
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUtilization of catecholic functionality in natural safrole and eugenol to synthesize mussel-inspired polymers†
Mouheddin T. Alhaffara,
Mohammad N. Akhtarb and
Shaikh A. Ali *a
*a
aChemistry Department, King Fahd University of Petroleum & Minerals, Dhahran 31261, Saudi Arabia. E-mail: shaikh@kfupm.edu.sa; Fax: +966 13 860 4277; Tel: +966 13 860 3830
bCenter for Refining and Petrochemicals, RI, King Fahd University of Petroleum & Minerals, Dhahran 31261, Saudi Arabia
First published on 9th July 2019
Abstract
Naturally occurring safrole I upon epoxidation gave safrole oxide II, which underwent ring opening polymerization using a Lewis acid initiator/catalyst comprising of triphenylmethylphosphonium bromide/triisobutylaluminum to afford new polyether III in excellent yields. Epoxy monomer II and allyl glycidyl ether IV in various proportions have been randomly copolymerized to obtain copolymer V. A mechanism has been proposed for the polymerization reaction involving chain transfer to the monomers. A strategy has been developed for the deprotection of the methylene acetal of V using Pb(OAc)4 whereby one of the methylene protons is replaced with a labile OAc group to give VI. The pendant allyl groups in VI have been elaborated via a thiol–ene reaction using cysteamine hydrochloride and thioglycolic acid to obtain cationic VII and anionic VIII polymers, both containing a mussel-inspired Dopa-based catechol moiety. During aqueous work up, the protecting group containing OAc was deprotected under mild conditions. Cationic VII and anionic VIII were also obtained via an alternate route using epoxide IX derived from 3,4-bis[tert-butyldimethylsilyloxy]allylbenzene. Monomer IX was homo- as well as copolymerized with IV using Lewis acid initiator/catalyst system to obtain homopolymer X and copolymer X1. Copolymer XI was then elaborated using a thiol–ene reaction followed by F− catalysed silyl deprotection to obtain mussel inspired polymers VII and VIII, which by virtue of having charges of opposite algebraic signs were used to form their coacervate.
1 Introduction
The performance of synthetic adhesive polymers in aquatic environments is most often frustrating because of the various adverse effects of water or moisture in inducing hydrolysis, swelling, interfacial wicking, etc.1,2 Rapid, strong and tough moisture-resistant adhesion to solid surfaces in the sea by the holdfast of marine mussels have lured many a researcher to mimic the essential features of the adhesive chemistry practiced by the mussels. Roughly 25–30 different adhesive proteins of varying molar masses used by the mussels contained a considerable proportion of randomly placed 3,4-dihydroxyphenyl-L-alanine (L-DOPA) (≈1–28 mol%) and 4-hydroxyarginine resulting from a post translational modification of amino acid tyrosine and arginine, respectively.1 To be an effective adhesive, it must exert more favourable interaction with the wet polar surfaces than a layer of water is able to offer. Polymers covalently bonded into the interfaces may mitigate the deleterious effects of moisture, but it is a costly process.3 As such the situation demanded a closer look at the way mussel's adhesive chemistry works. The byssus is a bundle of filaments of DOPA decorated proteins secreted by mussels that function to attach the mussels to a solid surface whereby the catecholic functionalities provide bidentate or covalent interfacial interactions. The proteins themselves gain cohesiveness through metal chelation, and covalent coupling.The cohesive and adhesive roles played by the catechol functionality of DOPA in byssal proteins and their effective adhesive performance in turbulent wet environments have now been firmly established. The biomimetic efforts have thus targeted the attractive mussel byssal proteins. However, the performance of biological adhesion, e.g., from mussels is much superior to biomimetic adhesives. Recent advances demonstrate that judicious biomimetic design incorporating key elements in the natural adhesive system is essential to replicate the wet adhesion of the marine organism.4,5 Several reviews deal with the recent progress of mussel-inspired underwater adhesives polymers having catechol-functional motifs for their potential applications in anti-biofouling, biological adhesives, and drug delivery.6,7 A review specifically provides an overview of the various applications of poly[2-(3,4-dihydroxyphenyl)ethylamine] (i.e. polydopamine (PDA)) in tumor targeted drug delivery systems and discusses the release behavior of the drug-loaded PDA-based nanocarriers.8
The synthesis of mussel mimetic polymers include DOPApolypeptides9,10 and DOPA and lysine polypeptide copolymers.11,12 Hydrogen bonding by phenolic hydroxyls of bidentate DOPA leads to its bridging adhesion to the polysiloxane surface of mica.13 The mussel mimicking adhesives has also been applied as complex coacervates,14,15 which are prepared using aqueous solutions of polyanions (e.g. DOPA-containing proteins having phosphoserine residues) and polycations (e.g. Dopa-containing proteins having 4-hydroxyarginine residues). With charge symmetry, the polymers of different algebraic signs undergo phase separation in a complex coacervation process, which is essential for wet adhesion. Synthesis of mussel mimetic polymers have been achieved through polymerization of catechol-based monomers.16,17 Dopamine methacrylamide and 2-methoxyethyl acrylate or poly(ethylene glycol) methyl ether methacrylate has been copolymerized to give adhesive polymers having divergent physical properties.18
A review article discusses the mussel-inspired chemistry in the surface engineering of polymer membranes to improve their performance. Catecholamines, deposited on the membrane surface, serve as a surface component for membrane modification or fabrication.19 Polydopamine capped graphene oxide sheets are crosslinked by polyethylenimine leading to ultrahigh modulus and high strength of macroscopic graphene oxide papers, which broaden the potential applications of graphene.20 The adhesive mechanism of mussel-inspired polymers has been investigated using the terpolymers of N-(3,4-dihydroxyphenethyl) methacrylamide (DMA), acrylic acid and butyl acrylate. The bulk adhesion was found to increase with a corresponding increase in DMA content reaching a maximum at around 40 mol%.21 Inspired by strong adhesion of mussel adhesive proteins, a high-performance nanocomposite is generated by assembling polydopamine-coated montmorillonite with corn starch.22 Underwater-superoleophobic materials have been successfully developed by a two-step dip-coating method with mussel-inspired coatings of polydopamine and subsequent zwitterionic sulfobetaine methacrylate grafting onto stainless steel meshes and used in oil/water separation.23 Two functional catechols were incorporated into backbones of polyacrylate and polyurethane, which showed excellent coatability on various surfaces.24
Homo- and copolymers of catecholic monomer 3,4-dihydroxystyrene with styrene or styrene sulfonate has been reported to give polymers having a wide range of physical properties.18,25 A complex coacervate adhesive has been synthesized from a mixture of catechol containing synthetic polyanion and a synthetic polycation.14,26 Coating of polyethylene glycol containing Dopa as an anchor has been demonstrated to prevent bacterial adhesion and biofilm formation on a variety of surfaces.27,28 One of the serious drawbacks of mussel proteins is the oxidation of DOPA at or above neutral pH to DOPA quinone which has been shown to be less sticky than DOPA. While the mussels have the talents29,30 to control the deleterious oxidation, it remains a challenge to the researchers to safeguard the integrity of the catecholic functionality in DOPA for facile bidentate surface binding. Even in the more adverse pH of 8.2 of seawater, the mussels secret their proteins at a low pH of 5–6 in the confined space of a reducing environment where the redox balance is provided by the thiol functionalities of a cysteine-rich protein.
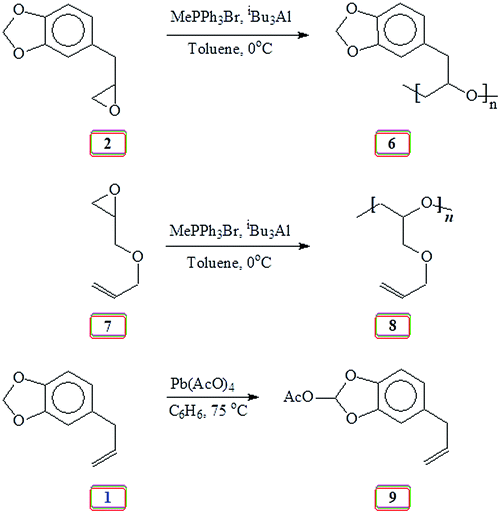
It is our intention to synthesize mussel-inspired polymers using ring opening polymerization of epoxide (oxirane). Several review articles summarize the developments in the ring opening polymerization of alkylene oxides via anionic, coordination and cationic polymerization using a variety of catalysts including metal-free organocatalysts to synthesize linear homo- and amphiphilic block as well as branched, hyperbranched, and dendrimer like polyethers.31–33 Herein we report the use of readily available naturally occurring safrole (1) (Scheme 1) or eugenol (10) (Scheme 5) to synthesize epoxy functionalized monomers safrole oxide (2) (Scheme 1) and eugenol-derived 14 (Scheme 5) and their ring opening homo- and copolymerization with allyl glycidyl ether (AGE) 7 (Scheme 2). The allyl pendants of the mussel mimicking polymers decorated with catechol functionalities would then offer the latitude of transformation to ionic polymer backbones of both algebraic signs. The work would thus pave the way to study these polymers from the perspective of coacervate adhesive.
