
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
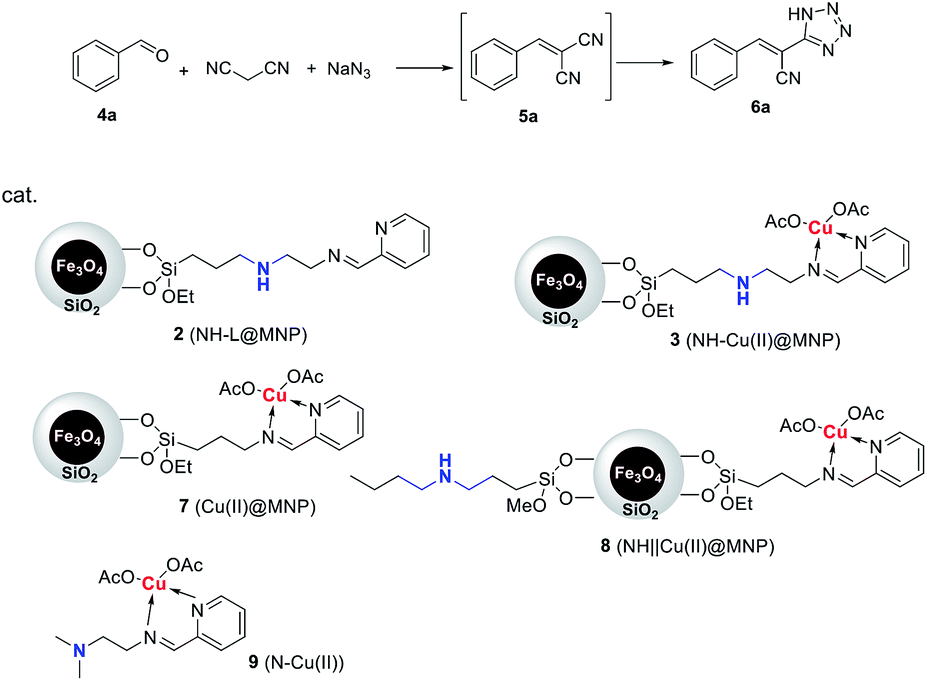
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn intramolecular relay catalysis strategy for Knoevenagel condensation and 1,3-dipolar cycloaddition domino reactions†
Xiaofeng Yuana,
Zijuan Wanga,
Qiang Zhang b and
Jun Luo*a
b and
Jun Luo*a
aSchool of Chemical Engineering, Nanjing University of Science and Technology, Nanjing 210094, China. E-mail: luojun@njust.edu.cn
bJiangsu Key Laboratory of Environmental Functional Materials, School of Chemistry, Biology and Material Engineering, Suzhou University of Science and Technology, Suzhou 215009, China
First published on 30th July 2019
Abstract
A relay catalysis strategy was established by using a bifunctional catalyst which was prepared by immobilization of organic chains containing secondary amine and Cu(II) complex onto silica-coated nano-Fe3O4. The simply prepared nanoparticles acted as efficient, intramolecular relays and magnetically recyclable base-metal bifunctional catalysts for Knoevenagel condensation and 1,3-dipolar cycloaddition domino reactions to prepare 5-substituted 1H-tetrazoles with excellent yields.
Introduction
Multifunctional catalysts for one-pot multistep reactions have attracted much attention recently owing to their high efficiency.1–12 Bifunctional catalysts with different catalytic centers on separate or the same supports are the dominant targets in this field.13–33 For example, Shylesh et al. used organic amines and sulfonic acid bifunctionalized porous organosilicas in one-pot deacetalization–nitroaldol cascade reactions.16 Shiju et al. prepared an efficient and robust bifunctional acid–base solid catalyst and evaluated the catalytic activities in deprotection–Henry domino reactions and deprotection–aldol reactions.17 Huang et al. prepared a bifunctional mesoporous silica nanoparticle with sulfonic acid moiety on its internal surface and organic amine moiety on its external surface and applied it in deacetalization–Henry cascade reactions.28 To sum up, the reported bifunctional supported catalysts were constructed by mainly two strategies: parallel immobilization of different catalytic moieties on the surface of the same support (Fig. 1(a)) or separate immobilization of different catalytic moieties on the internal and external surface respectively (Fig. 1(b)). The two design ideas are smart and attractive. However, for domino reactions, the mass transfer of intermediate products between two catalytic centers still needs time and energy. So, how can we make the transfer procedure more efficient? With this question in mind, we put forward a new strategy by sequentially connecting different catalytic moieties on the same organic chain and immobilizing it on the surface of one support (Fig. 1(c)).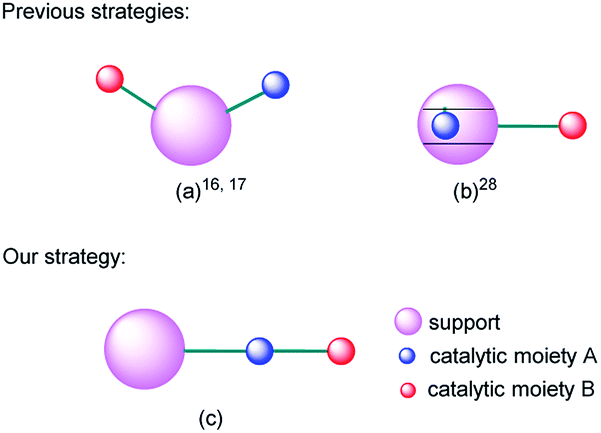 | ||
| Fig. 1 Strategies of nanoparticle-supported bifunctional catalysts. | ||
Magnetic nanoparticles (MNPs) are attractive catalyst supports because they have advantages of excellent dispersion, easy separation and high compatibility with numerous kinds of catalysts.34–42 Among them, silica coated MNPs attract increasing interests because they have a considerable amount of derivable hydroxyl groups and can be easily functionalized by catalyst-containing organosilicate esters.29,43–56 Our previous work speculated that the reactants would be adsorbed on the surface of the nano-support, so the local concentration of reactants around the catalytic sites would increase and then effectively elevate the reaction rate.48
The new relay catalysis using easily recyclable bifunctional nano-catalysts is illustrated in Fig. 2.
