
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Immunocapture sample clean-up in determination of low abundant protein biomarkers – a feasibility study of peptide capture by anti-protein antibodies†
Maren C. S.
Levernæs
a,
Bassem
Farhat
a,
Inger
Oulie
a,
Sazan S.
Abdullah
a,
Elisabeth
Paus
b,
Léon
Reubsaet
 a and
Trine G.
Halvorsen
*a
a and
Trine G.
Halvorsen
*a
aDepartment Pharmacy, University of Oslo, P. O. Box 1068 Blindern, 0316 Oslo, Norway. E-mail: trinegha@farmasi.uio.no; Fax: +47 22854402; Tel: +47 22855735
bDepartment of Medical Biochemistry, Norwegian Radium Hospital, Oslo University Hospital, Norway
First published on 29th October 2019
Abstract
Immunocapture in mass spectrometry based targeted protein analysis using a bottom-up workflow is nowadays mainly performed by target protein extraction using anti-protein antibodies followed by tryptic digestion. Already available monoclonal antibodies (mAbs) which were developed against intact target proteins (anti-protein antibodies) can capture proteotypic epitope containing peptides after tryptic digestion of the sample. In the present paper considerations when developing a method for targeted protein quantitation through capture of epitope containing peptides are discussed and a method applying peptide capture by anti-protein antibodies is compared with conventional immunocapture MS. The model protein used for this purpose was progastrin releasing peptide (ProGRP), a validated low abundant biomarker for Small Cell Lung Cancer with reference values in serum in the pg mL−1 range. A set of mAbs which bind linear epitopes of ProGRP are available, and after a theoretical consideration, three mAbs (E146, E149 and M18) were evaluated for extraction of proteotypic epitope peptides from a complex sample. M18 was the best performing mAb for peptide capture by anti-protein antibodies, matching the LOD (54 pg mL−1) and LOQ (181 pg mL−1) of the existing conventional immunocapture LC-MS/MS method for determination of ProGRP. Peptide and protein capture using the same mAb were also compared with respect to sample clean-up, and the peptide capture workflow yielded cleaner extracts and therewith less complex chromatograms. Analysis of five patient samples demonstrated that peptide capture by anti-protein antibodies can be used for the determination of various levels of endogenously present ProGRP.
Introduction
In recent years the proteomics society has shown a growing interest in targeted mass spectrometry (MS) based methods to quantify protein biomarkers.1 Despite its unique mass selectivity, sensitive detection of intact proteins by MS is limited. The protein biomarker is thus usually digested by a proteolytic enzyme to generate proteotypic peptides that are analyzed by LC-MS.2,3 The choice of the surrogate peptide, also known as the signature peptide or proteotypic peptide, used for the quantitation of the entire protein is crucial for the quality of the assay and should thus be unique to the targeted protein.2–4Among the most promising methods to ensure high sensitivity and selectivity in targeted analysis of low-abundant protein biomarkers is a combination of immunoaffinity enrichment (immunocapture), protein digestion and LC-MS/MS detection of signature peptides.1–6 Immunocapture can be performed both prior to and after the protein digestion step, and provides unique selectivity through the unique interaction of the target protein or signature peptide and the capture antibody.2 The antibodies used for immunocapture target either linear or conformational epitopes. Linear epitopes consist of continuously neighbouring amino acid residues along the protein sequence, while conformational epitopes consist of amino acid residues that are discontinuously arranged along the protein sequence, that are brought together through folding of the polypeptide chain.7 Thus antibodies targeting conformational epitopes can be used for the enrichment of the intact protein, whereas antibodies targeting linear epitopes can both capture intact protein and epitope containing peptides.5 Additionally, anti-peptide antibodies may be produced to target signature peptides (SISCAPA).3,5
The greatest advantage of choosing peptide immunocapture over protein immunocapture is that peptide samples are easier to handle, due to degradation, unfolding and solubility issues often associated with full-length proteins.5 Quantitative, multiplexed immuno-MRM assays targeting peptides rather than proteins are expected to produce cleaner eluates and thus less interfering peptides are introduced to the LC-MS.8 In addition, the proteolytic step is performed prior to extraction, creating the possibility that the antibodies can be re-used and thus the cost per sample is reduced. Approaches based on peptide capture are also less likely to be influenced by auto-antibodies, as these antibodies are degraded during the proteolytic step.9
The use of monoclonal antibodies (mAbs), unlike polyclonal antibodies with its batch to batch variation,8 is especially attractive in assay development for both peptide and protein enrichment as they are renewable reagents of which the production can be standardized. The availability of validated anti-peptide mAbs for immunoaffinity enrichment of proteotypic peptides is however very limited.10 The number of anti-protein mAbs, on the other hand, is huge and are often commercially available due to ongoing antibody screening in pharmaceutical industry.1 Hence, peptide capture by anti-protein antibodies might be a promising alternative for the enrichment of proteotypic peptides without the need for time-consuming and expensive development of anti-peptide antibodies. In 2004, Zhao et al. characterized the epitope of an anti-troponin antibody by epitope excision and the identified (missed cleavage) peptide was used as the surrogate peptide in the targeted protein quantitation.11 This approach is most easily applied to antibodies with known epitopes. However, as demonstrated by Schoenherr et al. it can also be applied without knowledge of the epitope configuration (overall success rate 14%).8 The strategy was also recently applied in an on-line set-up where mAbs were covalently immobilized on acrylate-monoliths coupled to nano-LC-MS, demonstrating the possibility of automated capture and analysis of proteotypic epitope peptides.12 In order to successfully develop a peptide capture assay based on anti-protein antibodies, the antibody should recognize a linear epitope, not disrupted by a cleavage site. Since the majority of the commercially available antibodies used for western blot analysis and ELISA assays have linear epitopes,13 this approach is predicted to be widely applicable.
In the study presented here, we thoroughly explore the potential of peptide enrichment using anti-protein antibodies for LC-MS based targeted protein determination by comparing it with conventional immunocapture MS strategy. The small cell lung cancer (SCLC) biomarker progastrin releasing peptide (ProGRP) was used as model compound and immunocapture MS of intact ProGRP as benchmark. The advantage of this model system is availability of well-characterized mAbs with known epitopes that could be used for comparison. The set-up that is compared with protein extraction utilizes the same antibody as applied in the on-line study described above. However, the present study thoroughly discusses the considerations necessary for peptide extraction using protein antibodies and compares the method with methods based on immunocapture of the intact protein followed by LC-MS/MS determination of signature peptides.14,15 In addition, to assess the feasibility of using the proteotypic epitope peptide as the surrogate peptide in the quantitation of proteins in real samples the low abundance model biomarker ProGRP was determined in serum; both in spiked samples and samples from patients diagnosed with SCLC.