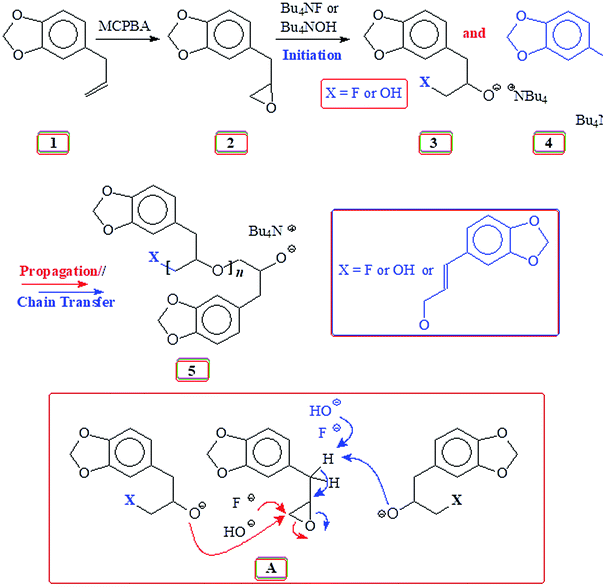 | ||
| Scheme 1 Base catalyzed polymerization of safrole oxide 2 using Bu4NF and Bu4NOH. | ||
 | ||
| Scheme 2 Lewis acid catalyzed polymerization of safrole oxide 2, AGE 7 and activation of methylene group. | ||
2 Experimental
2.1. Materials
m-Chloroperbenzoic acid (MCPBA), LiCl, tert-butylammonium fluoride (TBAF·3H2O), tert-butylammonium hydroxide (TBAH), 4-dimethylaminopyridine (DMAP), iBu3Al solution (25% in toluene), eugenol, 2,2-dimethoxy-2-phenylacetophenone (DMPA), t-butyldimethylsilyl chloride (TBDMSCl), thioglycolic acid, cysteamine and safrole were purchased from Sigma Aldrich. MePPh3Br and imidazole from Fluka, 1 M tert-butylammonium fluoride (TBAF) solution in THF from ChemCruz, and diphenylsilane and B(C6F5)3 from Alfa Aesar were used as received without any further purification. 4-Allylcatechol was prepared by reacting eugenol with LiCl in dimethyl formamide (DMF) as described.34 Allyl glycidyl ether (AGE) from Sigma Aldrich was dried over CaH2 and distilled. Pb(AcO)4 was freshly prepared.35 Membrane (Spectra/Por) with a MWCO of 6000–8000 Daltons was purchased from Spectrum Laboratories, Inc.2.2. Physical methods
Elemental analyses were performed on a PerkinElmer (Series II Model 2400). FTIR and NMR were recorded using a PerkinElmer 16F PC and a 500 MHz JEOL LA spectrometer, respectively. Tetramethylsilane (TMS) in CDCl3 and residual H in D2O at δ 4.65 ppm were used as internal standards. The 13C chemical shifts in D2O were referenced against 13C peak of external standard dioxane at δ 67.4 ppm. Gel Permeation Chromatographic analysis was performed at 30 °C using a PL-GPC 220 manufactured by Agilent Technologies having two detectors (i.e. Refractive Index & Light Scattering), equipped with two columns (PL aquagel-OH 8 µm; 300 × 7.5 mm). Deionized water with 0.02 wt% NaN3 was used as solvent for water soluble polymer samples. A sample solution of 5 mg/1.5 mL was prepared at ambient temperature in water having 0.02 wt% NaN3. The resulting solution (100 µl) was injected into the GPC columns and chromatographic data was analyzed using GPC/SEC software by Agilent. In case of THF soluble polymer samples, GPC was equipped with two columns (PLgel Olexis 300 × 7.5 mm). The sample solution was prepared using tetrahydrofuran (THF) containing 0.0125 wt% antioxidant, [2,6-di-tert-butyl-4-methyl phenol (BHT)]. The equipment was calibrated using polyethylene oxide (PEO) and polystyrene (PS) standards for water-soluble and THF-soluble polymers, respectively.2.3. Synthesis of safrole oxide (2) from safrole (1)
Safrole was converted to safrole oxide using a modified procedure.36,37 Safrole 1 (32.4 g) was added to a solution of MCPBA (54 g, 0.22 mol, 70% purity) in chloroform (400 mL) and stirred at 25 °C for 24 h. After adding additional MCPBA (20 g, 0.082 mol), the reaction mixture was stirred for a further 24 h and was washed with 10% sodium hydroxide solution (3 × 70 mL) and water (2 × 30 mL). The organic layer was dried (MgSO4) and concentrated. The residual liquid was purified by silica gel chromatography using hexane/ether as eluent to give safrole oxide (2) as a colorless oil (28.8 g, 67%). The oil was then distillated over calcium hydride to exclude any moisture bp0.8 mbarHg 92 °C. δH (CDCl3): 2.52 (1H, m), 2.83–3.73 (3H, m), 3.10 (1H, m), 5.91 (2H, s), 6.69 (1H, d, J 7.6 Hz), 6.74 (1H, s), 6.75 (1H, d, J 7.9 Hz); δC (CDCl3): 28.39, 46.77, 52.53, 100.87, 108.26, 109.43, 121.85, 130.79, 146.28, 147.66 (CDCl3 middle carbon: 77.00).2.4. Lewis acid catalyzed polymerization of safrole oxide 2 using iBu3Al38,39
As described in entry 10 (Table 1), methyltriphenylphosphonium bromide (11.9 mg, 0.0333 mmol) was placed in an RB flask and closed with a rubber septum. Safrole oxide 2 (445 mg, 2.5 mmol) and anhydrous toluene (1.6 mL) were added under Ar by a syringe. The mixture was cooled to 0 °C, then 1 M triisobutylaluminium solution (0.376 mL of 25 wt% in toluene, 0.403 mmol) was injected by a syringe under Ar. The polymerization was quenched after 2 h by adding 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeOH/H2O (v/v). After removal of the solvents, the residue was dissolved in CH2Cl2 (50 mL) and filtered over Celite 545. The filtrate was dried (over Na2SO4) and evaporated to obtain polymer 6.
:1 MeOH/H2O (v/v). After removal of the solvents, the residue was dissolved in CH2Cl2 (50 mL) and filtered over Celite 545. The filtrate was dried (over Na2SO4) and evaporated to obtain polymer 6.
| Entry | I (mmol) | C (mmol) | [SO]/[I] | [C]/[I] | Time (h) | Yieldc (%) | Mn,Theord | Mn,Expe | PDI |
|---|---|---|---|---|---|---|---|---|---|
| a Polymerization was carried out at 0 °C using 2.5 mmol of monomer 2 with 1.8 mL of additional toluene except in entry 4 where no additional toluene was added.b 25 wt% solution in toluene (≈1 M iBu3Al).c NMR indicates complete conversion to polymer where isolated yields are over 90%.d For entry 2: molar mass = [molar mass of SO] × [SO]/[I] = 178.19 × 216 = 38489 (assuming 100% conversion).e GPC using light scattering detector.f Carried out at 20 °C.g 1 M iBu3Al solution in hexane. |
|||||||||
| 1 | 0.0256 | 0.263 | 98 | 10.3 | 3 | 94 | 17500 |
||
| 2 | 0.0116 | 0.261 | 216 | 22.5 | 3 | 91 | 38500 |
14450 |
1.38 |
| 3 | 0.0458 | 0.264 | 55 | 5.76 | 6 | 95 | 9800 | 7500 | 1.45 |
| 4 | 0.0220 | 0.239 | 114 | 10.9 | 2 | 93 | 20300 |
11150 |
1.23 |
| 5 | 0 | 0.418 | — | — | 2 | 0 | |||
| 6f | 0 | 0.418 | — | — | 24 | 0 | |||
| 7g | 0.0331 | 0.416 | 76 | 12.6 | 3 | 69 | |||
| 8 | 0.0227 | 0.725 | 110 | 31.9 | 12 | 70 | |||
| 9 | 0.0222 | 0.235 | 113 | 10.6 | 12 | 65 | |||
| 10 | 0.0328 | 0.403 | 76 | 12.3 | 2 | 99 | 17000 |
9970 | 1.32 |
For purification, polymer 6 (Scheme 2) was dissolved in minimum amount of dichloromethane and then precipitated in methanol; the polymer was then separated by centrifuge. This process was repeated to remove the initiator and unreacted monomer (found: C 67.0; H 5.6%. C10H10O3 requires C 67.41; H 5.66%); νmax (KBr) (Fig. S7†): 3444, 3066, 2899, 2772, 1608, 1475, 1435, 1248, 1090, 1026, 742, and 693 cm−1.
2.5. Polymerization of AGE 7 (ref. 40 and 41)
A solution of AGE 7 (296 mg, 2.60 mmol), methyltriphenylphosphonium bromide (8.2 mg, 0.023 mmol) in anhydrous toluene (1.6 mL) under Ar was cooled by ice bath, then triisobutylaluminium (0.24 mL, 0.26 mmol of 25 wt% in toluene) was added by a syringe under Ar. The polymerization was stopped after 2 h by adding 4:1 MeOH/H2O (10 mL). After removing the solvents, the residue was dissolved in a CH2Cl2 (50 mL) and filtered over Celite 545. The filtrate was dried (Na2SO4) and evaporated to obtain polymer 8.
2.6. Activation of methylene acetal of safrole 1 as a model case42,43
Pb(OAc)4 (4.23 g, 9.5 mmol) was added under N2 to a solution of safrole 1 (1.05 g, 6.48 mmol) in benzene (35 mL) at 75 °C and stirred for 3 h. The reaction mixture was taken up in EtOAc (30 mL) and washed with H2O (3 × 25 mL). The organic layer was dried (Na2SO4) and concentrated. The residual liquid upon silica gel chromatography using EtOAc/hexane afforded 9 as a faint yellow oil (1.08 g, 82%) (found: C 65.2; H 5.4%. C12H12O4 requires C 65.45; H 5.49%); δH (CDCl3): 2.09 (3H, s), 3.34 (2H, d, J 6.7 Hz), 5.05–5.09 (2H, m), 5.89–5.96 (1H, m), 6.77 (1H, dd, J 7.9 Hz, J 1.6 Hz), 6.83 (1H, d, J 1.6 Hz), 6.88 (1H, d, J 7.9 Hz), 7.66 (1H, s); δC (CDCl3): 20.92, 39.80, 108.85, 109.64, 112.64, 115.92, 122.30, 134.88, 134.92, 137.16, 143.05, 144.83, 168.99, (CDCl3 middle carbon: 77.00); νmax (KBr) (Fig. S8†): 3079, 3005, 2979, 2914, 1838, 1768, 1639, 1495, 1445, 1376, 1255, 1216, 1181, 1100, 1009, 963, and 760 cm−1.2.7. Synthesis of [(4-allyl-1,2-phenylene)bis(oxy)]bis(tert-butyldimethylsilane) (13) (Scheme 5)44
TBDMSCl (25.0 g, 166 mmol) and DMAP (2.11 g, 17 mmol) were added under N2 to a mixture of allylcatechol 12 (10.86 g, 72.4 mmol) (Scheme 5) and imidazole (23.6 g, 347 mmol) in DMF (130 mL) at 0 °C. After stirring at 25 °C for 16 h, the reaction mixture was taken up in water (300 mL) and extracted with ether (2 × 150 mL). After washing with water (4 × 200 mL), the ether layer was dried (Na2SO4). The removal of the solvent followed by distillation (bp0.2 mbarHg122 °C) afforded 13 as a colorless oil (20.5 g, 75%) (found: C 66.2; H 10.0%. C21H38O2Si2 requires C 66.60; H 10.11%); δH (CDCl3): 0.17 (12H, s), 0.97 (18H, s), 3.24 (2H, d, J 6.7 Hz), 4.99–5.03 (2H, m), 4.99–5.03 (1H, m), 6.59 (1H, dd, J 1.8 Hz, J 8.3 Hz), 6.64 (1H, d, J 1.8 Hz), 6.72 (1H, d, J 8.3 Hz). δC (CDCl3): −4.10, 18.44, 25.97, 39.45, 115.33, 120.83, 121.31, 121.47, 133.07, 137.83, 145.02, 146.59 (CDCl3 middle carbon: 77.01). IR KBr (Fig. S9†): 2957, 2930, 2896, 2859, 1640, 1606, 1577, 1510, 1473, 1463, 1421, 1294, 1254, 1228, 1155, 1125, 986, 912, 839, and 781 cm−1.2.8. Synthesis of [(4-(oxiran-2-ylmethyl)-1,2-phenylene)bis(oxy)]bis(tert-butyldimethylsilane) (14)45 (Scheme 5)
MCPBA (19.8 g, 80 mmol, 70% purity, 30% water) was dissolved in 150 mL CH2Cl2, and aqueous layer was removed. Silyl derivative 13 (16.3 g, 43 mmol) was added to the MCPBA/dichloromethane solution at 0 °C and stirred at 25 °C for 24 h. The reaction mixture was washed with 10% NaOH solution (3 × 20 mL) and water (2 × 25 mL). The CH2Cl2 layer was dried (Na2SO4), concentrated and distilled (bp0.2 mbarHg127–130 °C) to obtain 14 (10.7 g, 63%) as a colorless liquid (found: C 63.7; H 9.6%. C21H38O3Si2 requires C 63.90; H 9.70%); δH (CDCl3): 0.17 (6H, s), 0.18 (6H, s), 0.966 (9H, s), 0.971 (9H, s), 2.49 (1H, q), 2.66 (1H, dd, J 14.7 Hz, J 5.5 Hz), 2.74–2.68 (2H, m), 3.06–3.09 (1H, m), 6.65 (1H, dd, J 8.3 Hz, J 2.2 Hz), 6.71 (1H, d, J 2.2 Hz), 6.74 (1H, d, J 8.3 Hz). δC (CDCl3): (−) 4.11, 18.43, 25.94, 37.99, 46.72, 52.57, 120.93, 121.82, 121.89, 130.14, 145.56, 146.65, (CDCl3 middle carbon: 77.02); νmax (KBr) (Fig. S10†): 3044, 2930, 2897, 2859, 1765, 1727, 1701, 1607, 1577, 1514, 1473, 1464, 1423, 1292, 1256, 1224, 1159, 1127, 988, 907, 842, and 783 cm−1.2.9. Lewis acid catalyzed polymerization of 14
As described in Table 2, methyltriphenylphosphonium bromide was taken in an RB flask under Ar and closed with a rubber septum. Epoxidized silyl derivative 14 and anhydrous toluene were injected under Ar by a syringe. The mixture was cooled to 0 °C, then triisobutylaluminium solution (25 wt% in toluene) was injected. After the specified time (Table 2), the reaction mixture was quenched by adding 4:1 MeOH/H2O mixture (10 mL). After extracting with CH2Cl2 (2 × 25 mL), the organic layer was dried (MgSO4), filtered over Celite 545 and concentrated to obtain polymer 15a (Scheme 5). The polymer was purified by dissolving in ether and precipitating in MeOH; the process was repeated three times (found: C 63.6; H 9.8%. C21H38O3Si2 requires C 63.90; H 9.70%); νmax (KBr) (Fig. S11†): 2930, 2896, 2859, 1607, 1578, 1518, 1473, 1427, 1422, 1362, 1305, 1254, 1224, 1160, 1128, 983, 852, 778, and 666 cm−1.