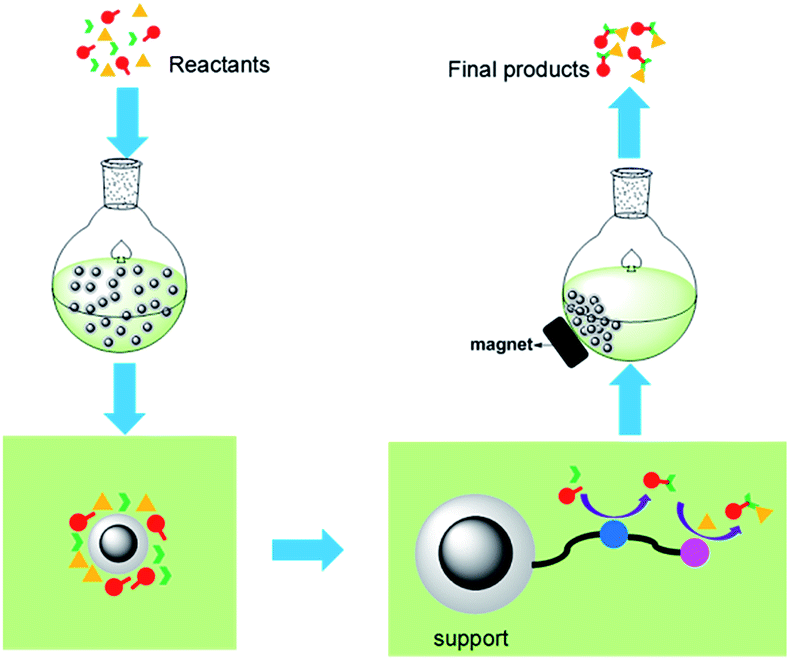 | ||
| Fig. 2 Ideal relay catalysis for domino reactions. | ||
5-Substituted 1H-tetrazoles have extensive applications in chemistry.57–65 The conventional synthesis of these compounds is [2 + 3] cycloaddition of nitriles with azide salts.66,67 A variety of catalysts for this transformation have been reported.68–81 However, the reported methods have one or more drawbacks such as use of highly toxic reagents, high cost, elevated reaction temperature, long reaction time or unsatisfactory recovery of catalyst. Therefore, the demand of efficient, economical and greener method for the preparation of 5-substituted 1H-tetrazoles is still demanded.
Herein, we reported a new relay catalysis system involving the establishment of a secondary amine/Cu(II) bifunctional magnetic nanoparticle (NH–Cu(II)@MNP) characteristic of sequentially connecting secondary amine and copper(II) complex on the same organic chain, and application of it as efficient, intramolecular relay and magnetically recyclable bifunctional nano-catalyst in the one-pot synthesis of 5-substituted 1H-tetrazoles through Knoevenagel condensation and 1,3-dipolar cycloaddition domino reactions.
Results and discussion
The procedure for the preparation of the nano-Fe3O4 supported secondary amine/Cu(II) bifunctional catalyst (NH–Cu(II)@MNP) is shown in Scheme 1. The support Fe3O4@SiO2 were prepared according to our previous work.48 Then [3-(2-aminoethyl)aminopropyl]triethoxysilane was introduced onto Fe3O4@SiO2 through a condensation reaction. After that, the secondary amine functionalized precursor NH-L@MNP (2) was prepared by a condensation reaction of compound 1 with pyridine-2-carbaldehyde. Meanwhile, the loading amount of the organic motif was determined to be 0.20 mmol g−1 by elemental analysis, which was also in accordance with thermogravimetric (TG) test. Ultimately, 2 was coordinated with Cu(OAc)2 in acetone at room temperature for 24 h to give the target catalyst 3.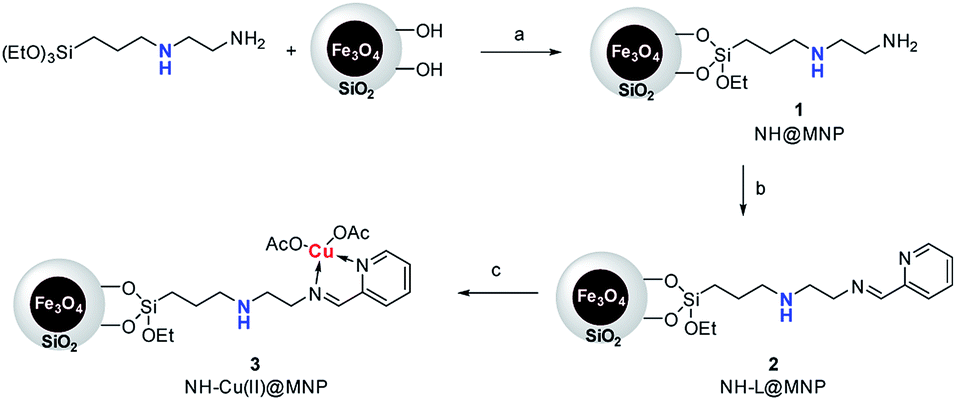 | ||
| Scheme 1 Synthesis of MNP-supported bifunctional catalyst NH–Cu(II)@MNP (3). Reaction conditions: (a) toluene, N2, reflux, 24 h; (b) pyridine-2-carbaldehyde, EtOH, N2, reflux, 4 h; (c) Cu(OAc)2·H2O, acetone, N2, RT, 24 h. | ||
The Fourier transform infrared (FT-IR) spectra of Fe3O4@SiO2 and catalyst 3 are demonstrated in Fig. 3. The spectrum of Fe3O4@SiO2 shows a moderate peak at 586 cm−1 (Fe–O vibration), a strong peak at 1087 cm−1 (Si–O–Si stretching vibration), a broad weak peak around 3263 cm−1 (O–H stretching vibration) and a weak peak at 1602 cm−1 (adsorbed water). All of these characteristic signals are observed in the spectrum of 3. Moreover, 3 shows typical bands at 1597 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N vibration) and 1446 cm−1 (CC vibration of aryl ring). These results indicate that organic moiety was successfully immobilized onto Fe3O4@SiO2.
N vibration) and 1446 cm−1 (CC vibration of aryl ring). These results indicate that organic moiety was successfully immobilized onto Fe3O4@SiO2.
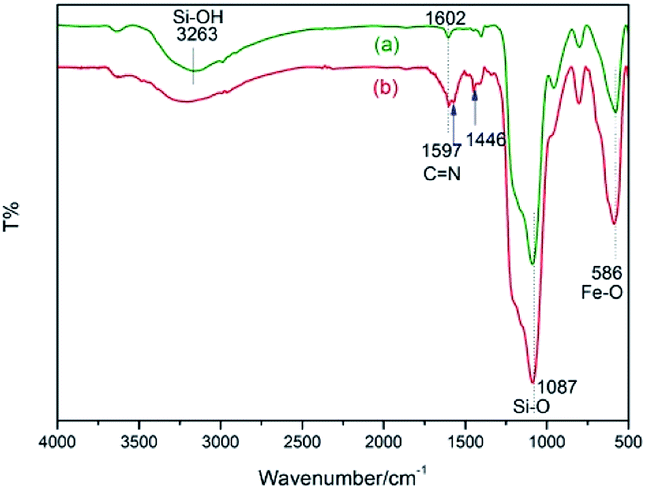 | ||
| Fig. 3 FT-IR spectra of (a) Fe3O4@SiO2 and (b) NH–Cu(II)@MNP. | ||
Fig. 4 shows the X-ray diffraction (XRD) patterns of Fe3O4@SiO2 and 3. Both samples have the diffraction peaks on the standard Fe3O4 sample (Fig. 4(a), JCPDS file no. 19-0629). The broad peak of 2θ = 20–30° belongs to silica layer. The pattern of 3 (Fig. 4(b)) is similar to that of Fe3O4@SiO2. But the intensity becomes weak in Fig. 4(b), indicating that the structure of Fe3O4@SiO2 remains unchanged after being functionalized. The average crystal size of the Fe3O4 cores is about 11.4 nm according to the Scherrer formula.
