
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Long non-coding RNA KCNQ1OT1 regulates cell proliferation, apoptosis and chemo-sensitivity through modulating the miR-186-5p/NCAM1 axis in acute myeloid leukemia cells
Jing Dai *,
Kai Wang,
Tao Liu,
Qiong Wang and
Yingxu Pang
*,
Kai Wang,
Tao Liu,
Qiong Wang and
Yingxu Pang
Department of Hematology, Zhoukou Central Hospital, No. 26, Renmin East Road, Zhoukou, 466000, Zhengzhou, China. E-mail: yangzhiming117@163.com; Tel: +86 394 8521603
First published on 7th November 2019
Abstract
Recent studies show that lncRNA KCNQ1OT1 and microRNA-186-5p (miR-186-5p) are involved in various human cancers. Moreover, it is reported that KCNQ1OT1 expression is upregulated in acute myeloid leukemia (AML). However, their roles in AML remain unknown. This study aimed to reveal the functional mechanism of KCNQ1OT1 and miR-186-5p in AML development. Quantitative real time polymerase chain reaction (qRT-PCR) was performed to detect the levels of genes. Cell proliferation and apoptosis were assessed by a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay and flow cytometry analysis respectively. A western blot assay was carried out to examine the protein levels. In addition, the interaction between miR-186-5p and KCNQ1OT1 or neural cell adhesion molecule 1 (NCAM1) was predicted by bioinformatics analysis tool starbase2.0 and confirmed by the dual luciferase reporter assay. KCNQ1OT1 and NCAM1 expressions were increased and miR-186-5p expression was decreased in AML samples and cells. The depletion of KCNQ1OT1 inhibited cell proliferation, and promoted apoptosis and chemo-sensitivity in AML. In addition, the upregulation of miR-186-5p suppressed AML cell proliferation, and induced apoptosis and chemo-sensitivity. Interestingly, KCNQ1OT1 directly downregulated miR-186-5p expression and miR-186-5p decreased NCAM1 expression by binding to the 3′ untranslated region (UTR) of NCAM1 mRNA. Furthermore, miR-186-5p knockdown or NCAM1 overexpression reversed the effects of KCNQ1OT1 depletion on AML cell progression. Our results firstly revealed a linear relationship between KCNQ1OT1, miR-186-5p, and NCAM1, and demonstrated that KCNQ1OT1 mediated AML cell progression via regulating the miR-186-5p/NCAM1 axis, revealing functional mechanisms of KCNQ1OT1 and miR-186-5p in AML development.
Introduction
Acute myeloid leukemia (AML) is a class of heterogeneous diseases that are characterized by the accumulation of abnormal blasts in the marrow.1 Present evidence suggests that the clinical outcomes for AML patients depend on cytogenetics, molecular genetic aberrations and clinical factors.2 According to statistics, there have been approximately 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 450 new cases of and 10920 deaths due to AML in the United States in 2019.3 Nowadays, different kinds of methods, including hematopoietic stem cell transplantation (HSCT) and chemotherapy, are used to treat AML patients.4 However, the outcome remains unsatisfactory. Therefore, the study of the molecular mechanism regarding AML development is essential for the treatment of AML patients.
450 new cases of and 10920 deaths due to AML in the United States in 2019.3 Nowadays, different kinds of methods, including hematopoietic stem cell transplantation (HSCT) and chemotherapy, are used to treat AML patients.4 However, the outcome remains unsatisfactory. Therefore, the study of the molecular mechanism regarding AML development is essential for the treatment of AML patients.
Long noncoding RNAs (LncRNAs) are a group of non-coding RNAs that have more than 200 nucleotides.5 In human cancers, lncRNAs modulate the levels of multiple genes that are involved in cell proliferation, apoptosis, autophagy, and chemo-sensitivity.6 For example, lncRNA KCNQ1OT1 mediates cell growth and drug resistance through affecting miR-211-5p expression in tongue cancer,7 and increases the chemo-resistance of oxaliplatin via regulating the miR-34a/ATG4B axis in colon cancer.8 In AML patients, KCNQ1OT1 expression is significantly increased, meaning it may be related to the regulation of AML.9 However, the functional mechanism of KCNQ1OT1 in AML development remains elusive.
MicroRNAs (miRNAs), with ∼22 nucleotides, are a class of small noncoding RNAs that mediate coding genes by regulating its expression or messenger RNA (mRNA) degradation.10 Accumulating evidence indicate that miRNAs are implicated with a lot of biological progresses, such as cell development, proliferation, apoptosis, and stem cell self-renewal.10,11 Furthermore, miRNAs exert these function in various human cancers and serve as a kind of biomarkers.12 MiR-186-5p, an endogenous miRNA, has been reported to exert function as a tumor suppressor in osteosarcoma and inhibits cell proliferation as well as metastasis through regulation of zinc finger E-box binding (ZEB) in human colorectal cancer.13 Besides, Lin et al. reported that microRNA-186-5p (miR-186-5p) induced chronic myeloid leukemia (CML) cell apoptosis and reduced cell survival.14 But, the study of miR-186-5p in AML is rare.
Neural cell adhesion molecule 1 (NCAM1), an important protein for cancer development, belongs to the immunoglobulin superfamily of adhesion molecules.15 Under the condition of activation, NCAM1 is involved in different signaling pathways, including FYN-focal adhesion kinase (FAK), mitogen-activated protein kinase (MAPK), and phosphatidylinositol 3-kinase (PI3K) pathways.16 It is reported that NCAM1 has high expression in natural killer (NK) cells and AML patients.17,18 Moreover, NCAM1 induces leukemogenesis and confers drug resistance to AML cells,18,19 revealing a role of NCAM1 as a biomarker for the therapy of AML patients. Therefore, NCAM1 plays crucial role in AML cells. However, the detail molecular mechanism is still poorly understood.
Here, we detected the expression levels of KCNQ1OT1, miR-186-5p, and NCAM1 in AML. Furthermore, the effects of miR-186-5p/NCAM1 on KCNQ1OT1 knockdown-mediated cell proliferation, promoted apoptosis and chemo-sensitivity were also analyzed.
Materials and methods
Tissues samples and cell culture
AML samples were obtained from patients following written informed consent in Zhoukou Central Hospital. The study was approved by the Ethics Committee of Zhoukou Central Hospital. This study was performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (NIH publication no. 85-23 rev. 1985) and was approved by the Institutional Animal Care and Use Committee of Zhoukou Central Hospital (Zhoukou, China).A total of 4 AML cell lines (HL-60, Kasumi-1, U937, THP1) and 1 normal cell line (HS-5) were provided by American Tissue Culture Collection (ATCC, Manassas, VA, USA). These cells were incubated in Dulbecco's modifed Eagle's medium (DMEM; Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10% fetal bovine serum (FBS, Thermo Fisher Scientific). All cells were cultured in an incubator humidified with 5% CO2 at 37 °C.
Firstly, Different concentrations of AraC were used to treat the cells when cell proliferation was performed. Then, 10 nM AraC was chosen for further experiments.
Plasmid and transfection
MiR-186-5p mimics, miR-186-5p inhibitor, small hairpin RNA against KCNQ1OT1 (sh-KCNQ1OT1), and their negative controls were purchased from Ribobio (GuangZhou, China). NCAM1 coding sequence was synthesized and inserted into pcDNA3.1 vector (Genepharma, Shanghai, China) for pcDNA-NCAM1 construction.Cell transfection assay was carried out by the Lipofectamine TM2000 (Invitrogen, Carlsbad, CA, USA).
