
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ramified derivatives of 5-(perylen-3-ylethynyl)uracil-1-acetic acid and their antiviral properties†
Ksenia A. Sapozhnikovaa,
Nikita A. Slesarchuk ab,
Alexey A. Orlovbcd,
Evgeny V. Khvatovac,
Eugene V. Radchenkob,
Alexey A. Chistova,
Alexey V. Ustinovaef,
Vladimir A. Palyulinb,
Liubov I. Kozlovskayacd,
Dmitry I. Osolodkin*bcd,
Vladimir A. Korshun*af and
Vladimir A. Brylevae
ab,
Alexey A. Orlovbcd,
Evgeny V. Khvatovac,
Eugene V. Radchenkob,
Alexey A. Chistova,
Alexey V. Ustinovaef,
Vladimir A. Palyulinb,
Liubov I. Kozlovskayacd,
Dmitry I. Osolodkin*bcd,
Vladimir A. Korshun*af and
Vladimir A. Brylevae
aShemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Miklukho-Maklaya 16/10, Moscow 117997, Russia. E-mail: v-korshun@yandex.ru
bDepartment of Chemistry, Lomonosov Moscow State University, Moscow 119991, Russia
cFSBSI "Chumakov FSC R&D IBP RAS", Poselenie Moskovsky, Moscow 108819, Russia
dSechenov First Moscow State Medical University, Moscow 119991, Russia. E-mail: dmitry_o@qsar.chem.msu.ru
eBiotech Innovations Ltd, Leninskie Gory 1 bd 75, Moscow 119992, Russia
fDepartment of Biology and Biotechnology, National Research University Higher School of Economics, Vavilova 7, Moscow 117312, Russia
First published on 20th August 2019
Abstract
The propargylamide of N3-Pom-protected 5-(perylen-3-ylethynyl)uracil acetic acid, a universal precursor, was used in a CuAAC click reaction for the synthesis of several derivatives, including three ramified molecules with high activities against tick-borne encephalitis virus (TBEV). Pentaerythritol-based polyazides were used for the assembly of molecules containing 2⋯4 antiviral 5-(perylen-3-ylethynyl)uracil scaffolds, the first examples of polyvalent perylene antivirals. Cluster compounds showed enhanced absorbance, however, their fluorescence was reduced due to self-quenching. Due to the solubility issues, Pom group removal succeeded only for compounds with one peryleneethynyluracil unit. Four compounds, including one ramified cluster 9f, showed remarkable 1⋯3 nM EC50 values against TBEV in cell culture.
Introduction
5-(Perylen-3-ylethynyl)-deoxy-uridine dUY11 (Fig. 1), prepared initially as a fluorescent nucleoside,1 had unexpectedly shown remarkably broad spectrum antiviral properties.2 Nucleoside derivative dUY11 and its arabino analog aUY11 effectively suppressed reproduction of a number of enveloped viruses in cell cultures, thus constituting a new class of antivirals with a non-nucleoside mechanism of action.2,3 The presumable target for these compounds is the lipid membrane bilayer of the virion envelope. Compounds dUY11 and aUY11 are rigid amphipathic fusion inhibitors (RAFIs)2c that target the viral envelope either by a shape-determined biophysical mechanism2c,3,4 or by photosensitization.5 Both shape-determined and photophysical properties of the conjugated aryl part of the molecule are probably crucial for the antiviral properties, and the carbohydrate part seems to be less important. Very recently,6 we prepared 5-(perylen-3-ylethynyl)uracil-1-acetic acid (cm1UY11, Fig. 1) with a carboxymethyl group replacing the pentose moiety. The acid showed a pronounced antiviral activity against tick-borne encephalitis virus (TBEV),6 herpes simplex virus type 1 (HSV-1),7 and African swine fewer virus (ASFV).8 Moreover, its 3-Pom-modified precursor cm1pUY11 (Fig. 1) and amides were also active against TBEV6 and HSV-1.7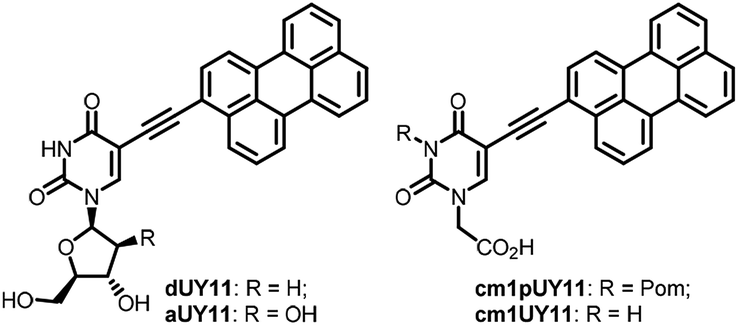 | ||
| Fig. 1 Structures of broad spectrum antiviral peryleneethynyluracil compounds. | ||
Thus, the 5-(perylen-3-ylethynyl)uracil scaffold seems to be responsible for the antiviral activity of perylene RAFIs. Lipophilic perylene residue is expected to reside in the lipid bilayer upon binding between a RAFI molecule and an enveloped virion. Therefore, one can expect cooperative enhancement of lipid membrane anchoring for compounds containing 2⋯4 perylene residues, or other effects of clustering perylenes.
The concept of clusterized/multivalent (or dendrimeric) antivirals has been developed for decades,9 both for specific and non-specific compounds and functional groups. Recent promising examples include polyanions,10 polycations,11 as well as dendrimers carrying amino acids,12 peptides,13 phenols,14 terpenes,15 mono-,16 oligosaccharides,17 and neuraminidase inhibitors.18 The approach, however, has been never applied to RAFIs.
To study the possible effects of combining several perylene cores in a single molecule on the antiviral activity, we synthesized small clusters of 5-(perylen-3-ylethynyl)-1-(carboxymethyl)uracil, quantified their fluorescence properties, and measured the efficiency of TBEV reproduction inhibition by these clusters in PEK cell culture.
Results and discussion
Synthesis of compounds
Recently, we used the azide–alkyne click reaction for the synthesis of dUY11 derivatives with enhanced activity.19 Here we report the application of Huisgen–Meldal–Sharpless reaction (Cu(I)-catalyzed alkyne–azide cycloaddition, CuAAC)20 for the synthesis of 5-(perylen-3-ylethynyl)-1-(carboxymethyl)uracil derivatives. First, we prepared a set of branched azides 2–5 (Scheme 1). The starting tetraol 1 was mesylated with a controlled excess of mesyl chloride (MsCl) in DCM in the presence of triethylamine as a base. Tuning the mesyl chloride/hydroxyl ratio leads to the desired distribution of products in the reaction. Earlier, we prepared azides 4 and 5 using 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 molar ratio of 1 to mesyl chloride21 and used them for the assembly of complex oligonucleotide conjugates.21,22 Here we report the synthesis of azides 2 and 3 as main products. Mesylation with 1:1.2 molar ratio (1: MsCl), and subsequent nucleophilic substitution with sodium azide affords compounds 2 and 3 in 32% and 27% yield, respectively, together with some amount of triazide 4.
:3 molar ratio of 1 to mesyl chloride21 and used them for the assembly of complex oligonucleotide conjugates.21,22 Here we report the synthesis of azides 2 and 3 as main products. Mesylation with 1:1.2 molar ratio (1: MsCl), and subsequent nucleophilic substitution with sodium azide affords compounds 2 and 3 in 32% and 27% yield, respectively, together with some amount of triazide 4.
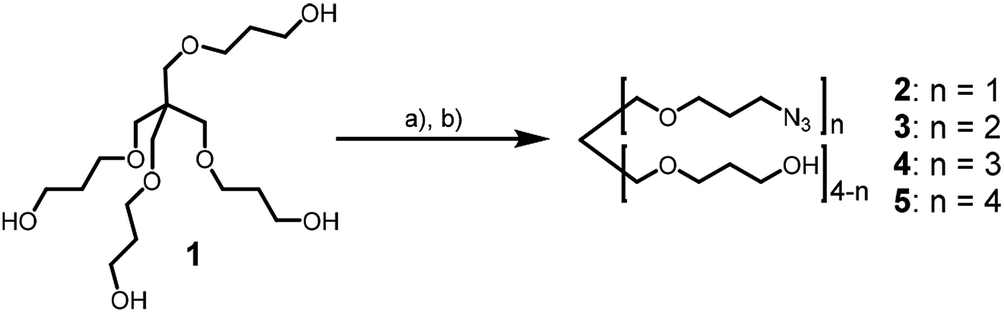 | ||
| Scheme 1 Synthesis of azides. Conditions: (a) methanesulfonyl chloride, NEt3, DCM; (b) NaN3, DMSO, rt, 12 h, 32% (2), 27% (3), 8% (4). | ||
We used pivaloyloxymethyl (Pom) protection for N3 position of the nucleobase for solubility reasons. 3-Pom-5-(perylen-3-ylethynyl)uracil acetic acid 6 was prepared from 5-iodouracil in several steps.6 It was coupled with propargylamine to yield alkyne precursor 7, which was conjugated with benzyl azide, azidoethanol, and pentaerythritol-based azides 2–5 (Scheme 2) via CuAAC to afford compounds 9.