| Entry | I (mmol) | C (mmol) | [SP]/[I] | [C]/[I] | Time (h) | Yieldc (%) | Mn,Theord | Mn,Expe | PDI |
|---|---|---|---|---|---|---|---|---|---|
| a Polymerization was carried out at 0 °C for 6 h using 2.5 mmol of monomer 14 with 1.8 mL of additional toluene except in entry 4 where no additional toluene was used.b 25 wt% solution in toluene (≈1 M iBu3Al).c Isolated and NMR yields are similar within 2%.d Mn for entry 2 = [molar mass of SP] × [SP]/[I] × % conversion/100 = 394.70 × 117 × 0.60 = 27708.e GPC using light scattering detector. |
|||||||||
| 1 | 0.0213 | 0.24 | 117 | 11.3 | 1 | 0 | — | — | |
| 2 | 0.0213 | 0.70 | 117 | 32.9 | 2 | 60 | 27700 |
16300 |
1.8 |
| 3 | 0.0495 | 0.53 | 50 | 10.7 | 20 | 90 | 17700 |
12900 |
1.5 |
| 4 | 0.0092 | 0.53 | 272 | 57.6 | 40 | 75 | 80200 |
11500 |
1.6 |
| 5 | 0.0370 | 0.53 | 68 | 14.3 | 18 | 85 | 22800 |
10300 |
1.8 |
2.10. Random copolymerization of safrole oxide 2 and allyl glycidyl ether 7
All the experiments were performed under argon atmosphere. As described in (Table 3), methyltriphenylphosphonium bromide was placed in an RB flask under Ar and closed with a rubber septum. Safrole oxide 2, allyl glycidyl ether 7 and anhydrous toluene were injected under Ar by a syringe. The mixture was cooled to 0 °C, and then triisobutylaluminium solution (25 wt% in toluene) was added under Ar in one portion by a syringe. The polymerization was stopped after complete polymerization (as indicated by 1H NMR spectrum) by adding 4:1 MeOH/H2O mixture (10 mL); the mixture was extracted with CH2Cl2, dried (MgSO4), and filtered over Celite 545. The filtrate upon evaporation afforded random copolymer 16 (Scheme 6). The following IR data belong to a 1:1 copolymer of 2 and 7. νmax (KBr) (Fig. S13†): 3076, 2869, 2773, 1646, 1608, 1500, 1443, 1353, 1249, 1121, 928, 808, and 774 cm−1 (found: C 65.1; H 6.7%. Repeat units 2 and 7 in 1:1 requires C 65.74; H 6.90%).
| Entry | SOc (mol%) | MePPh3Br (mmol) | Toluene (mL) | iBu3Al (mmol) | Temp. (°C) | Time (h) | Yield (%) |
|---|---|---|---|---|---|---|---|
| a Polymerization was carried out using a total of 5.0 mmol of monomer 2 and 7.b 25 wt% solution in toluene (≈1 M iBu3Al).c Mol% of the monomer in the mixture of the two monomers.d This reaction was run using a total of 35 mmol of 2 and 7, however, calculations are shown for the total of 5.0 mmol.e This reaction was run using a total of 50 mmol of 2 and 7, however, calculations are shown for the total of 5.0 mmol. | |||||||
| 1 | 50 | 0.063 | 2.5 | 0.835 | 20 | 20 | 91 |
| 2 | 50 | 0.065 | 2.0 | 0.860 | 20 | 24 | 93 |
| 3 | 50 | 0.025 | 1.0 | 0.484 | 0 | 2 | 89 |
| 4d | 50 | 0.042 | 3.0 | 0.513 | 0 | 18 | 92 |
| 5e | 15 | 0.040 | 2.5 | 0.534 | 0 | 20 | 93 |
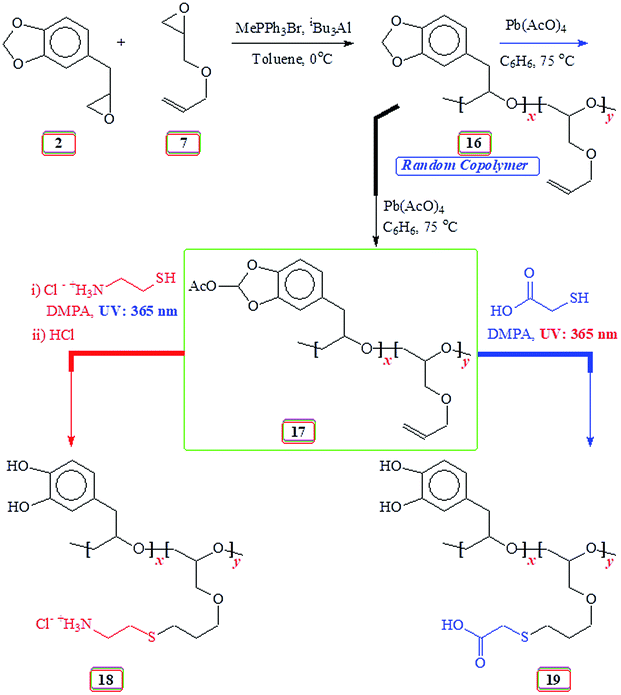
2.11. Conversion of copolymer 16 to 17 (Scheme 6) using lead tetraacetate
The procedure as described under Section 2.7 was followed.42,43 A solution of polymer 16 (containing 15 mol% safrole oxide 2 repeating unit) (1.1 g, 8.9 mmol of total repeating units and 1.33 mmol of safrole oxide units) in benzene (25 mL) was heated to 75 °C in a round bottom flask. After the addition of Pb(OAc)4 (0.90 g, 2.0 mmol) under N2, the mixture was stirred at 75 °C for 4 h. Then, it was cooled, diluted with EtOAc (30 mL) and washed with H2O (3 × 25 mL). The organic layer was dried (Na2SO4) and concentrated to give 17. For a copolymer having 1:1 ratio of the monomer units: νmax (KBr) (Fig. S14†): 3080, 3013, 2919, 2867, 1763, 1645, 1496, 1446, 1352, 1215, 1105, 923, 786, 758 cm−1. The polymers having various compositions gave satisfactory elemental analysis.
2.12. Synthesis of 18 (Scheme 6) via thiol–ene reaction
The copolymer 17 (prepared from entry 5, Table 3) (containing 85 mol% repeating unit of allyl glycidyl ether 7) (274 mg, 2.2 mmol, containing 1.9 mmol of alkene motifs) was dissolved in THF (2 mL) and methanol (1.5 mL). Cysteamine·HCl (1.4 g, 12 mmol) and photoinitiator 2,2-dimethoxy-2-phenylacetophenone (DMPA) (227 mg, 0.9 mmol) were added. The mixture was purged with N2 for 10 min, then irradiated with a 365 nm UV lamp until the completion of the reaction as revealed by 1H NMR spectrum. The reaction mixture was taken up in 1:1 MeOH/0.1 M HCl (5 mL) and dialyzed against water. The polymer solution upon freeze-drying afforded the deprotected polymer 18 (394 mg, 86%). (15 mol% catechol units) νmax (KBr) (Fig. S15†): 3469, 2960, 2923, 2857, 1624, 1494, 1384, 1259, 1105, 808, and 587 cm−1 (found: C 45.6; H 7.9; N, 5.5; S, 12.1%. Repeat units 2 (diol form) and 7 (after thiolene reaction) in 15:85 requires C, 44.79; H, 7.75; N, 5.45; S, 12.47%).
2.13. Synthesis of 19 (Scheme 6) via thiol–ene reaction
The copolymer 17 (prepared from entry 5, Table 3) (containing 85 mol% repeating unit of allyl glycidyl ether 7) (240 mg, 1.9 mmol, containing 1.6 mmol akene motifs) was dissolved in THF (3 mL). Thioglycolic acid (1.00, 11 mmol) and photoinitiator 2,2-dimethoxy-2-phenylacetophenone (DMPA) (200 mg, 0.8 mmol) were added. After purging with N2 for 10 min, the reaction mixture was irradiated with a 365 nm UV lamp until the completion of the reaction as revealed by 1H NMR spectrum. The product dissolved in methanol was precipitated in chloroform. The process was repeated to give 19 (292 mg, 81%). The polymer was found to be insoluble in water. (15 mol% catechol units) νmax (KBr) (Fig. S16†): 3500 (br), 2916, 2869, 1734, 1601, 1520, 1471, 1111, 682, and 523 cm−1 (found: C 48.1; H 6.5; S, 13.2%. Repeat units 2 (diol form) and 7 (after thiolene reaction) in 15:85 requires C, 48.88; H, 6.75; S, 13.61%).