RNA extraction and quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted from AML samples and cells using TRIzol (Invitrogen) based on the user's manual. Then, a PrimeScript™ RT Reagent kit (Takara Biotechnology Co., Ltd., Dalian, China) was supplied to obtain complementary DNA (cDNA). Next, qPCR was carried out using SYBR Premix Ex Taq Master mix (Takara Biotechnology Co, Ltd.) in line with the manufacturer's instructions. U6 and GAPDH were chosen as internal genes. The data were analyzed using the 2−ΔΔCt method. In our research, some primers were used, their sequences were listed as follows: KCNQ1OT1 forward, 5′-CTTTGCAGCAACCTCCTTGT-3′ and reverse, 5′-TGGGGTGAGGGATCTGAA-3′; miR-186-5p forward, 5′-AAGAATTCTCCTTTTGGGCT-3′ and reverse, 5′-GTGCGTGTCGTGGAGTCG-3′; NCAM1 forward, 5′- AACAAAGCATGATGGGTGAA-3′ and reverse, 5′-GTCTGTGGTGTTGGAAATGC-3′; U6 forward, 5′-TGCGGGTGCTCGCTTCGGCAGC-3′ and reverse, 5′-CCAGTGCAGGGTCCGAGGT-3′; GAPDH forward, 5′-CATGAGAAGTATGACAACAGCCT-3′ and reverse, 5′-AGTCCTTCCACGATACCAAAGT-3′.Cell proliferation assay
Cell proliferation ability was assessed by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay. Briefly, U937 or THP cells were transfected with vectors used in this study and incubated for 24 h, 48 h, or 72 h. Subsequently, MTT solution (5 mg mL−1, 20 μL) was added to cells that were plated in 96 well plate. The plate was transferred to an incubator with 37 °C for 4 h, and then 200 μL dimethylsulfoxide (Beyotime, Haimen, China) was supplied for dissolution of formazan precipitate. Finally, the absorbance was determined at 490 nm using an ELISA plate reader (Bio-Rad, Richmond, CA, USA).Cell apoptosis assay
In this research, apoptosis rate of U937 or THP cells was assessed by Propidium iodide (PI)/Annexin V-Fluorescein (FITC) Cell Apoptosis Kit (BD Biosciences, San Jose, CA, USA) based on the user's manual. Firstly, the U937 or THP cells were transfected with vectors and cultured for 48 h. Next, the cells were washed with PBS for 2 times, and then stained using Annexin V-FITC/PI for 15 min. Finally, cell apoptosis rate was measured using flow cytometry (BD Bioscience).Western blot assay
Total proteins in AML tumors or cells were obtained using RIPA buffer (Beyotime). 20 μg protein was separated by dodecyl sulfate, sodium salt-polyacrylamide gel electrophoresis (SDS-PAGE, 10%), electro-transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA), and then blocked using PBST containing 5% non-fat skim milk. Subsequently, the membranes were incubated by the primary antibody against NCAM1, Bcl-2, Bax, un-cleaved caspase-3, cleaved-caspase-3, and GAPDH (1:1000, Abcam, Cambridge, MA, USA) at 4 °C overnight. Next, the membranes were coated with corresponding secondary antibody (1:500) at room temperature for 1 h. Finally, FluorChem imaging system (Thermo Fisher Scientific) was used to measure protein signal.
The dual luciferase reporter assay
The interaction between miR-186-5p and KCNQ1OT1 or NCAM1 was predicted by online software starbase2.0 and verified by the dual luciferase reporter assay. Briefly, the 3′UTR sequence of wide type KCNQ1OT1/NCAM1 (WT-KCNQ1OT1/NCAM1 3′UTR-WT) or mutated wide KCNQ1OT1/NCAM1 (MUT-KCNQ1OT1/NCAM1 3′UTR-MUT) mutated at binding sites of miR-186-5p were amplified and cloned into pGL3 vector (Promega, Madison, WI, USA). Next, every vector was co-transfected into U937 and THP1 cells with miR-186-5p mimics or mimics-NC, respectively. Finally, the luciferase activity was measured by the Lmax multiwall luminometer (Molecular Devices, LLC, Sunnyvale, CA, USA).Statistical analysis