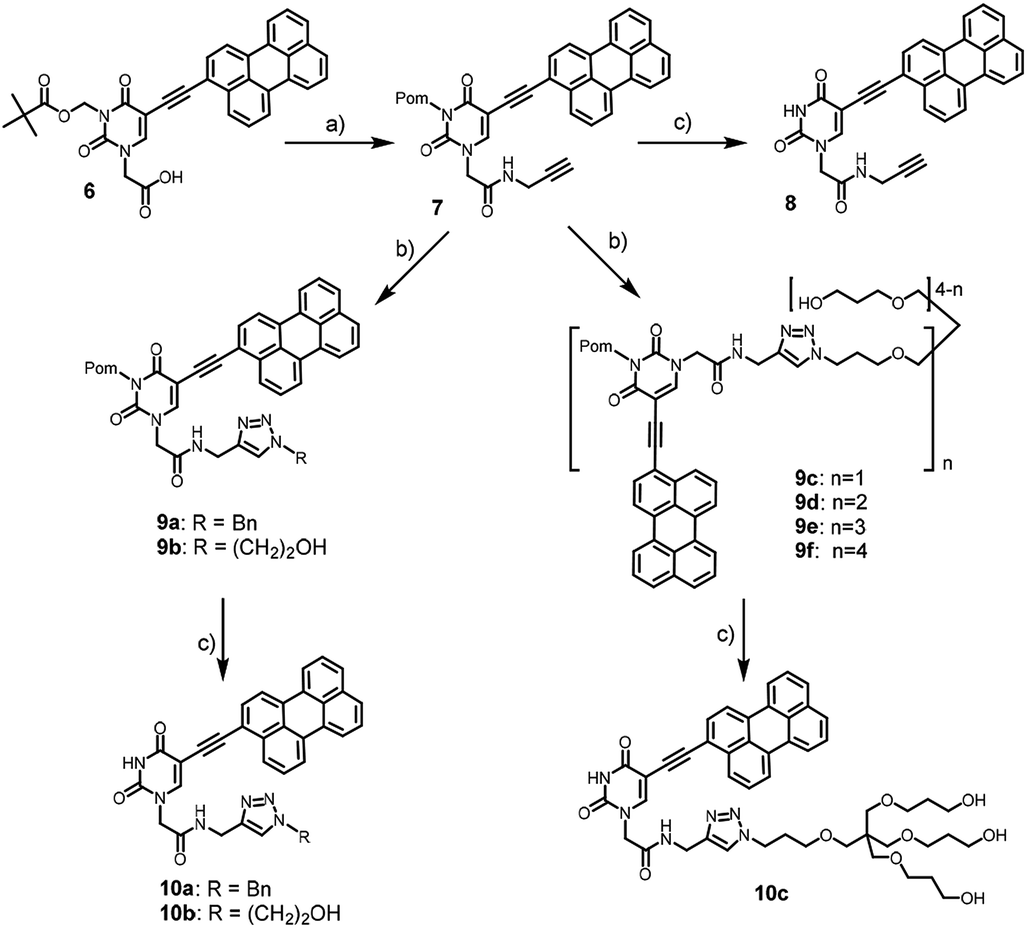 | ||
| Scheme 2 Synthesis of the model and target compounds. Conditions: (a) propargylamine, PyBOP, DIPEA, DMF, 0 °C, 55%; (b) CuSO4·5H2O, TBTA, aq. ascorbic acid, DMSO, rt, 12 h, azide (benzylazide, azidoethanol, or 2–5), 75% (9a); 67% (9b); 81% (9c), 70% (9d), 54% (9e), 38% (9f); (c) NaOH, DMSO, MeOH, rt, 30 min, 35% (10a), 40% (10b), 30% (10c). | ||
The alkaline removal of pivaloyloxymethyl (Pom) protection from compounds 7 and 9a–c yielded the corresponding amides 8, 10a–c (Scheme 2). However, the usual Pom group deprotection protocol with sodium methoxide or ammonia in methanol23 was inappropriate because both the starting compounds and the desired products were insoluble in alcohol. Therefore, we modified the deprotection protocol by choosing NaOH as the base, and DMSO/MeOH/H2O mixture as the reaction medium. As a result, full conversion of the starting material occurred in 30 min (TLC control).
While the conversions of compounds 9a–c into 10a–c by TLC were excellent, their isolated yields in deprotection steps (Scheme 2) turned out to be poor because of the losses during the purification using column chromatography. The products showed low solubility and chromatographic isolations were carried out in the overloading mode. Thus, a number of fractions containing the products were overlapping with some minor impurities, and they were excluded to obtain high quality samples for antiviral activity studies.
We failed to prepare and isolate Pom-deprotected compounds from 9d–f because of the incomplete deprotection, and low solubility of the desired products.
Spectral studies
Compounds 9d–f contain 2⋯4 chromophoric units linked with flexible spacers. To obtain some insight into the structure of the compounds in a solution and to reveal a possible influence of spectral properties on antiviral activity,2d we compared their spectra to each other, and to those of monomeric compounds 9c and 10c. UV-Vis spectra (Fig. 2) showed no significant difference between Pom-protected compound 9c and the deprotected derivative 10c; the shape of the spectrum, and the positions of maxima were always identical.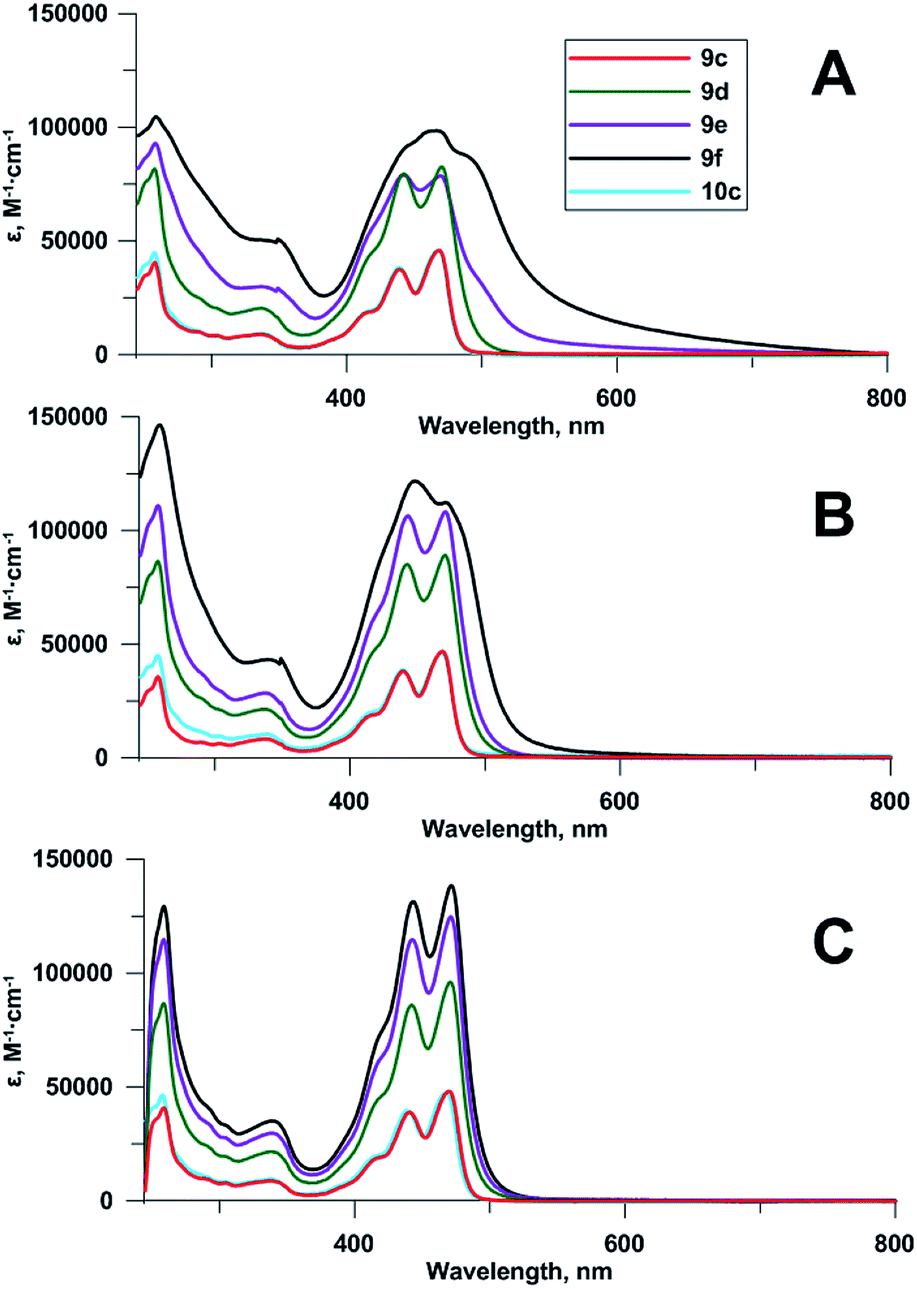 | ||
| Fig. 2 Absorbance spectra of compounds 9c–f, 10c at 5 μM concentrations: (A) 0.2% DMSO in 96% EtOH, (B) 2% DMSO in 96% EtOH, (C) 20% DMSO in 96% EtOH. | ||
The increase of the number of chromophoric units led to the corresponding increase of the molar attenuation coefficient in true solution (20% DMSO in 96% ethanol as a solvent). Upon the reduction of DMSO concentration to 2% and further to 0.2%, the bands in absorbance spectra of tetramer 9f, and then of trimer 9e became broader and less structured (Fig. 2). The effect, confirming the aggregation of chromophoric residues, was observed earlier for other perylene compounds.19,24
Fluorescence spectra of compounds 9c–f and 10c were recorded at 0.1 μM concentrations in 96% EtOH (Fig. 3). Due to self-quenching, fluorescence intensity decreases with the increase in the number of fluorophores comprising the 5-(perylen-3-ylethynyl)uracil groups. There were no drastic changes in fluorescence spectra for compounds 9c–f and 10c, except for relative enhancement of 550 nm band, and proximal long wavelength emission for polychromophore molecules 9d–f, suggesting weak excimer fluorescence from perylene residues located close to each other.