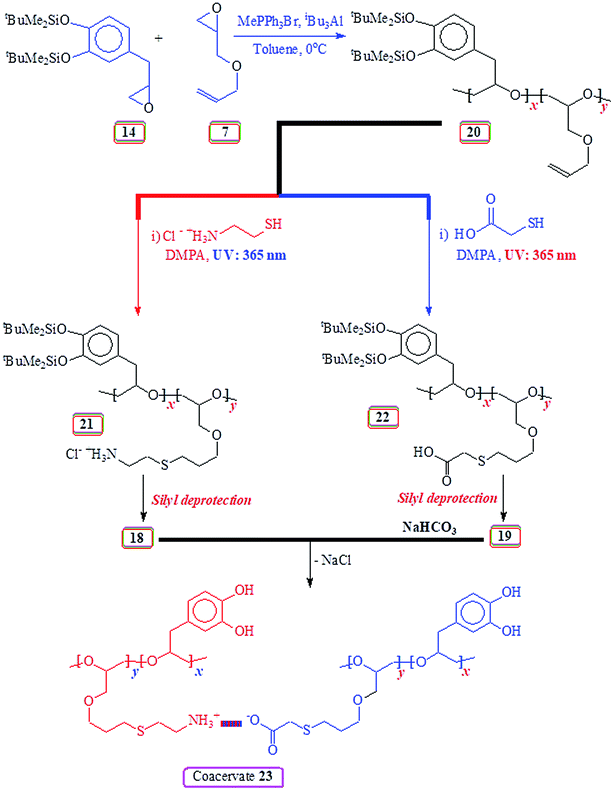
2.14. Copolymerization of silyl protected 14 and AGE 7
As described in (Table 4), silyl protected monomer 14, allyl glycidyl ether 7 and anhydrous toluene were added onto methyltriphenylphosphonium bromide contained in a RB flask under Ar. The mixture was cooled to 0 °C and polymerization was started with the addition of triisobutylaluminium solution (25 wt% in toluene). The polymerization was stopped (after its completion as indicated by 1H NMR spectrum) by adding 4:1 MeOH/H2O mixture (10 mL) and extracted with CH2Cl2 (25 mL). The organic layer was dried (MgSO4), filtered over Celite 545, and concentrated to obtain 20 (Scheme 8). The polymer was dissolved in diethyl ether, precipitated in methanol and separated by centrifuging. This process was repeated to obtain pure 20. (25 mol% silyl protected, entry 3, Table 4): νmax (KBr) (Fig. S17†): 3081, 3014, 2855, 1646, 1605, 1576, 1512, 1469, 1422, 1358, 1298, 1255, 1224, 1160, 984, 839, 782, 696, and 667 cm−1 (found: C 62.3; H 9.0%. Repeat units 14 and 7 in 25:75 requires C, 63.55; H, 9.30%).
| Entry | SPc (mol%) | I (mmol) | [M]d/[I] | C (mmol) | [C]/[I] | Time (h) | Conv.e,f (%) | Mn,Theorg | Mn,Exph | PDI |
|---|---|---|---|---|---|---|---|---|---|---|
| a Polymerization was carried out at 0 °C using a total of 5.0 mmol of monomer 14 and 7 with an additional amount toluene added (3.5 mL).b 25 wt% solution in toluene (≈1 M iBu3Al).c Mol% of the monomer the mixture of the two monomers.d Total monomers of silyl protected oxide 14 and allyl glycidyl ether 7 is 5 mmol.e % conversion monomer 14 as determined by 1H NMR, the number in parentheses belongs to % conversion of AGE 7.f Isolated yield was in the range 85–95%.g Mn for entry 4 = [molar mass of SP × 0.10 + molar mass of AGE × 0.90] × [M]/[I] = 142.2 × 111 = 15784 (assuming 100% conversion).h GPC with a light scattering detector.i This reaction was run using a total of 30 mmol of 14 and 7; however, calculation is based on a total of 5.0 mmol of monomers 14 and 7. |
||||||||||
| 1 | 50 | 0.045 | 111 | 0.76 | 17 | 3 | 88 (88) | |||
| 2 | 50 | 0.053 | 94 | 0.76 | 14 | 5 | 95 (99) | 23900 |
14300 |
1.5 |
| 3 | 25 | 0.045 | 111 | 0.50 | 11 | 12 | 99 (99) | 20500 |
12700 |
1.3 |
| 4 | 10 | 0.045 | 111 | 0.50 | 11 | 12 | 96 (99) | 15800 |
6100 | 2.0 |
| 5 | 10 | 0.092 | 54 | 0.50 | 5 | 3 | 84 (99) | |||
| 6 | 10 | 0.012 | 417 | 0.50 | 42 | 15 | 78 (93) | |||
| 7i | 10 | 0.032 in | 156 | 0.50 | 16 | 17 | 99 (99) | 22200 |
7900 | 1.8 |
| 8 | 5 | 0.045 | 111 | 0.50 | 11 | 12 | 98 (99) | 14200 |
5900 | 2.1 |
2.15. Silyl deprotection of polymer 15a
TBAF (1.0 M in THF) (1.26 mL, 1.26 mmol) was added to a solution of polymer 15a (entry 4, Table 2) (227 mg, 0.575 mmol) in THF (4 mL) under N2 at 0 °C. After stirring at 0 °C for 1 h, a 1:1 mixture of H2O/acetic acid (2 mL) was added to the reaction mixture. The work-up procedures included the evaporation of the solvents, redissolving the residue in MeOH and precipitation in water. The process was repeated two times to wash out remaining TBAF to obtain deprotected polymer 15b (Scheme 5), (yield: 72 mg, 75%). The fluoride ion could chelate with catechol moiety according to literature.46,47 The polymer was soluble in methanol but insoluble in H2O, Et2O and CH2Cl2 (found: C 64.7; H 5.8%. C9H10O3 requires C 65.05; H 6.07%); νmax (KBr) (Fig. S12†): 3445, 2921, 2870, 1608, 1521, 1446, 1363, 1285, 1190, 1113, 1052, 958, 871, 811, 788, and 754 cm−1.
2.16. Synthesis of 21 (Scheme 8) via thiol–ene reaction (10 mol% catechol units)48
The copolymer 20 (entry 7, Table 4) (690 mg, containing 4.32 mmol of alkene motifs) was dissolved in THF (18 mL) and methanol (4.5 mL). Cysteamine·HCl (3.6 g, 33 mmol) and photoinitiator 2,2-dimethoxy-2-phenylacetophenone (DMPA) (582 mg, 2.4 mmol) were added. After purging with N2 for 10 min, the mixture was irradiated with a 365 nm UV lamp at 25 °C until the reaction is completed. The reaction mixture was dialyzed against MeOH (to remove DMAP) for 30 min and then against deionized water (1 h). The resultant polymer solution was freeze-dried to obtain polymer 21 (1.11 g, 90%). For a sample of 21 derived from entry 2 (Table 4) (50% silyl): (found: C, 55.1; H, 8.9; N, 2.1; S, 4.8%. 21 in a 1:1 ratio of the repeating units requires C, 55.96; H, 9.07; N, 2.25; S, 5.15%); νmax (KBr) (Fig. S18†): 3430, 2962, 2927, 2857, 1637, 1511, 1467, 1422, 1385, 1301, 1256, 1221, 1162, 1100, 981, 910, 840, 778, 664, and 614 cm−1.
2.17. Synthesis of 22 (Scheme 8) via thiol–ene reaction (10 mol% catechol units)
The reaction was performed for polymer samples 20 having SP 14 and AGE 7 repeating units in mole ratios of 50:50 and 10:90 to obtain polymer 22 using UV light in dark room.
Thioglycolic acid (1.88 g, 20.4 mmol) and photoinitiator DMPA (522 mg, 2.1 mmol) were added to a solution of copolymer 20 (entry 7, Table 4) (618 mg, 4.05 mmol akene motifs) in THF (12 mL). After purging the mixture with N2 for 10 min, it was then irradiated with a 365 nm UV lamp at 25 °C until the reaction is completed. The THF-soluble product was precipitated with the addition of ether to the reaction mixture. The process of dissolving in THF followed by precipitation in ether was repeated three times to obtain 22 (885 mg, 84%). For a sample of 22 derived from entry 2 (Table 4) (50% silyl): (found: C, 57.2; H, 8.8; S, 5.1%. 22 in a 1:1 ratio of the repeating units requires C, 57.96; H, 8.72; S, 5.33%); IR for a 50:50 copolymer 22 is given: (50% silyl) νmax (KBr) (Fig. S19†): 3458, 2931, 2858, 2511, 1727, 1632, 1511, 1467, 1421, 1385, 1301, 1255, 1223, 1122, 983, 910, 841, 790, 669, and 552 cm−1.
2.18. Synthesis of 18 from 21 (catechol 10 mol%)
The copolymer 21 as obtained from 20 (1.0 mmol) (entry 7, Table 4) using procedure described under Section 2.16, was dissolved in MeOH/0.5 M HCl (6 mL) and stirred under N2 at 25 °C for 18 h. The reaction mixture was dialyzed against water and then freeze-dried to obtain polymer 18 in (90%) (found: C, 43.0; H, 8.0; N, 5.5; S, 12.7%. 18 containing 10 mol% catechol units requires C, 43.90; H, 7.82; N, 5.69; S, 13.02%). IR and NMR spectra of 18 (10 mol% catechol repeating unit) were found to be like the one prepared earlier from safrole-oxide under Section 2.14. Acid hydrolysis of silyl groups in 1:1 copolymer 21 (i.e. containing 50 mol% catechol units) to get polymer 18 could not be achieved because of solubility problem.
2.19. Synthesis of 19 from 22 (catechol 10 mol%)
TBAF (1.0 M in THF) (0.25 mL, 0.25 mmol) was added to a solution of polymer 22 (50 mg, 0.22 mmol) in THF (1.0 mL) under N2 at 0 °C and stirred for 12 h. After the elapsed time, a 2:1 mixture of H2O/acetic acid (2 mL) was added. The crude polymer was soluble in methanol but insoluble in H2O. Therefore, it was purified by dissolving in methanol and precipitating in water. The process was repeated two times to obtain polymer 19 (yield: 90%) (found: C, 47.5; H, 6.9; S, 13.9%. 19 containing 10 mol% catechol units requires C, 48.10; H, 6.78; S, 14.27%). Removal of the acid hydrolysis of silyl groups in 1:1 copolymer 22 (i.e. containing 50 mol% catechol units) to get polymer 19 could not be achieved because of solubility problem. IR and NMR spectra of 19 (10 mol% silyl) were found to be like the one prepared earlier from safrole-oxide under Section 2.15.
