
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
First total synthesis of ampullosine, a unique isoquinoline alkaloid isolated from Sepedonium ampullosporum, and of the related permethylampullosine†
Didier F. Vargas ,
Enrique L. Larghi* and
Teodoro S. Kaufman*
,
Enrique L. Larghi* and
Teodoro S. Kaufman*
Instituto de Química Rosario (IQUIR, CONICET-UNR), Facultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario, Suipacha 531, S2002LRK Rosario, Argentina. E-mail: Kaufman@iquir-conicet.gov.ar; larghi@iquir-conicet.gov.ar
First published on 16th October 2019
Abstract
A straightforward and convenient approach toward the first total synthesis of ampullosine, a structurally unique 3-methylisoquinoline alkaloid isolated from Sepedonium ampullosporum, is reported. Access to the related O-methyl ampullosine methyl ester from a common intermediate is also disclosed. The synthetic sequence toward the natural product comprised a Kolbe-type carboxylation of 3,5-dihydroxybenzoic acid and further esterification of the diacid, followed by masking of one of the phenols through selective ester reduction and subsequent acetonide formation. Installation of the three-carbon atom required for the 3-methylpyridine ring was performed by triflation of the remaining free phenol and a Pd-catalyzed Suzuki–Miyaura reaction with potassium E-propenyltrifluoroborate. Deprotection of the acetonide, followed by partial oxidation of the benzylic alcohol to the salicylaldehyde, O-methylation of the free phenol and hydrazonation of the resulting ortho-anisaldehyde derivative gave a hydrazone-based 1-azatriene. This was further subjected to 6π-azaelectrocyclization to afford permethylampullosine (11 steps, 14% overall yield), whereas exhaustive demethylation with AlI3 generated in situ gave ampullosine (12 steps, 3.2% global yield).
Introduction
The genus Sepedonium (Ascomycetes) is saproparasitic and comprises asexual states of fungi that parasitize on other fungi; more specifically basidiocarps of the mushroom order Boletales.1 They operate by first settling on their fleshy living hosts, which they kill to employ the organic matter as nutrients to grow and develop. These fungicolous species have a mould-like aspect, with thick-walled gold-yellow colored aleurioconidia and colorless thin-walled phialoconidia.The genus was established at the beginning of the XIX century. However, the seminal works of Petch and Pieschel2 described variations of S. chrysospermum, namely S. ampullosporum,3a S. chalcipori,3b and S. microspermum,3c which along with S. laevigatum ended up conforming autonomic species on the basis of sequence analysis of the internal transcribed spacer region of the nuclear ribosomal RNA genes.1
Fungi of the genus Sepedonium have been studied as producers of antibiotics and pigments, as well as for their properties as antagonist agents of various plant pathogens. Chemical investigations of Sepedonium spp. revealed that this species is an interesting source of various natural products with potential biopharmaceutical applications (Fig. 1), including peptaibols, uncommon peptides with a broad spectrum of biological activities,4 the unusual cyclic peptide chrysosporide,5 the azaphilone chrisodin (1),6 as well as some mono- and bis-anthraquinones (skyrin, rugulosin, chrysophanol)1,7 and tropolone derivatives (2–3),8 among others.5
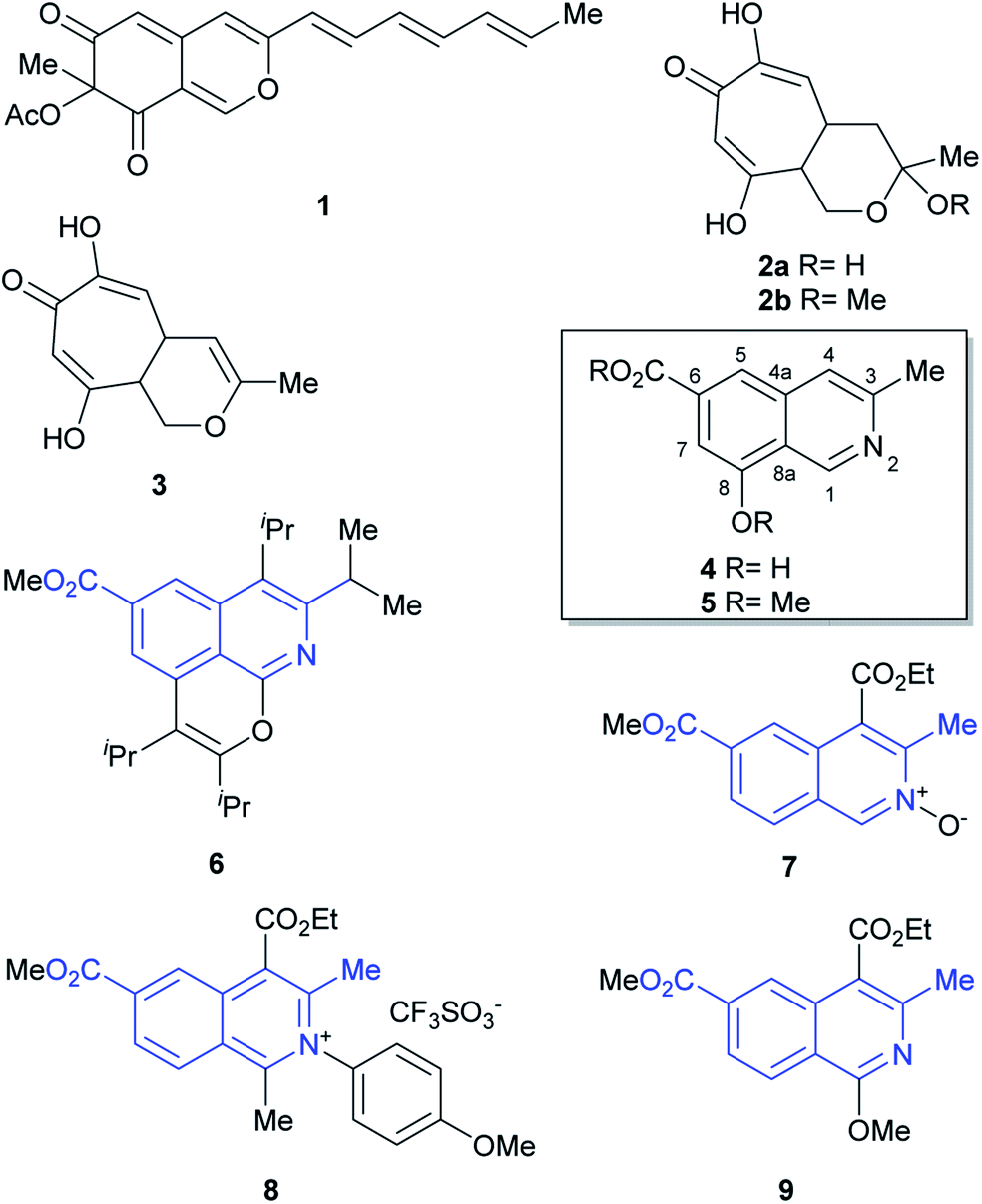 | ||
| Fig. 1 Chemical structures of some polyketides (1–3) and ampullosine (4), isolated from Sepedonium species, as well as the semisynthetic ampullosine derivative (5) and structurally related, synthetic 3-methyl 6-carboxymethyl isoquinolines (6–9). | ||
Infected organisms are rarely colonized by secondary fungal parasites, to compete with Sepedonium for host nutrients. This observation supports the speculation that Sepedonium species produce small defense molecules that prevent further infections.
Recently, the group of Arnold examined the culture extract of S. ampullosporum and isolated ampullosine (4).9 This alkaloid, responsible for the deep yellow color of its culture fluid, is the first and only isoquinoline isolated from the genus Sepedonium. Its structure was established by spectroscopic means.
In addition, methylation of the heterocycle afforded O-methyl ampullosine methyl ester (5), which contributed to the identification of the natural product. Employing LC/ESI, the authors were able to detect ampullosine in ten out of twelve strains of Sepedonium, belonging to eight different species. However, they observed that the two most phylogenetically distant species lacked the alkaloid.
The structure of ampullosine is unique. Despite some 3-methylisoquinolines have been synthesized, the combination of this motif with a 6-carboxylic acid moiety is rare; further, although they are seldom found in patents,10 a few and scattered examples started to appear in the literature just recently, favored by the flourishment of research into C–H activation reactions based on ruthenium (6)11 and rhodium (7–9)12 catalysis. In many cases, however, the current limitations of this novel chemistry still turn unfavorable its use to approach ampullosine.
We have been interested in the synthesis of structurally unique heterocyclic natural products13 and have used the 6π-electrocyclization of 1-azatrienes as an efficient and atom-economic strategy toward the synthesis of natural products or their analogs, containing the 3-methyl isoquinoline motif.14
In pursuit of these interests, herein we wish to report the first total synthesis of ampullosine (4) and the synthesis of O-methyl ampullosine methyl ester (5), from a common intermediate, using the easily available 3,5-dihydroxybenzoic acid and employing a 6π-azaelectrocyclization reaction as the key transformation, according to the retrosynthetic analysis depicted in Scheme 1.