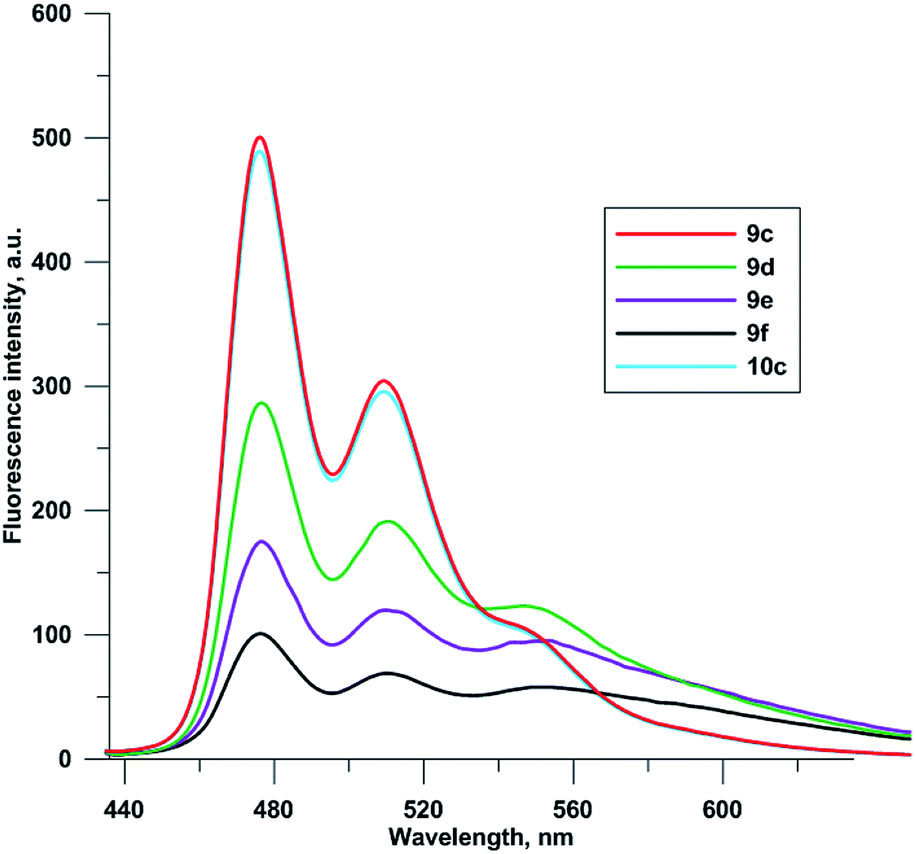 | ||
| Fig. 3 Fluorescence spectra of 9c–f, 10c (0.1 μM, 96% EtOH, λex 420 nm). | ||
Antiviral properties
All prepared compounds, containing one (7–9b, 10a–c) or several (9d–f) perylene residues, were tested for cytotoxicity and for ability to inhibit TBEV (strain Absettarov) reproduction in PEK cell culture as measured by plaque reduction assay (Table 1). All experimental conditions were the same as in our previous studies.2d,6,19,25| # | EC50, μM | CC50, μM | |
|---|---|---|---|
| 24 h | 7 d | ||
| a Data for compound 6 are from ref. 6.b Morphological changes of cell membranes were observed. | |||
| 6 | 0.00121 ± 0.00025a | >50 | >50 |
| 7 | 0.00097 ± 0.00015 | >50 | >50 |
| 8 | 0.002 ± 0.001 | >50 | <50 |
| 9a | 0.046 ± 0.017 | >50 | >50 |
| 9b | 0.0010 ± 0.0003 | >50 | >50 |
| 9c | 0.008 ± 0.001 | >50b | 18 |
| 9d | 0.024 ± 0.007 | >50b | <50 |
| 9e | 0.015 ± 0.001 | >50b | <50 |
| 9f | 0.0033 ± 0.0011 | >50 | >50 |
| 10a | 0.0081 ± 0.0017 | >50b | >50b |
| 10b | 0.066 ± 0.013 | >50 | >50 |
| 10c | 0.10 ± 0.05 | >50 | 37 ± 13 |
All the compounds showed little to no cytotoxicity on PEK cells and selectivity indices (SI = CC50/EC50) up to 50000. The alkyne precursor 7 appeared to be the most potent compound in the series, showing nanomolar EC50. Its unprotected analogue 8 and acid precursor 6 showed slightly lower activity, thus revealing low influence of small substituents in the uracil moiety, similarly to one of the previous studies,25a but contrary to another.6 Although cluster compounds 9d–f inhibited TBEV reproduction in one or two digit nanomolar concentrations, their single-perylene analogues were more potent. Remarkably, Pom deprotection of branched compound 9c, affording 10c, led to a considerable activity decrease.
Molecular modeling
While the abbreviation RAFI stands for Rigid Amphipathic Fusion Inhibitor, the compounds synthesized here contain a large number of rotatable bonds. Thus, their antiviral activity and membrane interactions may be largely defined by their conformational space. To obtain some insight into this conformational space, we performed a simple conformational analysis for molecular models of the compounds, seeking the global minima of the compound energies using the Confort method26 implemented in SYBYL-X 2.1.27 The optimized structures are shown in Fig. 4, along with the molecular surfaces colored by the hydrophobic potential.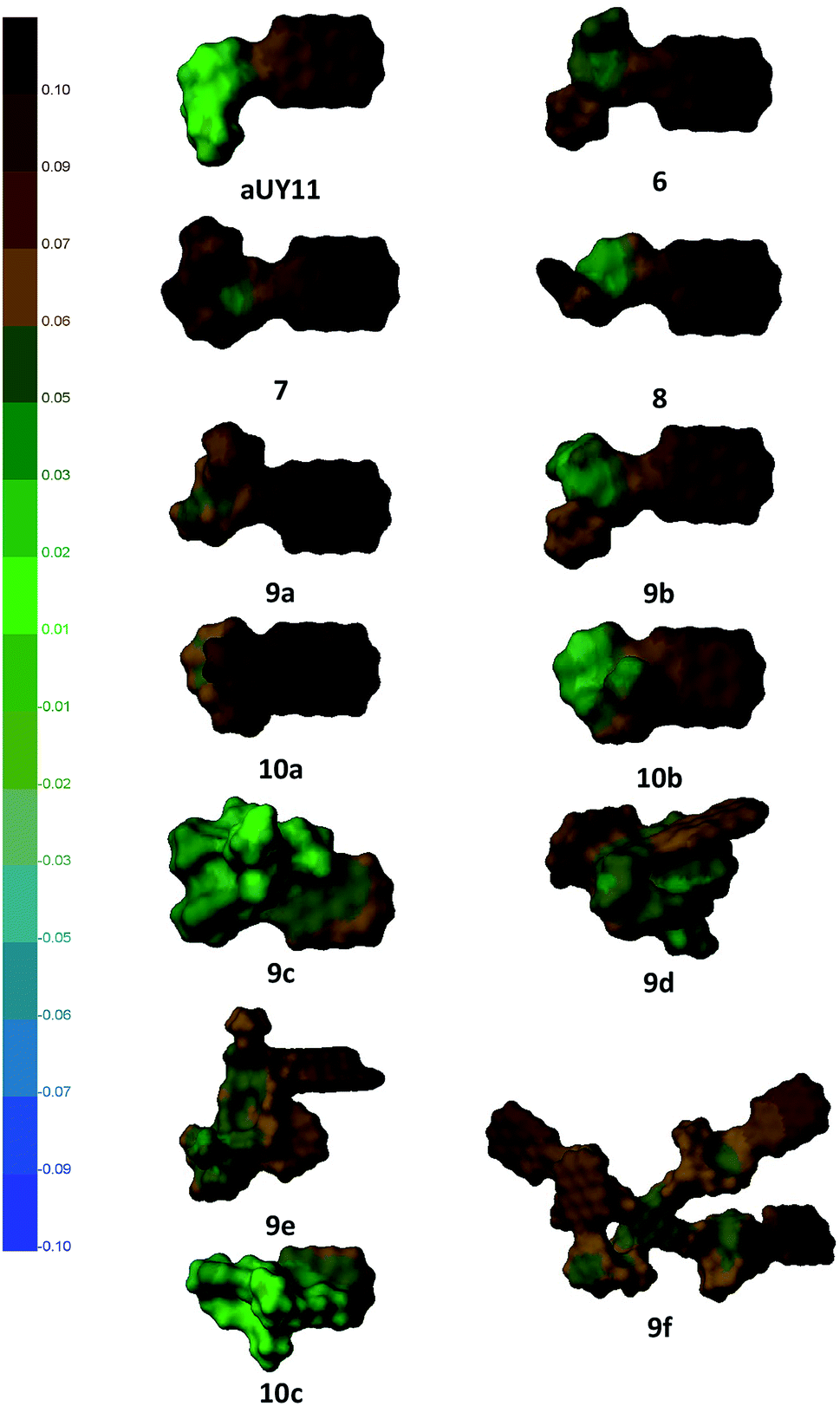 | ||
| Fig. 4 Structures of compounds optimized using SYBYL-X 2.1 software; molecular surfaces are colored by the hydrophobic potential (left scale). | ||
As the most important part of the RAFI molecules is the perylene moiety,2d,25a its exposure and ability to interact with the viral membrane should play a crucial role in the potency of the compounds. It can be seen that compounds 6–8, as well as aUY11, do not show substantial flexibility, and the perylene moiety of these compounds is completely exposed. Molecules 9 and 10, bearing longer and more flexible moieties, show less uniform antiviral activity, which is more strongly affected by possible conformational behavior. For example, the influence of Pom is opposite in the pairs 9a–10a and 9b–10b; in the former, the benzyl moiety may interact with perylene, whereas in the latter the hydroxyethyl group should prefer a more hydrophilic environment.