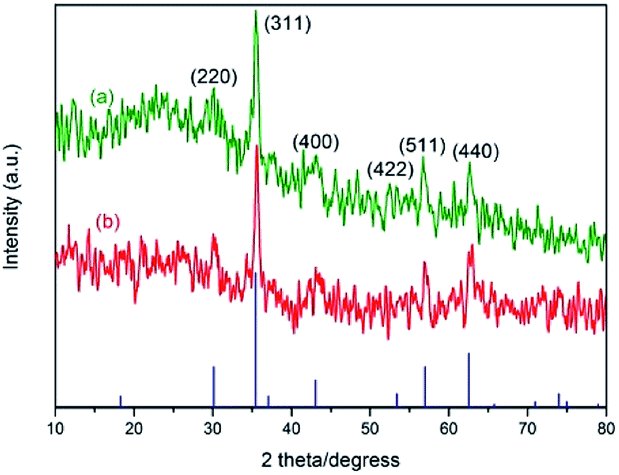 | ||
| Fig. 4 XRD patterns of (a) Fe3O4@SiO2 and (b) NH–Cu(II)@MNP catalyst 3. | ||
The transmission electron microscope (TEM) images of Fe3O4@SiO2 and 3 were recorded (Fig. 5). As shown in Fig. 5(a), the Fe3O4 cores are obviously surrounded by grey silica shells. The average size of the Fe3O4 cores and the support Fe3O4@SiO2 are about 8–12 nm and 20 nm, respectively. As shown in Fig. 5(b), the size of 3 is about 20–30 nm. And after being reused six times, the morphology of recovered 3 does not show an obvious change (Fig. 5(c)).
 | ||
| Fig. 5 TEM images of (a) Fe3O4@SiO2, (b) NH–Cu(II)@MNP and (c) catalyst after being reused six times. | ||
The surface composition of 3 was analyzed with X-ray photoelectron spectroscopy (XPS). As shown in Fig. 6(a), the peaks with binding energy at 24.9, 102.2, 154.8, 284.9, 399.1, 531.7, 933.1 and 953.3 eV were assigned to O 2s, Si 2p, Si 2s, C 1s, N 1s, O 1s, Cu 2p3/2 and Cu 2p1/2, which affirmed that the catalyst was made up of C, O, Si, N and Cu. However, the peaks with binding energy at 711 and 723 eV were distributed to Fe 2p3/2 and Fe 2p1/2, which indicated that there is a little Fe3O4 present on the surface of catalyst 3. The intense and broad peaks at 932.8 eV (Cu 2p3/2) and 952.5 eV (Cu 2p1/2) along with the presence of the characteristic shakeup satellite peaks suggest that the copper on the surface has an oxidation state of +2 (Fig. 6(b)).82,83 Furthermore, in the N 1s spectrum of 3, –N (398.7 eV) and –NH– (399.5 eV) were observed (Fig. 6(c)).84 In a word, it is clear that the secondary amine and Cu(II) were successfully anchored on the surface of the Fe3O4@SiO2 support.
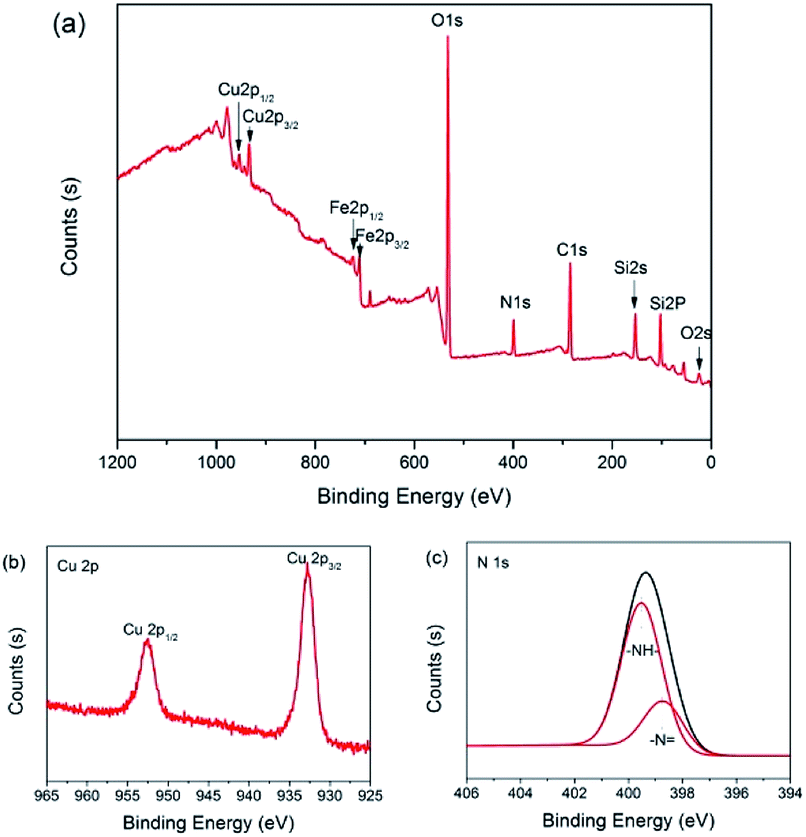 | ||
| Fig. 6 The XPS spectra of the NH–Cu(II)@MNP catalyst 3. (a) Survey scan; (b) Cu 2p and (c) N 1s. | ||
Besides, the elemental mapping images (see ESI†) also confirmed the existence of Fe, Si, N and Cu, and these elements species were equably distributed on the NH–Cu(II)@MNP. Furthermore, the distribution density of nitrogen and copper was less than that of iron and silicon. This was in accordance with the load ratio of copper and nitrogen on Fe3O4@SiO2 support.
The thermostability of 3 was investigated by thermogravimetric analysis (Fig. 7). The TG curve indicates an initial weight loss of 1.3% up to 100 °C, which is ascribed to the adsorbed water. A main weight loss of 6.6% is observed from 350 to 620 °C, which is attributed to the decomposition of organic structure. The peak in the derivative thermogravimetric (DTG) curve shows that the fastest loss of the organic moiety occurred at 480 °C. Therefore, the thermostability of catalyst 3 is excellent.