All data in this research were based on at least three independent biological reduplication. Student's t-test was performed for analysis of significant difference. Values were presented as the mean ± standard deviation (SD). Statistical difference was considered as P less than 0.05.Results
KCNQ1OT1 expression was increased in AML samples and cells
To explore the role of KCNQ1OT1 in AML, the expression level of KCNQ1OT1 was detected by qRT-PCR assay in AML samples and corresponding normal samples. The results showed that KCNQ1OT1 was upregulated in AML samples (Fig. 1A). Meanwhile, KCNQ1OT1 expression in AML cells was also detected. As shown in Fig. 1B, KCNQ1OT1 expression was significantly increased in AML cell lines (HL-60, Kasumi-1, U937, and THP1) compared with normal cell line (HS5). In addition, we also analyzed the correlation between KCNQ1OT1 expression and overall survival rate of AML patients after surgery, and found that patients that had high KCNQ1OT1 expression exhibited low overall survival rate (Fig. 1C). Therefore, KCNQ1OT1 might play crucial role in AML development.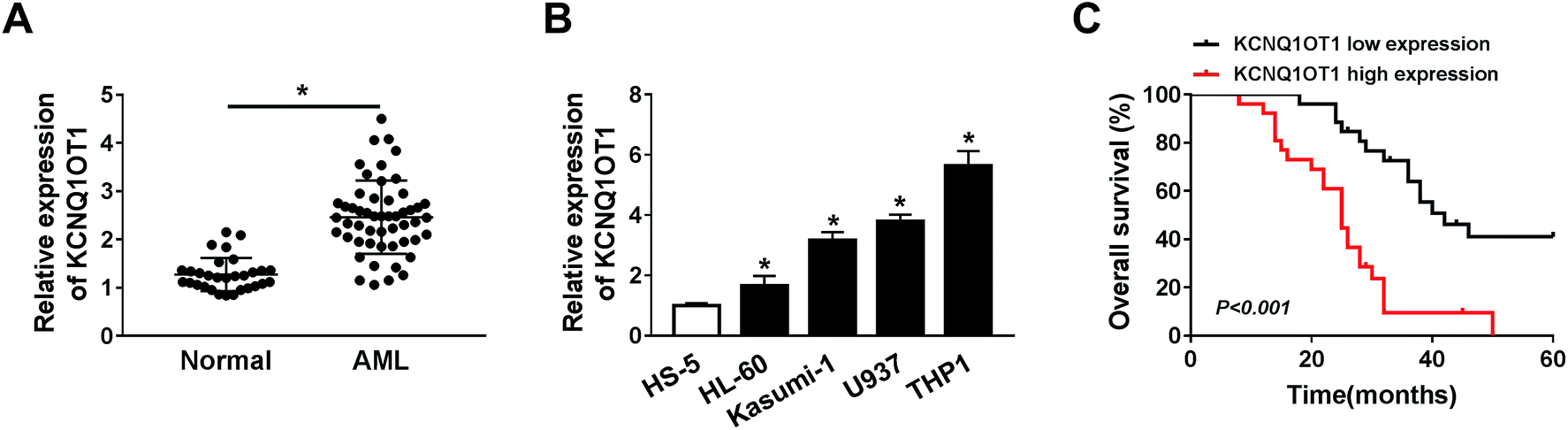 | ||
| Fig. 1 KCNQ1OT1 was upregulated in AML samples and cells, and related to poor prognosis in AML patients. (A and B) QRT-PCR assay was performed to detect the expression level of KCNQ1OT1 in AML and normal samples (A) as well as AML and normal cells (B). (C) Relationship between KCNQ1OT1 expression level and overall survival rate of AML patients after surgery was analyzed. *P < 0.05. | ||
The knockdown of KCNQ1OT1 suppressed cell proliferation, promoted apoptosis and chemo-sensitivity in AML cells
To further investigate the function of KCNQ1OT1 in AML cell progression, we depleted KCNQ1OT1 using two specific shRNAs against KCNQ1OT1. As indicated in Fig. 2A, the transfection of sh-KCNQ1OT1 (#1 or #2) dramatically decreased the expression level of KCNQ1OT1. Next, MTT assay was carried out to determine cell proliferation ability. The data suggested that the knockdown of KCNQ1OT1 significantly suppressed cell proliferation in both U937 (Fig. 2B) and THP1 (Fig. 2C) cell lines. Meanwhile, increased cell apoptosis rate was observed in KCNQ1OT1-depleted cells using flow cytometry analysis (Fig. 2D). Then, the expression levels of some proteins, including Bcl-2 that inhibited cell apoptosis as well as Bax and cleaved-caspase-3 that induced cell apoptosis, were examined by western blot assay. As shown in Fig. 2E and F, KCNQ1OT1 knockdown downregulated Bcl-2 expression, and upregulated Bax and cleaved-caspase-3 expressions in both U937 and THP1 cell lines. Besides, cytarabine (Ara-C) chemo-sensitivity of cell was also analyzed in this research. The results demonstrated that the downregulation of KCNQ1OT1 reduced cell viability in both U937 (Fig. 2G) and THP1 (Fig. 2H) cell lines after treatment of vary concentrations of AraC. From these data, the knockdown of KCNQ1OT1 suppressed cell progression in AML.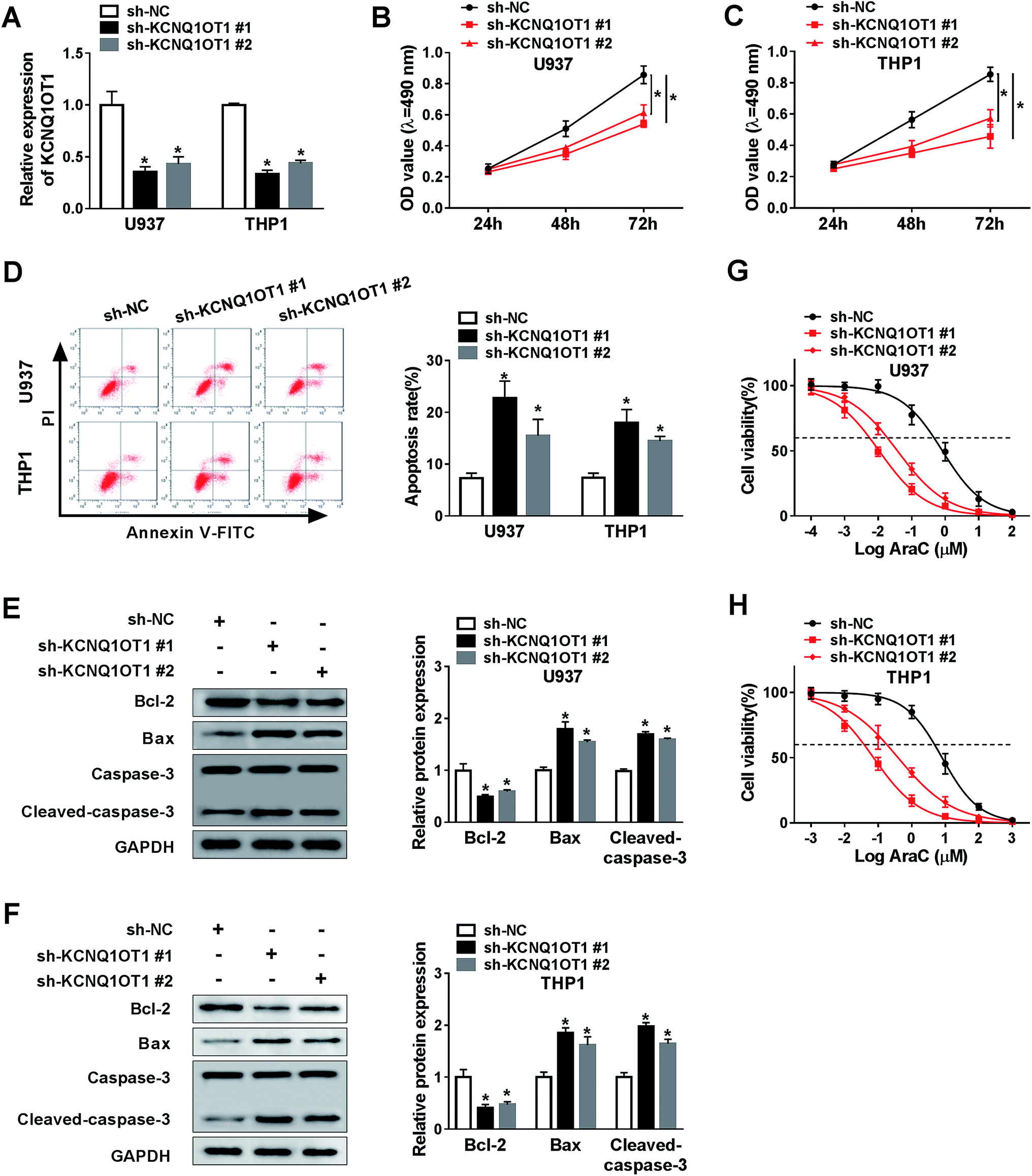 | ||