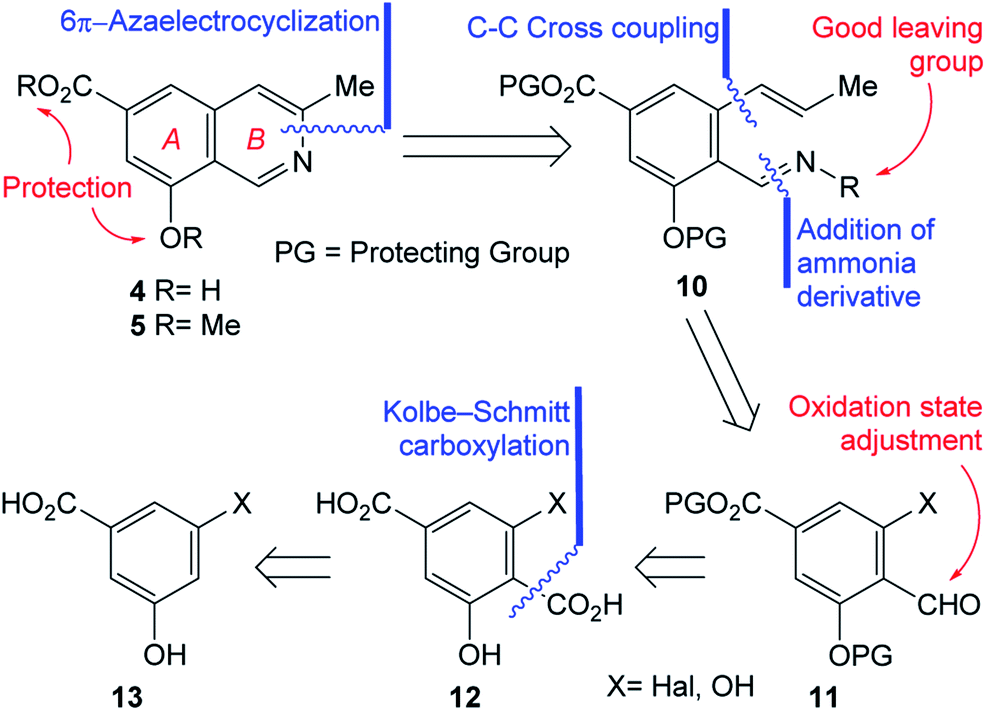 | ||
| Scheme 1 Retrosynthetic analyses of ampullosine (4) and its derivative 5. | ||
Results and discussion
The initial N–C3 disconnection of the analysis was performed on 4 and 5 and unveiled the polysubstituted benzenoid 10 as a fit key intermediate for the proposed 6π-azaelectrocyclization based ring closure toward both targets. The main features of 10 are a properly masked carboxyl group, a differentially protected phenolic moiety, according to the final targets and an ortho-disubstituted carbonyl condensed with an ammonia derivative which carries a suitable leaving group.A couple of additional disconnections were carried out on 10 to uncover the salicylaldehyde derivative 11. The reasoning was that condensation with an ammonia derivative (oxime, hydrazone, etc.) could provide the required activated nitrogen atom for the pyridine ring of the target; in addition, it was conjectured that a suitable cross-coupling reaction with an allyl/propenyl derivative15 could perform the substitution of a properly located halogen or activated phenol, enabling the installation of the three carbon atoms chain, required to build the heterocyclic ring.
Further analysis of 11 determined that its substituent pattern could be advantageously simplified towards a suitable starting material, by considering an oxidation state adjustment of the aldehyde. This placed terephthalic acid derivative 12 in the path toward the natural product. In turn, the ortho-disubstituted carboxylic acid was disconnected, to reveal compound 13 as an apt starting material, considering the availability of protocols to access salicylic acid derivatives from phenols.16
Interestingly, despite different 3-halobenzoic acids have been reported in the literature,17 they exhibited some accessibility drawbacks; among them, lack of suitable precursors or efficient transformations from commercial materials. Therefore, it was concluded that among the possible candidates to fulfil the odd structural requirements imposed by the retrosynthetic analysis, only 3,5-dihydroxybenzoic (14) is inexpensive, easily available and displays the most ideal substitution pattern.
On the other hand, despite various methodologies are available for the ortho formylation of phenols, some protocols such as Duff, Skattebøl, Reimer–Tiemann, Vilsmeier–Haack and Gattermann–Koch require activated substrates, while others (ortho metalation) demand protection of the sensitive groups, such as the carboxyl moiety, and all of them may afford mixtures of isomeric products.18 Furthermore, some literature precedents suggested that formylation of 13 may result in the undesired formyl derivative, and in low yield.19
Therefore, the synthesis commenced with 14 (Scheme 2), which was submitted to a Kolbe–Schmitt type carboxylation with KHCO3 at 180 °C, under a CO2 atmosphere (balloon).20 It was observed that the use of glycerol as reaction medium resulted in a deep red coloured gummy mass, which difficulted extraction and product purification, affording 15 in poorly reproducible yields, below 60%. Luckily, however, employing a minimum amount of propylene glycol (final concentration of 14 was ∼4 mol L−1) as solvent provided 15 as an easily recoverable pale yellow solid in better yield (80%).
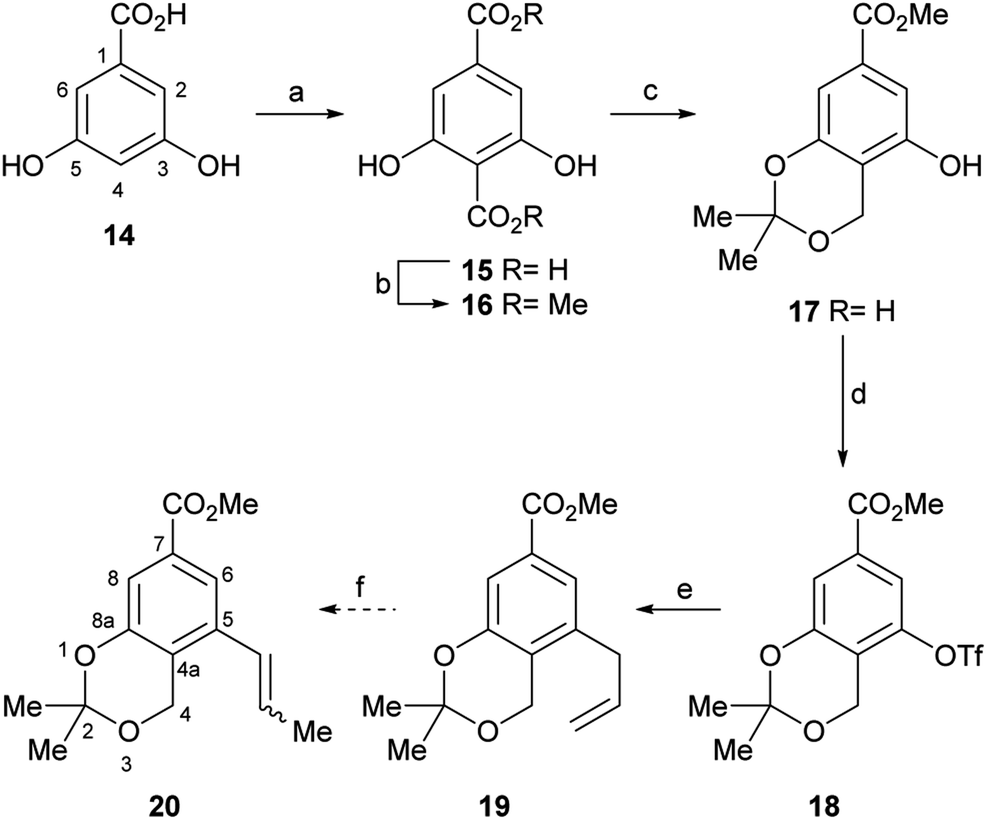 | ||
Scheme 2 Reagents and conditions: (a) CO2 (sealed tube), KHCO3, 1,2-propanediol, 180 °C, 6 h; (b) Me2SO4, KHCO3, acetone, reflux, 10 h, (70% overall); (c) (1) NaBH4, THF/H2O (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 0.1 M phosphate buffer pH 7.5, 0 °C→r.t., 1.5 h; (2) 2,2-DMP, TsOH, r.t., 5 h (75% overall); (d) PhNTf2, DMAP, NEt3, CH2Cl2, r.t. 16 h, (96%); (e) nBu3SnCH2CH :1), 0.1 M phosphate buffer pH 7.5, 0 °C→r.t., 1.5 h; (2) 2,2-DMP, TsOH, r.t., 5 h (75% overall); (d) PhNTf2, DMAP, NEt3, CH2Cl2, r.t. 16 h, (96%); (e) nBu3SnCH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, PdCl2(PPh3)2, PPh3, LiCl, diglyme, 125 °C, 15 h (19 and 20, 95:5, 70% global); (f) RuClH(CO)(PPh3)3 (cat.), PhMe, 80 °C. CH2, PdCl2(PPh3)2, PPh3, LiCl, diglyme, 125 °C, 15 h (19 and 20, 95:5, 70% global); (f) RuClH(CO)(PPh3)3 (cat.), PhMe, 80 °C. | ||
Furthermore, similar performances were observed when the reaction was carried out in a closed vessel, purged with CO2, a requirement of the reaction mechanism.21 Without purification, the resulting solid mass containing the diacid product 15 was submitted to selective esterification with Me2SO4 providing the dimethyl ester 16 (70% yield for the two steps).
In turn, the diester was selectively reduced with NaBH4, under anchimeric assistance provided by the neighbouring phenol moieties to the ortho-disubstituted C4 carboxylate, and the resulting benzyl alcohol was converted to the acetonide 17 in 75% overall yield.
In order to install the projected three carbon atom chain for the heterocyclic ring, compound 17 was next activated toward a Stille cross-coupling reaction, by triflate ester formation with PhNTf2. The latter reagent was elected over the more reactive Tf2O after considering previous failures of the anhydride in similar transformations.22a
Luckily, the reaction of 17 with N-phenyl triflimide, employing Et3N as base and DMAP as nucleophilic substitution catalyst, furnished 96% of the triflate 18.22b,c Interestingly chromatographic elution with hexane:CH2Cl2 mixtures proved critical to obtain the product free from N-phenyltriflamide, a by-product of the transformation.
In continuation of the sequence, the triflate was submitted to a Stille cross-coupling reaction with nBu3SnCH2CHCH2 in diglyme, in the presence of LiCl and under PdCl2(PPh3)2 catalysis. This afforded the expected allyl-substituted compound 19 in mixture (95:5) with the propenyl derivative E-20 in combined 70% yield, as observed in its 1H NMR spectrum. However, despite our experience, it was found comparatively difficult to obtain clean samples of 19, devoid of undesirable contamination with tin residues, which somehow hindered a proper development of the projected isomerization step toward 20.
Therefore, an alternative was sought. Fortunately for our needs, due to their improved air-stability, reduced cost and improved nucleophilicity, the use of organotrifluoroborate salts has emerged23 as an excellent alternative to the boronates and boronic acids, expensive conventional substrates for the Suzuki–Miyaura reactions.24 Their advantages are more evident when it is also considered that vinylboronic acids often require excess reagent to achieve better yields25 and suffer from stability issues, tending to polymerize.26
Therefore, the modification of the Suzuki–Miyaura reaction pioneered by Molander was implemented, and the cross-coupling of 18 was performed with potassium propenyl trifluoroborate (21)27 and Cs2CO3 as base. The reaction required optimization (Table 1), since the initial results with Pd(OAc)2 as the metal source and DavePhos as ligand afforded only moderate yields of 20 (53–58%) when the reaction was run in a toluene-water system (4:1, v/v) at 90–100 °C during 16–24 h, respectively (entries 1 and 2).