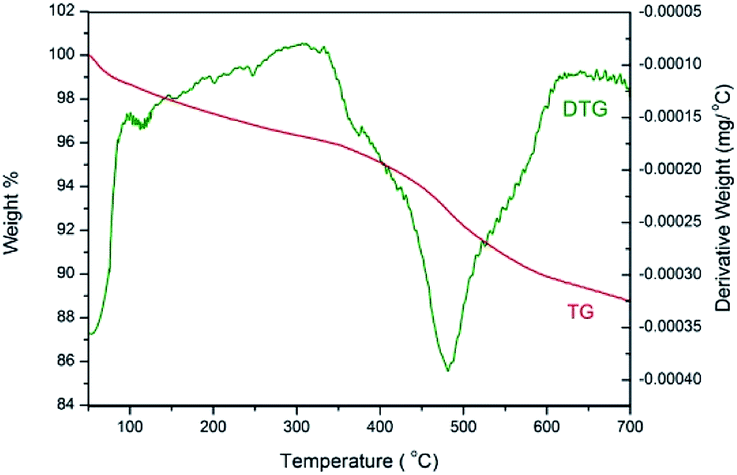 | ||
| Fig. 7 Thermogravimetric (TG) and derivative thermogravimetric (DTG) curves for NH–Cu(II)@MNP. | ||
The magnetic property was evaluated by vibrating sample magnetometer (VSM) test at room temperature (Fig. 8). The magnetization of samples could be completely saturated at high fields of up to 1.5 T. And the maximum magnetic saturation intensity of 3 is 37.6 emu g−1 (Fig. 8(b)). The VSM magnetization curve of the sample goes through the zero point, so the catalyst has no magnetic hysteresis phenomenon, which indicates that the as-prepared catalyst is superparamagnetic.
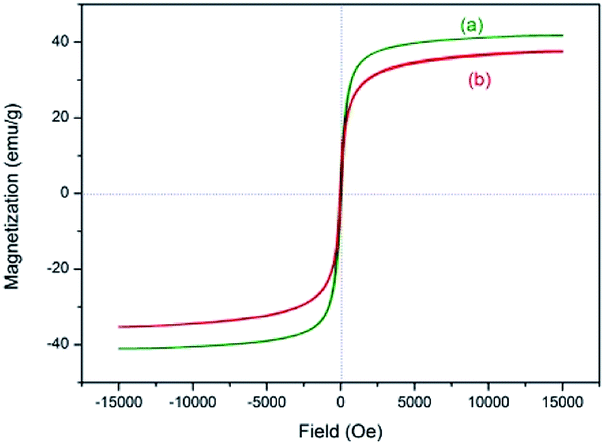 | ||
| Fig. 8 VSM magnetization curve of (a) Fe3O4@SiO2 and (b) NH–Cu(II)@MNP. | ||
The catalyst can be easily separated from the reaction system using an external magnet (Fig. 9).
 | ||
| Fig. 9 A reaction mixture in the absence (a) or presence (b) of a magnetic field. | ||
The bifunctional catalytic performance of 3 was firstly investigated in a one-pot synthesis of (E)-3-phenyl-2-(1H-tetrazole-5-yl) acrylonitrile (6a) using benzaldehyde, malononitrile and sodium azide as the reactants. The domino reaction sequence was composed of two steps: Knoevenagel condensation and 1,3-dipolar cycloaddition. To clarify the bifunctional catalytic activity of 3, the catalytic activities of a series of control samples were evaluated and the results are summarized in Table 1. For comparison, Cu(II)@MNP (7), NH‖Cu(II)@MNP (8, parallel immobilization of NH and Cu(II) on the same support like Fig. 1(b)) and N–Cu(II) (9) were also prepared (see ESI†).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | T (°C) | t (h) | 5ab (%) | 6ab (%) |
| a Reaction conditions: benzaldehyde (1.0 mmol), malononitrile (1.1 mmol), sodium azide (1.2 mmol), catalyst (1 mol%, 20 mg), EtOH (10 mL), 80 °C.b Isolated yield.c Only for the Knoevenagel condensation of benzaldehyde and malononitrile, i.e. in the absence of sodium azide.d Only for the 1,3-dipolar cycloaddition of 5a and sodium azide.e Cu(OAc)2 (20 mol%), diethylamine (20 mol%).f N–Cu(II) (1 mol%). | |||||
| 1c | 2 | 80 | 2.0 | 90 | — |
| 2c | 3 | 80 | 1.0 | 91 | — |
| 3c | 3 | 40 | 3.5 | 91 | — |
| 4c | None | 40 | 12.0 | 90 | — |
| 5d | 3 | 80 | 4.0 | — | 93 |
| 6d | 7 | 80 | 5.0 | — | 90 |
| 7 | None | 80 | 5.0 | 41 | 38 |
| 8 | Fe3O4@SiO2 | 80 | 5.0 | 35 | 43 |
| 9 | 2 | 80 | 5.0 | 26 | 50 |
| 10 | 7 | 80 | 5.0 | 17 | 69 |
| 11 | 3 | 80 | 5.0 | 5 | 92 |
| 12 | 2 + 7 | 80 | 5.0 | 15 | 72 |
| 13 | 8 | 80 | 5.0 | 13 | 80 |
| 14e | Cu(OAc)2 + Et2NH | 80 | 5.0 | 0 | 64 |
| 15f | 9 | 80 | 5.0 | 0 | 63 |
The model Knoevenagel condensation of benzaldehyde (4a) and malononitrile at 80 °C was conducted using NH-L@MNP (2) and NH–Cu(II)@MNP (3) as catalysts. Both of them showed excellent activity while the latter was a little beat better (Table 1, entries 1–3). As for the control experiment without any catalyst, much longer time was required to get comparative yield (Table 1, entry 4). Meanwhile, the model 1,3-dipolar cycloaddition of 5a and sodium azide catalyzed by 3 gave excellent yield (Table 1, entry 5). However, the control experiment using Cu(II)@MNP (7) as catalyst, longer reaction time was needed and lower yield was obtained (Table 1, entry 6). The domino reaction without any catalyst was conducted, both intermediate and final product were isolated but with low yields (Table 1, entry 7). Meanwhile, the support Fe3O4@SiO2 and 2 had almost negligible effect on the yield of 6a (Table 1, entries 8 and 9). But the product ratio (6a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5a) of the reaction changed. In the case of 7, the desired product was obtained with a much higher yield of 69% (Table 1, entry 10). To our delight, the sequentially bifunctionalized catalyst 3 could efficiently catalyze the domino reaction and gave an excellent yield of 92% (Table 1, entry 11). In comparison, when a mixture of the two monofunctionalized catalysts 2 and 7 were used, only 72% of 6a were obtained (Table 1, entry 12). Meanwhile, the parallelly bifunctionalized catalyst NH‖Cu(II)@MNP (8) afforded obviously lower yield (80%, Table 1, entry 13), which was in accordance with our hypothesis. That is, the sequentially bifunctionalized catalyst 3 exhibited superb catalytic activity and selectivity than parallelly bifunctionalized analogue, the reason must be that the relay catalytic process is in favor of the mass transfer of intermediate product. To explore the effect of support, the catalytic performance of a homogeneous catalyst N–Cu(II) (9) was tested, a significantly lower yield of 63% was obtained (Table 1, entry 15). The result showed that 9 displayed decreased catalytic efficiency. Moreover, we found that the physical mixture of Cu(OAc)2 and diethylamine exhibited lower catalytic activity too (Table 1, entry 14).