| Fig. 2 The knockdown of KCNQ1OT1 inhibited cell proliferation, promoted apoptosis and chemo-sensitivity in AML cells. (A) The expression level of KCNQ1OT1 was determined using qRT-PCR assay in both U937 and THP1 cell lines transfected with sh-NC, SH-KCNQ1OT1#1, and SH-KCNQ1OT1#2, respectively. (B and C) MTT assay was performed to assess cell proliferation in both U937 (B) and THP1 (C) cell lines. (D) Cell apoptosis rate was measured by flow cytometry analysis. (E and F) Western blot assay was carried out to detect apoptosis-related protein level in both U937 (E) and THP1 (F) cell lines. (G and H) Cell viability was examined by MTT assay in both U937 (G) and THP1 (H) cell lines after treatment of vary concentration of Ara-C. *P < 0.05. | ||
KCNQ1OT1 downregulated the expression level of miR-186-5p
Bioinformatics analysis website starBase2.0 predicted that miR-186-5p was a potential target gene of KCNQ1OT1 (Fig. 3A). In order to verify this prediction, the dual luciferase reporter assay was performed in this study. The data suggested that miR-186-5p mimics significantly diminished the luciferase activity of WT-KCNQ1OT1, but not MUT-KCNQ1OT1 in both U937 (Fig. 3B) and THP1 (Fig. 3C) cell lines, indicating that KCNQ1OT1 interacts with miR-186-5p. Next, the effect of KCNQ1OT1 on miR-186-5p expression was analyzed in cells transfected with sh-NC or sh-KCNQ1OT1. The results indicated that KCNQ1OT1 knockdown significantly increased miR-186-5p expression in both U937 and THP1 cell lines (Fig. 3D). Then, we detected the expression level of miR-186-5p in AML. As shown in Fig. 3E, decreased miR-186-5p expression was observed in AML samples. These results suggested that KCNQ1OT1 negatively regulated miR-186-5p expression. In addition, we also confirmed that KCNQ1OT1 expression was negatively related to the expression level of miR-186-5p (Fig. 3F). In addition, decreased miR-186-5p expression was observed in U937 and THP1 cells (Fig. 3G). Taken together, KCNQ1OT1 downregulated miR-186-5p expression through targeting it.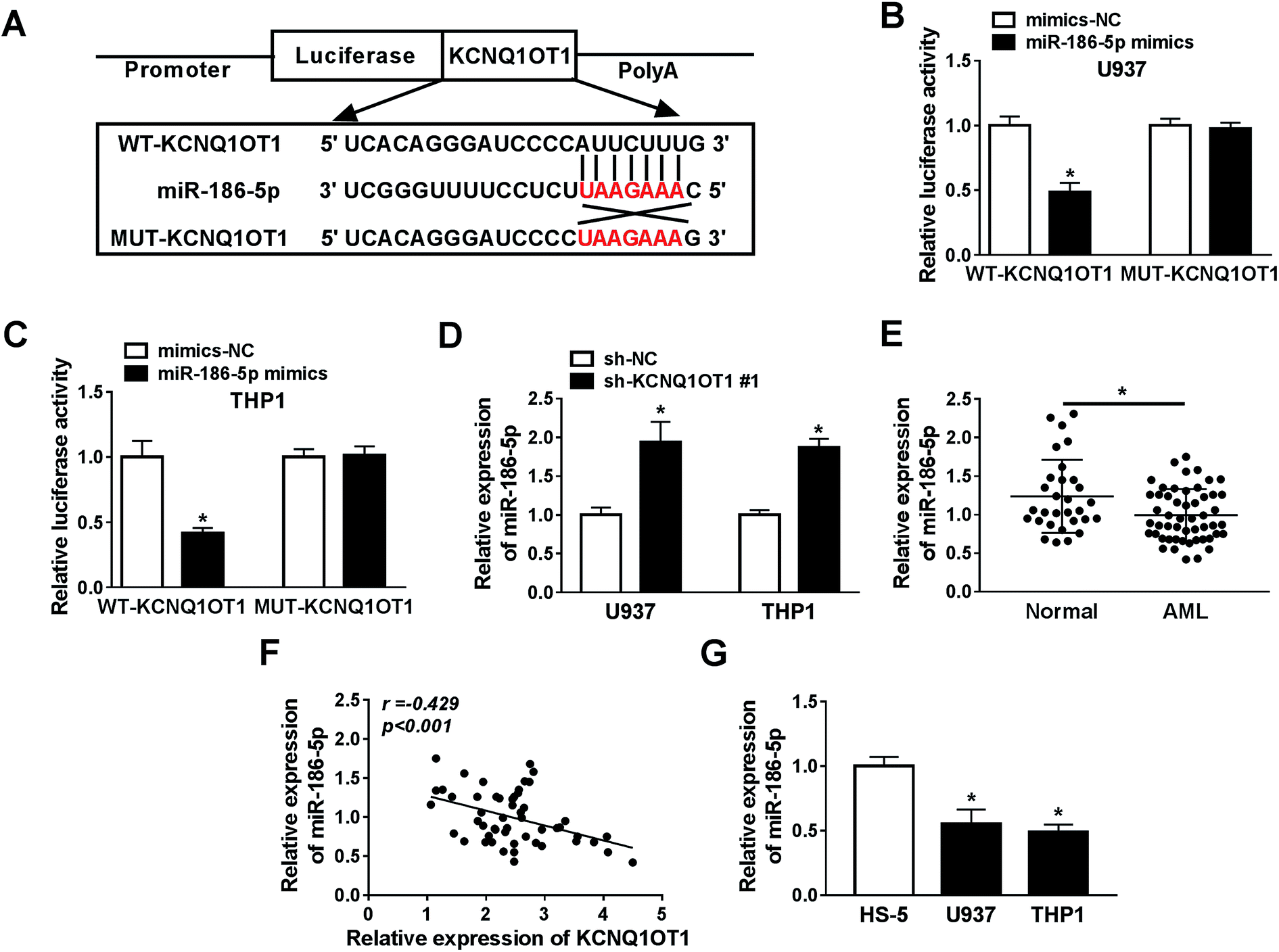 | ||
| Fig. 3 KCNQ1OT1 as a molecular sponge antagonized miR-186-5p availability. (A) Potential target gene of SH-KCNQ1OT1 was predicted by bioinformatics analysis software starbase2.0. Mutated sites of SH-KCNQ1OT1 were indicated in red color. (B and C) Relative luciferase activity was determined in both U937 (B) and THP1 (C) cell lines that were co-transfected with miR-186-5p mimics or mimics-NC, and WT-KCNQ1OT1 or MUT-KCNQ1OT1. (D) The expression level of miR-186-5p was measured in both U937 and THP1 cell lines transfected with sh-NC or sh-KCNQ1OT1#1. (E) QRT-PCR assay was performed to detect miR-186-5p expression in AML and normal samples. (F) Association between KCNQ1OT1 expression and miR-186-5p expression was analyzed using qRT-PCR assay. (G) The expression level of miR-186-5p was examined in AML cells (U937 and THP1). *P < 0.05. | ||
The upregulation of miR-186-5p inhibited cell proliferation, induced apoptosis and chemo-sensitivity in AML cells
To analyze the role of miR-186-5p in AML cell progression and chemo-sensitivity, U937 and THP1 cells were transfected with miR-186-5p mimics. Then, MTT assay was carried out to assess cell proliferation ability. Compared with control group, transfection of miR-186-5p mimics dramatically suppressed cell proliferation in both U937 (Fig. 4A) and THP1 (Fig. 4B) cell lines. In addition, Flow cytometry analysis showed that the upregulation of miR-186-5p promoted cell apoptosis (Fig. 4C). Meanwhile, cell apoptosis-related protein levels were also analyzed using western blot assay. As expected, the overexpression of miR-186-5p significantly decreased Bcl-2 level, and enhanced Bax and cleaved-caspase-3 levels in both U937 (Fig. 4D) and THP1 (Fig. 4E) cell lines. Besides, we checked miR-186-5p overexpression cell chemo-sensitivity. As shown in Fig. 4F and G, miR-186-5p overexpression decreased chemo-sensitivity of U937 and THP1 cells against AraC. These results indicated that the upregulation of miR-186-5p suppressed cell proliferation, and promoted apoptosis and chemo-sensitivity in AML cells.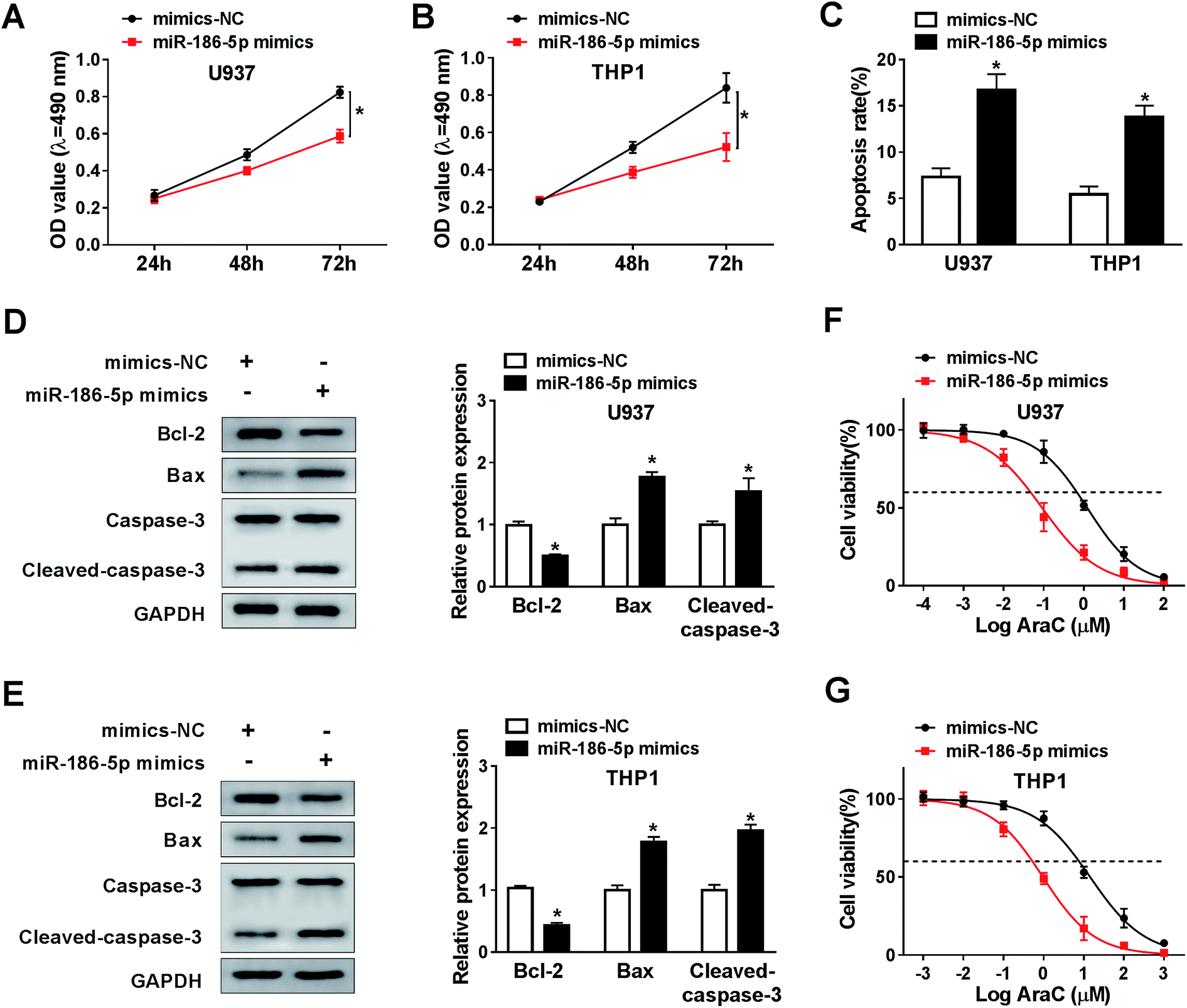 | ||