Experimental
Protein standards and chemicals
Cloned ProGRP isoform 1 and anti-ProGRP (mAbs E146, E149 and M18) were provided by the Central Laboratory, Norwegian Radium Hospital, Oslo University Hospital (Oslo, Norway). Trypsin (TPCK treated, from bovine pancreas, sequencing grade), Lys-C (from Lysobacter enzymogenes), and formic acid (FA) were purchased from Sigma Aldrich (St. Louis, MO, USA). All other chemicals used were of analytical grade. The stable isotopic labeled internal standard with sequence NH2-ALGNQQPSWDSEDSSNF(K*)–COOH (purity: >95%), where K* denotes Lys labeled with 13C/15N, was bought from Innovagen (Lund, Sweden).Serum samples
Human serum from healthy subjects was obtained from Oslo University Hospital, Ullevål (Oslo, Norway), and serum samples from cancer patients were supplied by the Norwegian Radium Hospital, Oslo University Hospital. All serum samples were stored at −30 °C. The use of both serum from healthy subjects and patient samples for our research purposes was performed in strict accordance to Norwegian law (“Lov om medisinsk og helsefaglig forskning (helseforskningsloven)”) and the use of patient samples was approved by the Norwegian Regional Committee for Medical Research Ethics (REK, http://helseforskning.etikkom.no). The research project is registered in the in the database for health related research at the Department of Pharmacy, University of Oslo (Oslo, Norway). Informed consent was obtained from all subjects. Methods used to analyze all serum samples were in accordance with relevant guidelines and regulations (mentioned above).Solutions
ProGRP isoform 1 (AA 1-125 + 8) was cloned from human cDNA (OriGene Technologies), expressed in Escherichia coli (Promega) using pGEX-6P-3 constructs (GE Healthcare) and purified as described elsewhere.16 The concentration of the ProGRP stock solution was determined by absorbance at 280 nm (A280). Working solutions were prepared by dilution with 50 mM ammonium bicarbonate solution (ABC solution) and stored at 4 °C.Initially, complex samples were prepared by adding Lys-C or trypsin digested ProGRP to trypsin digested human serum from healthy subjects. The digested standards were added to the digested serum immediately before performing the extraction. In later experiments, spiked serum samples were prepared by adding intact ProGRP immediately before digestion with trypsin beads.
Immobilization of trypsin to sepharose beads
Covalent immobilization of trypsin to sepharose beads was performed as described elsewhere.17 In brief, NHS-activated sepharose beads (NHS-activated Sepharose™ 4 Fast Flow, GE Healthcare, Uppsala, Sweden) were prewashed with 10 volumes of cold washing buffer (0.1 M phosphate buffer, pH 7.8). An equal volume of 20 mg mL−1 trypsin in coupling buffer (0.1 M ethanolamine, 0.25 M benzamidine and 0.1 M phosphate buffer, pH 7.8) was added and shaken (800 rpm) at room temperature for 25 min. Unbound trypsin was removed before modification buffer (0.2 M acetic acid N-hydroxysuccinimide (NHS)esther in 0.1 M phosphate buffer, pH 7.8) was added (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) and shaken (800 rpm) at room temperature for 20 min. The excess NHS was deactivated by blocking buffer (0.1 M ethanolamine in 0.1 M phosphate buffer, pH 8.0) (1:5 v/v) and shaken (800 rpm) at room temperature for 10 min. The trypsin coated beads were stored in storage buffer (50 mM Tris pH 8.2, 1 mM calcium chloride and 0.02% sodium azide) at 4 °C, protected from light. The final concentration of the immobilized trypsin beads are according to Freije et al.17 about 16.3 mg mL−1.
:1 v/v) and shaken (800 rpm) at room temperature for 20 min. The excess NHS was deactivated by blocking buffer (0.1 M ethanolamine in 0.1 M phosphate buffer, pH 8.0) (1:5 v/v) and shaken (800 rpm) at room temperature for 10 min. The trypsin coated beads were stored in storage buffer (50 mM Tris pH 8.2, 1 mM calcium chloride and 0.02% sodium azide) at 4 °C, protected from light. The final concentration of the immobilized trypsin beads are according to Freije et al.17 about 16.3 mg mL−1.
Enzymatic digestion
In-solution digestion of ProGRP isoform 1 diluted in freshly prepared ABC solution (50 mM) was initiated by adding freshly prepared protease (trypsin or Lys-C) to give an enzyme-to-protein ratio of 1:40 (w/w). On-beads digestion of spiked serum samples were initiated by adding freshly prepared trypsin to give an enzyme-to-antibody ratio of 1:5 (w/w). Samples were incubated over night at 800 rpm at 37 °C.
Digestion of protein precipitated serum samples were initiated by adding 30 μL trypsin beads. Samples were incubated for 2 h at 800 rpm at 37 °C.