:5a) of the reaction changed. In the case of 7, the desired product was obtained with a much higher yield of 69% (Table 1, entry 10). To our delight, the sequentially bifunctionalized catalyst 3 could efficiently catalyze the domino reaction and gave an excellent yield of 92% (Table 1, entry 11). In comparison, when a mixture of the two monofunctionalized catalysts 2 and 7 were used, only 72% of 6a were obtained (Table 1, entry 12). Meanwhile, the parallelly bifunctionalized catalyst NH‖Cu(II)@MNP (8) afforded obviously lower yield (80%, Table 1, entry 13), which was in accordance with our hypothesis. That is, the sequentially bifunctionalized catalyst 3 exhibited superb catalytic activity and selectivity than parallelly bifunctionalized analogue, the reason must be that the relay catalytic process is in favor of the mass transfer of intermediate product. To explore the effect of support, the catalytic performance of a homogeneous catalyst N–Cu(II) (9) was tested, a significantly lower yield of 63% was obtained (Table 1, entry 15). The result showed that 9 displayed decreased catalytic efficiency. Moreover, we found that the physical mixture of Cu(OAc)2 and diethylamine exhibited lower catalytic activity too (Table 1, entry 14).
The domino reaction of benzaldehyde, malononitrile, sodium azide was investigated to find the optimal conditions (Table 2). The reaction without any catalyst afforded the product with low yield (Table 2, entry 1). Along with the increasing load of catalyst, the yield ascended accordingly (Table 2, entries 2 and 3). The best result was obtained by applying 20 mg of 3 as catalyst and conducting the reaction at 80 °C (Table 2, entry 4). More catalyst demonstrated no significant effect on the yield (Table 2, entries 6 and 7). Prolonging the reaction time to 10 h resulted in almost unchanged yield (Table 2, entry 5). Lower reaction temperature was unfavourable to the reaction (Table 2, entries 4, 8 and 9).
|
||||
|---|---|---|---|---|
| Entry | 3 (mg) | T (°C) | t (h) | Yieldb (%) |
| a Reaction conditions: benzaldehyde (1.0 mmol), malononitrile (1.1 mmol), sodium azide (1.2 mmol), NH–Cu(II)@MNP, EtOH (10 mL), 5 h.b 6a (%), isolated yield. | ||||
| 1 | 0 | 80 | 5 | 39 |
| 2 | 5 | 80 | 5 | 66 |
| 3 | 15 | 80 | 5 | 90 |
| 4 | 20 | 80 | 5 | 92 |
| 5 | 20 | 80 | 10 | 91 |
| 6 | 50 | 80 | 5 | 91 |
| 7 | 100 | 80 | 5 | 89 |
| 8 | 20 | 50 | 5 | 75 |
| 9 | 20 | 20 | 5 | 13 |
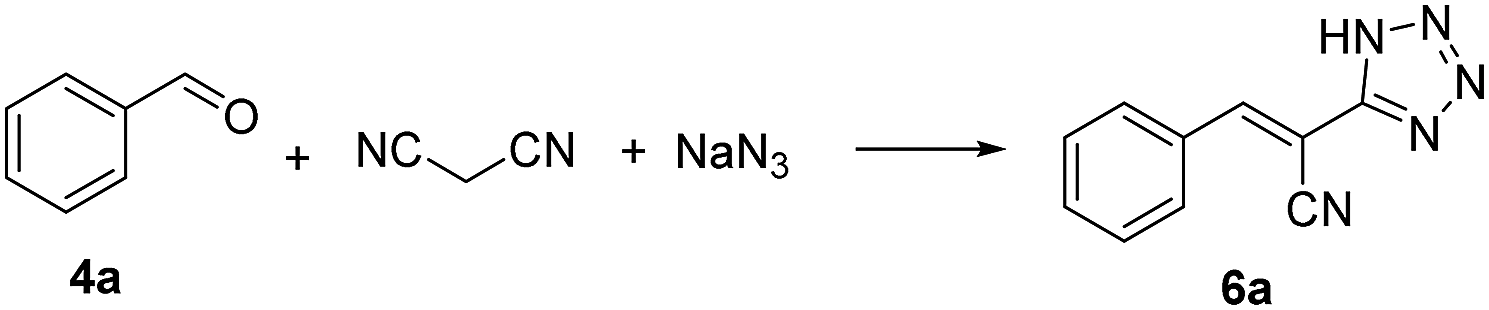
Thereafter, the above optimized reaction conditions were explored for the synthesis of a number of 5-substituted 1H-tetrazoles. As shown in Table 3, a variety of aldehydes reacted smoothly with malononitrile and sodium azide to afford the corresponding tetrazoles with excellent yields. meta-Methylbenzaldehyde (4c) resulted in a little higher yield than ortho-methylbenzaldehyde (4b) and para-methylbenzaldehyde (4d). The aldehydes containing electron-withdrawing groups like bromo, chloro, iodo, fluoro and cyano in para positions (4e–i) produced more satisfactory yields than those containing electron-donating groups like ethyl, isopropyl, methoxy and tert-butyl in para positions (4j–m). Heterocyclic aldehydes like thiophene-2-carbaldehydes (4n and 4o) also exhibited high reactivity and afforded the desired products with excellent yields. It is very strange that pyridine-2-carbaldehyde (4p) gave no desired product. When pyridine (1 equiv.) was added to the mixture of benzaldehyde, malonitrile, sodium azide and catalyst 3, the yield of tetrazole was only 36%. Based on this result, we supposed that the coordination of pyridine moiety and copper prevented the formation of the target product. From the above results, it is obvious that 3 is an efficient catalyst for the preparation of large spectrum of substituted tetrazoles with very high yields in ethanol.
A plausible mechanism is shown in Scheme 2. Initially, the substrates were absorbed on the surface of the catalyst by hydrogen bonds. Then active α-H of malonitrile is abstracted by the secondary amine moiety (A) and the carbanion formed attacks the carbonyl, activated by acidic proton in ammonium, of benzaldehyde (B). After that, a synergistic dehydration occurs (C) to produce the intermediate product 2-benzylidenemalononitrile (5a). Subsequently, the coordination of the cyano group in 2-benzylidenemalononitrile with Cu(II) accompanying with anions exchange (replacement of acetate anion with azide) occurs (D), then a 1,3-dipolar cycloaddition between the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N group and azide anion takes place readily to form the tetrazole ring (E). Another anions exchange occurs to afford the catalyst and sodium tetrazolate, gives target product by acidic workup.
N group and azide anion takes place readily to form the tetrazole ring (E). Another anions exchange occurs to afford the catalyst and sodium tetrazolate, gives target product by acidic workup.