| Fig. 4 MiR-186-5p suppressed cell proliferation, induced apoptosis and chemo-sensitivity in AML cells. (A and B) Cell proliferation was determined by MTT assay in both U937 (A) and THP1 (B) cell lines transfected with mimics or miR-186-5p mimics. (C) Flow cytometry analysis was used to measure cell apoptosis. (D and E) Cell apoptosis-related protein levels were detected using western blot assay in both U937 (D) and THP1 (E) cell lines. (F and G) Cell viability was analyzed in both U937 (F) and THP1 (G) cell lines after Ara-C treatment. *P < 0.05. | ||
MiR-186-5p negatively regulated NCAM1 expression
Bioinformatics analysis website starBase2.0 was used to predict downstream genes of miR-186-5p, the data showed that NCAM1 was a potential target gene of miR-186-5p (Fig. 5A). Then, the dual luciferase reporter assay suggested that U937 (Fig. 5B) and THP1 (Fig. 5C) cells co-transfected with miR-186-5p mimics and NCAM1 3′ UTR-WT, but not miR-186-5p mimics and NCAM1 3′ UTR-MUT, displayed lower luciferase activity compared to the control group. These data confirmed interaction between miR-186-5p and NCAM1. Furthermore, the effect of miR-186-5p on NCAM1 expression was analyzed. As indicated in Fig. 5D, miR-186-5p overexpression significantly decreased NCAM1 mRNA level and miR-186-5p knockdown dramatically increased NCAM1 mRNA level. Moreover, this regulated effect on NCAM1 protein level was also observed in both U937 (Fig. 5E) and THP1 (Fig. 5F) cell lines. Next, we determined the expression level of NCAM1 in AML, and found that NCAM1 was upregulated in AML samples (Fig. 5G). Meanwhile, the relationship between NCAM1 and miR-186-5p or KCNQ1OT1 was also analyzed in AML samples. As expected, NCAM1 expression was negatively correlated with miR-186-5p expression (Fig. 5H) and positively correlated with KCNQ1OT1 expression (Fig. 5I). In addition, the expression level of NCAM1 in AML cells was also examined. Consistent with that in AML samples, NCAM1 expression was significantly increased in AML U937 (Fig. 5J) and THP1 (Fig. 5K) cells. These results revealed that miR-186-5p negatively regulated NCAM1 expression via binding to 3′ UTR of NCAM1 mRNA.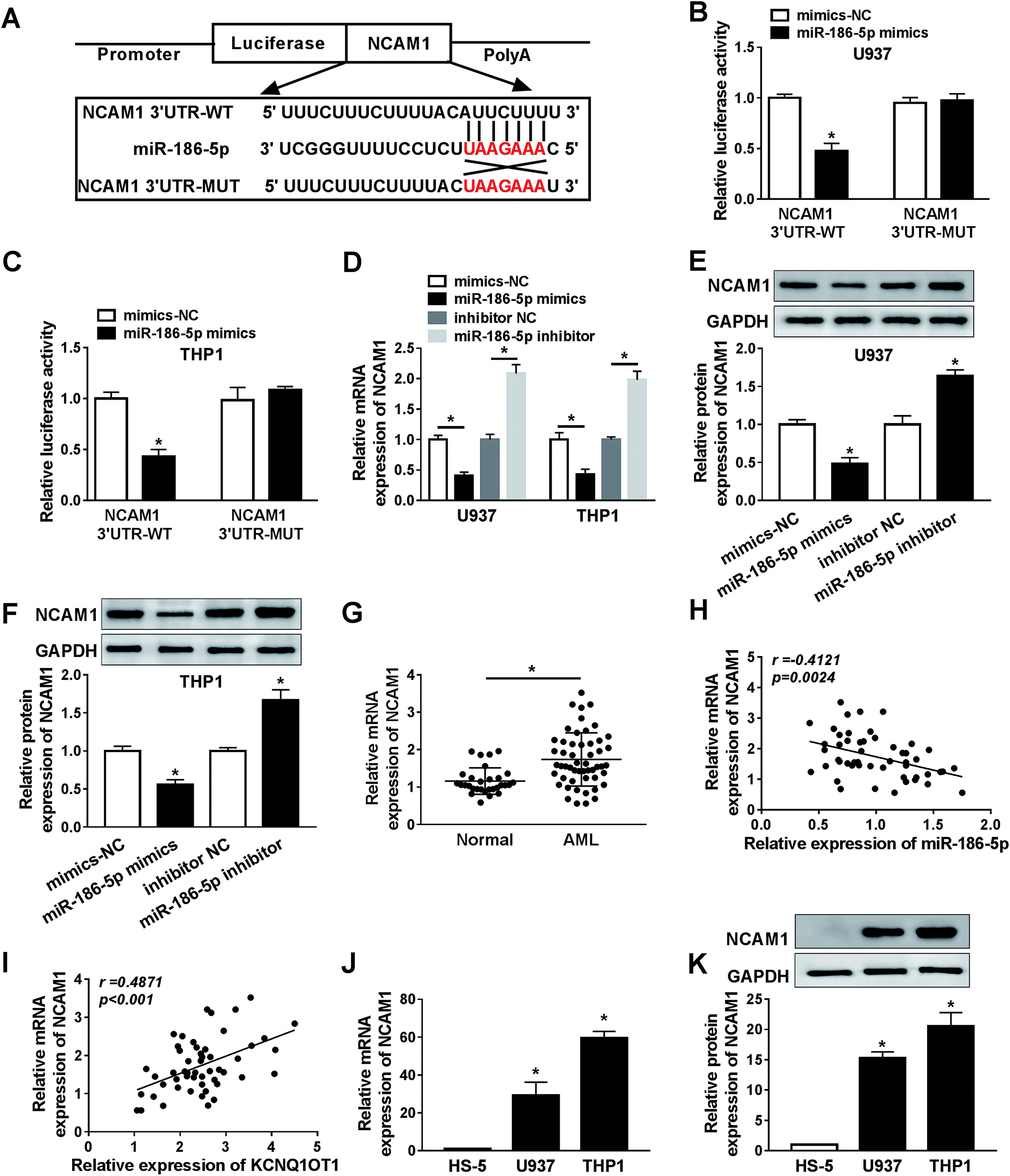 | ||
| Fig. 5 NCAM1 directly interacted with miR-186-5p. (A) Bioinformatics analysis software starbase2.0 was used to predict potential target genes of miR-186-5p. Mutated binding sites of miR-186-5p in NCAM1 were indicated in red color. (B and C) Relative luciferase activity was determined in both U937 (B) and THP1 (C) cell lines that were co-transfected with miR-186-5p mimics or mimics-NC and NCAM1 3′UTR-WT or NCAM1 3′UTR-MUT. (D, E, and F) The mRNA (D) and protein (E and F) levels of NCAM1 were examined in both U937 and THP1 cell lines transfected with mimics-NC, miR-186-5p mimics, inhibitor NC, and miR-186-5p inhibitor, respectively. (G) Relative mRNA expression of NCAM1 in AML and normal samples was detect by qRT-PCR assay. (H and I) Association between NCAM1 expression and KCNQ1OT1 expression or miR-186-5p expression was analyzed. (J and K) The mRNA (G) and protein (H) levels of NCAM1 were measured in AML cells (U937 and THP1). *P < 0.05. | ||