Using coloring by the hydrophobic potential, one can see the amphipathic nature of perylenylethynyluracil compounds. Perylenylethynyluracil units are probably responsible for lipid bilayer anchoring and for the general antiviral effect. The polar part of the molecules seems to have a modulating (within two orders of magnitude)6 effect on the inhibition of a viral replication. Remarkably, in the most active compounds, the polar part of the molecule has a small hydrophobic unit, Pom or benzyl, but not both together. Although neither the target, nor the mechanism of antiviral action of RAFIs was directly proven to date, this simple observation can be used in a future design of antivirals.
Experimental
General methods
Reagents and solvents were from commercial suppliers and used as received, except DMF, DMSO, CH2Cl2, CHCl3, EtOH and methanol were used freshly distilled from CaH2. Azides 4, 5,21 tetrakis(5-hydroxy-2-oxapentyl)methane21 and 3-Pom-5-(perylen-3-ylethynyl)uracil-1-acetic acid 66 were prepared as described. 500 MHz 1H and 125.7 MHz 13C NMR spectra were recorded on Bruker AMX-400 or Bruker Avance 500 spectrometers and referenced to DMSO-d6 (2.50 ppm for 1H and 39.5 ppm for 13C) or CDCl3 (7.26 ppm for 1H and 77.16 ppm for 13C). 1H NMR coupling constants are reported in hertz (Hz) and refer to apparent multiplicities. Electrospray ionization high resolution mass spectra (ESI HRMS) of low molecular weight compounds were recorded using a Thermo Scientific Orbitrap Exactive mass spectrometer (positive or negative ion mode). UV spectra were recorded on a Varian Cary 100 spectrophotometer. Fluorescence spectra were recorded using a PerkinElmer LS 55 fluorescence spectrometer. Analytical thin layer chromatography was performed on Kieselgel 60 F254 precoated aluminum plates (Merck). Silica gel column chromatography was performed using Merck Kieselgel 60 0.040–0.063 mm.Cell culture and viruses
Porcine embryo kidney (PEK) cell line was maintained at 37 °C in medium 199 (FSBSI "Chumakov FSC R&D IBP RAS", Russia) supplemented with 5% fetal bovine serum (Gibco). Tick-borne encephalitis virus strain Absettarov (GenBank access no. KU885457) was from the laboratory collection of FSBSI "Chumakov FSC R&D IBP RAS".Synthetic procedures
600), 470 (48000), λmin, nm (ε, M−1 cm−1): 452 (26800); fluorescence (96% EtOH, λem 530 nm, excitation λmax, nm): 334, 440, 467; (96% EtOH, λex 420 nm, emission λmax, nm): 476, 509. 1H NMR (500 MHz; DMSO-d6) δ 8.78 (t, 1H, J = 5.35 Hz), 8.43 (s, 1H), 8.38 (d, 1H, J = 7.55 Hz), 8.36–8.27 (m, 3H), 8.25 (d, 1H, J = 8.23 Hz), 7.82–7.77 (m, 2H), 7.68–7.62 (m, 2H), 7.53 (t, 2H, J = 7.68 Hz), 5.86 (s, 2H), 4.53 (s, 2H), 3.99–3.94 (m, 2H), 3.2–3.18 (m, 1H), 1.14 (s, 9H). 13C NMR (125 MHz; DMSO-d6) δ 176.6, 166.0, 160.5, 149.7, 149.3, 134.1, 133.7, 131.3, 131.0, 130.5, 130.1, 129.8, 128.6, 128.3, 127.8, 127.7, 127.6, 127.0, 126.9, 125.6, 121.6, 121.4, 121.2, 120.2, 119.1, 96.8, 90.9, 87.8, 80.6, 73.5, 65.1, 50.8, 38.3, 28.1, 26.7. HRMS (ESI, m/z): calcd for C37H29N3O5Na ([M + Na]+): 618.1999, found: 618.2000.Click reaction between alkyne 7 and mono-azides, a general procedure
Azide (0.30 mmol) and propargyl amide 7 (0.26 mmol) were dissolved in DMSO (10 mL) under vigorous magnetic stirring. The solution was flushed with argon for degassing. 0.1 M solution of CuSO4·5H2O and TBTA ligand (1:1) in 55% DMSO was added (260 μL). Then ascorbic acid (100 mg mL−1 in H2O; 91 μL) was quickly loaded and the flask was tightly closed. After 12 h TLC showed full consumption of the starting alkyne 7. Reaction mixture was quenched with brine (50 mL) and lumpy solid precipitate was isolated by centrifugation. The crude product was washed with distilled water (2 × 10 mL). The residue was applied on the silica gel using evaporation of the acetone solution with small portion of the sorbent. The obtained powder was carefully spilled on the column filled with silica gel in CH2Cl2. The following compounds were isolated.