Immobilization of monoclonal antibodies to magnetic beads
The monoclonal antibodies were covalently immobilized to tosylactivated magnetic beads (Dynabeads M280 tosylactivated, Invitrogen, Thermo Fisher Scientific, Oslo, Norway) using 1 mg of antibody to 50 mg of magnetic beads. To ensure the right orientation of the antibodies on the beads, the antibodies were added hydrochloric acid (HCl) to pH 2.5 and incubated for 1 h on ice,18 before neutralization by the addition of sodium hydroxide to pH 7. Coupling of the antibody to the beads was performed at pH 9.5 overnight at room temperature. The coupling volume was 1 g beads to 50 mL solution; 1/5 of final volume was 0.5 M borate buffer and added coupling buffer (50 mM trizma base, 100 mM sodium chloride, and 7.7 mM sodium azide in mqH2O, pH 7.5) to final volume. To remove any unbound antibody, the beads were washed twice with storage buffer (2.5 M sodium chloride, 60 mM sodium dihydrogen phosphate monohydrate, and 7.7 mM sodium azide in milliQ-H2O, pH 6.7) for 2 h, and once overnight, at room temperature. Antibody coated magnetic beads were stored in storage buffer (pH 6.7) at 4 °C.Peptide extraction
To remove any unbound anti-ProGRP the antibody-coated magnetic beads were prewashed as described elsewhere:14 The desired volume of beads was washed with 1 mL PBS containing 0.05% Tween 20, and re-dissolved in PBS, yielding a solution with the initial bead concentration, ready for use.The immunoaffinity extraction was performed as follows using magnetic beads: Protein LoBind Eppendorf tubes containing the sample (an in-solution digest of the protein standard or a protein precipitated, diluted and digested serum sample) were added 20 μL of prewashed antibody-coated magnetic beads. To capture the peptides the Eppendorf tubes were rotated and shaken for 1 h on a HulaMixer (Invitrogen), to facilitate the epitope–antibody interaction. The Eppendorf tubes were then placed in the magnetic rack (DynaMag-2 from Invitrogen) to collect the beads and remove the solution. The beads were then washed with 500 μL of PBS containing 0.05% Tween 20, 500 μL of PBS, 300 μL of Tris–HCl (pH 7.4), and 300 μL of 50 mM ABC solution, prior to elution.
Elution of extracted peptides
After immunocapture, the elution step was used for enrichment by adding 15 μL of freshly prepared 2% formic acid (FA) to the washed beads. Samples were shaken at room temperature for 5 min, placed in the magnetic rack and the supernatant containing eluted peptides was transferred to new Protein LoBind Eppendorf tubes. Additional 15 μL of 2% FA was added to the beads, shaken for 5 min and the two supernatants were collected in the same tube. The combined eluates were directly injected into the LC-MS system.Preparation of serum samples
Fifty μL human serum was protein precipitated using cold acetonitrile (−32 °C) in a ratio of 1:0.7 (v/v) with subsequent vortex mixing for 1 min and centrifugation at 10000 rpm for 10 min. Due to its size (13705 Da), ProGRP does not precipitate but remains in the supernatant.19 The supernatant was then transferred to new Protein LoBind Eppendorf tubes and diluted 1:40 with ABC solution (50 mM) to ensure optimal digestion conditions. Digestion was performed by adding 30 μL trypsin beads solution and the samples were incubated for 2 h at 37 °C. The samples were then centrifuged to sediment the beads and the solution was transferred to new Protein LoBind Eppendorf tubes. The internal standard was added prior to extraction. Epitope peptide extraction and elution of bound peptides were then performed as described above.
Nano LC-MS/MS analysis
Two different nano LC-MS systems were used: an LTQ Discovery Orbitrap MS and a TSQ Quantiva, both from Thermo Fischer (Rockford, IL, US). The same columns and mobile phases were used for both systems. The samples were trapped on a C18 Acclaim PepMap 100 enrichment column (300 μm i.d. × 5 mm, 5 μm; Thermo Fischer) and further separated on a C18 Acclaim PepMap 100 analytical column (75 μm i.d. × 15 cm, 3 μm; Thermo Fischer). The loading buffer consisted of 20 mM FA:MeCN (97:2, v/v), and the mobile phases consisted of A: 20 mM FA:MeCN (95:5, v/v) and B: 20 mM FA:MeCN (5:95, v/v). An automatic filtration of the samples were also performed prior to trapping as described elsewhere using a steel filter (replacement screen, 1/16′′ from Teknolab, Norway).20
000 over a mass range between m/z 300–2000 Da with charge state disabled. Up to six of the most intense ions per scan were fragmented by collision induced dissociation (CID) at 35% relative collision energy, activation time of 30 ms, and analysed in the linear ion trap. The fragmented m/z values were dynamically excluded for 15 s to minimize the extent of repeat sequencing of peptides and to fragment lower intensity m/z values.
Data interpretation
The MS raw files were processed with Proteome Discoverer 1.4 (Thermo Fischer), using the SEQUEST algorithm, searching against a ProGRP database generated from the sequence obtained from UniProt (January, 2015). Up to five missed cleavages were considered using trypsin, Lys-C and chymotrypsin as enzymes. Methionine oxidation was chosen as variable modification. The initial parent and fragment ion maximum mass deviation was set to 10 ppm and 0.8 Da, respectively.The Thermo Scientific Xcalibur software version 2.1 (Thermo Fischer) was used to manually extract ion chromatograms (XICs), peak area and signal intensities of selected tryptic peptides.
The extraction yield was calculated from the analysis of both the bound (eluate) and unbound (supernatant) fraction of the epitope peptide.
Evaluation of statistical significance was performed using a paired sample t-test, with a cut-off value of 0.05.
In silico digestion and similarity search
The sequence for ProGRP isoform 1 was acquired through The National Center for Biotechnology Information (NCBI) database (accession number NP_002082.2). In silico digests (Protein Prospector version 5.18.1) were performed using the following parameters: trypsin or Lys-C digest, zero missed cleavages, no modifications, peptide mass 400–2000, and minimum peptide length of five amino acids. The results were used to determine which protease should be used to generate a zero missed cleavage peptide containing the intact epitope of mAb E146 and mAb E149/M18.To investigate if the epitope containing zero missed cleavage peptides solely originated from ProGRP, an NCBI BLAST (National Center for Biotechnology Information, Basic Local Alignment Search Tool) sequence similarity search was performed and used to ensure specificity of the proteotypic epitope peptides. BLASTP 2.6.0+ was used as an algorithm with protein sequences from NCBI's Reference Sequence Project (RefSeq) as the database of choice. In addition to Homo sapiens, the BLAST searches were performed on Mus musculus and Bos taurus, because the antibodies and the proteases used were derived from these organisms respectively.