KCNQ1OT1 mediated AML cell progression by regulation of miR-186-5p/NCAM1 axis
Based on the above on results, it was speculated that KCNQ1OT1 regulated AML cell progression by mediating miR-186-5p and NCAM1. To confirm this hypothesis, U937 and THP1 cells were transfected with sh-NC, sh-KCNQ1OT1#1, sh-KCNQ1OT1#1 + miR-186-5p inhibitor, and sh-KCNQ1OT1#1 + pcDNA-NCAM1 respectively. Then, NCAM1 expression was detected in these conditions. As shown in Fig. 6A and B, the mRNA and protein levels of NCAM1 were decreased by KCNQ1OT1 knockdown, and then rescued by miR-186-5p depletion or NCAM1 overexpression. Subsequently, MTT assay data showed that KCNQ1OT1 knockdown inhibited cell proliferation, and the depletion of miR-186-5p or the upregulation of NCAM1 reversed this inhibition in both U937 (Fig. 6C) and THP1 (Fig. 6D) cell lines. Meanwhile, we found that miR-186-5p knockdown or NCAM1 overexpression weakened the effect of KCNQ1OT1 knockdown on cell apoptosis (Fig. 6E). Besides, the effect of KCNQ1OT1 knockdown on cell apoptosis-related protein was reversed by the depletion of miR-186-5p or the upregulation of NCAM1 in both U937 (Fig. 6F) and THP1 (Fig. 6G) cell lines. Similarly, cell chemo-sensitivity was also determined in this part. The results demonstrated that miR-186-5p downregulation or NCAM1 upregulation attenuated the effect of KCNQ1OT1 knockdown on cell chemo-sensitivity in both U937 (Fig. 6H) and THP1 (Fig. 6I) cell lines. From these data, KCNQ1OT1 mediated cell proliferation, apoptosis, and chemo-sensitivity through regulating miR-186-5p/NCAM1 axis in AML cells.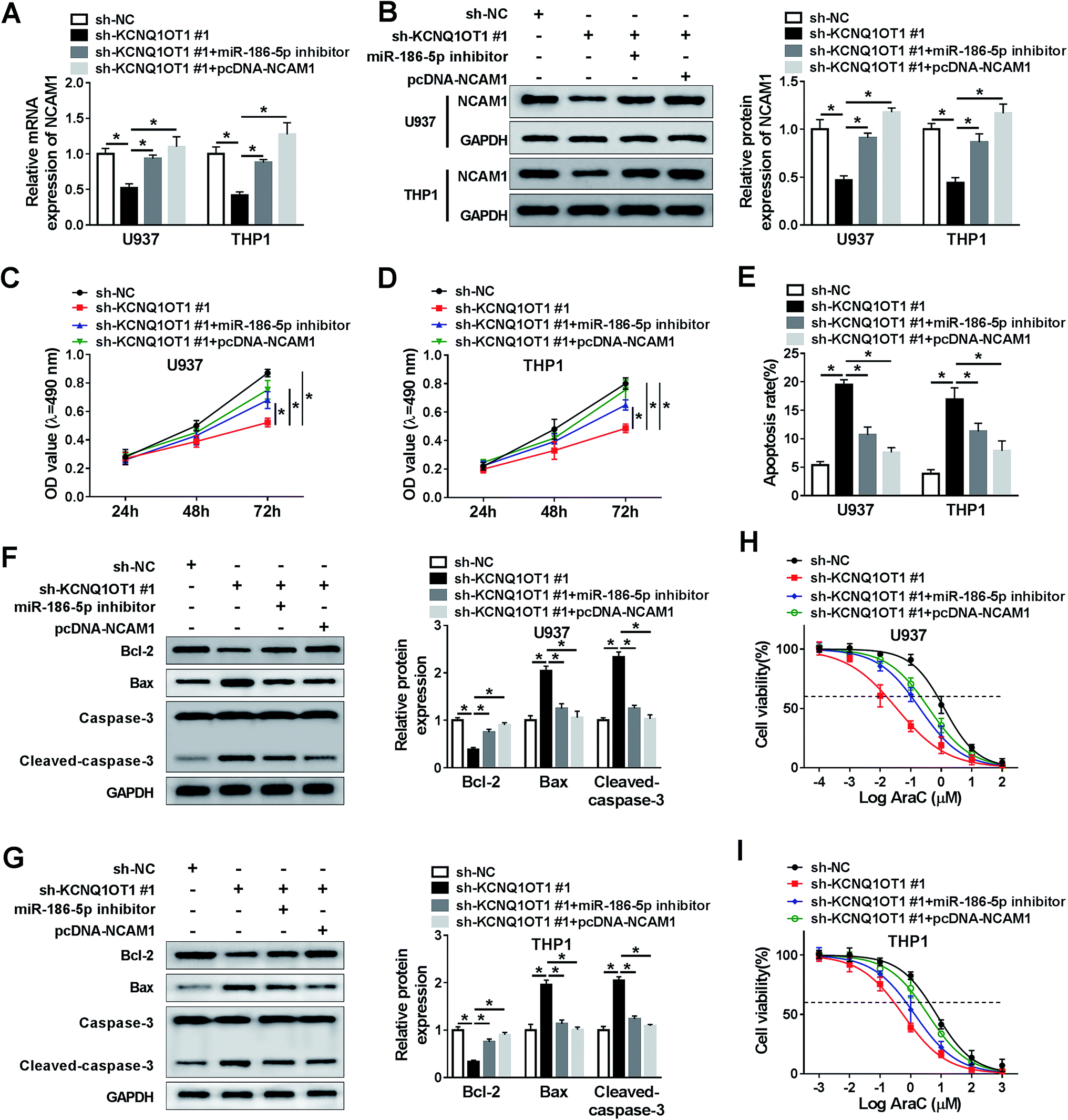 | ||
| Fig. 6 KCNQ1OT1 mediated cell proliferation, apoptosis and chemo-sensitivity through regulating miR-186-5p/NCAM1 axis in AML cells. (A) The mRNA level of NCAM1 was determined by qRT-PCR assay in both U937 and THP1 cells transfected with sh-NC, sh-KCNQ1OT1#1, sh-KCNQ1OT1#1 + miR-186-5p inhibitor, and sh-KCNQ1OT1#1 + pcDNA-NCAM1, respectively. (B) Western blot assay was performed to detect the protein level of NCAM1. (C and D) Cell proliferation ability was assessed by MTT assay in both U937 (C) and THP1 (D) cell lines. (E) Flow cytometry analysis was used to analyze cell apoptosis rate. (F and G) Cell apoptosis-related protein levels were measured in both U937 (F) and THP1 (G) cell lines. (H and I) Cell viability was examined in both U937 (H) and THP1 (I) cell lines after Ara-C treatment. *P < 0.05. | ||
Discussion
Acute myeloid leukaemia (AML) is the most common type of acute leukaemia among adults worldwide.20 LncRNAs and miRNAs have been reported to exert function in human cancer development. In this study, we confirmed that KCNQ1OT1 mediated AML cell progression through regulating miR-186-5p/NCAM1 axis, providing theoretical basis for the treatment of AML patients.KCNQ1OT1 was identified as an lncRNA that played important role in cancer development. Previous results suggested that KCNQ1OT1 was highly expressed in AML patients compared with that in control.9 The association between KCNQ1OT1 expression and AML was also analyzed in our research. Increased KCNQ1OT1 expression was observed in AML samples and cells, this results was consistent with previous data. Though the effect of KCNQ1OT1 on AML cell progression is unclear yet, Chen et al. confirmed that KCNQ1OT1 promoted proliferation through mediating the expression level of SMAD family member 4 (SMAD4) in lens epithelial cells21 and Xian et al. suggested that KCNQ1OT1 increased the methotrexate resistance of cell in colorectal cancer.22 These results confirmed that KCNQ1OT1 positively regulated cell progression. Consistence with these data, our data indicated that the KCNQ1OT1 depletion suppressed cell proliferation as well as promoted apoptosis and cell chemo-sensitivity to AraC in AML. Therefore, KCNQ1OT1 played crucial role in AML development.