600), 470 (48000), λmin, nm (ε, M−1 cm−1): 452 (26800). Fluorescence (96% EtOH, λem 530 nm, excitation λmax, nm): 334, 440, 467; (96% EtOH, λex 420 nm, emission λmax, nm): 476, 509. 1H NMR (500 MHz, DMSO-d6) δ 8.79–8.73 (m, 1H), 8.42–8.38 (m, 2H), 8.38–8.24 (m, 4H), 7.99 (s, 1H), 7.83–7.78 (m, 2H), 7.7–7.63 (m, 2H), 7.54 (t, 2H, J = 7.62 Hz), 7.39–7.28 (m, 6H), 5.88 (s, 2H), 5.58 (s, 2H), 4.54 (s, 2H), 3.3–3.27 (m, 2H), 1.14 (s, 9H). 13C NMR (125 MHz, DMSO-d6) δ 176.6, 166.1, 160.5, 149.7, 149.4, 144.5, 136.0, 134.2, 133.7, 131.3, 131.0, 130.5, 130.0, 129.8, 128.7, 128.7, 128.3, 128.1, 128.0, 127.9, 127.7, 127.6, 127.0, 126.9, 125.6, 123.1, 121.6, 121.4, 121.3, 120.3, 119.1, 96.8, 90.9, 87.9, 65.1, 52.8, 51.0, 38.3, 34.4, 26.7. HRMS (ESI, m/z): calcd for C44H36N6O5Na ([M + Na]+): 751.2639, found: 751.2629.600), 470 (48000), λmin, nm (ε, M−1 cm−1): 452 (26800). Fluorescence (96% EtOH, λem 530 nm, excitation λmax, nm): 334, 440, 468; (96% EtOH, λex 420 nm, emission λmax, nm): 476, 509. 1H NMR (500 MHz, DMSO-d6) δ 8.85–8.8 (m, 1H), 8.47–8.43 (m, 2H), 8.42–8.33 (m, 3H), 8.27 (t, 1H, J = 8.23 Hz), 7.93 (s, 1H), 7.86–7.8 (m, 2H), 7.72–7.66 (m, 2H), 7.56 (t, 2H, J = 7.82 Hz), 5.86 (s, 2H), 4.54 (s, 2H), 4.41–4.36 (m, 4H), 3.78 (t, 2H, J = 4.8 Hz), 1.14 (s, 9H). 13C NMR (125 MHz, DMSO-d6) δ 176.6, 166.1, 160.5, 149.7, 149.5, 143.9, 134.2, 133.7, 131.3, 131.1, 130.5, 130.1, 129.8, 128.7, 128.4, 127.9, 127.8, 127.6, 127.0, 127.0, 125.6, 123.4, 121.7, 121.5, 121.3, 120.3, 119.2, 96.7, 90.9, 87.9, 65.1, 59.9, 52.2, 50.9, 38.3, 34.4, 26.7; HRMS (ESI, m/z): calcd for C39H34N6O6Na ([M + Na]+): 705.2432, found: 705.2422.600), 470 (48000), λmin, nm (ε, M−1 cm−1): 452 (26800). Fluorescence (96% EtOH, λem 530 nm, excitation λmax, nm): 334, 440, 468; (96% EtOH, λex 420 nm, emission λmax, nm): 476, 509. 1H NMR (400 MHz; DMSO-d6) δ 8.78 (t, 1H, J = 5.48 Hz), 8.48–8.26 (m, 6H), 7.94 (s, 1H), 7.86–7.8 (m, 2H), 7.73–7.65 (m, 2H), 7.57 (t, 2H, J = 7.87 Hz), 5.87 (s, 2H), 4.54 (s, 2H), 4.42–4.35 (m, 4H), 4.32 (t, 3H, J = 5.09 Hz), 3.48–3.41 (m, 6H), 3.39 (t, 6H, J = 6.36 Hz), 3.34 (t, 2H, J = 6.04 Hz), 3.3–3.25 (m, 8H), 2.07–1.99 (m, 2H), 1.62 (quint, 6H, J = 6.36 Hz), 1.14 (s, 9H). 13C NMR (100 MHz, DMSO-d6) δ 176.5, 166.0, 160.4, 149.7, 149.3, 144.0, 134.1, 133.7, 131.3, 131.0, 130.5, 130.0, 129.8, 128.6, 128.3, 127.8, 127.8, 127.6, 127.0, 126.9, 125.6, 122.9, 121.6, 121.4, 121.3, 120.3, 119.1, 96.7, 90.8, 87.9, 69.3, 69.1, 67.9, 67.3, 65.0, 57.9, 50.9, 46.6, 44.9, 38.3, 34.4, 32.6, 29.9, 26.6. HRMS (ESI, m/z): calcd for C54H64N6O12Na ([M + Na]+): 1011.4474, found: 1011.4458.Click reaction between alkyne 7 and polyazides 3–5, general procedure
Azide (0.025 mmol) and propargyl amide 7 with 5% excess for each azido group (0.53, 0.79 and 1.05 mmol respectively for cases 9d, 9e, 9f) were dissolved in DMSO (3 mL) under vigorous magnetic stirring. The solution was flushed with argon for degassing and 0.1 M solution of CuSO4·5H2O and TBTA ligand (1:1) in 55% DMSO were added (80 μL). Then ascorbic acid (100 mg mL−1 in H2O; 30 μL) was quickly loaded and the flask was tightly closed. After 12 h the reaction mixture was quenched with brine (6 mL) and the solid precipitated was isolated using centrifugation. The brine was decanted and the crude product was washed with distilled water (2 × 3 mL). The residue was applied on the silica gel by evaporation of the acetone solution with small portion of the sorbent. The obtained powder was carefully spilled on the column filled with silica gel in CH2Cl2. The following compounds were obtained.
000), 470 (96100), λmin, nm (ε, M−1 cm−1): 454 (66800). Fluorescence (96% EtOH, λem 530 nm, excitation λmax, nm): 336, 441, 469; (96% EtOH, λex 420 nm, emission λmax, nm): 477, 510. 1H NMR (400 MHz, DMSO-d6) δ 8.78 (t, 2H, J = 5.48 Hz), 8.43–8.22 (m, 12H), 7.93 (s, 2H), 7.83–7.77 (m, 4H), 7.7–7.62 (m, 4H), 7.58–7.5 (m, 4H), 5.85 (s, 4H), 4.54 (s, 4H), 4.41–4.31 (m, 10H), 3.48–3.3 (m, 12H), 3.3–3.25 (m, 8H), 2.07–1.98 (m, 4H), 1.66–1.58 (m, 4H), 1.12 (s, 18H). 13C NMR (100 MHz, DMSO-d6) δ 176.6, 166.1, 160.5, 149.7, 149.3, 144.1, 134.2, 133.7, 131.3, 131.0, 130.5, 130.1, 129.8, 128.7, 128.3, 127.9, 127.8, 127.6, 127.0, 126.9, 125.6, 122.9, 121.6, 121.4, 121.3, 120.3, 119.1, 96.8, 90.9, 87.8, 69.2, 69.0, 68.0, 67.4, 65.1, 57.9, 51.0, 46.7, 45.0, 38.3, 34.4, 32.6, 29.9, 26.6. HRMS (ESI, m/z): calcd for C91H93N12O16 ([M + H]+): 1609.6827, found: 1609.6813.700), 471 (124700), λmin, nm (ε, M−1 cm−1): 455 (91300). Fluorescence (96% EtOH, λem 530 nm, excitation λmax, nm): 335, 440, 468; (96% EtOH, λex 420 nm, emission λmax, nm): 477, 511. 1H NMR (400 MHz, DMSO-d6) δ 8.77 (t, 3H, J = 5.48 Hz), 8.43–8.22 (m, 18H), 7.93 (s, 3H), 7.83–7.77 (m, 6H), 7.68–7.62 (m, 6H), 7.57–7.5 (m, 6H), 5.85 (s, 6H), 4.53 (s, 6H), 4.4–4.34 (m, 12H), 4.32 (t, 1H, J = 5.17 Hz), 3.43–3.34 (m, 10H), 3.3–3.26 (m, 8H), 2.06–1.97 (m, 6H), 1.65–1.57 (m, 2H), 1.12 (s, 27H). 13C NMR (100 MHz, DMSO-d6) δ 176.5, 166.1, 160.4, 149.7, 149.3, 144.1, 134.1, 133.7, 131.3, 131.0, 130.5, 130.0, 129.7, 129.5, 128.6, 128.3, 127.8, 127.7, 127.6, 127.0, 126.9, 125.5, 122.9, 121.6, 121.3, 121.2, 120.2, 119.1, 96.8, 90.9, 87.8, 69.1, 68.0, 67.4, 65.0, 57.9, 50.9, 46.6, 44.9, 38.3, 34.4, 32.6, 29.9, 26.6. HRMS (ESI, m/z): calcd for C128H120N18O20Na ([M + Na]+): 2251.8818, found: 2251.8766.500), 471 (138300), λmin, nm (ε, M−1 cm−1): 455 (107200). Fluorescence (96% EtOH, λem 530 nm, excitation λmax, nm): 419, 440, 464; (96% EtOH, λex 420 nm, emission λmax, nm): 452, 477, 509. 1H NMR (400 MHz, DMSO-d6) δ 8.79–8.75 (m, 4H), 8.41–8.21 (m, 24H), 7.93 (s, 4H), 7.81–7.76 (m, 8H), 7.66–7.6 (m, 8H), 7.55–7.48 (m, 8H), 5.84 (s, 8H), 4.53 (s, 8H), 4.4–4.33 (m, 16H), 3.38–3.32 (m, 8H), 3.28 (s, 8H), 2.06–1.97 (m, 8H), 1.11 (s, 36H). 13C NMR (100 MHz, DMSO-d6) δ 176.5, 166.0, 160.4, 149.7, 149.2, 144.1, 134.1, 133.7, 131.2, 131.0, 130.4, 130.0, 129.7, 128.6, 128.3, 127.8, 127.7, 127.5, 126.9, 126.9, 125.5, 122.9, 121.5, 121.3, 121.2, 120.2, 119.1, 96.8, 90.9, 87.8, 68.9, 67.3, 65.0, 50.9, 46.6, 38.2, 34.4, 29.8, 28.9, 26.6. HRMS (ESI, m/z): calcd for C165H148N24O24Na ([M + Na]+): 2872.0991, found: 2872.0181.Pom group removal, a general procedure