Results and discussion
Considerations in peptide capture by anti-protein antibody method development
For the model system applied in the present paper, several anti-protein mAbs (Nordlund et al.21) which recognise and bind to different regions of our model protein ProGRP (Fig. 1) are already available. To investigate if an epitope containing zero missed cleavage peptide could be generated for the available mAbs, in silico digests with different enzymes (trypsin and Lys-C) were performed in Protein Prospector. This search resulted in one proteotypic epitope peptide candidate for E146, E172, M11 and M16 using Lys-C and one for E149, M7, M8, M9, M15, M18 and M19 using trypsin or Lys-C, for M37 no epitope peptide candidate was available for trypsin and Lys-C. The following BLAST query showed that the only source for the two peptides was ProGRP. Finally, SEQUEST identified the proteotypic epitope peptide candidates performed on data from FT-Orbitrap analysis on in-solution digests of ProGRP. Thus, the Lys-C generated peptide QQLREYIRWEEAARNLLGLIEAK and the trypsin (or Lys-C) generated peptide ALGNQQPSWDSEDSSNFK could be used as proteotypic epitope peptides for mAb E146, E172, M11 and M16 and mAbs E149, M7, M8, M9, M15, M18 and M19, respectively.
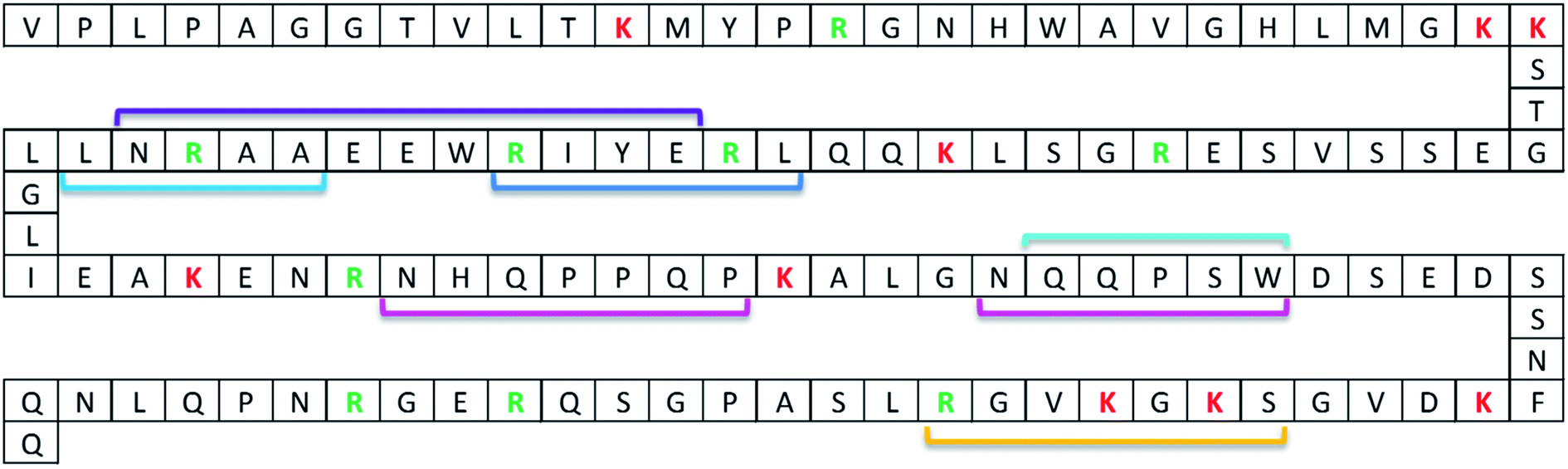 | ||
| Fig. 1 ProGRP isoform 1 sequence with binding epitopes of available anti-ProGRP mAbs. The binding epitopes have been determined by pepscan analysis. Linear epitopes: E172 (purple), E146 (dark blue), M11 and M16 (light blue), E149, M7, M8, M9, M15, M18 and M19 (turquoise), and M37 (yellow). Non-linear epitopes: E168 (pink). Trypsin cleavages sites are marked with red (lysine) and green (arginine) letters. | ||
The effect of residual enzyme on the mAbs in the present study was investigated by subjecting the antibody coated magnetic beads to various amount of proteolytic enzyme prior to extraction of intact ProGRP. The antibody coated beads E146 and M18/E149 were incubated with Lys-C (6.25 and 250 ng mL−1) or trypsin (6.25 ng mL−1, 250 ng mL−1 and 50 mg mL−1), respectively, for 2 h at room temperature. After enzyme treatment, the antibody containing beads were washed before being used in the extraction of intact ProGRP from spiked samples (250 ng mL−1). After extraction, on-beads trypsin digestion was carried out overnight. The quantitative yield of the proteotypic peptides generated after digestion were compared to those generated from an extraction of intact ProGRP by antibody containing beads not exposed to active enzyme prior to extraction (control).
There was, as shown in Fig. 2, a significant decrease in the quantitative yield of ProGRP after the extraction by antibodies exposed to the proteolytic enzyme compared to the control (P-value < 0.05), except for the E146 beads which were more resistant towards low amounts of proteolytic enzyme (or Lys-C less efficient in digesting the antibodies) and showed no significant difference in the quantitative yield compared to the control (P-value > 0.05). When exposed to the amount of enzyme needed to digest a protein precipitated serum sample (50 mg mL−1), the antibodies still worked, but their performance (monitored by signal intensity of proteotypic peptides) was reduced to about 20–30%. The observed decrease is most likely due to digestion of the antibody. When targeting low abundant protein biomarkers, high extraction yield is needed in order to reach sufficient detection and quantitation limits. Thus, alternative digestion procedures would be beneficial to preserve the antibodies performance.
