
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePerformance of metal–organic frameworks for the adsorptive removal of potentially toxic elements in a water system: a critical review
Sammani Ramanayakaa,
Meththika Vithanage *a,
Ajit Sarmahb,
Taicheng Anc,
Ki-Hyun Kimd and
Yong Sik Ok*e
*a,
Ajit Sarmahb,
Taicheng Anc,
Ki-Hyun Kimd and
Yong Sik Ok*e
aEcosphere Resilience Research Center, Faculty of Applied Sciences, University of Sri Jayewardenepura, Nugegoda, Sri Lanka. E-mail: meththika@sjp.ac.lk; kkim61@hanyang.ac.kr
bDepartment of Civil & Environmental Engineering, Faculty of Engineering, The University of Auckland, Auckland, 1142, New Zealand
cGuangzhou Key Laboratory Environmental Catalysis and Pollution Control, Guangdong Key Laboratory of Environmental Catalysis and Health Risk Control, School of Environmental Science and Engineering, Institute of Environmental Health and Pollution Control, Guangdong University of Technology, Guangzhou 510006, China
dAir Quality & Materials Application Lab, Department of Civil & Environmental Engineering, Hanyang University, Seoul 04763, South Korea
eKorea Biochar Research Center, O-Jeong Eco-Resilience Institute (OJERI), Division of Environmental Science and Ecological Engineering, Korea University, Seoul 02841, South Korea. E-mail: yongsikok@korea.ac.kr
First published on 24th October 2019
Abstract
Elevated levels of potentially toxic elements (PTEs) in aqueous environments have drawn attention recently due to their presence and toxicity to living beings. There have been numerous attempts to remove PTEs from aqueous media. The potential of metal–organic frameworks (MOFs) in removing PTEs from aqueous media has been recognized due to their distinctive advantages (e.g., increased removal capability, large surface area, adjustable porosity, and recyclability). Because of the poor stability of MOFs in water, pre and post synthetic modification and functionalization of MOFs have also been developed for water treatment investigations. This review addresses the performance and mechanisms of PTE removal in various modified MOFs in detail. In order to compare the performance of MOFs, here we used partition coefficient (PC) instead of maximum adsorption capacity, which is sensitively influenced by initial loading concentrations. Therefore, the PC of each material was used to evaluate the adsorption performance of different MOFs and to compare with other sorbents. Furthermore, it discusses the scale-up issues and forthcoming pathway for the research and development needs of MOFs for effective PTE removal. This review further elucidates the main removal mechanisms of PTEs by MOFs. Commercial or domestic water treatment systems or water filters can utilize engineered MOFs to treat water by adsorptive removal. However, marketable products have yet to be investigated thoroughly due to limitations of the large-scale synthesis of MOFs.
1. Introduction
The importance of water quality in sustainable development is acknowledged in the Agenda for Sustainable Development. Sustainable Development Goal (SDG) 6 includes two targets for water quality achievements by 2030. SDG 6.1 seeks to “achieve universal and equitable access to safe and affordable drinking water for all,” and SDG 6.3 seeks to “improve water quality by reducing pollution, eliminating dumping and minimizing release of hazardous chemicals and materials, halving the proportion of untreated wastewater, and substantially increasing recycling and safe reuse globally”.1 Water pollution has become a global challenge due to industrial, agricultural, domestic, and other anthropogenic activities of billions of human inhabitants on the planet. Ineffective wastewater treatment and management have resulted in widespread pollution of water systems, while global consideration predominantly emphasized on water scarcity, efficacy of water usage, and allocation matters.2 Continuous water quality degradation around the world has been observed to worsen global water scarcity.Among the various types of contaminants in water, potentially toxic elements (PTEs) that include both metals and non-metals have received recent attention due to their high concentrations in water sources in the environment3,4 (Table 1). Environmental significance is determined based on the concentration levels of such contaminants in water. Some PTEs like Zn, Cu, and Ni are also classified as micronutrients, which are necessary dietary components in minimal quantities. However, they also become toxic to living organisms at higher concentrations. Receive PTEs are brought into the environmental waters via both natural and anthropogenic pathways. Among various sources of PTEs into the environment, mining, agricultural practices, and industrial activities are considered to be the prominent.5,6,3 PTEs are persistent in the environment and can cause chronic diseases in exposed plants and animals to alarm the need for their removal from water systems.7