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Run no. | Catalyst | Ligand | Solvent | Temp. (°C) | Time (h) | Yield (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 18 | 19 | 20 | 22 | 23 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a The experiments were carried out at a final substrate concentration of 0.1 M in the mixture of solvents, employing 1.6 equiv. of the trifluoroborate salt, 3 equiv. Cs2CO3, 5 mol% of the Pd catalyst and 10 mol% of the ligand.b A PhMe/H2O 4:1 (v/v) medium was employed.c The reaction was carried in a dioxane/H2O 10:1 (v/v) medium.d A THF/H2O 4:1 (v/v) medium was used.e The reaction was carried out under microwave irradiation (250 W).f LiCl (0.4 equiv.) was added. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | Pd(OAc)2 | DavePhos | PhMe/H2Ob | 100 | 16 | — | 13 | 53 | 4 | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | Pd(OAc)2 | DavePhos | PhMe/H2Ob | 90 | 24 | — | 9 | 58 | 3 | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | PdCl2(PPh3)2 | DPPF | PhMe/H2Ob | 100 | 15 | — | — | 36 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | PdCl2(PPh3)2 | DavePhos | Dioxane/H2Oc | 100 | 15 | — | 3 | 47 | 3 | 2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | Pd(OAc)2 | DavePhos | THF/H2Od | 70 | 15 | 10 | — | 56 | — | 12 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | PdCl2(PPh3)2 | DavePhos | PhMe/H2Ob | 100 | 15 | — | 4 | 85 | 4 | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | PdCl2(PPh3)2 | tBuPhos | PhMe/H2Ob | 100 | 15 | 4 | 3 | 68 | 4 | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | PdCl2(PPh3)2 | DavePhos | PhMe/H2Ob,e | 100 | 0.7 | — | 5 | 69 | 6 | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | PdCl2(PPh3)2 | DavePhos | THF/H2Od,f | 80 | 24 | — | — | 95 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
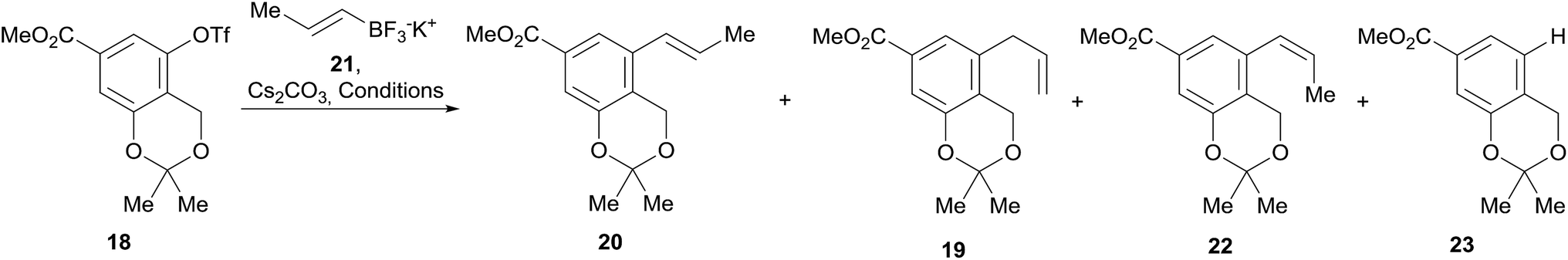
Furthermore, under these conditions, the required product was observed in mixture with minor amounts of the Z-isomer 22 (3–4%) and with the allyl derivative 19 (9–13%). This was somewhat surprising, since alkenyltrifluoroborates are known to retain the double bond geometry with a high degree of fidelity.24b
Changing the palladium source to PdCl2(PPh3)2 did not met with much better success (entries 3 and 4) when DPPF was employed as ligand (36% yield) or in the presence of DavePhos in dioxane/H2O (10:1, v/v). Similar moderate results (53–56% yield) were obtained with the use of Pd(OAc)2 and DavePhos in THF/H2O (4:1, v/v) at 70 °C for 15 h (entry 5), where some unreacted starting material (10%) was concomitantly isolated, along with the proto-deoxygenated product 23 (12%), recognized through the diagnostic signals in its 1H NMR spectrum [δ 7.55 (dd, J = 1.5 and 7.8, H-2), 7.50 (d, J = 1.5, H-6) and 7.03 (d, J = 7.8, H-3)].
Employing a toluene/water solvent in association with PdCl2(PPh3)2 was found to afford significantly improved results, attaining 85% yield of 20 (entry 6). This performance could not be surpassed by the use of tBuPhos as ligand (entry 7) nor by microwave irradiation (entry 8), which gave 20 in 68–69% yield. In all these cases, mixtures of 19, 20 and 22 were observed.
Examining the whole set of results, it was noticed that the deoxygenated product 23 was obtained only in ethereal solvents. It was conjectured that this outcome may be the result of the lack of reactivity or stability of some reaction intermediate. Thus, it was thought that addition of LiCl may improve the results.
It has been shown that LiCl is a required additive when the transformation is run in ethereal solvents, because it may act as a source of chloride ligand, stabilizing the Pd intermediates and making the cross-coupling stage more efficient.28 It has also been demonstrated that the lithium salt turns the Pd catalyst more active towards transmetallation and increases its proneness to undergo oxidative addition, resulting in a powerful accelerant of the reaction rate. Furthermore, LiCl enhances both the polarity of the solvent, and the leaving ability of anionic ligands.
Therefore, LiCl was added to a THF/H2O mixture of the starting triflate, Cs2CO3, PdCl2(PPh3)2 and DavePhos. Rewardingly, the LiCl supplemented reaction cleanly afforded 95% yield of the expected derivative 20, free of isomers and under milder conditions (80 °C), as shown in entry 9. Next, the acetonide moiety of 20 was smoothly hydrolysed by exposure to p-toluenesulfonic acid in THF/H2O, affording the benzylic alcohol 24 in almost quantitative yield (Scheme 3).
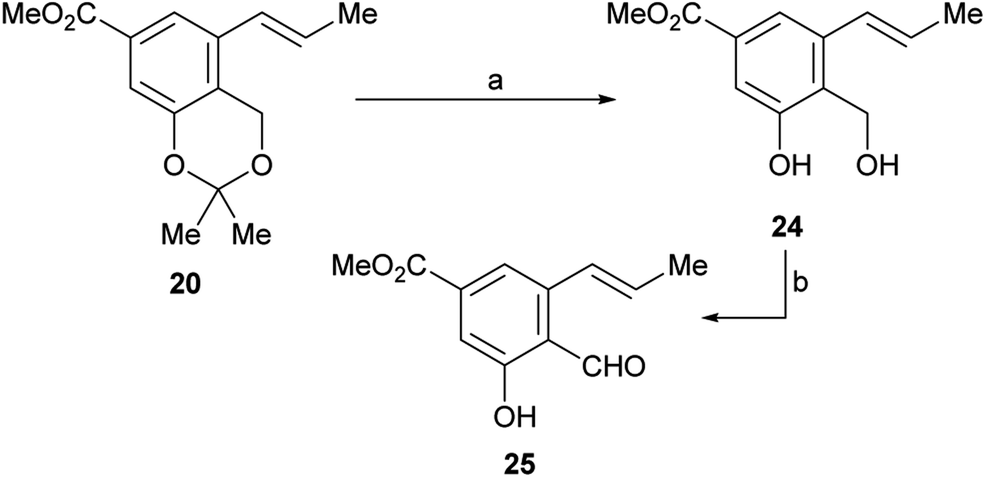 | ||
| Scheme 3 Reagents and conditions: (a) TsOH (cat.), THF/H2O, 60 °C, 48 h, (98%) or 1 M HCl, THF, 45 °C, 18 h, (90%); (b) see Table 2. | ||
However, oxidation of the latter to the corresponding aldehyde 25, which is a key step of the synthesis, proved to be tricky. The starting alcohol demonstrated to exhibit some instability, probably favoured by an easy benzylic protonation, followed by dehydration and further quinomethide formation; on the other hand, the tested oxidants were also able to oxidize the neighbouring phenol moiety to the related quinones.
As shown in Table 2, a series of trial and error experiments with PDC,29 IBX30 and activated MnO2 (ref. 31) were executed. After initial discouraging results (entries 1–3) where 25 was obtained (20–34% yield) along with unidentifiable products, presumable resulting from overoxidation,32 it was found that MnO2 provided a significantly better yield when the solvent was changed from CH2Cl2 to EtOAc (entry 4).
| Run no. | Oxidizing agent | Solvent | Temperature (°C) | Time (h) | Yield (%) |
|---|---|---|---|---|---|
| 1 | IBX | EtOAc | 50 | 1.5 | 23 |
| 2 | PDC | CH2Cl2 | r.t. | 2 | 34 |
| 3 | MnO2 | CH2Cl2 | r.t. | 16 | 20 |
| 4 | MnO2 | EtOAc | r.t. | 16 | 50 |
| 5 | MnO2 | EtOAc | 50 | 5 | 54 |
| 6 | MnO2 | EtOAc | 78 | 3 | 55 |
Additionally, it was observed that warming to 50 °C improved the performance to 54% yield (entry 5), also shortening the reaction time from 16 h to 5 h, whereas the best results (55% yield) were achieved in boiling EtOAc for 3 h (entry 6), when it was observed complete consumption of the starting material.
After having secured a satisfactory access to the key propenyl salicylaldehyde 25, the latter was submitted to our recently developed one-pot hydrazonation/6π-azaelectrocyclization protocol.33 Thus, 25 was exposed to 1,1-dimethylhydrazine in PhCF3 with the addition of AcOH as a promoter, to afford the hydrazone intermediate 26 (Scheme 4). Without purification, the hydrazone was heated at 160 °C for 45 min in the same medium, under microwave irradiation. Unexpectedly, however, the process resulted in recovery of 50% of the starting aldehyde 25, and no signs of the expected product 27 could be detected.