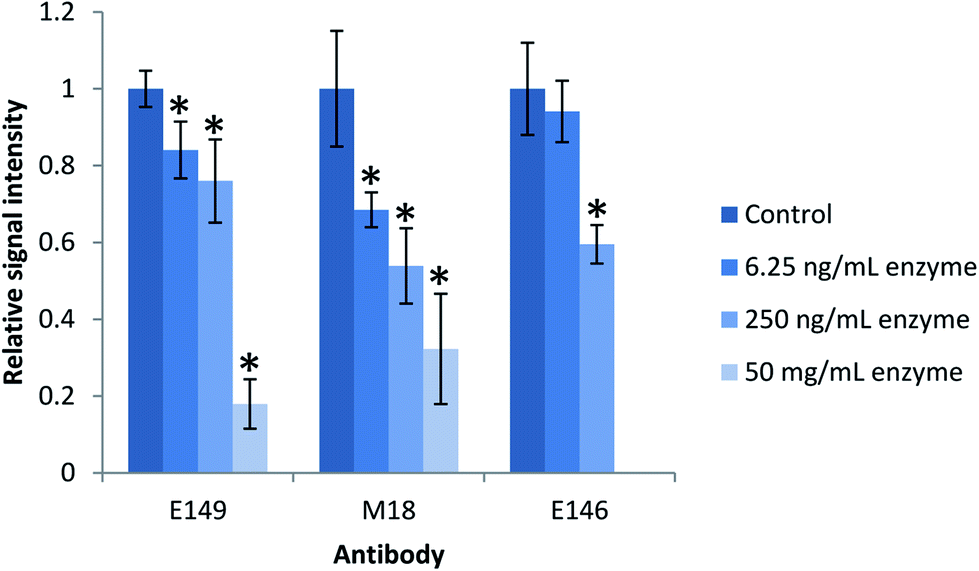 | ||
| Fig. 2 Effect of protease on antibody performance. The antibodies were incubated with active enzyme in an enzyme-to-antibody ratio of 1:1280 (6.25 ng), 1:32 (250 ng) and 6250:1 (50 mg) for 2 h prior to extraction of intact ProGRP (250 ng mL−1, n = 3). mAbs E149 and M18 were incubated with trypsin while mAb E146 was incubated with Lys-C. Antibody performance was evaluated by immunocapture of intact ProGRP followed by digestion on-beads overnight and subsequent LC-MS/MS analysis. Error bars represents the standard deviation. *Significantly different from the control (P-value < 0.05). | ||
In the research presented here we circumvent the interference of residual proteolytic enzyme activity during extraction by using trypsin immobilized on beads. This allowed us to remove trypsin from the sample prior to extraction.29
Proof-of-concept and evaluation of peptide capture from complex samples
A proof-of-concept study was performed for all three mAbs (E146/E149/M18) after immunocapture of an in-solution digest of ProGRP. These experiments are described in detail in the ESI Section† “Evaluation of anti-protein mAbs for peptide extraction”. Although all three antibodies were capable of extracting their proteotypic epitope peptide, M18 was superior with an extraction yield of 95%.Subsequent to this, peptide capture was evaluated from complex samples. These experiments are described in detail in ESI Section† “Peptide extraction from complex samples”. All three antibodies were capable of extracting digested ProGRP from a complex sample (protein precipitated and digested serum). However as preparation of Lys-C immobilized beads were too expensive, and M18 outperformed E149 with respect to extraction efficiency, the following method optimization (described in detail in ESI Section† “Method optimization of peptide capture”) was performed using M18 coated magnetic beads only.
Method evaluation
A brief evaluation of the peptide enrichment method was performed using M18 as capturing antibody and subsequent triple quadrupole mass spectrometry determination demonstrating the linearity, limit of detection (LOD) and limit of quantitation (LOQ). Serum was spiked with ProGRP concentrations ranging from 500 pg mL−1 to 50 ng mL−1 (6 concentration levels, n = 3), and stable isotopic labeled peptide was used as internal standard (added after digestion and prior to peptide enrichment). A linear curve (1/X) with acceptable correlation value (R2 = 0.96) was produced for the proteotypic epitope peptide (ALGNQQPSWDSEDSSNFK). The % RSD values were 13.7% or lower for all six concentration levels, using internal standard correction. This is generally considered satisfactory for bioanalytical methods.30 From the signal-to-noise ratio of the lowest concentration of the curve, the LOD for the ProGRP determination was estimated to be 54 pg mL−1 (S/N = 3), and the LOQ was estimated to be 181 pg mL−1 (S/N = 10). These results show the potential for a reliable detection and quantitation of the protein biomarker based on peptide capture by anti-protein antibodies.Comparison of peptide extraction with protein extraction
To further evaluate peptide capture by anti-protein antibodies as alternative for protein quantitation, a comparison of protein and peptide extraction with M18 was performed with emphasis on extraction efficiency, general clean-up efficiency, detection and quantitation limits.The observed background was, as expected, considerably higher after protein extraction and on-beads digestion compared to the extraction of the peptide (Fig. 3). One might expect this to be due to the digestion of the antibodies; however, only 11 out of the 111 unique peptides identified originated from the antibody M18. As also shown for on-beads digestion previously,31 the major contribution to the increased background was the detection of 100 unique peptides from 19 human proteins. In comparison, only three unique peptides from serum albumin were identified in addition to the epitope peptide after peptide extraction. Similar findings were done in earlier studies of protein extraction followed by elution of intact protein prior to digestion:31 using mAb E146 for intact protein extraction 115 unique peptides from 32 human proteins were detected after on-beads digestion while elution followed by subsequent digestion resulted in identification of 29 unique peptides from five human proteins. The observed difference may be due to the assumption that smaller proteolytic peptides to a lesser degree bind non-specific to the antibody and/or the magnetic beads compared to intact proteins. Nevertheless, these results demonstrate that peptide extraction provides the highest degree of sample clean-up. Cleaner extracts are expected to provide less matrix effects, which further will improve the sensitivity. Peptide extraction may thus be an advantage in multiplexed assays where several biomarkers are determined at once.