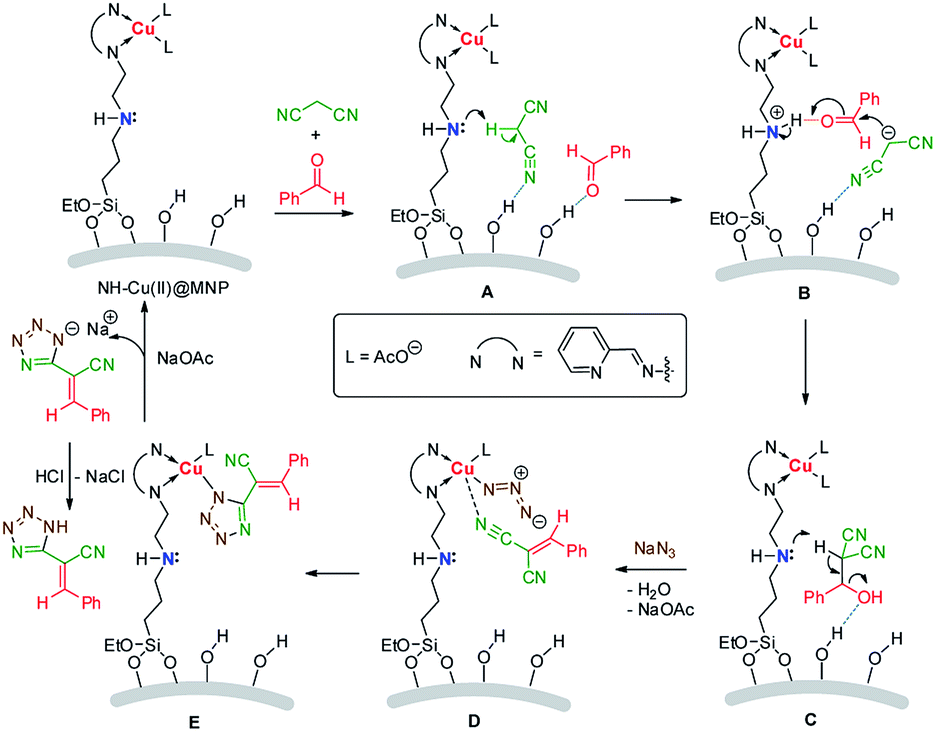 | ||
| Scheme 2 Plausible reaction mechanism. | ||
One of the most significant features of a catalyst is its recyclability. Thus, the recyclability of 3 were tested in the model reaction under the optimized reaction conditions. The catalyst was separated from the reaction mixture by an external magnet and then washed with ethanol. Then the recovered catalyst was used in the next run. For all the six cycles, the catalyst could be reused without significant loss of catalytic activity (Fig. 10).
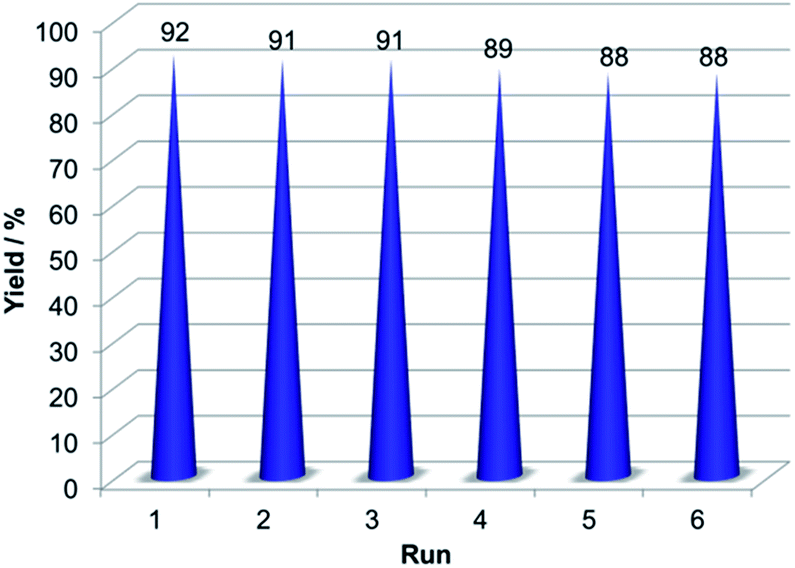 | ||
| Fig. 10 Recycling experiment of the NH–Cu(II)@MNP. | ||
Conclusions
In summary, we have successfully established a relay catalysis strategy for one-pot domino reactions. A silica-coated nano-Fe3O4-supported bifunctional amine/Cu(II) catalyst was prepared and used in the synthesis of 5-substituted 1H-tetrazoles via a domino Knoevenagel condensation/1,3-dipolar cycloaddition pathway. Furthermore, the catalyst could be easily recovered from the reaction system by an external magnetic field and reused several times without loss of catalytic activity.Experimental section
Materials
Toluene was dehydrated according to standard operation and stored on 4 Å molecular sieves. Other solvents and all of the reagents were commercially available and were used without further purification. The core–shell Fe3O4@SiO2 nanoparticles were prepared according to our previous work.48Synthesis of NH@MNP (1)
Fe3O4@SiO2 (1.0 g) was dispersed in anhydrous toluene (50 mL) by sonication for 1 h. [3-(2-Aminoethyl)aminopropyl]triethoxysilane (0.529 g, 2.0 mmol) was then added, and the reaction mixture was refluxed for 24 h under nitrogen atmosphere. After being cooled to room temperature, 1 was recycled by a permanent magnet, washed with dry toluene and acetone to remove unreacted species and then dried under vacuum overnight.Synthesis of NH-L@MNP (2)
1 (1.0 g) was dispersed in anhydrous ethanol (40 mL) by sonication for 30 min. Pyridine-2-carbaldehyde (0.214 g, 2.0 mmol) was then added, and the resulting mixture was stirred under reflux for 4 h under nitrogen atmosphere. Product 2 was recycled by a permanent magnet, washed with extra dry ethanol and then dried under vacuum overnight.Synthesis of NH–Cu(II)@MNP (3)
Copper sulfate monohydrate (0.399 g, 2.0 mmol) were dissolved in acetone (40 mL) and the solution was stirred at room temperature for 20 min, then 2 (1.0 g) was added. The final mixture was stirred at room temperature for 24 h under nitrogen atmosphere. The catalyst 3 was collected by a permanent magnet, washed several times with acetone and dried under vacuum overnight. The loading content of Cu was 1.3% as determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES).General procedure for the synthesis of tetrazoles 6
A mixture of aldehyde (1 mmol), malononitrile (1.1 mmol), sodium azide (1.2 mmol) and 3 (20 mg, 1.0 mol%) in EtOH (10 mL) was stirred at 80 °C for set time. After completion of the reaction, as indicated by TLC, the reaction mixture was cooled to ambient temperature. The catalyst 3 was recovered by the same procedure mentioned above and used for the next run. To the reaction mixture was added 2 N HCl (30 mL) under vigorous stirring to precipitate the product. Then the mother liquor was extracted by ethyl acetate (3 × 50 mL). The combined organic layers were dried over anhydrous Na2SO4 and evaporated under reduced pressure to obtain the crude product, the pure products were achieved by column chromatography on silica gel.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was sponsored by the Natural Science Foundation of Jiangsu Province of China (no. BK2010485) and Qing Lan Project of Jiangsu Province, P. R. China (2014).Notes and references
- Y. Yang, X. Liu, X. Li, J. Zhao, S. Bai, J. Liu and Q. Yang, Angew. Chem., Int. Ed., 2012, 51, 9164–9168 CrossRef CAS PubMed
.