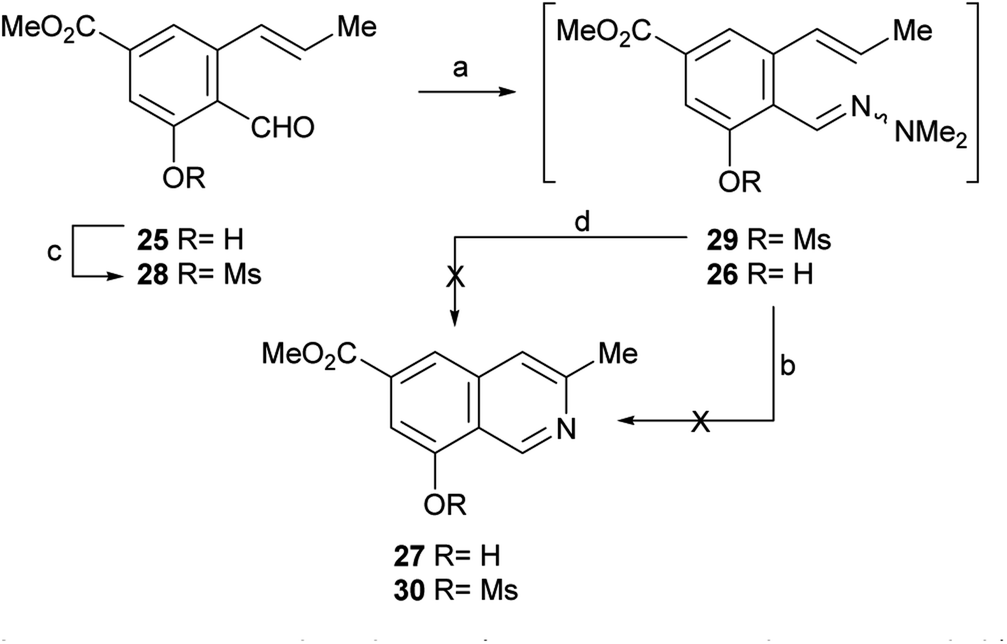 | ||
| Scheme 4 Reagents and conditions: (a) Me2N–NH2, AcOH, PhCF3, r.t., 1.5 h; (b) MW, 160 °C, 45 min (25, 50% recovered); (c) MsCl, Et3N, CH2Cl2, 0 °C→r.t., 24 h (55%); (d) MW, 160 °C, 45 min. | ||
Under the hypothesis that the presence of a free phenolic hydroxyl group could have caused the observed outcome, 25 was protected with a readily removable group. Therefore, 25 was treated with MeSO2Cl and Et3N and DMAP in CH2Cl2, to afford 55% yield of the unstable mesylate ester 28. The latter exhibited diagnostic signals of the methanesulfonate group, as a singlet resonating at δH 3.32 ppm and a resonance at δC 38.6 ppm.
Unfortunately, however, application of the one-pot hydrazonation/6π-azaelectrocyclization protocol also met with failure; neither the expected mesylate 30 nor the isoquinoline 27 were found.33
In view of these discouraging results, an alternate protecting group was sought. Considering our previous results with the benzyl ether and MOM as protecting groups,33 and taking into account our objective, to synthetically access permethylampullosine (5), the intermediate 25 was O-methylated with MeI in DMF at 0 °C, employing K2CO3 as base, to give 31 in 85% yield (Scheme 5). Then, the latter was submitted to our hydrazonation/6π-azaelectrocyclization/elimination protocol,33 affording 5 in 64% overall yield, through the intermediacy of 32a, which was not isolated because of its poor hydrolytic stability.
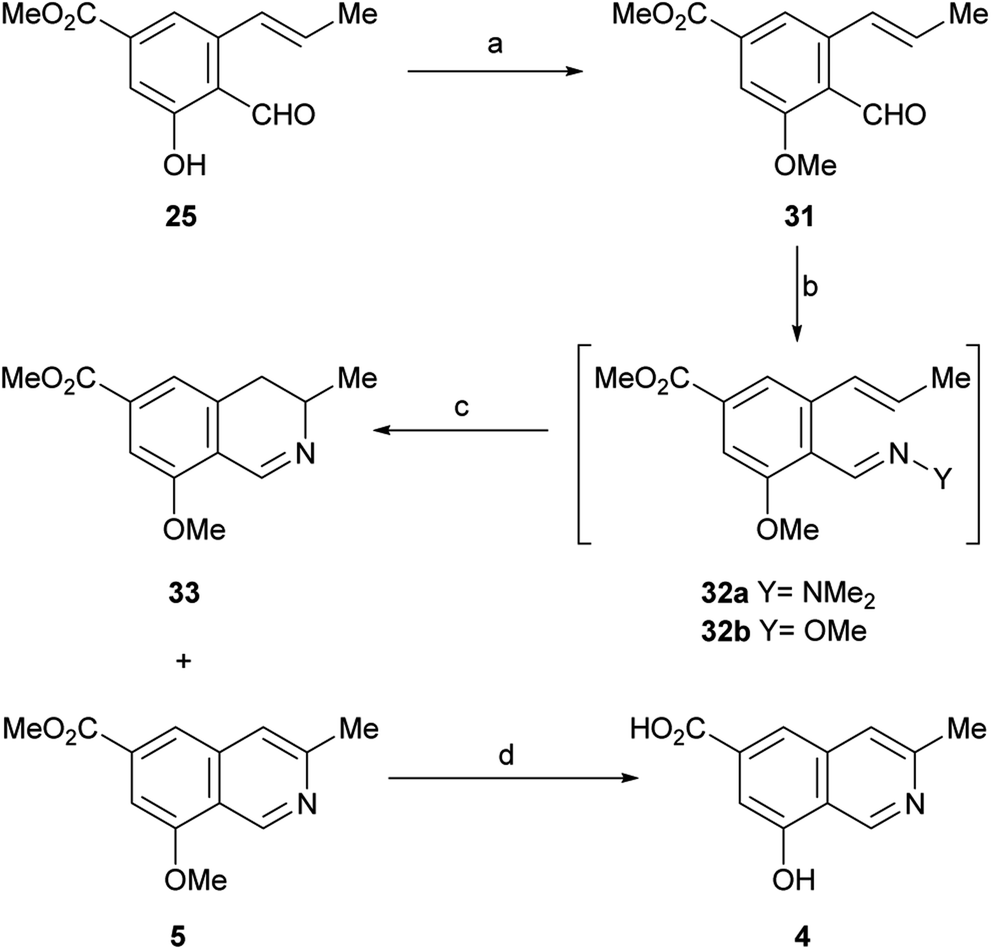 | ||
| Scheme 5 Reagents and conditions: (a) MeI, K2CO3, DMF, 0→t.a., 1.5 h (85%); (b) Me2N–NH2, AcOH, PhCF3, r.t., 1.5 h or MeO–NH2, EtOH, r.t., 1 h; (c) MW, 160 °C (via 32a: 1 h, 5, 64%; 33, 26%; via 32b; 45 min, 5, 50%; 33, 15%); (d) Al0, I2, DMSO, MeCN, 80 °C, 20 h (23%). | ||
Interestingly, the 3,4-dihydroisoquinoline 33 was isolated concomitantly in 26% yield. The latter was recognized by its pattern of signals in the 1H NMR spectrum, related to H-3 (m), H-4 (dd) and Me-3 (d). A reaction mechanism to explain the generation of 3,4-dihydroisoquinolines as side products of 6π-azaelectrocyclization of similar heterocyclic systems has been proposed.33
It has been shown that, in aqueous solution, aliphatic hydrazones are 102–103 times more sensitive to hydrolysis than the analogous oximes.34 Therefore, conjecturing that the use of a more stable ammonia derivative for condensation with 31 may improve the performance of the 6π-azaelectrocyclization, 31 was converted into the methoxime intermediate 32b by reaction with methoxylamine in absolute EtOH. After replacing the solvent with PhCF3, compound 32b was heated under microwave irradiation at 180 °C for 45 min to undergo the 6π-azaelectrocyclization, furnishing 5 in 50% yield, accompanied by the related 3,4-dihydroisoquinoline 33 (15% yield).
With these results it was concluded that the hydrazone pathway should be preferred to the methoxime alternative. The 1H and 13C NMR spectroscopic data of the synthetic compound 5 fully matched those reported by the group of Arnold for their semisynthetic O-methyl ampullosine methyl ester.9
It was also found that treatment of 5 with aluminium and iodine (which generates in situ the powerful Lewis acid AlI3),35 effected the simultaneous removal of both methyl groups, affording ampullosine (4) in 23% yield.
Experimental
General information
All the reactions were carried out under dry argon atmospheres, using oven-dried glassware and freshly distilled anhydrous solvents. Anhydrous CH2Cl2 was obtained from an M. Braun solvent purification and dispenser system. Anhydrous EtOAc was obtained by a 3 h reflux of the PA grade product over P2O5 and further distillation of the liquid. Anhydrous DMF was obtained by heating the PA grade product over BaO for 4 h, followed by distillation under reduced pressure. Anhydrous Et3N was prepared by distillation of the commercial product from CaH2. The anhydrous solvents were stored in dry Young ampoules. All other solvents and reagents were used as received.The reactions were monitored by TLC (silica gel 60 GF254) run in different hexanes:EtOAc mixtures. The chromatographic spots were revealed by exposure to UV light (254 and 365 nm) and spraying with ethanolic p-anisaldehyde/sulfuric acid reagent, followed by careful heating to improve selectivity; to detect the isoquinolines, the Dragendorff reagent (Munier and Macheboeuf modification)36a was used. The flash column chromatographies were performed under conditions that entailed a modification of known procedures.36b–d They were run under positive pressure with slurry-packed silica gel 60 H for thin layer chromatography (particle size < 55 μm), employing gradient of solvent polarity techniques with hexane:EtOAc, hexane:CH2Cl2 and EtOAc:EtOH, as required. The column size (height and diameter), amount of silica gel, solvent polarity variation (gradient) step, solvent volume per step and fraction volume were adjusted for each case (compound and reaction), taking into account mainly the amount of sample, its polarity and the presence of impurities, especially if their Rf value in the corresponding TLC analysis was close to that of the product. The chromatographic variables were adjusted aiming to elute the product approximately after the 15th tube, when the solvent polarity was equivalent to that yielding a Rf ≈ 0.50 in the TLC plate. The chromatographies were continued after elution of the expected product, in order to ensure recovery of all the relevant compounds.