2.20. Coacervate formation (10% catechol unit)
A sample of 19 (514 mg) was dissolved in MeOH and dialyzed against NaHCO3 solution. After 1 h, the polymer became soluble and the dialysis was continued against water for 24 h; the polymer remained soluble. A solution of 18 (494 mg) in water was added to the dialyzed solution; the resultant turbid mixture was dialyzed against water for 24 h. The turbid solution was freeze-dried to obtain the 23 (Scheme 8), the coacervate of 18 and 19 (1.00 g). νmax (KBr) (Fig. S20†): 3456, 2919, 2869, 1733 (w), 1620 (overlapping), 1578, 1461, 1384, 1280, 1226, 1110, 780, 900 and 701 cm−1.3 Results and discussion
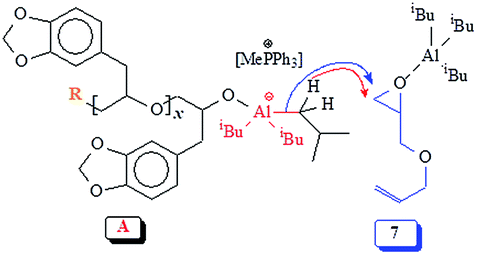
Natural safrole 1 upon oxidation with MCPBA was converted to its epoxide 2 (Scheme 1). Fig. 1a and 2a displays the 1H and 13C NMR spectra of 2. Numerous attempts were made to polymerize safrole oxide 2 using various basic catalyst (see ESI†). The results are given in (Table S1†). The ring opening polymerization monomer 2 as neat or in solvent THF was carried out under argon using TBAF (1–6 mol%) or TBAH (2–5 mol%) or NaOtBu as initiators at 50–100 °C for 2–72 h. Under the reaction conditions, the monomer was recovered unreacted in the case of initiator NaOtBu (Table S1,† entry 14). TBA-initiated polymerization also failed to give any polymer (entries 1 and 2). In other cases, we were unable to obtain the expected polymer 5 cleanly; extensive base catalysed elimination reaction occurred. Use of TBAH as the initiator led to the formation of polymer in high yields in the temperature range 70–100 °C; however, the proton NMR analysis revealed the presence of alkene motifs (≈10 mol%) on the polymer terminal (Fig. S1b†). Here, the F− or OH− or the ring opened alkoxide ion (RO−) is involved in a competing reaction between nucleophilic ring opening and base catalysed elimination reaction via abstraction of labile benzylic protons as depicted in Scheme 1. The elimination process was very extensive with F− as confirmed by appearance of 1H signal at δ 6.2, 6.5 and 4.1 ppm attributed to the –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CH2–O motifs (Fig. S1b†). For the sake of comparison, the 1H NMR spectrum of cinnamyl alcohol is shown in Fig. S1c.† The signals for the protons marked ‘a’, ‘b’ and ‘c’ are readily identifiable for comparison, thereby confirming the base catalysed chain transfer to monomer 2 by abstraction of benzylic protons leading to alkene motifs akin to the motifs of cinnamyl alcohol. With TBAH, ring opening polymerization happened accompanied by chain transfer via elimination process as depicted by A (Scheme 1). The extensive chain transfer to monomer led us to pursue this important polymerization using other protocols with the objective of minimizing the chain transfer, which puts a limit on the maximum molar mass possible for the polymers.
CH–CH2–O motifs (Fig. S1b†). For the sake of comparison, the 1H NMR spectrum of cinnamyl alcohol is shown in Fig. S1c.† The signals for the protons marked ‘a’, ‘b’ and ‘c’ are readily identifiable for comparison, thereby confirming the base catalysed chain transfer to monomer 2 by abstraction of benzylic protons leading to alkene motifs akin to the motifs of cinnamyl alcohol. With TBAH, ring opening polymerization happened accompanied by chain transfer via elimination process as depicted by A (Scheme 1). The extensive chain transfer to monomer led us to pursue this important polymerization using other protocols with the objective of minimizing the chain transfer, which puts a limit on the maximum molar mass possible for the polymers.
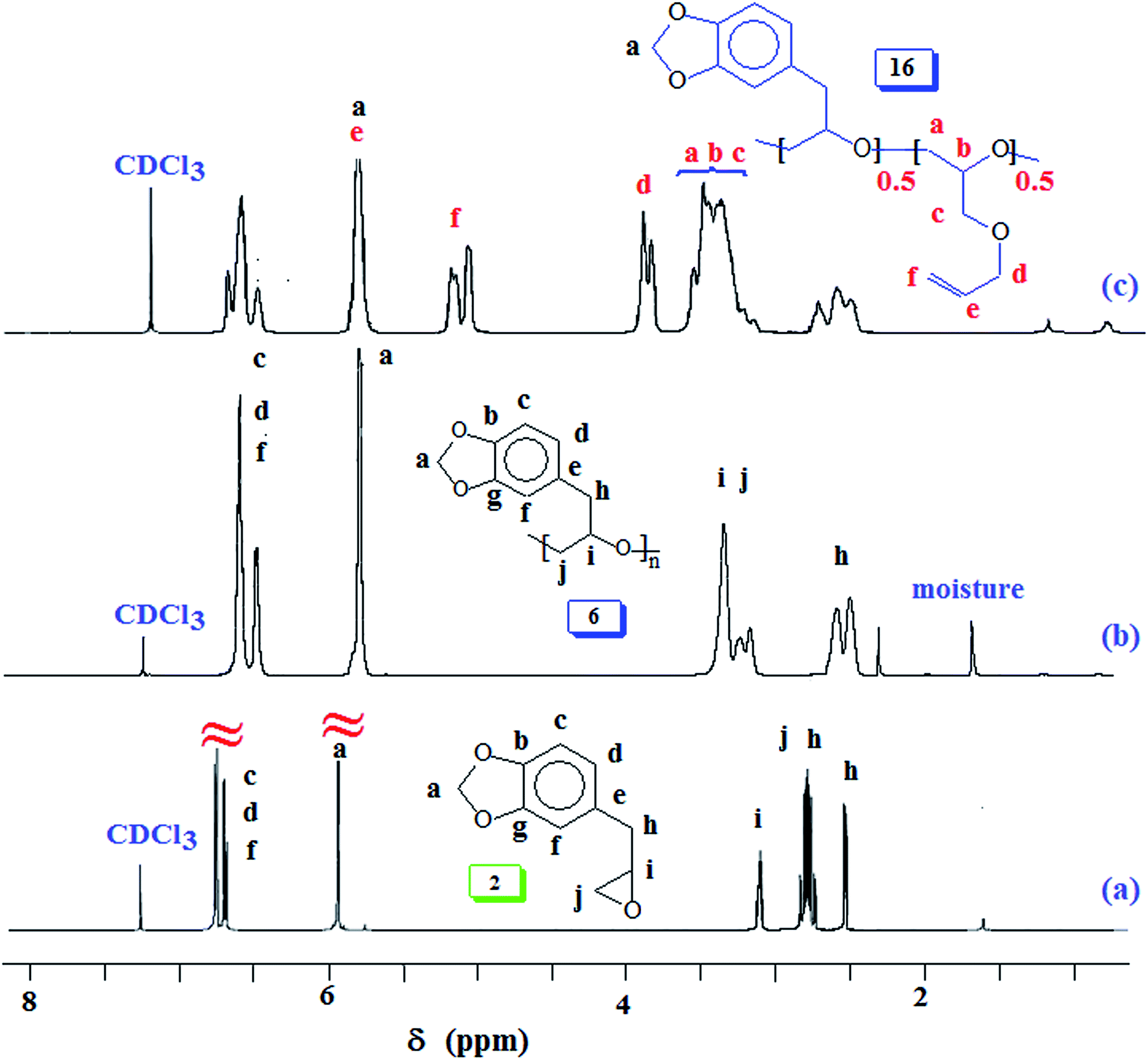 | ||
| Fig. 1 1H NMR spectra of (a) safrole oxide 2, (b) polymer 6, and (c) copolymer 16. | ||
 | ||
| Fig. 2 13C NMR spectra of (a) safrole oxide 2 and (b) polymer 6 in CDCl3. | ||
In the precedent literature, considerable efforts have been exerted to control the living character of the polymerization of propylene oxide (PO). In most cases, alkali metal alkoxides and hydroxides are widely used as anionic polymerization initiators.49–51 The high basicity of propagating species (akin to an alkoxide, RO−) leads to the abstraction of proton from methyl group of the PO, thereby constituting a chain transfer to the monomer. This process also occurred in the current work involving the base catalysed polymerization of safrole oxide (SO) 2, thereby resulting in the extensive formation of SO oligomers possessing a terminal cinnamylic unsaturation (Scheme 1). One significant problem inherent in SO is the much higher acidity of the benzylic protons than that of CH3 protons in PO; the labile H in SO is, therefore, much more prone to base catalysed abstraction than the methyl protons in PO.
Polymerization reaction of 2 using Lewis acid triisobutylaluminium as a catalyst and methyltriphenyphosphonium bromide (I) as an initiator38,39,52 gave ring opened polymer 6 in excellent yields (Scheme 2), (Table 1). The 1H NMR spectra (Fig. 1c) revealed the formation of ring opened polymer without any indication of chain transfer reaction via abstraction of allylic proton.
Experimental evidence suggests the requirement of [AlR3]/[I] ≥ 1 for successful polymerization. Trialkylaluminum participates in the formation of an aluminate complex as depicted by ‘A’ (Scheme 3) which by itself is not reactive enough to effectively initiate and/or propagate the current SO polymerization. An excess of iBu3Al ensures a fast polymerization at 0 °C owing to activating effect of the “free” triisobutylaluminum derivative as depicted by B. Note that the experimental molar masses of poly(safrole oxide) (PSO) are not close to theoretical values based on the formation of one polymer chain per Ph3MeP+Br− (see Table 1). In the context of molar masses by GPC, it is worth mentioning that the chemical structures of the standards (PEO or PS) and the polymers/copolymers are different. As such (for a note of caution), the investigated polymers/copolymers and standards may not have the same hydrodynamic radii for the same molar mass. However, using the same standards, GPC has been extensively used to determine the molar masses of polymers/copolymers derived from epoxide containing a range of substituents.38,39,53,54 The measured molar masses have then been compared with the theoretical values as described for the current polymers.
 | ||
| Scheme 3 Mechanism of Lewis acid catalyzed polymerization. | ||
Thus, the living-like polymerization of SO is jeopardized by a significant contribution of the chain transfer process to the monomer. For the PO polymerization, it has been reported, however, that the number of PPO chains remains identical to the number of Ph3MeP+Br− molecules.38
The polymerization mechanism may thus involve the electrophilic complex B which is inserted into the nucleophilic species C (Scheme 3). The involvement of reactive complex B minimizes the transfer process to monomer SO, as observed in the cases involving alkali metal alkoxide initiators. The greater electron-withdrawing effect of R3Al in B imparts greater positive charges on the ring-carbons involved in the ring opening process, than on the benzyl hydrogens involved in the transfer process to monomer (Scheme 1). Also, note that the basicity of alkoxide species in alkoxy aluminate complexes B or C is greatly reduced as compared to alkali metal alkoxide species, thereby inhibiting the proton abstraction reaction leading to chain transfer to the monomer.
