. Deng and W. A. Goddard III, J. Phys. Chem. B, 2004, 108, 8614–8621 CrossRef CAS.
- R. A. Marcus, Annu. Rev. Phys. Chem., 1964, 15, 155–196 CrossRef CAS.
- R. A. Marcus, Angew. Chem., Int. Ed., 1993, 32, 1111–1121 CrossRef.
- X. Yang, L. Wang, C. Wang, W. Long and Z. Shuai, Chem. Mater., 2008, 20, 3205–3211 CrossRef CAS.
- X. Yang, Q. Li and Z. Shuai, Nanotechnology, 2007, 18, 424029 CrossRef.
- C. Wang, F. Wang, X. Yang, Q. Li and Z. Shuai, Org. Electron., 2008, 9, 635–640 CrossRef CAS.
- S.-H. Wen, A. Li, J. Song, W.-Q. Deng, K.-L. Han and W. A. Goddard III, J. Phys. Chem. B, 2009, 113, 8813–8819 CrossRef CAS PubMed.
- K. Senthilkumar, F. Grozema, F. Bickelhaupt and L. Siebbeles, J. Chem. Phys., 2003, 119, 9809–9817 CrossRef CAS.
- J. P. Perdew, J. Chevary, S. Vosko, K. A. Jackson, M. R. Pederson, D. Singh and C. Fiolhais, Phys. Rev. B, 1992, 46, 6671 CrossRef CAS.
- G. T. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- C. F. Guerra, J. Snijders, G. Te Velde and E. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 Search PubMed.
- ADF2014 SCM Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http://www.scm.com Search PubMed.
- Materials Studio, San Diego, CA, 2005 Search PubMed.
- G. Day, W. Motherwell, H. Ammon, S. Boerrigter, R. Della Valle, E. Venuti, A. Dzyabchenko, J. Dunitz, B. Schweizer and B. Van Eijck, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 511–527 CrossRef CAS PubMed.
- R. Jin and Y. Chang, Phys. Chem. Chem. Phys., 2015, 17, 2094–2103 RSC.
- W.-J. Chi and Z.-S. Li, Phys. Chem. Chem. Phys., 2015, 17, 5991–5998 RSC.
- S. S. Reddy, K. Gunasekar, J. H. Heo, S. H. Im, C. S. Kim, D. H. Kim, J. H. Moon, J. Y. Lee, M. Song and S. H. Jin, Adv. Mater., 2016, 28, 686–693 CrossRef CAS PubMed.
- S. Tze Chien, C. Shi, X. Guichuan, L. Xinfeng and W. Bo, Nanotechnology, 2015, 26, 342001 CrossRef.
- D. A. Egger, S. Weissman, S. Refaely-Abramson, S. Sharifzadeh, M. Dauth, R. Baer, S. Kummel, J. B. Neaton, E. Zojer and L. Kronik, J. Chem. Theory Comput., 2014, 10, 1934–1952 CrossRef CAS.
- J. M. Ho, A. Klamt and M. L. Coote, J. Phys. Chem. A, 2010, 114, 13442–13444 CrossRef CAS.
- Y. Zhang, Y. Y. Li, C. Chen, L. Wang and J. L. Zhang, Org. Electron., 2017, 49, 255–261 CrossRef CAS.
- H. Liu and X. Liu, J. Mater. Chem. C, 2018, 6, 6816–6822 RSC.
- A. Zhugayevych, O. Postupna, R. C. Bakus II, G. C. Welch, G. C. Bazan and S. Tretiak, J. Phys. Chem. C, 2013, 117, 4920–4930 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1–S4, Fig. S1–S6. See DOI: 10.1039/c9ra03408k |
| This journal is © The Royal Society of Chemistry 2019 |