Pom-protected compound (66 μmol) was dissolved in the mixture containing DMSO (1.2 mL) and methanol (600 μL). To this solution 10 M NaOH (60 μL) was added under vigorous stirring. After 30 min TLC showed full consumption of the starting material. The reaction mixture was poured into 10% citric acid (6 mL). The precipitate was centrifuged thoroughly and washed with water (4 × 3 mL). The residue was applied on the silica gel using evaporation of the acetone solution with small portion of the sorbent. The obtained powder was carefully spilled on the column filled with silica gel in CHCl3. Elution with gradient of EtOH in CHCl3 from 5 to 30% and evaporation gave pure compound as orange solid. The following compounds were isolated.800), 468 (47520), λmin, nm (ε, M−1 cm−1): 450 (28200). Fluorescence (96% EtOH, λem 530 nm, excitation λmax, nm): 440, 466; (96% EtOH, λex 420 nm, emission λmax, nm): 450, 475, 508. 1H NMR (500 MHz, DMSO-d6) δ 11.83 (br s, 1H), 8.82–8.78 (m, 1H), 8.43 (d, 1H, J = 7.32 Hz), 8.4–8.3 (m, 3H), 8.27 (d, 1H, J = 8.24 Hz), 7.84–7.79 (m, 2H), 7.71–7.63 (m, 2H), 7.58–7.52 (m, 2H), 7.47 (s, 2H), 3.97–3.91 (m, 2H), 3.18 (t, 1H, J = 2.44 Hz). 13C NMR (100 MHz, DMSO-d6) δ 166.4, 162.1, 150.0, 149.9, 134.2, 133.7, 131.1, 131.0, 130.4, 130.1, 129.8, 128.6, 128.3, 127.8, 127.7, 127.6, 127.1, 127.0, 125.7, 121.6, 121.4, 121.3, 120.3, 119.5, 97.4, 90.6, 88.6, 80.7, 73.5, 49.8, 28.1. HRMS (ESI, m/z): calcd for C31H20N3O3 ([M + H]+): 482.1499, found: 482.1489.800), 468 (47520), λmin, nm (ε, M−1 cm−1): 450 (28200). Fluorescence (96% EtOH, λem 530 nm, excitation λmax, nm): 440, 466; (96% EtOH, λex 420 nm, emission λmax, nm): 475, 508. 1H NMR (400 MHz, DMSO-d6) δ 11.78 (br s, 1H), 8.73 (t, 1H, J = 5.48 Hz), 8.45 (d, 1H, J = 7.47 Hz), 8.43–8.33 (m, 3H), 8.31–8.26 (m, 2H), 8.0 (s, 1H), 7.87–7.81 (m, 2H), 7.73–7.65 (m, 2H), 7.57 (t, 2H, J = 7.79 Hz), 7.41–7.28 (m, 5H), 5.58 (s, 2H), 4.45 (s, 2H), 4.37 (d, 2H, J = 5.56 Hz). 13C NMR (100 MHz, DMSO-d6) δ 166.4, 162.1, 150.0, 149.8, 144.5, 136.0, 134.2, 133.7, 131.1, 131.0, 130.4, 130.1, 129.8, 128.7, 128.6, 128.3, 128.1, 127.9, 127.8 (two overlapped signals), 127.6, 127.0, 127.0, 125.7, 123.1, 121.6, 121.4, 121.3, 120.3, 119.5, 97.3, 90.5, 88.6, 52.7, 49.9, 34.3. HRMS (ESI, m/z): calcd for C38H27N6O3 ([M + H]+): 615.2139, found: 615.2135.600), 470 (66000), λmin, nm (ε, M−1 cm−1): 452 (37600). Fluorescence (96% EtOH, λem 530 nm, excitation λmax, nm): 440, 466; (96% EtOH, λex 420 nm, emission λmax, nm): 475, 508. 1H NMR (400 MHz, DMSO-d6) δ 11.79 (br s, 1H), 8.75 (t, 1H, J = 5.64 Hz), 8.45 (d, 1H, J = 7.47 Hz), 8.43–8.33 (m, 3H), 8.33–8.25 (m, 2H), 7.91 (s, 1H), 7.87–7.8 (m, 2H), 7.74–7.64 (m, 2H), 7.57 (t, 2H, J = 7.47 Hz), 5.01 (t, 1H, J = 5.25 Hz), 4.47 (s, 2H), 4.42–4.34 (m, 4H), 3.78 (q, 2H, J = 5.35 Hz). 13C NMR (100 MHz, DMSO-d6) δ 166.4, 162.1, 150.0, 149.9, 144.0, 134.2, 133.7, 131.1, 131.0, 130.4, 130.1, 129.8, 128.6, 128.3, 127.8, 127.8, 127.6, 127.0, 127.0, 125.7, 123.3, 121.6, 121.4, 121.3, 120.3, 119.5, 97.3, 90.5, 88.6, 59.8, 52.1, 49.9, 34.3. HRMS (ESI, m/z): calcd for C33H25N6O4 ([M + H]+): 569.1932, found: 569.1926.800), 468 (47520), λmin, nm (ε, M−1 cm−1): 450 (28200). Fluorescence (96% EtOH, λem 530 nm, excitation λmax, nm): 440, 467; (96% EtOH, λex 420 nm, emission λmax, nm): 476, 508. 1H NMR (400 MHz, DMSO-d6) δ 11.78 (br s, 1H), 8.73 (t, 1H, J = 5.48 Hz), 8.45 (d, 1H, J = 7.47 Hz), 8.43–8.33 (m, 3H), 8.32–8.26 (m, 2H), 7.94 (s, 1H), 7.87–7.81 (m, 2H), 7.72–7.65 (m, 2H), 7.57 (t, 2H, J = 7.63 Hz), 4.46 (s, 2H), 4.41–4.35 (m, 4H), 4.32 (t, 3H, J = 5.17 Hz), 3.48–3.41 (m, 6H), 3.38 (t, 6H, J = 6.36 Hz), 3.36–3.31 (m, 2H), 3.3–3.24 (m, 8H), 2.07–1.98 (m, 2H), 1.62 (quint, 6H, J = 6.36 Hz). 13C NMR (100 MHz, DMSO-d6) δ 166.4, 162.1, 150.0, 149.8, 144.1, 134.2, 133.7, 131.1, 131.0, 130.4, 130.1, 129.8, 128.6, 128.3, 127.8, 127.6, 127.0, 126.9, 125.7, 122.9, 121.6, 121.4, 121.3, 120.3, 119.5, 97.3, 90.6, 88.6, 69.3, 69.1, 67.9, 67.3, 57.9, 49.9, 46.6, 44.9, 34.3, 32.6, 29.9. HRMS (ESI, m/z): calcd for C48H55N6O10 ([M + H]+): 875.3974, found: 875.3973.Cell toxicity assay
A cytotoxicity test in porcine embryo kidney (PEK) cells was performed as described previously.2a,6,19,25 In brief, PEK cells were seeded and incubated for 72 h at 37 °C. Stock solutions of the compounds with the concentration of 5 mM were prepared in 100% DMSO (Sigma). Two-fold dilutions of studied compounds were prepared in medium 199 on Earle solution to obtain final concentrations starting from 50 μM. Equal volumes of compound dilutions were added in four replicates to the cells. Cell control was treated with the same sequential concentrations of DMSO as in compounds dilutions in four replicates. After incubation at 37 °C on days 1 or 7 CC50 values were calculated according to the Kerber method.28Antiviral activity assays
Plaque reduction test was performed as previously described2a,6,19,25 on tick-borne encephalitis virus strain Absettarov. Stock solutions of the compounds with concentration of 5 mM were prepared in 100% DMSO (Sigma). Compounds were added to PEK cells simultaneously with virus and incubated at 37 °C for 1 h with gentle shaking every 15–20 min. Then, each well was overlaid with 1 mL of 1.26% methylcellulose (Sigma) containing 2% FBS (Gibco). After incubation at 37 °C in CO2 incubator for 6 days cells were fixed with 96% ethanol. Plaques were stained with 0.4% gentian violet. EC50 values were calculated according to Reed-and-Muench method.29Statistical analysis
Data are expressed as means ± standard deviations. The statistical significance was analyzed using Student's t test for at least three independent experiments.Molecular modelling
The molecules were drawn in InstantJChem30 and transferred to SYBYL-X 2.1 (ref. 27) in MOL format. MMFF94 charges31 were assigned to the atoms and the Powell32 optimization (10000 steps, gradient termination, threshold 0.05 kcal (mol−1 × Å)) was performed in MMFF94s force field.31b Then Confort26 global minimization was performed, sampling 2000 conformations (program maximum) and optimizing them with termination by negative change. Precision parameter was set to 0.001. Electrostatics was considered. The number of acyclic rotors concurrently sampled was set to 200, rotors in cycles were not sampled due to the aromaticity of all the studied cycles. Compound 9f contains more atoms than can be treated by Confort and thus was not optimized using this method. Fast Connolly surfaces33 for the optimized structures were calculated by MOLCAD in SYBYL-X 2.1 and colored according to hydrophobic potential.
Conclusions
In summary, we used pentaerythritol-based azides for the preparation of clusters containing up to four residues of 5-(perylen-3-ylethynyl)uracil, an antiviral scaffold. UV-Vis, fluorescence, and anti-TBEV properties of compounds were studied. For the first time, the antiviral activity of RAFI clusters was demonstrated. Four compounds, including tetramer 9f, showed one-digit nanomolar activity against TBEV, being among the most potent anti-TBEV molecules to date. Some structural features of the most active compounds can be used in further design of antivirals.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The research was supported by Russian Science Foundation (project no. 15-15-00053; synthesis of compounds, spectral studies, and, in part, antiviral studies). The virus for this work was kindly provided by Prof. G. G. Karganova. BSL-3 facilities, virus and cell collection maintenance, molecular modeling, and antiviral activity assessment were supported by the State research funding for FSBSI "Chumakov FSC R&D IBP RAS".References
- (a) V. A. Korshun, E. V. Manasova, K. V. Balakin, A. D. Malakhov, A. V. Perepelov, T. A. Sokolova and Y. A. Berlin, Nucleosides Nucleotides, 1998, 17, 1809–1812 CrossRef CAS; (b) M. V. Skorobogatyi, A. D. Malakhov, A. A. Pchelintseva, A. A. Turban, S. L. Bondarev and V. A. Korshun, ChemBioChem, 2006, 7, 810–816 CrossRef CAS PubMed.