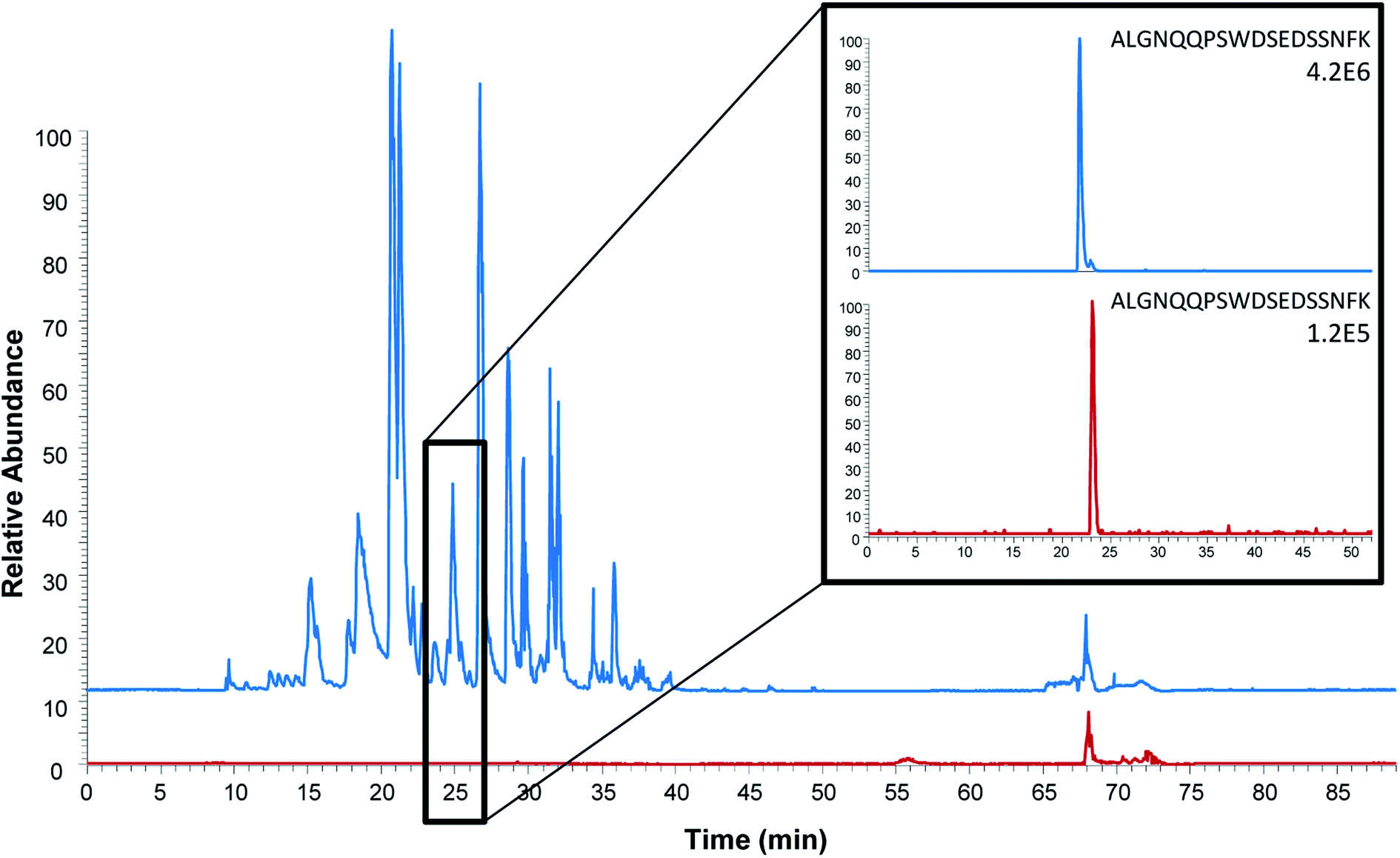 | ||
| Fig. 3 Comparison of base peak chromatograms (full scan Orbitrap analysis) after intact protein extraction (blue) and proteotypic epitope peptide extraction (red). Extracted ion chromatograms of the proteotypic epitope peptide (ALGNQQPSWDSEDSSNFK, m/z 1005.45) are shown on the right. Serum spiked with 150 ng mL−1 ProGRP was used as sample. | ||
Another method for determination of ProGRP in serum using immunocapture of intact protein, digestion and LC-MS/MS has previously been described.14 This method is also capable of determining ProGRP at its reference level in serum (estimated LOD and LOQ of 48 pg mL−1 and 162 pg mL−1). The obtained LOD and LOQ of the peptide capture method is hence comparable to this while the LOD and LOQ of protein capture performed as described above is not (approximately 10-fold higher LOD and LOQ). However, the methods are not directly comparable due to several differences. First, the antibody used for protein capture differs, mAb E146 is used in the previously published method while mAb M18 is used in the work described above. Secondly, two different signature peptides are monitored, NLLGLIEAK in the previously published method vs. ALGNQQPSWDSEDSSNFK in the present method, and it is previously seen that the signal intensity of NLLGLIEAK is higher than for ALGNQQPSWDSEDSSNFK after a digest (i.e. better ionization or better digestion outcome).12 Thirdly, the sample volume differs, 1000 μL in previously published method vs. 50 μL in the present method. Finally, two different LC-MS/MS systems are used the previously published methods employs an older triple quadrupole (TSQ Quantum Access, Thermo Scientific) coupled to a micro-LC while in the present paper a newer triple quadrupole (TSQ Quantiva) coupled to a nano-LC is used.
Analysis of patient samples
Even though the purpose of this study was to evaluate the applicability of anti-protein mAbs for immunocapture of proteotypic epitope peptides and not to develop a new assay for the determination of ProGRP, it was important to demonstrate the applicability of peptide extraction in real samples. Serum samples from both healthy individuals and five patients diagnosed with SCLC (in the range 0.45 ng mL−1 to 2.6 ng mL−1) were thus analyzed using the optimized workflow. The proteotypic epitope peptide was detected in serum from both healthy individuals (Fig. 4B) and patients suffering from SCLC (Fig. 4C), demonstrating that the approach can be used for detection of a wide range of ProGRP levels. Compared to the routinely used immunoassays (TR-IFMA), the ProGRP levels estimated using the peptide capture and LC-MS/MS method was approximately 10 times higher. There may be several reasons for this difference; however, due to the small number of samples no real method comparison can be performed. Nevertheless, these results demonstrated that peptide capture by anti-protein antibodies was successful also in real samples from patients suffering from SCLC.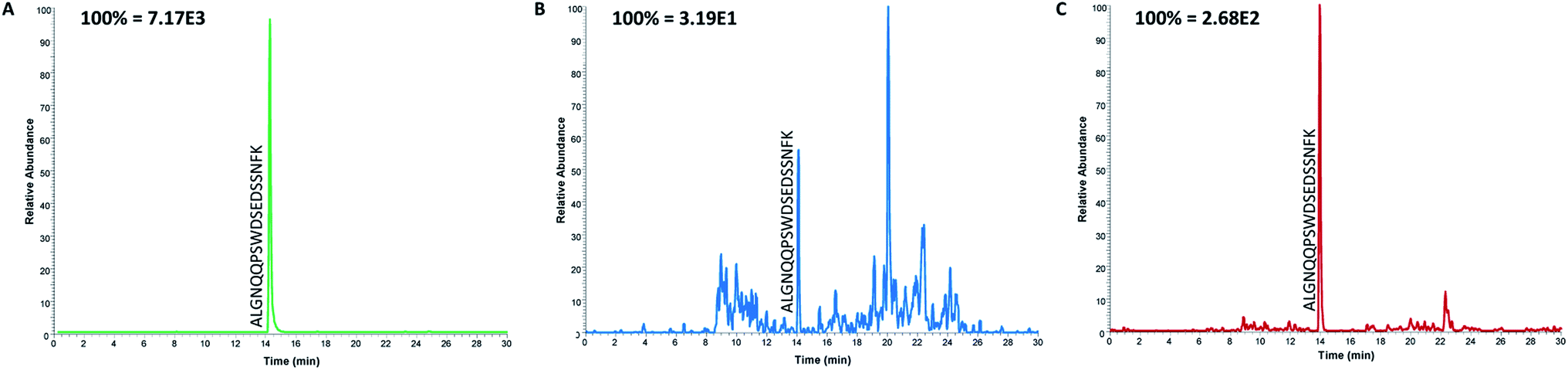 | ||
| Fig. 4 Extracted ion chromatograms of the proteotypic epitope peptide after the analysis of serum samples from a spiked standard containing 100 ng mL−1 ProGRP (A), a healthy individual (B) and from a patient with SCLC (C). | ||
Conclusions
In this study we have demonstrated that peptide capture of proteotypic epitope peptides by anti-protein antibodies after protein digestion of target protein is a promising workflow for targeted biomarker determination.Compared to a previously validated LC-MS/MS method for the immunocapture of intact protein the results demonstrated that peptide extraction provided cleaner extracts, similar sensitivity and hence most likely less matrix effects compared to the extraction of intact protein. These qualities will make peptide extraction an attractive method for multiplexed assays where several biomarkers are determined at once. The developed peptide capture method targeting the proteotypic epitope peptide of the SCLC marker ProGRP was successfully applied to the analysis of the five patient samples, and proved that the assay was able to detect different levels of endogenous ProGRP.