Present data proved that lncRNAs were related to gene regulation and impaired the function of miRNAs by sponging miRNAs.23 Using bioinformatics analysis website starbase2.0, we found that miR-186-5p was a potential target gene of KCNQ1OT1. Next, this prediction was confirmed by the dual luciferase reporter assay. Moreover, our data also showed that KCNQ1OT1 decreased miR-186-5p expression. Therefore, it was speculated that miR-186-5p expression was lower in AML compared with that in control. As expected, our results indicated that miR-186-5p level was decreased in AML samples and cells. This result was in agreement with previous data in CML.14 Accumulating evidence demonstrated that miR-186-5p negatively modulated tumor development in various human cancers, including non-small cell lung cancer,24 gastric cancer,25 liver cancer,26 and ovarian cancer.27 But, the role of miR-186-5p in AML was not reported. Here, we confirmed that miR-186-5p upregulation suppressed cell proliferation and promoted apoptosis as well as chemo-sensitivity in AML. These data confirmed that miR-186-5p played a negative role in AML.
In human cancers, miRNAs regulate gene expression by directly binding to 3′ UTR of downstream genes that are related to tumorigenesis.28 Therefore, we predicted the potential target genes of miR-186-5p using starbase2.0, and found that miR-186-5p likely interacted with NCAM1. Subsequently, this interaction was verified by the dual luciferase reporter assay. Moreover, we also confirmed that miR-186-5p downregulated NCAM1 expression in AML. Previous results showed that NCAM1 displayed high expression level in AML patients.18 Similarly, increased NCAM1 expression was observed in AML samples and cells in our study. Taken together, it was possible that KCNQ1OT1 mediated miR-186-5p/NCAM1 axis to regulate AML cell progression. Next, this hypothesis was confirmed by our results revealing that KCNQ1OT1 knockdown suppressed cell proliferation and induced apoptosis as well as chemo-sensitivity through regulation of miR-186-5p/NCAM1 axis.
In conclusion, our data revealed that KCNQ1OT1 regulated AML cell progression via affecting miR-186-5p/NCAM1 axis, providing potential value for the treatment of AML patients.
Conflicts of interest
The authors declare that they have no financial conflicts of interest.References
- E. H. Estey, Am. J. Hematol., 2013, 88, 318–327 CrossRef CAS.
- U. Krug, C. Rollig, A. Koschmieder, A. Heinecke, M. C. Sauerland, M. Schaich, C. Thiede, M. Kramer, J. Braess, K. Spiekermann, T. Haferlach, C. Haferlach, S. Koschmieder, C. Rohde, H. Serve, B. Wormann, W. Hiddemann, G. Ehninger, W. E. Berdel, T. Buchner and C. Muller-Tidow, Lancet, 2010, 376, 2000–2008 CrossRef CAS; C. H. Tsai, H. A. Hou, J. L. Tang, C. Y. Liu, C. C. Lin, W. C. Chou, M. H. Tseng, Y. C. Chiang, Y. Y. Kuo, M. C. Liu, C. W. Liu, L. I. Lin, W. Tsay, M. Yao, C. C. Li, S. Y. Huang, B. S. Ko, S. C. Hsu, C. Y. Chen, C. T. Lin, S. J. Wu and H. F. Tien, Leukemia, 2016, 30, 1485–1492 CrossRef.