- (a) V. L. Andronova, M. V. Skorobogatyi, E. V. Manasova, Y. A. Berlin, V. A. Korshun and G. A. Galegov, Russ. J. Bioorg. Chem., 2003, 29, 262–266 CrossRef CAS; (b) M. V. Skorobogatyi, A. V. Ustinov, I. A. Stepanova, A. A. Pchelintseva, A. L. Petrunina, V. L. Andronova, G. A. Galegov, A. D. Malakhov and V. A. Korshun, Org. Biomol. Chem., 2006, 4, 1091–1096 RSC; (c) M. R. St. Vincent, C. C. Colpitts, A. V. Ustinov, M. Muqadas, M. A. Joyce, N. A. Barsby, R. F. Epand, R. M. Epand, S. A. Khramyshev, O. A. Valueva, V. A. Korshun, D. L. J. Tyrrell and L. M. Schang, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17339–17344 CrossRef CAS PubMed; (d) A. A. Orlov, A. A. Chistov, L. I. Kozlovskaya, A. V. Ustinov, V. A. Korshun, G. G. Karganova and D. I. Osolodkin, Med. Chem. Commun., 2016, 7, 495–499 RSC.
- C. C. Colpitts, A. V. Ustinov, R. F. Epand, R. M. Epand, V. A. Korshun and L. M. Schang, J. Virol., 2013, 87, 3640–3654 CrossRef CAS PubMed.
- L. M. Schang, Future Virol., 2014, 9, 283–299 CrossRef CAS.
- (a) F. Vigant, A. Hollmann, J. Lee, N. C. Santos, M. E. Jung and B. Lee, J. Virol., 2014, 88, 1849–1854 CrossRef PubMed; (b) F. Vigant, N. C. Santos and B. Lee, Nat. Rev. Microbiol., 2015, 13, 426–437 CrossRef CAS PubMed.
- A. A. Chistov, A. A. Orlov, P. P. Streshnev, N. A. Slesarchuk, I. O. Aparin, B. Rathi, V. A. Brylev, S. V. Kutyakov, I. V. Mikhura, A. V. Ustinov, G. Westman, V. A. Palyulin, N. Jain, D. I. Osolodkin, L. I. Kozlovskaya and V. A. Korshun, Eur. J. Med. Chem., 2019, 171, 93–103 CrossRef CAS PubMed.
- S. Speerstra, A. A. Chistov, G. V. Proskurin, A. A. Aralov, E. A. Ulashchik, P. P. Streshnev, V. V. Shmanai, V. A. Korshun and L. M. Schang, Antiviral Res., 2018, 149, 164–173 CrossRef CAS PubMed.
- A. Hakobyan, I. Galindo, A. Nañez, A. Arabyan, Z. Karalyan, A. A. Chistov, P. P. Streshnev, V. A. Korshun, C. Alonso and H. Zakaryan, J. Gen. Virol., 2018, 99, 148–156 CrossRef CAS PubMed.
- (a) A. Gazumyan, B. Mitsner and G. A. Ellestad, Curr. Pharm. Des., 2000, 6, 525–546 CrossRef CAS PubMed; (b) U. Boas and P. M. H. Heegaard, Chem. Soc. Rev., 2004, 33, 43–63 RSC; (c) A. R. Borges and C.-L. Schengrund, Curr. Drug Targets: Infect. Disord., 2005, 5, 247–254 CrossRef CAS; (d) T. D. McCarthy, P. Karellas, S. A. Henderson, M. Giannis, D. F. O'Keefe, G. Heery, J. R. A. Paull, B. R. Matthews and G. Holan, Mol. Pharm., 2005, 2, 312–318 CrossRef CAS PubMed; (e) D. Sepúlveda-Crespo, R. Ceña-Díez, J. L. Jiménez and M. A. Muñoz-Fernández, Med. Res. Rev., 2017, 37, 149–179 CrossRef PubMed; (f) Y. Kim, E. J. Park and D. H. Na, Arch. Pharmacal Res., 2018, 41, 571–582 CrossRef CAS PubMed.
- (a) Y. Gong, B. Matthews, D. Cheung, T. Tam, I. Gadawski, D. Leung, G. Holan, J. Raff and S. Sacks, Antiviral Res., 2002, 55, 319–329 CrossRef CAS PubMed; (b) D. Tyssen, S. A. Henderson, A. Johnson, J. Sterjovski, K. Moore, J. La, M. Zanin, S. Sonza, P. Karellas, M. P. Giannis, G. Krippner, S. Wesselingh, T. McCarthy, P. R. Gorry, P. A. Ramsland, R. Cone, J. R. A. Paull, G. R. Lewis and G. Tachedjian, PLoS One, 2010, 5, e12309 CrossRef PubMed; (c) J. Sánchez-Rodríguez, L. Díaz, M. Galán, M. Maly, R. Gómez, F. J. de la Mata, J. L. Jiménez and M. A. Muñoz-Fernández, J. Biomed. Nanotechnol., 2015, 11, 1783–1798 CrossRef; (d) D. Sepúlveda-Crespo, J. L. Jiménez, R. Gómez, F. J. De La Mata, P. L. Majano, M. A. Muñoz-Fernández and P. Gastaminza, Nanomedicine, 2017, 13, 49–58 CrossRef PubMed; (e) C. Guerrero-Beltrán, R. Ceña-Diez, D. Sepúlveda-Crespo, J. De la Mata, R. Gómez, M. Leal, M. A. Muñoz-Fernández and J. L. Jiménez, Nanoscale, 2017, 9, 17263–17273 RSC; (f) S. Moreno, D. Sepúlveda-Crespo, F. J. de la Mata, R. Gómez and M. A. Muñoz-Fernández, Antiviral Res., 2017, 146, 54–64 CrossRef CAS PubMed; (g) C. Guerrero-Beltran, I. Rodriguez-Izquierdo, M. J. Serramia, I. Araya-Durán, V. Márquez-Miranda, R. Gomez, F. J. de la Mata, M. Leal, F. González-Nilo and M. A. Muñoz-Fernández, Bioconjugate Chem., 2018, 29, 1584–1594 CrossRef CAS PubMed; (h) M. R. Kandi, J. Mohammadnejad, M. S. Ardestani, R. Zabihollahi, S. Soleymani, M. R. Aghasadeghi and K. Baesi, Mol. Biol. Rep., 2019, 46, 143–149 CrossRef CAS PubMed.
- S. Asaftei and E. De Clercq, J. Med. Chem., 2010, 53, 3480–3488 CrossRef CAS PubMed.
- (a) B. Martínez-Gualda, L. Sun, E. Rivero-Buceta, A. Flores, E. Quesada, J. Balzarini, S. Noppen, S. Liekens, D. Schols, J. Neyts, P. Leyssen, C. Mirabelli, M. J. Camarasa and A. San-Félix, Antiviral Res., 2017, 139, 32–40 CrossRef PubMed; (b) B. Martínez-Gualda, L. Sun, O. Martí-Marí, C. Mirabelli, L. Delang, J. Neyts, D. Schols, M. J. Camarasa and A. San-Félix, Antiviral Res., 2019, 168, 210–214 CrossRef PubMed.
- (a) A. Luganini, A. Giuliani, G. Pirri, L. Pizzuto, S. Landolfo and G. Gribaudo, Antiviral Res., 2010, 85, 532–540 CrossRef CAS PubMed; (b) A. Luganini, S. F. Nicoletto, L. Pizzuto, G. Pirri, A. Giuliani, S. Landolfo and G. Gribaudo, Antimicrob. Agents Chemother., 2011, 55, 3231–3239 CrossRef CAS PubMed; (c) K. Hatano, T. Matsubara, Y. Muramatsu, M. Ezure, T. Koyama, K. Matsuoka, R. Kuriyama, H. Kori and T. Sato, J. Med. Chem., 2014, 57, 8332–8339 CrossRef CAS PubMed.
- A. Flores, M. J. Camarasa, M. J. Pérez-Pérez, A. San-Félix, J. Balzarini and E. Quesada, Org. Biomol. Chem., 2014, 12, 5278–5294 RSC.
- (a) S. L. Xiao, L. L. Si, Z. Y. Tian, P. X. Jiao, Z. B. Fan, K. Meng, X. S. Zhou, H. Wang, R. Y. Xu, X. Han, G. Fu, Y. M. Zhang, L. H. Zhang and D. M. Zhou, Biomaterials, 2016, 78, 74–85 CrossRef CAS PubMed; (b) Z. Y. Tian, L. L. Si, K. Meng, X. S. Zhou, Y. M. Zhang, D. M. Zhou and S. L. Xiao, Eur. J. Med. Chem., 2017, 134, 133–139 CrossRef CAS PubMed; (c) Y. Yang, H.-J. He, H. Chang, Y. Yu, M.-B. Yang, Y. He, Z.-C. Fan, S. S. Iyer and P. Yu, Eur. J. Med. Chem., 2018, 143, 1723–1731 CrossRef CAS.