Based on these results, it is expected that the approach successfully can be applied to other protein biomarkers. The huge variety of and easy access to commercially available anti-protein antibodies allows for a wide application area. Peptide extraction using monoclonal anti-protein antibodies targeting linear epitopes has great potential in targeted proteomic approaches as it might facilitate the enrichment of proteotypic (or signature) peptides without the need for time-consuming and expensive development of anti-peptide antibodies, independently from batch-to-batch variations.
Data availability statement
The data sets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.Author contributions
The manuscript was written through contributions of all authors. All authors (Maren C. S. Levernæs, Bassem Farhat, Inger Oulie, Sazan S. Abdullah, Elisabeth Paus, Léon Reubsaet, and Trine G. Halvorsen) have given approval to the final version of the manuscript.Conflicts of interest
The authors declare that there are no conflicts to declare.Acknowledgements
This work has been performed within DIATECH@UiO, a strategic research initiative at the Faculty of Mathematics and Natural Sciences, University of Oslo, Oslo, Norway.References
- B. L. Ackermann and M. J. Berna, Coupling Immunoaffinity Techniques with MS for Quantitative Analysis of Low-Abundance Protein Biomarkers, Expert Rev. Proteomics, 2007, 4, 175–186 CrossRef CAS PubMed.
- E. N. Fung, P. Bryan and A. Kozhich, Techniques for Quantitative LC-MS/MS Analysis of Protein Therapeutics: Advances in Enzyme Digestion and Immunocapture, Bioanalysis, 2016, 8, 847–856 CrossRef CAS PubMed.
- J. R. Whiteaker, L. Zhao, S. E. Abbatiello, M. Burgess, E. Kuhn, C. Lin, M. E. Pope, M. Razavi, N. L. Anderson and T. W. Pearson, Evaluation of Large Scale Quantitative Proteomic Assay Development Using Peptide Affinity-Based Mass Spectrometry, Mol. Cell. Proteomics, 2011, 10, M110.005645 CrossRef PubMed.
- J. Dittrich, S. Becker, M. Hecht and U. Ceglarek, Sample Preparation Strategies for Targeted Proteomics via Proteotypic Peptides in Human Blood using Liquid Chromatography Tandem Mass Spectrometry, Proteomics: Clin. Appl., 2015, 9, 5–16 CAS.
- T. Boström, J. O. Takanen and S. Hober, Antibodies as Means for Selective Mass Spectrometry, J. Chromatogr. B, 2016, 1021, 3–13 CrossRef PubMed.
- A. G. Madian, N. S. Rochelle and F. E. Regnier, Mass-Linked Immuno-Selective Assays in Targeted Proteomics, Anal. Chem., 2012, 85, 737–748 CrossRef PubMed.
- M. H. V. Van Regenmortel, What Is a B-Cell Epitope?, in Epitope Mapping Protocols, ed. M. Schutkowski, and U. Reineke, Humana Press, Totowa, NJ, 2nd edn, 2009, DOI:10.1007/978-1-59745-450-6_1.
- R. M. Schoenherr, L. Zhao, R. G. Ivey, U. J. Voytovich, J. Kennedy, P. Yan, C. Lin, J. R. Whiteaker and A. G. Paulovich, Commercially Available Antibodies can be Applied in Quantitative Multiplexed Peptide Immunoaffinity Enrichment Targeted Mass Spectrometry Assays, Proteomics, 2016, 16, 2141–2145 CrossRef CAS PubMed.
- T. G. Halvorsen and L. Reubsaet, Antibody Based Affinity Capture LC-MS/MS in Quantitative Determination of Proteins in Biological Matrices, TrAC, Trends Anal. Chem., 2017, 95, 132–139 CrossRef CAS.
- R. M. Schoenherr, R. G. Saul, J. R. Whiteaker, P. Yan, G. R. Whiteley and A. G. Paulovich, Anti-Peptide Monoclonal Antibodies Generated for Immuno-Multiple Reaction Monitoring-Mass Spectrometry Assays have a High Probability of Supporting Western Blot and ELISA, Mol. Cell. Proteomics, 2015, 14, 382–398 CrossRef CAS PubMed.
- C. Zhao, B. Trudeau, H. Xie, J. Prostko, J. Fishpaugh and C. Ramsay, Epitope Mapping and Targeted Quantitation of the Cardiac Biomarker Troponin by SID-MRM Mass Spectrometry, Proteomics, 2014, 14, 1311–1321 CrossRef CAS PubMed.