Equipment
The melting points (uncorrected) were measured on an Ernst Leitz Wetzlar model 350 hot-stage microscope. The FT-IR spectra were recorded on a Shimadzu Prestige 21 spectrophotometer, as solid dispersions in KBr disks (solid samples). The NMR spectra were recorded in CDCl3 unless informed otherwise with a Bruker Avance 300 NMR spectrometer (300.13 MHz for 1H, 75.48 MHz for 13C and 282.38 MHz for 19F). In addition, some spectra were acquired with a Bruker Avance 400 MHz spectrometer (400.13 MHz for 1H, 100.61 MHz for 13C). The chemical shifts are reported in ppm on the δ scale, and TMS was used as the internal standard (δ = 0.0 ppm); the residual solvent peaks of CDCl3 (δH = 7.26 ppm, δC = 77.16 ppm), DMSO-d6 (δH = 2.50 ppm, δC = 39.52 ppm) and CD3OD (δH = 3.31 ppm, δC = 49.00 ppm) as well as the signal of CFCl3 for (δF = 0.00 ppm), were used as internal reference. The magnitudes of the coupling constant (J) values are given in hertz. To complete the elucidation, NOE and 2D NMR experiments (HSQC and HMBC) were employed. HRMS was obtained with a Bruker MicroTOF-Q II instrument from UMyMFOR (Buenos Aires, Argentina). The microwave-assisted reactions were carried out in a CEM Discover microwave reactor. The UV-Vis spectrum of ampullosine (4) was acquired in an Agilent model 8453 spectrophotometer equipped with a diode-array detector, in the range 205–700 nm and matched quartz cells (10 mm optical path). The sample was dissolved in MeOH, and the spectrum was taken against a blank of the solvent.:0 → 2:8 v/v) to afford the desired product 16 (394 mg, 70%, two steps), as a white solid, Rf: 0.53 (hexane:EtOAc, 6:4 v/v), mp: 146–148 °C (lit. 20a and 20d 146–149 °C); 1H NMR δ: 9.67 (br s, w1/2 = 10.2, 2H, ArOH), 7.11 (s, 2H, H-3 and H-5), 4.11 (s, 3H, 1-CO2Me) and 3.90 (s, 3H, 4-CO2Me); 13C NMR δ: 169.6 (1-CO2Me), 165.8 (4-CO2Me), 160.8 (C-2 and C-6), 137.3 (C-4), 109.2 (C-3 and C-5), 102.9 (C-1), 53.4 (1-CO2Me) and 52.6 (4-CO2Me).:EtOAc, 1:0 → 1:1 v/v), furnishing 17 (307 mg, 75%, two steps), as a white solid, Rf: 0.50 (hexane:EtOAc 7:3 v/v), mp: 141–142 °C; IR (KBr, ![[small nu, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e1dd.gif) ): 3327, 2995, 2846, 1697, 1595, 1431, 1365, 1276, 1149, 1060, 950, 862, 763 and 642 cm−1; 1H NMR δ: 7.15 (d, J = 1.2, 1H, H-8), 7.11 (d, J = 1.2, 1H, H-6), 5.92 (s, 1H, ArOH), 4.86 (s, 2H, H-4), 3.89 (s, 3H, CO2Me) and 1.55 (s, 6H, Me-2); 13C NMR δ: 167.2 (CO2Me), 152.5 (C-8a), 152.1 (C-5), 130.0 (C-7), 112.7 (C-4a), 111.0 (C-6), 107.7 (C-8), 99.8 (C-2), 58.1 (C-4), 52.5 (CO2Me) and 24.7 (Me-2); HRMS (ESI-TOF): found m/z: 261.0729; C12H14NaO5 ([M + Na]+) requires m/z: 261.0733.:CH2Cl2, 1:0 → 3:2 v/v), furnishing 18 (451 mg, 96%), as a white solid, Rf: 0.20 (hexane:CH2Cl2, 7:3 v/v), mp: 63–64 °C; IR (KBr, ): 3086, 2997, 2962, 1724, 1629, 1583, 1423, 1328, 1253, 1138, 1029, 925, 829, 767 and 638 cm−1; 1H NMR δ: 7.55 (d, J = 1.4, 1H, H-6), 7.51 (d, J = 1.4, 1H, H-8), 4.93 (s, 2H, H-4), 3.92 (s, 3H, CO2Me) and 1.56 (s, 6H, Me-2); 13C NMR δ: 165.1 (CO2Me), 153.1 (C-8a), 145.1 (C-5), 131.3 (C-7), 118.7 (q, J = 320, CF3), 118.6 (C-6), 118.0 (C-4a), 113.7 (C-8), 100.8 (C-2), 57.4 (C-4), 52.8 (CO2Me) and 24.6 (Me-2); 19F NMR δ: −73.3 (s, CF3SO3Ar); HRMS (ESI-TOF): found m/z: 393.0226; C13H13F3NaO7S ([M + Na]+) requires m/z: 393.0226.CH2 (31 μL, 0.10 mmol), the reaction tube was sealed and the mixture was heated at 125 °C for 15 h. After cooling to room temperature, the reaction mixture was diluted with Et2O (15 mL), a saturated solution of KF (10 mL) was added and the mixture was stirred for 20 min in order to quench the organotin-derivatives. The aqueous phase was separated and the organic layer was washed with brine (3 × 8 mL). The organic solvent was filtered through a short pad of Florisil and Celite (1:1 w/w) and the filtrate was dried over MgSO4 prior to concentration under reduced pressure. Column chromatography (hexane:EtOAc, 1:0 → 8:2 v/v) of the residue gave the corresponding allyl substituted compound 19 as a 95:5 mixture with the related β-propenyl derivative E-20 (14.6 mg, 70%), as a colour5 0.47 (hexane:EtOAc, 9:1 v/v); 1H NMR δ: 7.43 (d, J = 1.4, 1H, H-6), 7.39 (d, J = 1.4, 1H, H-8), 5.90 (ddt, J = 17.0, 10.1 and 6.2, 1H, ArCH2CHCH2), 5.10 (ddt, J = 10.1, 1.4 and 1.4, 1H, ArCH2CHCH2cis), 5.00 (ddt, J = 17.0, 1.6 and 1.6, 1H, ArCH2CHCH2trans), 4.83 (s, 2H, H-4), 3.88 (s, 3H, CO2Me), 3.25 (dt, J = 6.2 and 1.6, 2H, ArCH2CHCH2) and 1.52 (s, 6H, Me-2); 13C NMR δ: 166.9 (CO2Me), 151.5 (C-8a), 136.2 (C-5), 135.1 (ArCH2CHCH2), 130.0 (C-7), 123.2 (C-4a), 122.3 (C-6), 116.8 (ArCH2CHCH2), 116.7 (C-8), 99.3 (C-2), 59.7 (C-4), 52.2 (CO2Me), 36.2 (ArCH2CHCH2) and 24.7 (Me-2).:EtOAc, 1:0 → 8:2 v/v), to afford the desired product 20 (67 mg, 95%), as a white solid, Rf: 0.47 (hexane:EtOAc, 9:1 v/v), mp: 70–71 °C; IR (KBr, ): 3030, 2999, 2850, 1722, 1577, 1431, 1338, 1224, 1141, 1012, 904, 819 and 767 cm−1; 1H NMR δ: 7.67 (d, J = 1.4, 1H, H-6), 7.36 (d, J = 1.4, 1H, H-8), 6.20–6.33 (m, 2H, ArCHCHMe and ArCHCHMe), 4.88 (s, 2H, H-4), 3.89 (s, 3H, CO2Me), 1.91 (d, J = 4.7, 3H, ArCHCHMe) and 1.53 (s, 6H, Me-2); 13C NMR δ: 167.0 (CO2Me), 151.4 (C-8a), 135.2 (C-5), 130.2 (ArCHCHMe), 130.0 (C-7), 125.6 (ArCHCHMe), 121.2 (C-4a), 118.8 (C-8), 116.6 (C-6), 99.2 (C-2), 60.2 (C-4), 52.2 (CO2Me), 24.7 (Me-2) and 18.9 (ArCHCHMe); HRMS (ESI-TOF): found m/z: 285.1098; C15H18NaO4 ([M + Na]+) requires m/z: 285.1097.
): 3327, 2995, 2846, 1697, 1595, 1431, 1365, 1276, 1149, 1060, 950, 862, 763 and 642 cm−1; 1H NMR δ: 7.15 (d, J = 1.2, 1H, H-8), 7.11 (d, J = 1.2, 1H, H-6), 5.92 (s, 1H, ArOH), 4.86 (s, 2H, H-4), 3.89 (s, 3H, CO2Me) and 1.55 (s, 6H, Me-2); 13C NMR δ: 167.2 (CO2Me), 152.5 (C-8a), 152.1 (C-5), 130.0 (C-7), 112.7 (C-4a), 111.0 (C-6), 107.7 (C-8), 99.8 (C-2), 58.1 (C-4), 52.5 (CO2Me) and 24.7 (Me-2); HRMS (ESI-TOF): found m/z: 261.0729; C12H14NaO5 ([M + Na]+) requires m/z: 261.0733.:CH2Cl2, 1:0 → 3:2 v/v), furnishing 18 (451 mg, 96%), as a white solid, Rf: 0.20 (hexane:CH2Cl2, 7:3 v/v), mp: 63–64 °C; IR (KBr, ): 3086, 2997, 2962, 1724, 1629, 1583, 1423, 1328, 1253, 1138, 1029, 925, 829, 767 and 638 cm−1; 1H NMR δ: 7.55 (d, J = 1.4, 1H, H-6), 7.51 (d, J = 1.4, 1H, H-8), 4.93 (s, 2H, H-4), 3.92 (s, 3H, CO2Me) and 1.56 (s, 6H, Me-2); 13C NMR δ: 165.1 (CO2Me), 153.1 (C-8a), 145.1 (C-5), 131.3 (C-7), 118.7 (q, J = 320, CF3), 118.6 (C-6), 118.0 (C-4a), 113.7 (C-8), 100.8 (C-2), 57.4 (C-4), 52.8 (CO2Me) and 24.6 (Me-2); 19F NMR δ: −73.3 (s, CF3SO3Ar); HRMS (ESI-TOF): found m/z: 393.0226; C13H13F3NaO7S ([M + Na]+) requires m/z: 393.0226.CH2 (31 μL, 0.10 mmol), the reaction tube was sealed and the mixture was heated at 125 °C for 15 h. After cooling to room temperature, the reaction mixture was diluted with Et2O (15 mL), a saturated solution of KF (10 mL) was added and the mixture was stirred for 20 min in order to quench the organotin-derivatives. The aqueous phase was separated and the organic layer was washed with brine (3 × 8 mL). The organic solvent was filtered through a short pad of Florisil and Celite (1:1 w/w) and the filtrate was dried over MgSO4 prior to concentration under reduced pressure. Column chromatography (hexane:EtOAc, 1:0 → 8:2 v/v) of the residue gave the corresponding allyl substituted compound 19 as a 95:5 mixture with the related β-propenyl derivative E-20 (14.6 mg, 70%), as a colour5 0.47 (hexane:EtOAc, 9:1 v/v); 1H NMR δ: 7.43 (d, J = 1.4, 1H, H-6), 7.39 (d, J = 1.4, 1H, H-8), 5.90 (ddt, J = 17.0, 10.1 and 6.2, 1H, ArCH2CHCH2), 5.10 (ddt, J = 10.1, 1.4 and 1.4, 1H, ArCH2CHCH2cis), 5.00 (ddt, J = 17.0, 1.6 and 1.6, 1H, ArCH2CHCH2trans), 4.83 (s, 2H, H-4), 3.88 (s, 3H, CO2Me), 3.25 (dt, J = 6.2 and 1.6, 2H, ArCH2CHCH2) and 1.52 (s, 6H, Me-2); 13C NMR δ: 166.9 (CO2Me), 151.5 (C-8a), 136.2 (C-5), 135.1 (ArCH2CHCH2), 130.0 (C-7), 123.2 (C-4a), 122.3 (C-6), 116.8 (ArCH2CHCH2), 116.7 (C-8), 99.3 (C-2), 59.7 (C-4), 52.2 (CO2Me), 36.2 (ArCH2CHCH2) and 24.7 (Me-2).:EtOAc, 1:0 → 8:2 v/v), to afford the desired product 20 (67 mg, 95%), as a white solid, Rf: 0.47 (hexane:EtOAc, 9:1 v/v), mp: 70–71 °C; IR (KBr, ): 3030, 2999, 2850, 1722, 1577, 1431, 1338, 1224, 1141, 1012, 904, 819 and 767 cm−1; 1H NMR δ: 7.67 (d, J = 1.4, 1H, H-6), 7.36 (d, J = 1.4, 1H, H-8), 6.20–6.33 (m, 2H, ArCHCHMe and ArCHCHMe), 4.88 (s, 2H, H-4), 3.89 (s, 3H, CO2Me), 1.91 (d, J = 4.7, 3H, ArCHCHMe) and 1.53 (s, 6H, Me-2); 13C NMR δ: 167.0 (CO2Me), 151.4 (C-8a), 135.2 (C-5), 130.2 (ArCHCHMe), 130.0 (C-7), 125.6 (ArCHCHMe), 121.2 (C-4a), 118.8 (C-8), 116.6 (C-6), 99.2 (C-2), 60.2 (C-4), 52.2 (CO2Me), 24.7 (Me-2) and 18.9 (ArCHCHMe); HRMS (ESI-TOF): found m/z: 285.1098; C15H18NaO4 ([M + Na]+) requires m/z: 285.1097.Method A. A stirred solution of the acetonide 20 (187 mg, 0.71 mmol) and p-TsOH.H2O (26 mg, 0.14 mmol) in a THF
:H2O mixture (1:1 v/v, 14 mL) was heated at 60 °C for 48 h. The reaction was cooled to 0 °C, quenched with saturated aqueous NaHCO3 (30 mL), and the resulting mixture was extracted with EtOAc (3 × 50 mL). The combined organic layers were dried over MgSO4, and concentrated in vacuo. The crude residue was purified by column chromatography (hexane:EtOAc, 1:0 → 4:6 v/v), to give 24 (154 mg, 98%), as a white solid, Rf: 0.25 (hexane:EtOAc, 7:3 v/v), mp: 95–97 °C; IR (KBr, ): 3502, 3385, 3169, 2953, 2850, 1689, 1581, 1427, 1340, 1247, 1099, 1001, 964, 883 and 761 cm−1; 1H NMR δ: 8.12 (br s, 1H, W1/2 = 15.3 Hz, ArOH), 7.54 (d, J = 1.6, 1H, H-6), 7.35 (d, J = 1.6, 1H, H-2), 6.48 (dq, J = 15.5 and 1.7, 1H, ArCHCHMe), 6.08 (dq, J = 15.5 and 6.6, 1H, ArCHCHMe), 4.96 (s, 2H, ArCH2OH), 3.88 (s, 3H, CO2Me), 3.04 (br s, 1H, W1/2 = 21.8 Hz, ArCH2OH) and 1.87 (dd, J = 6.6 and 1.7, 3H, ArCHCHMe); 13C NMR δ: 167.3 (CO2Me), 156.4 (C-3), 138.2 (C-5), 130.7 (ArCHCHMe), 130.3 (C-4), 127.2 (ArCHCHMe), 126.5 (C-1), 120.0 (C-6), 115.8 (C-2), 60.1 (ArCH2OH), 52.4 (CO2Me) and 18.8 (ArCHCHMe); HRMS (ESI-TOF): found m/z: 245.0782; C12H14NaO4 ([M + Na]+) requires m/z: 245.0784.
Method B. A stirred solution of 20 (98 mg, 0.37 mmol) in THF (4.5 mL) was treated with 1 M HCl (3 mL) and the system was heated at 45 °C for 18 h. The reaction was cooled to 0 °C and quenched with saturated NaHCO3 (10 mL); the organic products were extracted with EtOAc (3 × 15 mL). The combined organic layers were dried over MgSO4, and concentrated in vacuo. The crude residue was purified by column chromatography to give 24 (74 mg, 90%). The spectroscopic data of the product were in full agreement with those of the product synthesized according to Method A.
:EtOAc, 1:0 → 7:3 v/v), to afford 25 (27 mg, 55%), as a yellow solid, Rf: 0.50 (hexane:EtOAc, 8:2 v/v), mp: 140–141 °C; IR (KBr, ): 3043, 2990, 2848, 1716, 1647, 1558, 1417, 1348, 1247, 1174, 1097, 999, 887 and 766 cm−1; 1H NMR δ: 11.74 (s, 1H, ArOH), 10.36 (s, 1H, CHO), 7.53 (d, J = 1.4, 1H, H-6), 7.46 (d, J = 1.4, 1H, H-2), 6.88 (dq, J = 15.5 and 1.7, 1H, ArCHCHMe), 6.21 (dq, J = 15.5 and 6.6, 1H, ArCHCHMe), 3.93 (s, 3H, CO2Me) and 1.97 (dd, J = 6.6 and 1.7, 3H, ArCHCHMe); 13C NMR δ: 195.8 (CHO), 165.9 (CO2Me), 162.5 (C-3), 143.8 (C-5), 137.3 (C-1), 135.0 (ArCHCHMe), 125.2 (ArCHCHMe), 119.5 (C-4), 119.4 (C-6), 117.4 (C-2), 52.7 (CO2Me) and 19.1 (ArCHCHMe); HRMS (ESI-TOF): found m/z: 243.0633; C12H12NaO4 ([M + Na]+) requires m/z: 243.0628.:EtOAc, 1:0 → 2:8 v/v) to afford 28 (27 mg, 55%; 77% br s m), as an unstable colorless oil Rf: 0.56 (hexane:EtOAc, 1:1 v/v); 1H NMR δ: 10.43 (s, CHO), 8.13 (d, J = 1.4, 1H, H-2), 7.88 (d, J = 1.4, 1H, H-6), 7.06 (dd, J = 15.6 and 1.7, 1H, ArCHCHMe), 6.30 (dq, J = 15.6 and 6.6, 1H, ArCHCHMe), 3.96 (s, 3H, CO2Me), 3.32 (s, 3H, MeSO3−) and 1.93 (dd, J = 6.6 and 1.7, 3H, ArCHCHMe); 13C NMR δ: 189.7 (CHO), 164.9 (CO2Me), 149.3 (C-3), 142.5 (C-5), 134.8 (C-1), 134.4 (ArCHCHMe), 129.0 (ArCHCHMe), 127.6 (C-4), 126.6 (C-6), 122.1 (C-2), 52.8 (CO2Me), 38.6 (MeSO3−) and 18.9 (ArCHCHMe).:EtOAc, 1:0 → 7:3 v/v) to afford 31 (48 mg, 85%), as a yellowish-white solid Rf: 0.52 (hexane:EtOAc, 8:2, v/v), mp: 72–73 °C; IR (KBr, ): 3086, 2954, 2873, 2785, 1718, 1681, 1566, 1404, 1330, 1238, 1103, 1010, 964, 819 and 765 cm−1; 1H NMR δ: 10.61 (d, J = 0.5, CHO), 7.77 (ddd, J = 1.4, 0.7 and 0.7, 1H, H-6), 7.48 (d, J = 1.4, 1H, H-2), 7.20 (dq, J = 15.5 and 1.7, 1H, ArCHCHMe), 6.30 (dq, J = 15.5 and 6.6, 1H, ArCHCHMe), 3.95 (s, 3H, ArOMe), 3.95 (s, 3H, CO2Me) and 1.93 (dd, J = 6.6 and 1.7, 3H, ArCHCHMe); 13C NMR δ: 192.3 (CHO), 166.3 (CO2Me), 162.3 (C-3), 141.2 (C-5), 134.8 (C-1), 131.7 (ArCHCHMe), 128.5 (ArCHCHMe), 124.6 (C-4), 120.8 (C-6), 110.1 (C-2), 56.3 (ArOMe), 52.7 (CO2Me) and 18.9 (ArCHCHMe); HRMS (ESI-TOF): found m/z: 235.0960; C13H15O4 ([M + H]+) requires m/z: 235.0965.Method A. Methyl 4-formylbenzoate 31 (22 mg, 0.09 mmol), glacial AcOH (5 μL, 0.09 mmol) and 1,1-dimethylhydrazine (7 μL, 0.09 mmol) were added to a microwave tube and diluted with PhCF3 (1 mL). The mixture was purged with argon and stirred at room temperature until TLC analysis indicated complete consumption of the starting aldehyde (∼1.5 h). Then, the vessel was capped and irradiated in the microwave reactor (160 °C, ca. 250 W, 1 h). After cooling to room temperature, the solvent was recovered by careful distillation under atmospheric pressure, and the residue was purified by column chromatography (hexane/EtOAc, 1
:0 → 0:1 v/v), to afford 5 (13.3 mg, 64%), as a yellowish solid, Rf: 0.60 (hexane/EtOAc, 6:4 v/v), mp: 126–129 °C; IR (KBr, ): 3068, 2947, 2848, 1716, 1633, 1570, 1433, 1354, 1238, 1122, 1035, 927, 825 and 762 cm−1; 1H NMR (300 MHz, CDCl3) δ: 9.56 (s, 1H, H-1), 8.03 (s, 1H, H-5), 7.50 (s, 1H, H-4), 7.37 (d, J = 1.2, 1H, H-7), 4.07 (s, 3H, ArOMe), 3.99 (s, 3H, CO2Me) and 2.71 (s, 3H, Me-3); 13C NMR (75 MHz, CDCl3) δ: 166.9 (CO2Me), 156.9 (C-8), 153.5 (C-3), 147.4 (C-1), 137.2 (C-4a), 132.2 (C-6), 121.2 (C-5), 120.6 (C-8a), 119.1 (C-4), 103.6 (C-7), 56.0 (ArOMe), 52.7 (CO2Me) and 24.3 (Me-3); 1H NMR (400 MHz, CD3OD) δ: 9.31 (s, 1H, H-1), 7.94 (s, 1H, H-5), 7.58 (s, 1H, H-4), 7.34 (s, 1H, H-7), 4.05 (s, 3H, ArOMe), 3.97 (s, 3H, CO2Me) and 2.63 (s, 3H, Me-3); 13C NMR (100 MHz, CD3OD) δ: 167.8 (CO2Me), 158.0 (C-8), 153.7 (C-3), 147.5 (C-1), 138.6 (C-4a), 134.0 (C-6), 121.8 (C-5), 121.5 (C-8a), 121.0 (C-4), 104.7 (C-7), 56.5 (ArOMe), 53.1 (CO2Me) and 23.5 (Me-3); HRMS (ESI-TOF): found m/z: 254.0798; C13H13NNaO3 ([M + Na]+) requires m/z: 254.0788. The 1H and 13C NMR spectral data of compound 5 were in agreement with the literature.9Increasing solvent polarity afforded 33 (5.5 mg, 26%), as an amber solid, mp: 99–101 °C; IR (KBr, ): 3005, 2951, 2837, 1724, 1618, 1570, 1446, 1319, 1222, 1112, 1001, 968, 864 and 767 cm−1; 1H NMR (300 MHz, CDCl3) δ: 8.72 (d, J = 2.6, 1H, H-1), 7.45 (s, 1H, H-5), 7.42 (s, 1H, H-7), 3.93 (s, 6H, ArOMe and CO2Me), 3.60–3.70 (m, 1H, H-3), 2.78 (dd, J = 16.2 and 5.6, 1H, HB-4), 2.49 (dd, J = 16.2 and 12.1, 1H, HB-4) and 1.39 (d, J = 6.9, 3H, Me-3); 13C NMR (75 MHz, CDCl3) δ: 166.6 (CO2Me), 156.9 (C-8), 154.1 (C-1), 138.3 (C-4a), 133.1 (C-6), 121.2 (C-7), 120.2 (C-8a), 110.6 (C-5), 55.9 (ArOMe), 52.5 (CO2Me), 52.0 (C-3), 32.5 (C-4) and 21.6 (Me-3); HRMS (ESI-TOF): found m/z: 256.0949; C13H15NNaO3 ([M + Na]+) requires m/z: 256.0944.