- (a) F. Lasala, E. Arce, J. R. Otero, J. Rojo and R. Delgado, Antimicrob. Agents Chemother., 2003, 47, 3970–3972 CrossRef CAS PubMed; (b) G. Tabarani, J. J. Reina, C. Ebel, C. Vivès, H. Lortat-Jacob, J. Rojo and F. Fieschi, FEBS Lett., 2006, 580, 2402–2408 CrossRef CAS PubMed; (c) R. Clayton, J. Hardman, C. C. LaBranche and K. D. McReynolds, Bioconjugate Chem., 2011, 22, 2186–2197 CrossRef CAS PubMed; (d) J. Luczkowiak, A. Muñoz, M. Sánchez-Navarro, R. Ribeiro-Viana, A. Ginieis, B. M. Illescas, N. Martín, R. Delgado and J. Rojo, Biomacromolecules, 2013, 14, 431–437 CrossRef CAS PubMed; (e) A. Muñoz, D. Sigwalt, B. M. Illescas, J. Luczkowiak, L. Rodríguez-Pérez, I. Nierengarten, M. Holler, J.-S. Remy, K. Buffet, S. P. Vincent, J. Rojo, R. Delgado, J.-F. Nierengarten and N. Martín, Nat. Chem., 2016, 8, 50–57 CrossRef; (f) S. Brument, C. Cheneau, Y. Brissonnet, D. Deniaud, F. Halary and S. G. Gouin, Org. Biomol. Chem., 2017, 15, 7660–7671 RSC; (g) B. M. Illescas, J. Rojo, R. Delgado and N. Martín, J. Am. Chem. Soc., 2017, 139, 6018–6025 CrossRef CAS PubMed; (h) A. Muñoz, B. M. Illescas, J. Luczkowiak, F. Lasala, R. Ribeiro-Viana, J. Rojo, R. Delgado and N. Martín, J. Mater. Chem. B, 2017, 5, 6566–6571 RSC; (i) L. Rodríguez-Pérez, J. Ramos-Soriano, A. Pérez-Sánchez, B. M. Illescas, A. Muñoz, J. Luczkowiak, F. Lasala, J. Rojo, R. Delgado and N. Martín, J. Am. Chem. Soc., 2018, 140, 9891–9898 CrossRef PubMed.
- (a) S. Sattin, A. Daghetti, M. Thépaut, A. Berzi, M. Sánchez-Navarro, G. Tabarani, J. Rojo, F. Fieschi, M. Clerici and A. Bernardi, ACS Chem. Biol., 2010, 5, 301–312 CrossRef CAS PubMed; (b) J. Luczkowiak, S. Sattin, I. Sutkevičiūte, J. J. Reina, M. Sánchez-Navarro, M. Thépaut, L. Martínez-Prats, A. Daghetti, F. Fieschi, R. Delgado, A. Bernardi and J. Rojo, Bioconjugate Chem., 2011, 22, 1354–1365 CrossRef CAS PubMed; (c) A. Berzi, J. J. Reina, R. Ottria, I. Sutkeviciute, P. Antonazzo, M. Sanchez-Navarro, E. Chabrol, M. Biasin, D. Trabattoni, I. Cetin, J. Rojo, F. Fieschi, A. Bernardi and M. Clerici, AIDS, 2012, 26, 127–137 CrossRef CAS PubMed; (d) N. Varga, I. Sutkeviciute, R. Ribeiro-Viana, A. Berzi, R. Ramdasi, A. Daghetti, G. Vettoretti, A. Amara, M. Clerici, J. Rojo, F. Fieschi and A. Bernardi, Biomaterials, 2014, 35, 4175–4184 CrossRef CAS PubMed; (e) S. Tang, W. B. Puryear, B. M. Seifried, X. H. Dong, J. A. Runstadler, K. Ribbeck and B. D. Olsen, ACS Macro Lett., 2016, 5, 413–418 CrossRef CAS; (f) S. J. Kwon, D. H. Na, J. H. Kwak, M. Douaisi, F. Zhang, E. J. Park, J. H. Park, H. Youn, C. S. Song, R. S. Kane, J. S. Dordick, K. B. Lee and R. J. Linhardt, Nat. Nanotechnol., 2017, 12, 48–54 CrossRef CAS; (g) M. Nagao, T. Matsubara, Y. Hoshino, T. Sato and Y. Miura, Bioconjugate Chem., 2019, 30, 1192–1198 CrossRef CAS PubMed; (h) W. Lu, W. Du, V. J. Somovilla, G. Yu, D. Haksar, E. de Vries, G.-J. Boons, R. P. de Vries, C. A. M. de Haan and R. J. Pieters, J. Med. Chem., 2019, 62, 6398–6404 CrossRef CAS PubMed.
- (a) Z.-L. Yang, X.-F. Zeng, H.-P. Liu, Q. Yu, X. Meng, Z.-L. Yan, Z.-C. Fan, H.-X. Xiao, S. S. Iyer, Y. Yang and P. Yu, Tetrahedron Lett., 2016, 57, 2579–2582 CrossRef CAS; (b) T.-F. Zhao, H.-J. Qin, Y. Yu, M.-B. Yang, H. Chang, N. Guo, Y. He, Y. Yang and P. Yu, J. Carbohydr. Chem., 2017, 36, 235–246 CrossRef CAS; (c) Z.-L. Yan, A.-Y. Liu, X.-X. Wei, Z. Zhang, L. Qin, Q. Yu, P. Yu, K. Lu and Y. Yang, Carbohydr. Res., 2019, 477, 32–38 CrossRef CAS PubMed.
- G. V. Proskurin, A. A. Orlov, V. A. Brylev, L. I. Kozlovskaya, A. A. Chistov, G. G. Karganova, V. A. Palyulin, D. I. Osolodkin, V. A. Korshun and A. V. Aralov, Eur. J. Med. Chem., 2018, 155, 77–83 CrossRef CAS PubMed.
- (a) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed; (b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- A. I. Ponomarenko, V. A. Brylev, K. A. Sapozhnikova, A. V. Ustinov, I. A. Prokhorenko, T. S. Zatsepin and V. A. Korshun, Tetrahedron, 2016, 72, 2386–2391 CrossRef CAS.
- V. M. Farzan, E. A. Ulashchik, Y. V. Martynenko-Makaev, M. V. Kvach, I. O. Aparin, V. A. Brylev, T. A. Prikazchikova, S. Y. Maklakova, A. G. Majouga, A. V. Ustinov, G. A. Shipulin, V. V. Shmanai, V. A. Korshun and T. S. Zatsepin, Bioconjugate Chem., 2017, 28, 2599–2607 CrossRef CAS.
- (a) M. Rasmussen and N. J. Leonard, J. Am. Chem. Soc., 1967, 89, 5439–5445 CrossRef CAS; (b) D. Dhanak and C. B. Reese, J. Chem. Soc., Perkin Trans. 1, 1986, 2181–2186 RSC.
- X. Dong, X. Guo, G. Liu, A. Fan, Z. Wang and Y. Zhao, Chem. Commun., 2017, 53, 3822–3825 RSC.
- (a) A. V. Aralov, G. V. Proskurin, A. A. Orlov, L. I. Kozlovskaya, A. A. Chistov, S. V. Kutyakov, G. G. Karganova, V. A. Palyulin, D. I. Osolodkin and V. A. Korshun, Eur. J. Med. Chem., 2017, 138, 293–299 CrossRef CAS PubMed; (b) L. I. Kozlovskaya, A. D. Golinets, A. A. Eletskaya, A. A. Orlov, V. A. Palyulin, S. N. Kochetkov, L. A. Alexandrova and D. I. Osolodkin, ChemistrySelect, 2018, 3, 2321–2325 CrossRef CAS.
- Confort, Certara, L.P., St. Louis, MO, USA, 2013 Search PubMed.
- Sybyl-X Molecular Modeling Software Packages, Version 2.1, Certara, L.P., St. Louis, MO, USA, 2013 Search PubMed.
- WHO, Polio laboratory manual, WHO, Geneva, 4th edn, 2004, WHO/IVB/04/10 Search PubMed.
- L. J. Reed and H. Muench, Am. J. Hyg., 1938, 27, 493–497 Search PubMed.
- Instant JChem 17.3.27.0, ChemAxon, 2017, http://www.chemaxon.com Search PubMed.
- (a) T. A. Halgren, J. Comput. Chem., 1996, 17, 490–519 CrossRef CAS; (b) T. A. Halgren, J. Comput. Chem., 1999, 20, 720–729 CrossRef CAS.
- M. J. D. Powell, Mathematical Programming, 1977, 12, 241–254 CrossRef.
- (a) M. L. Connolly, Science, 1983, 221, 709–713 CrossRef CAS PubMed; (b) M. L. Connolly, J. Appl. Crystallogr., 1983, 16, 548–558 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C NMR and HRMS spectra. See DOI: 10.1039/c9ra06313g |
| This journal is © The Royal Society of Chemistry 2019 |