It is reported that the initiation process involved the attack by not only bromide but also by the isobutyl group and hydride as shown in Scheme 3,39,52 thereby leading to three polymers having bromide, isobutyl, and hydride end groups.52 iBu3Al can work both as a hydride as well as an isobutyl anion in the ring-opening reaction.55,56
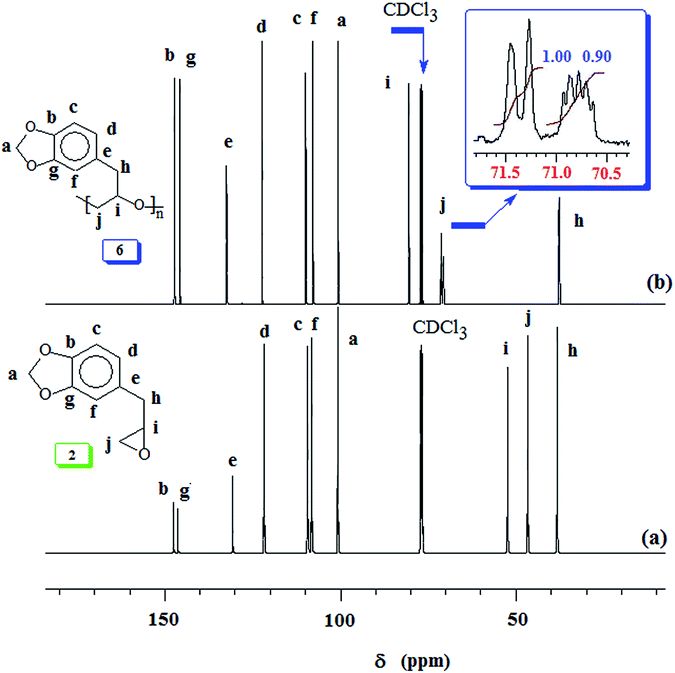
Generally, propagation via nucleophilic attacks on the less hindered methylene carbon of epoxides leads to regioregular polymers.57,58 Optically pure (i.e. enantiopure) epoxides are thus expected to provide an easy access to isotactic polymers since the methine carbon retains its configuration (Scheme 4). However, for a racemic epoxide, formation of an atactic polymer is expected with equal percentages of mm, mr, rm and rr triads (Scheme 4). 13C NMR MR spectrum revealed the regioregularity and stereochemistry of monomer insertion. The observed methine and methylene carbon signals ensure the exclusive head-to-tail (HT) chain propagation.59 In the current work, while the methine carbon marked ‘i’ appeared as single signal, the methylene carbon marked ‘j’ resolved into triads and diads (Fig. 2b). The 13C NMR spectrum thus showed the current polymer 6 as atactic (Scheme 4).38,59,60
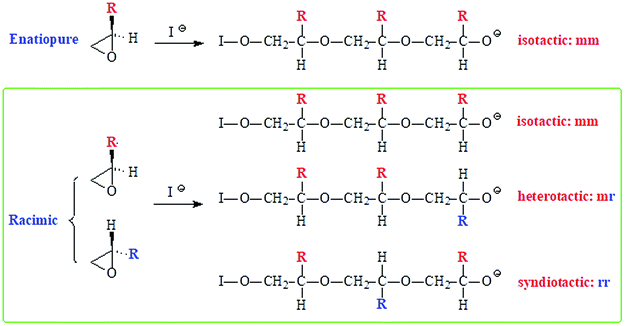 | ||
| Scheme 4 Regioregular ring opening polymerization of enantiopure and racemic epoxide. | ||
Our research plan includes the synthesis of copolymers of monomers safrole oxide 2 and allyl glycidyl ether 7 (Scheme 2). In this context, triisobutylaluminium-methyltriphenyphosphonium bromide catalyst-initiator system was used to homopolymerize 7 to obtain polymer 8 (Scheme 2). The 1H and 13C NMR spectra of 7 and 8 are shown in respective Fig. S2 and S3.† The spectral data is consistent with the regioregular ring opening to give 8. As in the case of safrole oxide polymer 6, the splitting of carbon marked ‘a’ in the 13C NMR spectrum (Fig. S3b†) pointed toward the formation of an atactic microstructure.
At this stage, we were apprehensive about the deprotection of methylene acetal protecting group in PSO 6. Methylene acetal is indeed a robust protective group which does not respond to acid catalyzed deprotection. As a model case, safrole 1 was used for examining the deprotection aspect using lead tetraacetate.42,61 To our relief, safrole 1 on treatment with the oxidizing agent afforded 9 having labile acetoxy group (Scheme 2). In fact, the similar acetoxy derivatives are usually removed during aqueous work up leading to catechol motifs. The 1H and 13C NMR spectra of 1 and 9 are shown in respective Fig. 3 and S4.† The methylene protons of 1 marked ‘a’ (Fig. 3a) is shifted downfield in the spectrum of 9 (Fig. 3b) owing to the presence of electron withdrawing AcO substituent. Similar downfield shift is observed for the carbon marked ‘j’ in the 13C NMR spectrum (Fig. S4b†).
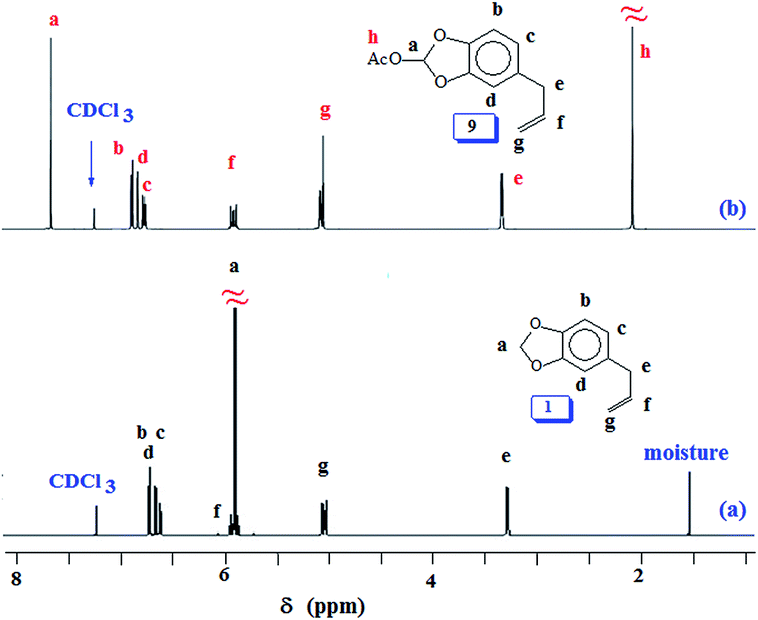 | ||
| Fig. 3 1H NMR spectra of (a) safrole oxide 1 and (b) acetoxy safrole 9 in CDCl3. | ||
Unaware of the AcO group's ability in 9 (Scheme 2) to survive during subsequent chemical transformation, we set out to explore the related chemistry using silyl protecting group. In this context, natural product eugenol 10 was demethylated and protected by reacting with diphenylsilane in the presence of catalyst tris-pentafluorotriphenylborane to give 11 (Scheme 5). To our dismay, silyl protected 11 was found to be extremely moisture sensitive; it broke down during silica gel chromatography to give allylcatechol 12 quantitatively. Thereafter, we decided to prepare 12 via demethylation using LiCl. The ‘OH’ groups in 12 were then protected to give 13 by reacting with tert-butyldimethylsilyl chloride in the presence of imidazole. Epoxidation using m-chloroperbenzoic acid transformed 13 to 14 which was then subjected to Lewis acid catalysed polymerization to afford 15a. The results of the polymerization reaction are given in (Table 2). The tert-butyldimethylsilyl groups in 15a were then deprotected using tetrabutylammonium fluoride to obtain 15b containing the catechol motifs.
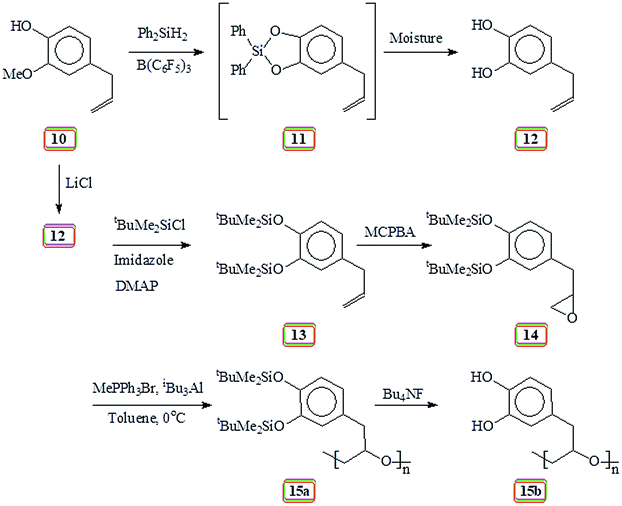 | ||
| Scheme 5 Lewis acid catalyzed polymerization of silylated epoxide 14. | ||
The 1H and 13C NMR spectra of 14, 15a and 15b are shown in respective Fig. 4 and S5.† The spectra confirmed the structures of the monomer and polymers. The silyl protons and carbons marked ‘a’, ‘b’ and ‘c’ in Fig. 4a, b, S5a and b† disappeared upon deprotection of the silyl groups as indicated by the absence of the corresponding signals in Fig. 4c and S5c.† However, it was very difficult to remove the F− as it is known to bind strongly with catechol motifs via H-bonding.46,47 In such a scenario, the ammonium counterion remained with the polymer sample as can be seen as minor peaks in the region around δ 1 ppm (Fig. 4c). Minor carbon signals are also observed in Fig. S5c.†
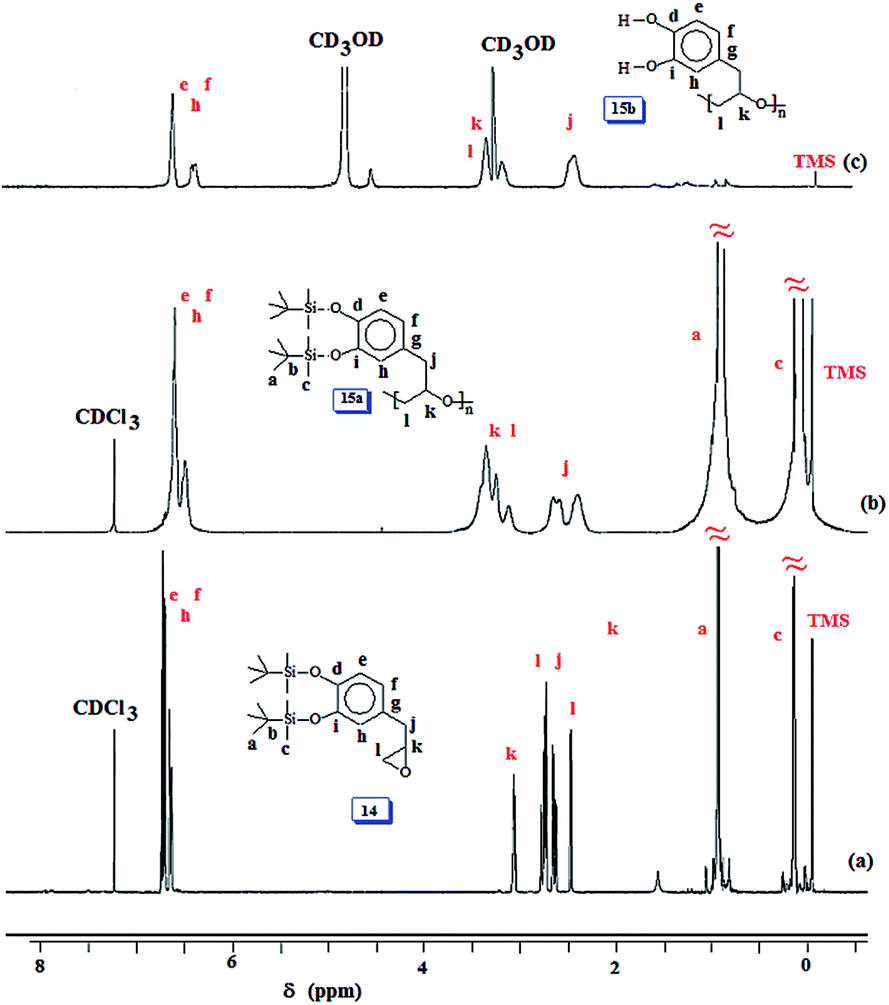 | ||
| Fig. 4 1H NMR spectra of (a) 14 and (b) 15a in CDCl3; and (c) 15b in CD3OD. | ||
Next, we focussed our attention to Lewis acid catalysed copolymerization reaction of SO (2) and AGE 7 as outlined in Scheme 6. Numerous attempts to obtain block copolymers by sequential addition of the monomers resulted in failures. At every instance, the reaction resulted in the formation of a mixture of homopolymers which were separated as explained in the Experimental section. A plausible rationale is illustrated in Scheme 7 where the hydride or butyl transfer to the second monomer might be able to initiate new chain thereby resulting in the formation of two homopolymers. We, therefore, shifted our attention to obtain the random copolymer from monomers 2 and 7 as shown in Scheme 6. Lewis acid catalysed polymerization of 2 and 7 afforded random copolymer 16 in excellent yields. The results are given in (Table 3). The 1H spectra of homopolymer 6 and the 1:1 2/7 random copolymer 16 are displayed in Fig. 1b and c. The feed ratio of the monomers matched with incorporated ratio of the corresponding repeating units as determined by integration of several non-overlapping proton signals Fig. 1c. The finding thus implies that the two monomers have equal reactivity ratio.