Method B. A mixture of O-methylhydroxylamine hydrochloride (16 mg, 0.19 mmol) and NaOAc (17 mg, 0.21 mmoL) in absolute EtOH (1.5 mL) was stirred at room temperature for 30 min under argon, when the resulting suspension was allowed to settle. An aliquot of the solution (1 mL) was transferred to a microwave tube containing the methyl 4-formylbenzoate 31 (23 mg, 0.1 mmol) and the resulting mixture was stirred at room temperature for 1 h under argon. Then, the solvent was removed under reduced pressure and the residue was dissolved in PhCF3 (1 mL). The tube was purged with Argon and the mixture was irradiated in the microwave reactor (180 °C, ca. 250 W, 45 min). After cooling to room temperature, the solvent was recovered by careful distillation under atmospheric pressure, and the residue was purified through a short column chromatography, to afford a separable mixture of 5 (11.5 mg, 50%) and 33 (3.5 mg, 15%). Their spectroscopic data were in full agreement with those of the corresponding products, synthesized employing Method A.
:EtOH, 1:0 → 2:8 v/v), furnishing 4 (3.7 mg, 23%), as a yellow solid, Rf: 0.40 (EtOAc:EtOH, 1:1 v/v). UV-Vis (MeOH): 227 nm (logε = 4.3) and 356 nm (logε = 3.5). 1H NMR (300 MHz, DMSO-d6) δ: 10.88 (bs, 1H, OH), 9.37 (s, 1H, H-1), 7.85 (s, 1H, H-5), 7.64 (s, 1H, H-4), 7.45 (s, 1H, H-7), 3.35 (s, 1H, OH) and 2.59 (s, 3H, Me-3); 13C NMR (100 MHz, DMSO-d6) δ 168.3 (6-CO2H), 154.8 (C-8), 152.2 (C-3), 147.2 (C-1), 137.3 (2C, C-4a and C-6), 119.5 (C-8a), 119.1 (C-4), 118.3 (C-5), 109.2 (C-7) and 24.2 (3-Me). HRMS (ESI-TOF): found m/z: 204.0659; C11H10NO3 ([M + H]+) requires m/z: 204.0655; found m/z: 226.0471; C11H9NNaO3 ([M + Na]+) requires m/z: 226.0475. The UV-Vis, 1H and 13C NMR spectral data were in agreement with the literature.9Conclusion
We have developed a straightforward and convenient approach toward the first total synthesis of ampullosine, a structurally unique 6-carboxy-3-methylisoquinoline. The heterocycle is the first and only alkaloid isolated from the saproparasitic fungus Sepedonium ampullosporum. The synthesis of O-methyl ampullosine methyl ester, from a common intermediate, is also reported.The synthesis of the natural product used 3,5-dihydroxybenzoic acid as starting material and its key steps included a Kolbe–Schmitt type carboxylation for introduction of C1 of the heterocyclic ring, a Molander cross-coupling with potassium propenyl trifluoroborate to install the remaining three carbon atoms, a carbonyl hydrazonation to add the heterocyclic nitrogen and a final 6π-azaelectrocyclization to build the isoquinoline system.
The total synthesis of ampullosine was achieved in 12 steps and 3.2% overall yield, whereas that of its permethyl derivative was accomplished in 14% overall yield, after 11 steps. Hydrolytic demethylation of O-methyl ampullosine methyl ester also afforded ampullosine. To the best of our knowledge, this is the first report of an electrocyclization reaction toward isoquinolines bearing a 6-carboxylic acid derivative.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET, PUE IQUIR 2016) and Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT, PICT 2017-0149) for financial support. D. F. V. also thanks CONICET for his Doctoral fellowship. We are also indebted to the ASaCTeI for an institutional grant (AC 2015-0005) to acquire a 400 MHz NMR spectrometer, and dedicate the paper to the memories of Prof. Dr Edmundo A. Rúveda (1934–2018), founder and first Director of IQUIR and of our friend BSc. María V. Méndez (1987–2019).Notes and references
- T. Sahr, H. Ammer, H. Besl and M. Fischer, Mycologia, 1999, 91, 935–943 CrossRef.
- (a) T. Petch, Trans. Br. Mycol. Soc., 1938, 21, 243–305 CrossRef; (b) E. Pieschel, Mykol. Mitteilungsblatt, 1962, 6, 62–68 Search PubMed.
- (a) S. C. Damon, Mycologia, 1952, 44, 86–96 CrossRef; (b) M. S. Foster, G. F. Bills and G. M. Mueller, Biodiversity of Fungi: Inventory and Monitoring Methods, Academic Press, New York, USA, 2004 Search PubMed; (c) H. Besl, A. Hagn, A. Jobst and U. Lange, Z. Mykol., 1998, 64, 45–52 Search PubMed.
- (a) T. Neuhof, A. Berg, H. Besl, T. Schwecke, R. Dieckmann and H. von Döhren, Chem. Biodiversity, 2007, 4, 1103–1115 CrossRef CAS; (b) A. Otto, A. Laub, L. Wendt, A. Porzel, J. Schmidt, G. Palfner, J. Becerra, D. Krüger, M. Stadler, L. Wessjohann, B. Westermann and N. Arnold, J. Nat. Prod., 2016, 79, 929–938 CrossRef CAS; (c) M. Kronen, P. Kleinwächter, B. Schlegel, A. Härtl and U. Gräfe, J. Antibiot., 2001, 54, 175–178 CrossRef CAS; (d) M. I. Mitova, A. Murphy, G. Lang, J. W. Blunt, A. L. J. Cole, G. Ellis and M. H. G. Munro, J. Nat. Prod., 2008, 71, 1600–1603 CrossRef CAS; (e) T. Degenkolb, J. Kirschbaum and H. Brückner, Chem. Biodiversity, 2007, 4, 1052–1067 CrossRef CAS.
- M. I. Mitova, B. G. Stuart, G. H. Cao, J. W. Blunt, A. L. J. Cole and M. H. G. Munro, J. Nat. Prod., 2006, 69, 1481–1484 CrossRef CAS.
- (a) A. Closse and D. Hauser, Helv. Chim. Acta, 1973, 56, 2694–2698 CrossRef CAS; (b) H. Haraguchi, M. Taniguchi, K. Motoba, K. Shibata, S. Oi and K. Hashimoto, Agric. Biol. Chem., 1990, 54, 2167–2168 CAS.
- S. Shibata, J. Shoji, A. Ohta and M. Watanabe, Chem. Pharm. Bull., 1957, 5, 380–382 CrossRef CAS.
- (a) P. V. Divekar, H. Raistrick, T. A. Dobson and L. C. Vining, Can. J. Chem., 1965, 43, 1835–1848 CrossRef CAS; (b) K. Nagao, N. Yoshida, K. Iwai, T. Sakai, M. Tanaka and T. Miyahara, Environ. Sci., 2006, 13, 251–256 CAS; (c) J. L. C. Wright, A. G. McInnes, D. G. Smith and L. C. Vining, Can. J. Chem., 1970, 48, 2702–2708 CrossRef CAS.
- D. N. Quang, J. Schmidt, A. Porzel, L. Wessjohann, M. Haid and N. Arnold, Nat. Prod. Commun., 2010, 5, 869–872 CrossRef CAS.
- (a) A. McDonald, WO Pat., 2015103317, 2015; (b) D. A. Claremon, L. W. Dillard, Y. Fan, L. Jia, S. B. Singh, C. M. Tice, Z. Xu, J. Yuan and L. Zhuang, WO Pat., 201787608, 2017.
- M. Shankar, K. Ghosh, K. Mukherjee, R. K. Rit and A. K. Sahoo, Org. Lett., 2018, 20, 5144–5148 CrossRef CAS.
- (a) J. Wang, S. Zha, K. Chen, F. Zhang and J. Zhu, Org. Biomol. Chem., 2016, 14, 4848–4852 RSC; (b) Z. Shi, D. C. Koester, M. Boultadakis−Arapinis and F. Glorius, J. Am. Chem. Soc., 2013, 135, 12204–12207 CrossRef CAS; (c) M. Tian, G. Zheng, X. Fan and X. Li, J. Org. Chem., 2018, 83, 6477–6488 CrossRef CAS.