- M. C. S. Levernæs, O. K. Brandtzaeg, S. F. Amundsen, L. Reubsaet, E. Lundanes, T. G. Halvorsen and S. R. Wilson, Selective Fishing for Peptides with Antibody-Immobilized Acrylate Monoliths, Coupled Online with NanoLC-MS, Anal. Chem., 2018, 90, 13860–13866 CrossRef PubMed.
- K. F. Opuni, M. Al-Majdoub, Y. Yefremova, R. F. El-Kased, C. Koy and M. O. Glocker, Mass Spectrometric Epitope Mapping, Mass Spectrom. Rev., 2018, 229–241 CrossRef CAS PubMed.
- S. B. Torsetnes, M. S. Nordlund, E. Paus, T. G. Halvorsen and L. Reubsaet, Digging Deeper into the Field of the Small Cell Lung Cancer Tumor Marker ProGRP: A Method for Differentiation of its Isoforms, J. Proteome Res., 2012, 12, 412–420 CrossRef PubMed.
- S. B. Torsetnes, M. S. Levernæs, M. N. Broughton, E. Paus, T. G. Halvorsen and L. Reubsaet, Multiplexing Determination of Small Cell Lung Cancer Biomarkers and their Isovariants in Serum by Immunocapture LC-MS/MS, Anal. Chem., 2014, 86, 6983–6992 CrossRef CAS PubMed.
- M. S. Nordlund, C. Fermer, O. Nilsson, D. J. Warren and E. Paus, Production and Characterization of Monoclonal Antibodies for Immunoassay of the Lung Cancer Marker ProGRP, Tumor Biol., 2007, 28, 100–110 CrossRef CAS PubMed.
- J. R. Freije, P. P. Mulder, W. Werkman, L. Rieux, H. A. Niederlander, E. Verpoorte and R. Bischoff, Chemically Modified, Immobilized Trypsin Reactor with Improved Digestion Efficiency, J. Proteome Res., 2005, 4, 1805–1813 CrossRef CAS PubMed.
- J. D. Conradie, M. Govender and L. Visser, Elisa Solid Phase: Partial Denaturation of Coating Antibody Yields a More Efficient Solid Phase, J. Immunol. Methods, 1983, 59, 289–299 CrossRef CAS PubMed.
- C. Rossetti, M. A. Świtnicka-Plak, T. Grønhaug Halvorsen, P. A. G. Cormack, B. Sellergren and L. Reubsaet, Automated Protein Biomarker Analysis: On-Line Extraction of Clinical Samples by Molecularly Imprinted Polymers, Sci. Rep., 2017, 7, 44298 CrossRef CAS PubMed.
- K. O. Svendsen, H. R. Larsen, S. A. Pedersen, I. Brenna, E. Lundanes and S. R. Wilson, Automatic Filtration and Filter Flush for Robust Online Solid-Phase Extraction Liquid Chromatography, J. Sep. Sci., 2011, 34, 3020–3022 CrossRef CAS PubMed.
- M. S. Nordlund, D. J. Warren, J. K. Laerdahl and E. Paus, Studies on Multiple Forms of Progrp in Serum from Small Cell Lung Cancer Patients, Tumor Biol., 2009, 30, 265–275 CrossRef CAS PubMed.
- M. S. Nordlund, D. J. Warren, K. Nustad, J. Bjerner and E. Paus, Automated Time-Resolved Immunofluorometric Assay for Progastrin-Releasing Peptide, Clin. Chem., 2008, 54, 919–922 CrossRef CAS PubMed.
- M. Razavi, L. E. Frick, W. A. LaMarr, M. E. Pope, C. A. Miller, N. L. Anderson and T. W. Pearson, High-Throughput SISCAPA Quantitation of Peptides from Human Plasma Digests by Ultrafast, Liquid Chromatography-Free Mass Spectrometry, J. Proteome Res., 2012, 11, 5642–5649 CrossRef CAS PubMed.
- N. L. Anderson, M. Razavi, T. W. Pearson, G. Kruppa, R. Paape and D. Suckau, Precision of Heavy–Light Peptide Ratios Measured by MALDI-TOF Mass Spectrometry, J. Proteome Res., 2012, 11, 1868–1878 CrossRef CAS PubMed.
- N. L. Anderson, N. G. Anderson, L. R. Haines, D. B. Hardie, R. W. Olafson and T. W. Pearson, Mass Spectrometric Quantitation of Peptides and Proteins Using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA), J. Proteome Res., 2004, 3, 235–244 CrossRef CAS PubMed.
- J. R. Whiteaker, L. Zhao, L. Anderson and A. G. Paulovich, An Automated and Multiplexed Method for High Throughput Peptide Immunoaffinity Enrichment and Multiple Reaction Monitoring Mass Spectrometry-Based Quantification of Protein Biomarkers, Mol. Cell. Proteomics, 2010, 9, 184–196 CrossRef CAS PubMed.
- Q. Xu, M. Zhu, T. Yang, F. Xu, Y. Liu and Y. Chen, Quantitative Assessment of Human Serum Transferrin Receptor in Breast Cancer Patients Pre-and Post-Chemotherapy Using Peptide Immunoaffinity Enrichment Coupled with Targeted Proteomics, Clin. Chim. Acta, 2015, 448, 118–123 CrossRef CAS PubMed.
- M. Razavi, N. L. Anderson, M. E. Pope, R. Yip and T. W. Pearson, High Precision Quantification of Human Plasma Proteins Using the Automated SISCAPA Immuno-MS Workflow, New Biotechnol., 2016, 33, 494–502 CrossRef CAS PubMed.
- L. Switzar, M. Giera and W. M. Niessen, Protein Digestion: An Overview of the Available Techniques and Recent Developments, J. Proteome Res., 2013, 12, 1067–1077 CrossRef CAS PubMed.
- Food and Drug Administration, Center for Drug Evaluation and Research/Center for Veterinary Medicine, Bioanalytical Method Validation - Guidance for Industry, U.S. Department of Health and Human Services, 2018 Search PubMed.
- M. C. S. Levernæs, M. N. Broughton, L. Reubsaet and T. G. Halvorsen, To Elute or Not to Elute in Immunocapture Bottom-up LC–MS, J. Chromatogr. B, 2017, 1055–1056, 51–60 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05071j |
| This journal is © The Royal Society of Chemistry 2019 |