 | ||
| Scheme 6 Lewis acid catalyzed copolymerization of epoxides 2/7 and thiol–ene reaction. | ||
 | ||
| Scheme 7 A plausible hydride or isobutyl transfer to activated monomer 7. | ||
The 1:1 random copolymer 16 was then subjected to lead tetraacetate oxidation thereby giving copolymer 17 containing acetoxy group (vide supra, Scheme 6). The 1H and spectra of 16 and 17 are displayed in Fig. 1c and 5a, respectively. The proton marked ‘a’ (Fig. 1c) is shifted downfield at ‘c’ (Fig. 5a) and the new CH3CO protons marked ‘d′’ appeared at δ 2 ppm as expected. The 13C NMR spectrum (Fig. S6†) also revealed the formation of acetoxy derivative 17. The acetoxy carbons marked ‘r′’ and ‘q′’ appeared at δ 20.5 and 170.3 ppm, respectively (Fig. S6b†). Also note that carbon marked ‘a′’ in 17 is shifted downfield as compared to ‘a’ of 16 respectively (Fig. S6a†).
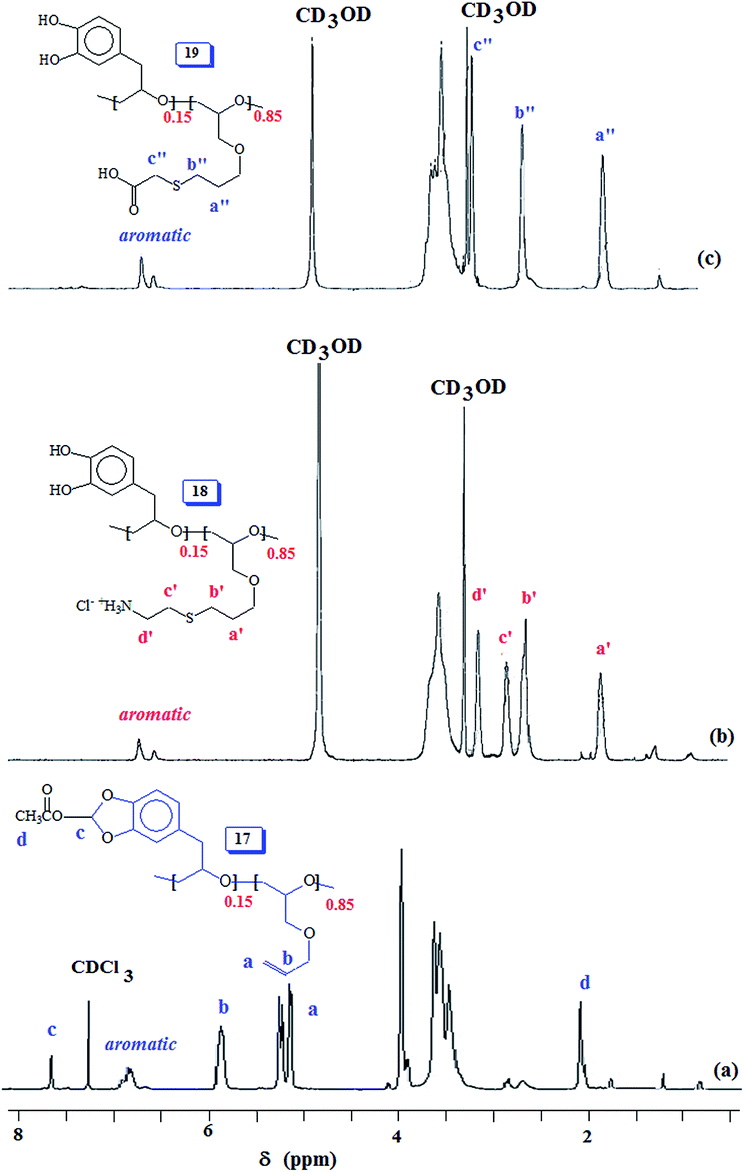 | ||
| Fig. 5 1H NMR spectra of 15:85 copolymer (a) 17 in CDCl3, (b) 18 and (c) 19 in CD3OD. | ||
The acetoxy derivative 17 was subjected to thiol–ene reaction using photoinitiator 4-dimethylaminopyridine (DMAP) using UV light (λ = 365 nm). The addition of cystemine·HCl (H3N+CH2CH2SH Cl−) and thioglycolic acid (HO2CCH2SH) to 17 with a 15:85 ratio of SO 2/AGE 7 repeating units converted 17 to 18 and 19, respectively (Scheme 6). The 1H NMR spectra of 17, 18 and 19, displayed in Fig. 5 confirms the successful transformation. The acetoxy proton signal (marked ‘d’, Fig. 5a) disappeared in the spectra of 18 and 19 (Fig. 5b and c) as a result of hydrolysis of the functional motifs during aqueous workup under mild acidic condition. This is further confirmed by the absence of the signal (for proton marked ‘c’, Fig. 5a) in the spectra of 18 and 19 (Fig. 5b and c). The spectra are consistent with the addition of the thiol motifs into the alkene double bond as confirmed the absence of alkene proton signals in Fig. 5b and c. The aromatic protons are visible; chemical shifts of some of the readily identifiable protons are assigned. The work presented in the previous paragraph are repeated using silyl protected epoxide 14 which was copolymerized with allyl glycidyl ether 7 (Scheme 8) to give copolymer 20 having various proportions of the x/y units. The results of the polymerization are given in (Table 4). The silyl protected copolymer was then elaborated using thiol–ene reaction as discussed before to give 21 and 22, which on treatment with HCl and Bu4NF respectively afforded 18 and 19 after deprotection of the silyl groups. The spectral analysis revealed the identical nature of the polymers to the polymers derived via acetoxy protective groups (vide supra). The 1H NMR spectra of random copolymer 20 with SP/AGE ratio of 1:1 and 0.10:0.90 are shown in Fig. 6a and b, respectively. Fig. 6c and d display the spectra for 21 and 22; the absence of alkene protons in the range δ 5–6 ppm clearly confirms the addition of the thio group onto the double bonds.
 | ||
| Scheme 8 Lewis acid catalyzed copolymerization of epoxides 14/7 and thiol–ene reaction. | ||
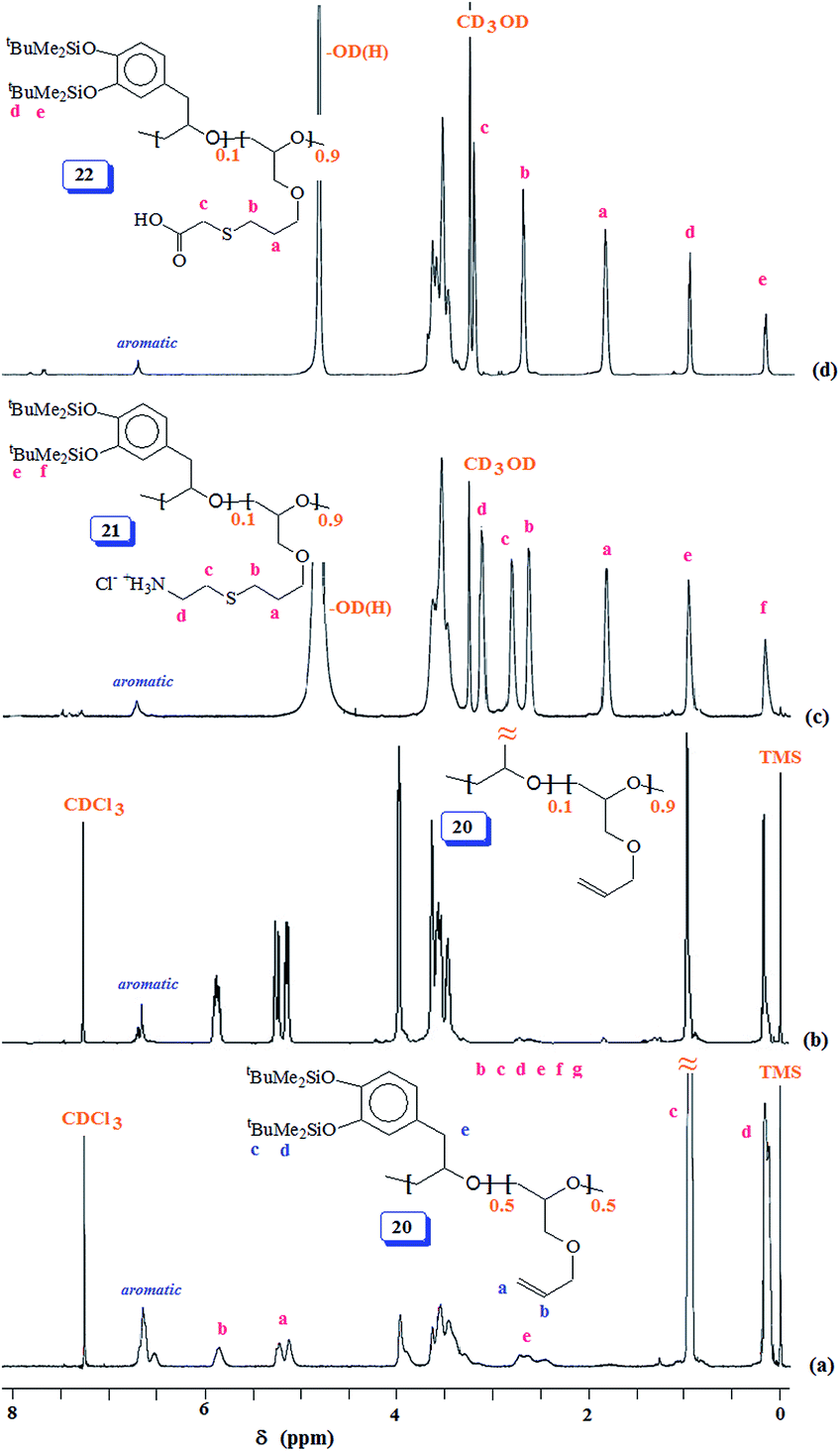 | ||
| Fig. 6 1H NMR spectra of (a) 50:50 copolymer 20 in CDCl3, (b) 10:90 copolymer 20 in CDCl3, (c) 10:90 copolymer 21 in CD3OD, and (d) 10:90 copolymer 22 in CD3OD. | ||
Coacervate 23, obtained by treating aqueous solutions of HCl salt 18 and Na-salt 19, was found to be insoluble in water (Scheme 8). The strong interaction leading to the 23 is evinced by inability of various concentration of NaCl to dislodge the component polymers from the coacervate.