- (a) J. L. Pergomet, A. B. J. Bracca and T. S. Kaufman, Org. Biomol. Chem., 2017, 15, 7040–7049 RSC; (b) S. O. Simonetti, E. L. Larghi and T. S. Kaufman, Org. Biomol. Chem., 2016, 14, 2625–2636 RSC; (c) M. V. Méndez, D. A. Heredia, E. L. Larghi, A. B. J. Bracca and T. S. Kaufman, RSC Adv., 2017, 7, 28298–28307 RSC; (d) J. L. Pergomet, E. L. Larghi, T. S. Kaufman and A. B. J. Bracca, RSC Adv., 2017, 7, 5242–5250 RSC; (e) D. F. Vargas, E. L. Larghi and T. S. Kaufman, Synthesis, 2019, 51, 2030–2038 CrossRef CAS.
- (a) D. A. Heredia, E. L. Larghi and T. S. Kaufman, Eur. J. Org. Chem., 2016, 1397–1404 CrossRef CAS; (b) C. C. Silveira, E. L. Larghi, S. R. Mendes, A. B. J. Bracca, F. Rinaldi and T. S. Kaufman, Eur. J. Org. Chem., 2009, 4637–4645 CrossRef CAS; (c) D. F. Vargas, E. L. Larghi and T. S. Kaufman, Nat. Prod. Rep., 2019, 36, 354–401 RSC; (d) I. Cortés, C. M. Borini Etichetti, J. E. Girardini, A. B. J. Bracca and T. S. Kaufman, Synthesis, 2019, 51, 433–440 CrossRef.
- (a) C. D. Campbell, R. L. Greenaway, O. T. Holton, P. R. Walker, H. A. Chapman, C. A. Russell, G. Carr, A. L. Thomson and E. A. Anderson, Chem.–Eur. J., 2015, 21, 12627–12639 CrossRef CAS; (b) A. Barbero, P. Cuadrado, I. Fleming, A. M. González and F. J. Pulido, J. Chem. Soc., Chem. Commun., 1992, 351–353 RSC; (c) H. Tanaka, Y. Tokumaru and S. Torii, Synlett, 1999, 774–776 CrossRef CAS.
- I. Bonneau-Gubelmann, M. Michel, B. Besson, S. Ratton and J.-R. Desmurs, Carboxylation of Hydroxy Aromatic Compounds. In: Industrial Chemistry Library, Elsevier, Amsterdam, The Netherlands, 1996, vol. 8, pp. 116–128 Search PubMed.
- (a) J. Gràcia, M. A. Buil, J. Castro, P. Eichhorn, M. Ferrer, A. Gavaldà, B. Hernández, V. Segarra, M. D. Lehner, I. Moreno, L. Pagès, R. S. Roberts, J. Serrat, S. Sevilla, J. Taltavull, M. Andrés, J. Cabedo, D. Vilella, E. Calama, C. Carcasona and M. Miralpeix, J. Med. Chem., 2016, 59, 10479–10497 CrossRef; (b) T. Inghardt, A. Johansson and A. Svensson, US Pat., 200419033, 2004; (c) A.-C. Roth-Rosendahl and E. Svernhage, WO Pat., 2003101956, 2003; (d) R. Dominique, R. A. Goodnow Jr, A. Kowalczyk, Q. Qiao, A. Sidduri and J. W. Tilley, US Pat., 2010240678, 2010; (e) J. Gracia Ferrer, M. A. Buil Albero, I. M. Moreno Mollo, L. M. Pages Santacana, R. S. Roberts, S. Sevilla and J. Taltavull Moll, EP Pat., 20102196465, 2010; (f) R. E. Counsell, T. D. Smith, V. V. Ranade, O. P. D. Noronha and P. Desai, J. Med. Chem., 1973, 16, 684–687 CrossRef CAS.
- M. B. Smith, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley, New York, USA, 7th edn, 2013 Search PubMed.
- (a) W. S. Leal, Y. Kuwahara and T. Suzuki, Agric. Biol. Chem., 1990, 54, 2593–2597 CAS; (b) N. Ueberschaar, Z. Xu, K. Scherlach, M. Metsä-Ketelä, T. Bretschneider, H.-M. Dahse, H. Görls and C. Hertweck, J. Am. Chem. Soc., 2013, 135, 17408–17416 CrossRef CAS; (c) M. Sato, Y. Kuwahara, S. Matsuyama and T. Suzuki, Biosci., Biotechnol., Biochem., 1993, 57, 1299–1301 CrossRef CAS.
- (a) K. Watanabe, J. Sakurai, H. Abe and T. Katoh, Chem. Commun., 2010, 46, 4055–4057 RSC; (b) C. M. Harris, J. J. Kibby, J. R. Fehlner, A. B. Raabe, T. A. Barber and T. M. Harris, J. Am. Chem. Soc., 1979, 101, 437–445 CrossRef CAS; (c) X. Wu, J. Zhou and B. B. Snider, Angew. Chem., Int. Ed., 2009, 48, 1283–1286 CrossRef CAS; (d) J. Sakurai, T. Kikuchi, O. Takahashi, K. Watanabe and T. Katoh, Eur. J. Org. Chem., 2011, 2948–2957 CrossRef CAS.
- Z. Markovic, S. Markovic, N. Manojlovic and J. Predojevic−Simovic, J. Chem. Inf. Model., 2007, 47, 1520–1525 CrossRef CAS.
- (a) Y.-S. Yao and S.-J. Yao, J. Org. Chem., 2008, 73, 5221–5225 CrossRef CAS; (b) H. Morita, Y. Tomizawa, J. Deguchi, T. Ishikawa, H. Arai, K. Zaima, T. Hosoya, Y. Hirasawa, T. Matsumoto, K. Kamata, W. Ekasari, A. Widyawaruyanti, T. S. Wahyuni, N. C. Zaini and T. Honda, Bioorg. Med. Chem., 2009, 17, 8234–8240 CrossRef CAS; (c) S. O. Simonetti, E. L. Larghi, A. B. J. Bracca and T. S. Kaufman, Org. Biomol. Chem., 2012, 10, 4124–4134 RSC.
- (a) G. A. Molander and N. Ellis, Acc. Chem. Res., 2007, 40, 275–286 CrossRef CAS PubMed; (b) H. Doucet, Eur. J. Org. Chem., 2008, 2013–2030 CrossRef CAS; (c) G. A. Molander and M. Rodríguez Rivero, Org. Lett., 2002, 4, 107–109 CrossRef CAS PubMed.
- (a) E. Alacid and C. Nájera, J. Org. Chem., 2009, 74, 8191–8195 CrossRef CAS; (b) G. A. Molander, T. Barcellos and K. M. Traister, Org. Lett., 2013, 15, 3342–3345 CrossRef CAS.
- (a) Y. Duan, J. Zhang, J. Yang and M. Deng, Chin. J. Chem., 2009, 27, 179–182 CrossRef CAS; (b) Z.-Y. Peng, J.-P. Wang, J. Cheng, X.-M. Xie and Z. Zang, Tetrahedron, 2010, 66, 8238–8241 CrossRef CAS.
- D. S. Matteson, J. Am. Chem. Soc., 1960, 82, 4228–4233 CrossRef CAS.
- M. D. Hammers, L. Singh, L. A. Montoya, A. D. Moghaddam and M. D. Pluth, Synlett, 2016, 27, 1349–1353 CrossRef CAS.
- (a) M. Kosugi and K. Fugami, J. Organomet. Chem., 2002, 19, 10–16 Search PubMed; (b) V. Farina, V. Krishnamurthy and W. J. Scott, The Stille Reaction, Wiley, New York, USA, 2004 Search PubMed.
- C. Wei, R. Wang, C. Zhang, G. Xu, Y. Li, Q.-Z. Zhang, L.-Y. Li, L. Yi and Z. Xi, Chem.–Asian J., 2016, 11, 1376–1381 CrossRef CAS.
- (a) D. H. Dethe and G. Murhade, Org. Lett., 2013, 15, 429–431 CrossRef CAS; (b) D. H. Dethe, B. D. Dherange, S. Ali and M. M. Parsutkar, Org. Biomol. Chem., 2017, 15, 65–68 RSC.
- (a) K. C. Nicolaou, T. Montagnon, G. Vassilikogiannakis and C. J. N. Mathison, J. Am. Chem. Soc., 2005, 127, 8872–8888 CrossRef CAS; (b) K. S. MacMillan, T. Nguyen, I. Hwang and D. L. Boger, J. Am. Chem. Soc., 2009, 131, 1187–1194 CrossRef CAS; (c) M. Moreno, W. A. M. Elgaher, J. Herrmann, N. Schläger, M. M. Hamed, S. Baumann, R. Müller, R. W. Hartmann and A. Kirschning, Synlett, 2015, 26, 1175–1178 CrossRef CAS.
- H. Cong, T. Yamato and Z. Tao, New J. Chem., 2013, 37, 3778–3783 RSC.
- D. F. Vargas, E. L. Larghi and T. S. Kaufman, Eur. J. Org. Chem., 2018, 5605–5614 CrossRef CAS.
- J. Kalia and R. T. Raines, Angew. Chem., Int. Ed., 2008, 47, 7523–7526 CrossRef CAS.
- (a) J. Tian, H. Yue, P. Yang and D. Sang, ChemistrySelect, 2019, 4, 38–41 CrossRef CAS; (b) D. Sang, X. Tu, J. Tian, Z. He and M. Yao, ChemistrySelect, 2018, 3, 10103–10107 CrossRef CAS; (c) D. Sang, C. Yi, Z. He, J. Wang, J. Tian, M. Yao and H. Shi, Tetrahedron Lett., 2018, 59, 1469–1472 CrossRef CAS.
- (a) H. Wagner, S. Bladt and V. Rickl, Drug Analysis: A Thin Layer Chromatography Atlas, Springer, Heidelberg, Germany, 2nd edn. 2009, p. 360 Search PubMed; (b) W. C. Still, M. Kahn and A. Mitra, J. Org. Chem., 1978, 43, 2923–2925 CrossRef CAS; (c) J. D. Fair and C. M. Kormos, J. Chromatogr. A, 2008, 1211, 49–54 CrossRef CAS; (d) D. F. Taber, J. Org. Chem., 1982, 47, 1352–1354 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of the intermediates and final products. See DOI: 10.1039/c9ra06839b |
| This journal is © The Royal Society of Chemistry 2019 |