- P. Li, C. Y. Cao, Z. Chen, H. Liu, Y. Yu and W. G. Song, Chem. Commun., 2012, 48, 10541–10543 RSC
- A. V. Biradar, V. S. Patil, P. Chandra, D. S. Doke and T. Asefa, Chem. Commun., 2015, 51, 8496–8499 RSC
- Y. Z. Chen, Y. X. Zhou, H. Wang, J. Lu, T. Uchida, Q. Xu, S. H. Yu and H. L. Jiang, ACS Catal., 2015, 5, 2062–2069 CrossRef CAS
- L. C. Lee, J. Lu, M. Weck and C. W. Jones, ACS Catal., 2016, 6, 784–787 CrossRef CAS
- Z. Jia, K. Wang, B. Tan and Y. Gu, ACS Catal., 2017, 7, 3693–3702 CrossRef CAS
- U. Diaz, D. Brunel and A. Corma, Chem. Soc. Rev., 2013, 42, 4083–4097 RSC
- P. Zhao, Y. Zhang, Y. Wang, H. Cui, F. Song, X. Sun and L. Zhang, Green Chem., 2018, 20, 1551–1559 RSC
- P. Li, Y. Yu, H. Liu, C. Y. Cao and W. G. Song, Nanoscale, 2014, 6, 442–448 RSC
- Z. Miao, Y. Luan, C. Qi and D. Ramella, Dalton Trans., 2016, 45, 13917–13924 RSC
- H. Wang, Y. Wang, Y. Guo, X. K. Ren, L. Wu, L. Liu, Z. Shi and Y. Wang, Catal. Today, 2019, 330, 124–134 CrossRef CAS
- B. Lai, F. Mei and Y. Gu, Chem.–Asian J., 2018, 13, 2529–2542 CrossRef CAS PubMed
- B. Voit, Angew. Chem., Int. Ed., 2006, 45, 4238–4240 CrossRef CAS PubMed
- K. Motokura, N. Fujita, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2005, 127, 9674–9675 CrossRef CAS PubMed
- R. K. Zeidan, S. J. Hwang and M. E. Davis, Angew. Chem., Int. Ed., 2006, 45, 6332–6335 CrossRef CAS PubMed
- S. Shylesh, A. Wagener, A. Seifert, S. Ernst and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 184–187 CrossRef CAS PubMed
- N. R. Shiju, A. H. Alberts, S. Khalid, D. R. Brown and G. Rothenberg, Angew. Chem., Int. Ed., 2011, 50, 9615–9619 CrossRef CAS PubMed
- K. Motokura, M. Tada and Y. Iwasawa, J. Am. Chem. Soc., 2009, 131, 7944–7945 CrossRef CAS PubMed
- F. Shang, J. Sun, H. Liu, C. Wang, J. Guan and Q. Kan, Mater. Res. Bull., 2012, 47, 801–806 CrossRef CAS
- F. Shang, J. Sun, S. Wu, Y. Yang, Q. Kan and J. Guan, Microporous Mesoporous Mater., 2010, 134, 44–50 CrossRef CAS
- E. Merino, E. Verde-Sesto, E. M. Maya, M. Iglesias, F. Sánchez and A. Corma, Chem.
Mater., 2013, 25, 981–988 CrossRef CAS
- Y. Zhang, B. Li and S. Ma, Chem. Commun., 2014, 50, 8507–8510 RSC
- B. Li, Y. Zhang, D. Ma, L. Li, G. Li, G. Li, Z. Shi and S. Feng, Chem. Commun., 2012, 48, 6151–6153 RSC
- F. Zhang, H. Jiang, X. Li, X. Wu and H. Li, ACS Catal., 2014, 4, 394–401 CrossRef CAS
- F. Zhang, H. Jiang, X. Wu, Z. Mao and H. Li, ACS Appl. Mater. Interfaces, 2015, 7, 1669–1677 CrossRef CAS PubMed
- S. Shylesh, A. Wagner, A. Seifert, S. Ernst and W. R. Thiel, Chem.–Eur. J., 2009, 15, 7052–7062 CrossRef CAS PubMed
- J. Gao, X. Zhang, Y. Lu, S. Liu and J. Liu, Chem.–Eur. J., 2015, 21, 7403–7407 CrossRef CAS PubMed
- Y. Huang, S. Xu and V. S. Lin, Angew. Chem., Int. Ed., 2011, 50, 661–664 CrossRef CAS PubMed
- L. Liu, Y. Ai, D. Li, L. Qi, J. Zhou, Z. Tang, Z. Shao, Q. Liang and H. B. Sun, ChemCatChem, 2017, 9, 3131–3137 CrossRef CAS
- Z. Weng, T. Yu and F. Zaera, ACS Catal., 2018, 8, 2870–2879 CrossRef CAS
- R. Kardooni and A. R. Kiasat, J. Taiwan Inst. Chem. Eng., 2018, 87, 241–251 CrossRef CAS
- P. Chandra, A. M. Jonas and A. E. Fernandes, ACS Catal., 2018, 8, 6006–6011 CrossRef CAS
- L. Xiong, H. Zhang, Z. He, T. Wang, Y. Xu, M. Zhou and K. Huang, New J. Chem., 2018, 42, 1368–1372 RSC
- S. Shylesh, V. Schunemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428–3459 CrossRef CAS PubMed
- S. Yang, C. Cao, Y. Sun, P. Huang, F. Wei and W. Song, Angew. Chem., Int. Ed., 2015, 54, 2661–2664 CrossRef CAS PubMed
- Z. S. Wu, S. Yang, Y. Sun, K. Parvez, X. Feng and K. Mullen, J. Am. Chem. Soc., 2012, 134, 9082–9085 CrossRef CAS PubMed
- J. Dai, H. Zou, R. Wang, Y. Wang, Z. Shi and S. Qiu, Green Chem., 2017, 19, 1336–1344 RSC
- A. Mohammadinezhad and B. Akhlaghinia, Green Chem., 2017, 19, 5625–5641 RSC
- W. Gu, X. Deng, X. Jia, J. Li and E. Wang, J. Mater. Chem. A, 2015, 3, 8793–8799 RSC
- R. B. Baig and R. S. Varma, Chem. Commun., 2013, 49, 752–770 RSC
- Q. Deng, Y. Shen, H. Zhu and T. Tu, Chem. Commun., 2017, 53, 13063–13066 RSC
- V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J. M. Basset, Chem. Rev., 2011, 111, 3036–3075 CrossRef CAS PubMed
- M. J. Jin and D. H. Lee, Angew. Chem., Int. Ed., 2010, 49, 1119–1122 CrossRef CAS PubMed
- Q. Yue, J. Li, Y. Zhang, X. Cheng, X. Chen, P. Pan, J. Su, A. A. Elzatahry, A. Alghamdi, Y. Deng and D. Zhao, J. Am. Chem. Soc., 2017, 139, 15486–15493 CrossRef CAS PubMed