4 Conclusions
The ring opening polymerization of safrole oxide 2, derived from naturally occurring safrole 1, using basic catalyst led to the formation of intractable materials as a result of extensive chain transfer reaction. However, safrole oxide 2, for the first time, underwent homo- as well as copolymerization with AGE 7 using the Lewis acid initiator/catalyst comprising of triphenylmethylphosphonium bromide/triisobutylaluminum. A mechanism has been proposed for the chain transfer reaction to monomer and the inability to form block copolymers. The random copolymer 16 of various compositions (from SO 2/AGE 7), obtained in excellent yields, were elaborated by activation of methylene acetal with acetylation to give 17 followed by incorporating anionic (CO2−) and cationic (NH3+) group on the terminal of the pendants by thiol–ene reaction to generate. The polymer backbones of 18 and 19 having charges of opposite algebraic signs was used for the formation of complex coacervate by electrostatic attraction. Such complex is expected to be adsorbed on a surface where the catechol motifs would stabilize the adhesion. A way has been found to remove the acetal protective group under mild conditions using lead tetraacetate to generate catechol moieties on the polymer backbone. Use of tert-butyldimethyl groups as a protective group for the catechol motifs has also been explored.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to acknowledge the support provided by King Abdulaziz City for Science and Technology (KACST) through the Science & Technology Unit at King Fahd University of Petroleum & Minerals (KFUPM) for funding this work through project no. 12-ADV2397 as part of the National Science, Technology and Innovation Plan. The authors would like to acknowledge the valuable guidance of Prof. Craig Hawker in conducting this research work.References
- J. Comyn, The relationship between joint durability and water diffusion, in Developments in Adhesives, ed. A. J. Kinloc, Appl. Sci., Barking UK, 1981, vol. 2, pp. 279–313 Search PubMed.
- P. Lee Bruce, P. B. Messersmith, J. N. Israelachvili and J. H. Waite, Annu. Rev. Mater. Res., 2011, 41, 99–132 CrossRef CAS.
- A. V. Pocius, Adhesives and Adhesives Technology, Hanser/Gardner, Cincinnati, 1997 Search PubMed.
- B. K. Ahn, J. Am. Chem. Soc., 2017, 139, 10166–10171 CrossRef CAS.
- K. Zhang, F. Zhang, Y. Song, J. B. Fan and S. Wang, Chin. J. Chem., 2017, 35, 811–820 CrossRef CAS.
- K. Zhang, F. Zhang, Y. Song, J. Fan and S. Wang, Chin. J. Chem., 2017, 35, 811–820 CrossRef CAS.
- N. K. Kaushik, N. Kaushik, S. Pardeshi, J. G. Sharma, S. H. Lee and E. H. Choi, Mar. Drugs, 2015, 13, 6792–6817 CrossRef CAS.
- Z. Wang, Y. Duan and Y. Duan, J. Controlled Release, 2018, 290, 56–74 CrossRef CAS.
- H. Yamamoto, S. Yamauchi and S. Ohara, Biomimetics, 1992, 1, 219–238 CAS.
- H. Tatehata, A. Mochizuki, T. Kawashima, S. Yamashita and H. Yamamoto, J. Appl. Polym. Sci., 2000, 76, 929–937 CrossRef CAS.
- M. Yu, J. Hwang and T. J. Deming, J. Am. Chem. Soc., 1999, 121, 5825–5826 CrossRef CAS.
- M. Yin, Y. Yuan, C. Liu and J. Wang, Biomaterials, 2009, 30, 2764–2773 CrossRef CAS.
- T. H. Anderson, J. Yu, A. Y. Estrada, M. U. Hammer, J. H. Waite and J. N. Israelachvili, Adv. Funct. Mater., 2010, 20, 4196–4205 CrossRef CAS.
- H. Zhao, C. J. Sun, R. J. Stewart and J. H. Waite, J. Biol. Chem., 2005, 280, 42938–42944 CrossRef CAS.
- H. Shao and R. J. Stewart, Adv. Mater., 2010, 22, 729–733 CrossRef CAS.
- H. Lee, B. P. Lee and P. B. Messersmith, Nature, 2007, 448, 338–341 CrossRef CAS.
- B. P. Lee, K. Huang, F. N. Nunalee, K. R. Shull and P. B. Messersmith, J. Biomater. Sci., Polym. Ed., 2004, 15, 449–464 CrossRef CAS.
- G. Westwood, T. N. Horton and J. J. Wilker, Macromolecules, 2007, 40, 3960–3964 CrossRef CAS.
- H. C. Yang, J. Luo, Y. Lv, P. Shen and Z. K. Xu, J. Membr. Sci., 2015, 483, 42–59 CrossRef CAS.
- Y. Tian, Y. Cao, Y. Wang, W. Yang and J. Feng, Adv. Mater., 2013, 25, 2980–2983 CrossRef CAS PubMed.
- F. Zhang, S. Liu, Y. Zhang, Y. Wei and J. Xu, RSC Adv., 2012, 2, 8919–8921 RSC.
- J. Li, M. Zhou, F. Cheng, Y. Lin and P. Zhu, Carbohydr. Polym., 2019, 221, 113–119 CrossRef CAS PubMed.
- X. Chen, Y. Zhai, X. Han, H. Liu and Y. Hu, Appl. Surf. Sci., 2019, 483, 399–408 CrossRef CAS.
- J. Duan, W. Wu, Z. Wei, D. Zhu, H. Tu and A. Zhang, Green Chem., 2018, 20, 912–920 RSC.
- X.-D. Pan, Z. Qina, Y.-Y. Yana and P. Sadhukhana, Polymer, 2010, 51, 3453–3461 CrossRef CAS.
- H. Shao, K. N. Bachus and R. J. Stewart, Macromol. Biosci., 2009, 9, 464–471 CrossRef CAS PubMed.
- R. Ko, P. A. Cadieux, J. L. Dalsin, B. P. Lee, C. N. Elwood and H. Razvi, J. Endourol., 2008, 22, 1153–1160 CrossRef.
- B. P. Lee, J. L. Dalsin and P. B. Messersmith, Biomacromolecules, 2002, 3, 1038–1047 CrossRef CAS.
- J. Yu, W. Wei, E. Danner, R. K. Ashley, J. N. Israelachvili and J. H. Waite, Nat. Chem. Biol., 2011, 7, 588–590 CrossRef CAS.
- X. X. Qin, K. J. Coyne and J. H. Waite, J. Biol. Chem., 1997, 272, 32623–32627 CrossRef CAS.
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170–2243 CrossRef CAS.
- Y. Sarazin and J. F. Carpentier, Chem. Rev., 2015, 115, 3564–3614 CrossRef CAS.
- A. L. Brocas, C. Mantzaridis, D. Tunc and S. Carlotti, Prog. Polym. Sci., 2013, 38, 845–873 CrossRef CAS.
- P. Kraft and W. Eichenberger, Eur. J. Org. Chem., 2003, 2003, 3735–3743 CrossRef.
- J. C. Bailar Jr, W. C. Fernelius and H. A. Skinner, Inorg. Synth., 1939, 1, 47–49 Search PubMed.
- L.-C. Shen, S.-Y. Chiang, I.-T. Ho, K.-Y. Wu and W.-S. Chung, Eur. J. Org. Chem., 2012, 4, 792–800 CrossRef.
- Z. Baoxiang, W. Dawei, Z. Hua, Q. Guangtao and M. Junying, Chin. J. Org. Chem., 2003, 23, 1026–1028 Search PubMed.
- C. Billouard, S. Carlotti, P. Desbois and A. Deffieux, Macromolecules, 2004, 37, 4038–4043 CrossRef CAS.
- K. Sakakibara, K. Nakano and K. Nozaki, Macromolecules, 2007, 40, 6136–6142 CrossRef CAS.
- A. L. Brocas, G. Cendejas, S. Caillol, A. Deffieux and S. Carlotti, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2677–2684 CrossRef CAS.
- B. F. Lee, M. J. Kade, J. A. Chute, N. Gupta, L. M. Campos, G. H. Fredrickson, E. J. Kramer, N. A. Lynd and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4498–4504 CrossRef CAS.
- K. C. Nicolaou, J. Wang, Y. Tang and L. Botta, J. Am. Chem. Soc., 2010, 132, 11350–11363 CrossRef CAS.
- K. C. Nicolaou, Y. Tang and J. Wang, Angew. Chem., Int. Ed., 2009, 48, 3449–3453 CrossRef CAS.
- S. Strych and D. Trauner, Angew. Chem., Int. Ed., 2013, 52, 9509–9512 CrossRef CAS.
- B. K. Ahn, D. W. Lee, J. N. Israelachvili and J. H. Waite, Nat. Mater., 2014, 13, 867–872 CrossRef CAS PubMed.
- X. Wu, X.-X. Chen, B.-N. Song, Y.-J. Huang, W.-J. Ouyang, Z. Li, T. D. James and Y.-B. Jiang, Chem. Commun., 2014, 50, 13987–13989 RSC.
- K. J. Winstanley, A. M. Sayer and D. K. Smith, Org. Biomol. Chem., 2006, 4, 1760–1767 RSC.
- L. M. Campos, K. L. Killops, R. Sakai, J. M. J. Paulusse, D. Damiron, E. Drockenmuller, B. W. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063–7070 CrossRef CAS.
- S. Boileau, in Comprehensive Polymer Science, Chain Polymerization, ed. G. C. Eastmond, A. Ledwith, S. Russo and P. Sigwalt, Pergamon Press, Oxford, 1989, vol. 3, Part 1, pp. 467–487 Search PubMed.
- S. D. Gagnon, in Encyclopedia of Polymer Science and Engineering, ed. R. P. Quirk, Wiley-Interscience, New York, 1985, vol. 6, pp. 273–307 Search PubMed.
- R. P. Quirk and G. M. Lizarraga, Macromol. Chem. Phys., 2000, 201, 1395–1404 CrossRef CAS.
- K. Sakakibara, K. Nakano and K. Nozaki, Chem. Commun., 2006, 57, 3334–3336 RSC.
- S. S. Müller, C. Moers and H. Frey, Macromolecules, 2014, 47, 5492–5500 CrossRef.
- J. Herzberger and H. Frey, Macromolecules, 2015, 48, 8144–8153 CrossRef CAS.
- G. Boireau, D. Abenhaim and E. Henry-Basch, Tetrahedron, 1980, 36, 3061–3070 CrossRef CAS.
- J. J. Eisch, Z. R. Liu and M. Singh, J. Org. Chem., 1992, 57, 1618–1621 CrossRef CAS.
- T. Hagiwara, Y. Terasaki, H. Hamana, T. Narita, J. Umezawa and K. Furuhashi, Makromol. Chem. Rapid Commun., 1992, 13, 363–370 CrossRef CAS.
- K. Maruoka and T. Ooi, Chem.–Eur. J., 1999, 5, 829–833 CrossRef CAS.
- B. Antelmann, M. H. Chisholm, S. S. Iyer, J. C. Huffman, D. Navarro-Llobet, M. Pagel, W. J. Simonsick and W. Zhong, Macromolecules, 2001, 34, 3159–3175 CrossRef CAS.
- M. H. Chisholm and D. Navarro-Llobet, Macromolecules, 2002, 35, 2389–2392 CrossRef CAS.
- M. Lee, W. Zhang and B. C. Noll, Angew. Chem., Int. Ed., 2009, 48, 3449–3453 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04719k |
| This journal is © The Royal Society of Chemistry 2019 |