- R. L. Siegel, K. D. Miller and A. Jemal, CA Cancer J. Clin., 2019, 69, 7–34 CrossRef PubMed.
- H. Dohner, E. Estey, D. Grimwade, S. Amadori, F. R. Appelbaum, T. Buchner, H. Dombret, B. L. Ebert, P. Fenaux, R. A. Larson, R. L. Levine, F. Lo-Coco, T. Naoe, D. Niederwieser, G. J. Ossenkoppele, M. Sanz, J. Sierra, M. S. Tallman, H. F. Tien, A. H. Wei, B. Lowenberg and C. D. Bloomfield, Blood, 2017, 129, 424–447 CrossRef; H. Dohner, D. J. Weisdorf and C. D. Bloomfield, N. Engl. J. Med., 2015, 373, 1136–1152 CrossRef PubMed.
- T. Derrien, R. Johnson, G. Bussotti, A. Tanzer, S. Djebali, H. Tilgner, G. Guernec, D. Martin, A. Merkel, D. G. Knowles, J. Lagarde, L. Veeravalli, X. Ruan, Y. Ruan, T. Lassmann, P. Carninci, J. B. Brown, L. Lipovich, J. M. Gonzalez, M. Thomas, C. A. Davis, R. Shiekhattar, T. R. Gingeras, T. J. Hubbard, C. Notredame, J. Harrow and R. Guigo, Genome Res., 2012, 22, 1775–1789 CrossRef CAS.
- E. Malek, S. Jagannathan and J. J. Driscoll, Oncotarget, 2014, 5, 8027–8038 CrossRef CAS; Z. Xu, Y. Yan, L. Qian and Z. Gong, Oncol. Rep., 2017, 37, 1359–1366 CrossRef; B. Yue, D. Cai, C. Liu, C. Fang and D. Yan, Mol. Ther., 2016, 24, 2064–2077 CrossRef.
- S. Zhang, H. Ma, D. Zhang, S. Xie, W. Wang, Q. Li, Z. Lin and Y. Wang, Cell Death Discovery, 2018, 9, 742 CrossRef.
- Y. Li, C. Li, D. Li, L. Yang, J. Jin and B. Zhang, OncoTargets Ther., 2019, 12, 2649–2660 CrossRef.
- Z. W. Jia, Y. Li, G. R. Cui, H. B. Zhao, P. Y. Li and J. M. Luo, Zhongguo Shiyan Xueyexue Zazhi, 2018, 26, 653–657 Search PubMed.
- D. P. Bartel, Cell, 2004, 116, 281–297 CrossRef CAS; D. P. Bartel, Cell, 2009, 136, 215–233 CrossRef.
- J. Contreras and D. S. Rao, Leukemia, 2011, 26, 404 CrossRef.
- S. I. Negoita, D. Sandesc, A. F. Rogobete, M. Dutu, O. H. Bedreag, M. Papurica, M. F. Ercisli, S. E. Popovici, R. Dumache, M. Sandesc, A. Dinu, A. M. Sas, D. Serban and D. Corneci, Clin. Lab., 2017, 63, 1315–1321 Search PubMed; K. B. Reddy, Cancer Cell Int., 2015, 15, 38 CrossRef CAS.
- J. Li, L. Xia, Z. Zhou, Z. Zuo, C. Xu, H. Song and J. Cai, Arch. Biochem. Biophys., 2018, 640, 53–60 CrossRef CAS; Z. Zhang, W. Zhang, J. Mao, Z. Xu and M. Fan, Cell. Physiol. Biochem., 2019, 52, 553–564 CrossRef.
- J. Lin, J. C. Ma, J. Yang, J. Y. Yin, X. X. Chen, H. Guo, X. M. Wen, T. J. Zhang, W. Qian, J. Qian and Z. Q. Deng, Oncogene, 2018, 37, 2432–2443 CrossRef CAS.
- B. A. Cunningham, J. J. Hemperly, B. A. Murray, E. A. Prediger, R. Brackenbury and G. M. Edelman, Science, 1987, 236, 799–806 CrossRef CAS.
- D. K. Ditlevsen, L. B. Kohler, M. V. Pedersen, M. Risell, K. Kolkova, M. Meyer, V. Berezin and E. Bock, J. Neurochem., 2003, 84, 546–556 CrossRef CAS PubMed; G. Paratcha, F. Ledda and C. F. Ibanez, Cell, 2003, 113, 867–879 CrossRef; R. S. Schmid, R. D. Graff, M. D. Schaller, S. Chen, M. Schachner, J. J. Hemperly and P. F. Maness, J. Neurobiol., 1999, 38, 542–558 CrossRef.
- M. A. Caligiuri, Blood, 2008, 112, 461–469 CrossRef CAS.
- S. M. Sykes, Blood, 2019, 133, 2247–2248 CrossRef CAS.
- D. Sasca, J. Szybinski, A. Schuler, V. Shah, J. Heidelberger, P. S. Haehnel, A. Dolnik, O. Kriege, E. M. Fehr, W. H. Gebhardt, G. Reid, C. Scholl, M. Theobald, L. Bullinger, P. Beli and T. Kindler, Blood, 2019, 133, 2305–2319 CrossRef CAS.
- E. Estey and H. Dohner, Lancet, 2006, 368, 1894–1907 CrossRef.
- B. Chen, J. Ma, C. Li and Y. Wang, Mol. Med. Rep., 2018, 18, 16–24 CAS.
- D. Xian and Y. Zhao, J. Cell. Mol. Med., 2019, 23, 3808–3823 CrossRef CAS.
- R. Kurokawa, Prog. Mol. Subcell. Biol., 2011, 51, 29–41 CrossRef CAS PubMed; Y. Tay, J. Rinn and P. P. Pandolfi, Nature, 2014, 505, 344–352 CrossRef.
- H. Li, C. Yin, B. Zhang, Y. Sun, L. Shi, N. Liu, S. Liang, S. Lu, Y. Liu, J. Zhang, F. Li, W. Li, F. Liu, L. Sun and Y. Qi, Carcinogenesis, 2013, 34, 2145–2155 CrossRef CAS.
- C. Cao, D. Sun, L. Zhang and L. Song, Oncotarget, 2016, 7, 79956–79963 Search PubMed.
- T. Ruan, X. He, J. Yu and Z. Hang, Oncol. Lett., 2016, 11, 2941–2945 CrossRef CAS.
- X. Zhu, H. Shen, X. Yin, L. Long, C. Xie, Y. Liu, L. Hui, X. Lin, Y. Fang, Y. Cao, Y. Xu, M. Li, W. Xu and Y. Li, Oncogene, 2016, 35, 323–332 CrossRef CAS.
- A. Min, C. Zhu, S. Peng, S. Rajthala, D. E. Costea and D. Sapkota, BioMed Res. Int., 2015, 2015, 186904 Search PubMed.
| This journal is © The Royal Society of Chemistry 2019 |