- I. Elsayed, M. Mashaly, F. Eltaweel, M. A. Jackson and E. B. Hassan, Fuel, 2018, 221, 407–416 CrossRef CAS
- N. Azgomi and M. Mokhtary, J. Mol. Catal. A: Chem., 2015, 398, 58–64 CrossRef CAS
- R. Gupta, M. Yadav, R. Gaur, G. Arora and R. K. Sharma, Green Chem., 2017, 19, 3801–3812 RSC
- Q. Zhang, H. Su, J. Luo and Y. Wei, Green Chem., 2012, 14, 201–208 RSC
- Q. Zhang, H. Su, J. Luo and Y. Wei, Catal. Sci. Technol., 2013, 3, 235–243 RSC
- S. Wang, Z. Zhang, B. Liu and J. Li, Catal. Sci. Technol., 2013, 3, 2104–2112 RSC
- N. Mir, A. Heidari, H. Beyzaei, S. Mirkazehi-Rigi and P. Karimi, Chem. Eng. J., 2017, 327, 648–655 CrossRef CAS
- Q. Zhou, Z. Wan, X. Yuan and J. Luo, Appl. Organomet. Chem., 2016, 30, 215–220 CrossRef CAS
- S. Wang, J. Tang, H. Zhao, J. Wan and K. Chen, J. Colloid Interface Sci., 2014, 432, 43–46 CrossRef CAS PubMed
- J. Zhou, Y. Li, H.-b. Sun, Z. Tang, L. Qi, L. Liu, Y. Ai, S. Li, Z. Shao and Q. Liang, Green Chem., 2017, 19, 3400–3407 RSC
- Y. Ai, Z. Hu, Z. Shao, L. Qi, L. Liu, J. Zhou, H. Sun and Q. Liang, Nano Res., 2018, 11, 287–299 CrossRef CAS
- T. Zeng, L. Yang, R. Hudson, G. Song, A. R. Moores and C. J. Li, Org. Lett., 2011, 13, 442–445 CrossRef CAS PubMed
- V. Rama, K. Kanagaraj and K. Pitchumani, J. Org. Chem., 2011, 76, 9090–9095 CrossRef CAS PubMed
- M. Zarghani and B. Akhlaghinia, RSC Adv., 2016, 6, 31850–31860 RSC
- M. Massi, S. Stagni and M. I. Ogden, Coord. Chem. Rev., 2018, 375, 164–172 CrossRef CAS
- T. A. Kuz'menko, A. S. Morkovnik, L. N. Divaeva, G. S. Borodkin and V. V. Kuz'menko, Chem. Heterocycl. Compd., 2015, 50, 1575–1585 CrossRef
- D. C. Zhong, Y. Q. Wen, J. H. Deng, X. Z. Luo, Y. N. Gong and T. B. Lu, Angew. Chem., Int. Ed., 2015, 54, 11795–11799 CrossRef CAS PubMed
- S. Khaghaninejad, M. M. Heravi, T. Hosseinnejad, H. A. Oskooie and M. Bakavoli, Res. Chem. Intermed., 2016, 42, 1593–1610 CrossRef CAS
- R. P. Singh, R. D. Verma, D. T. Meshri and J. M. Shreeve, Angew. Chem., Int. Ed., 2006, 45, 3584–3601 CrossRef CAS PubMed
- A. L. Chandgude and A. Domling, Green Chem., 2016, 18, 3718–3721 RSC
- Z. N. Tisseh, M. Dabiri, M. Nobahar, H. R. Khavasi and A. Bazgir, Tetrahedron, 2012, 68, 1769–1773 CrossRef CAS
- F. Himo, Z. P. Demko, L. Noodleman and K. B. Sharpless, J. Am. Chem. Soc., 2002, 124, 12210–12216 CrossRef CAS PubMed
- F. Himo, Z. P. Demko, L. Noodleman and K. B. Sharpless, J. Am. Chem. Soc., 2003, 125, 9983–9987 CrossRef CAS PubMed
- D. Habibi, M. Nasrollahzadeh and Y. Bayat, Synth. Commun., 2011, 41, 2135–2145 CrossRef CAS
- S. Kumar, S. Dubey, N. Saxena and S. K. Awasthi, Tetrahedron Lett., 2014, 55, 6034–6038 CrossRef CAS
- D. Kong, Y. Liu, J. Zhang, H. Li, X. Wang, G. Liu, B. Li and Z. Xu, New J. Chem., 2014, 38, 3078–3083 RSC
- D. R. Patil, Y. B. Wagh, P. G. Ingole, K. Singh and D. S. Dalal, New J. Chem., 2013, 37, 3261–3266 RSC
- P. Mani, A. K. Singh and S. K. Awasthi, Tetrahedron Lett., 2014, 55, 1879–1882 CrossRef CAS
- F. Dehghani, A. R. Sardarian and M. Esmaeilpour, J. Organomet. Chem., 2013, 743, 87–96 CrossRef CAS
- M. Abdollahi-Alibeik and A. Moaddeli, New J. Chem., 2015, 39, 2116–2122 RSC
- M. Esmaeilpour, J. Javidi and S. Zahmatkesh, Appl. Organomet. Chem., 2016, 30, 897–904 CrossRef CAS
- M. Esmaeilpour, J. Javidi, F. Nowroozi Dodeji and M. Mokhtari Abarghoui, J. Mol. Catal. A: Chem., 2014, 393, 18–29 CrossRef CAS
- M. Khanmoradi, M. Nikoorazm and A. Ghorbani-Choghamarani, Appl. Organomet. Chem., 2017, 31, e3693 CrossRef
- M. Abdollahi-Alibeik and A. Moaddeli, J. Chem. Sci., 2016, 128, 93–99 CrossRef CAS
- F. Taghavi, M. Gholizadeh, A. S. Saljooghi and M. Ramezani, MedChemComm, 2017, 8, 1953–1964 RSC
- A. Ghorbani-Choghamarani, L. Shiri and G. Azadi, RSC Adv., 2016, 6, 32653–32660 RSC
- G. Azadi, A. Ghorbani-Choghamarani and L. Shiri, Transition Met. Chem., 2017, 42, 131–136 CrossRef CAS
- L. Liao, Y. Liu, Z. Li, J. Zhuang, Y. Zhou and S. Chen, RSC Adv., 2016, 6, 94976–94988 RSC
- L. Chen, B. Li and D. Liu, Catal. Lett., 2014, 144, 1053–1061 CrossRef CAS
- B. Huang, Y. Liu, B. Li, S. Liu, G. Zeng, Z. Zeng, X. Wang, Q. Ning, B. Zheng and C. Yang, Carbohydr. Polym., 2017, 157, 576–585 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04081a |
| This journal is © The Royal Society of Chemistry 2019 |