| Common PTEs (those with most significant environmental significance are indicated with *) | Anthropogenic uses | Maximum Contaminant Levels (MCL) in water (μg L−1), EPA** |
|---|---|---|
| a EPA** Environmental potential agency. | ||
| Antimony (Sb) | Semiconductor manufacturing alloy preparation, batteries, fewer abrasion metals, flame-retarding materials, paints, glass and pottery | 6 |
| Arsenic (As)* | Glass and ceramics, medications, agrochemicals, feed additives | 10 |
| Beryllium (Be) | Aircraft and missiles production, communication satellites, windshield frames, brake discs, support beams, nuclear energy production | 4 |
| Cadmium (Cd)* | Battery manufacturing, electrochemical industry, solar cells, pigments, electrical appliances, nuclear reactors | 5 |
| Chromium (Cr)* | Electrochemical industry, stainless steel, dyes, tannery, textiles, photography, pigment industry | 10 |
| Copper (Cu)* | Electrical appliances, alloys, pesticides | 1300 |
| Fluorine (F)* | High-temperature plastics, electrical equipment nuclear energy and chemical industry | 2000 |
| Gold (Au) | Electronics, jewelry, computers, dentistry, aerospace engineering | 6 |
| Lead (Pb)* | Anti-knock agents, battery industry, paints, ammunition, glass, ceramicware, rubber manufacturing | 15 |
| Mercury (Hg)* | Catalyst manufacturing, electrical appliances, batteries, fluorescent lights, felt production, thermometers, and barometers | 2 |
| Molybdenum (Mo)* | Alloying agent in steel, heat, and corrosion-resistant materials used in the chemical industry, lubricants | 40 |
| Nickel (Ni)* | Alloys battery industry, electrical appliances, electrochemical industry, paint and pigment manufacturing | 100 |
| Rubidium (Ru) | Vacuum tubes, photocells, space crafts, thin-film batteries | 2 |
| Selenium (Se)* | Micronutrients, pharmaceuticals | 5 |
| Silver (Ag) | Jewelry, electrical utilities, digital imaging, clothing, soaps, photochromic lenses | 100 |
| Thallium (TI) | Photocells, glassware | 2 |
| Uranium (U) | Nuclear reactors, military purposes | 30 |
| Vanadium (V) | Alloys, superconducting materials, vehicle spare parts | 50 |
| Zinc (Zn)* | Alloy manufacturing, galvanizing, rubber industry, paper production, paints, enamel and plastic products, fertilizer industry, feed additives, drugs, cosmetics | 5000 |
An array of water treatment technologies with rigorous norms has been implemented globally over the last few decades to remediate PTEs in water. These technologies vary in their efficiency and effectiveness, such as chlorination, ozonation, photocatalytic oxidation, adsorption, electrochemical oxidation, and coagulation–flocculation. Among these options, adsorption is often taken to be a preferable method to treat water.3 Adsorption is user-friendly, environmentally sound, and cost-effective.9 However, most adsorption methods have inherent limitations.8
Numerous materials such as biochar, activated carbon, chitosan composites, plant products, fly-ash, nanoparticles, graphene oxide, natural minerals, and clay/polymer composites have been used as adsorbent for the removal of PTE from water. Biochar and its modifications have gained recent attention for their activated carbon-negative nature and high surface area compared to some other adsorbents, which may improve their ability to treat various contaminants in water.9 Some common materials that have been explored extensively during the last decade for adsorption of heavy metals are natural and nano metal oxides (NMOs). Characteristics of NMOs are the considerable surface area and extraordinary reactivity.10 Nanoparticles tend to aggregate due to their nanoscale size, and a subsequent decrease of adsorption efficiency is observed.11 At the same time, impregnation of NMOs onto natural or synthetic porous structures has advanced the stability of NMOs.11,12 Zeolites are another class of efficient adsorbents that can be readily synthesized; hence, they are commonly used for the removal of PTEs.13 Some of these materials are high in production cost, due to comparatively low adsorption capacity, leaching, and bed clogging, which make them impractical to use for adsorptive removal.8 Many adsorbents are active only in a particular pH range, or physicochemical instability, low regeneration/recycling capacity, high selectivity, low surface area, and high susceptibility to interference effects. All these properties can limit the adsorbent's capacity to remove PTEs from aqueous media.
Recent adsorbent research has focused on metal–organic frameworks (MOFs), highly ordered, crystalline, and porous materials, known as for their extraordinary performances in remediating various contaminants.14–17 Similar to nanomaterials, MOFs are useful in many different applications. However, MOFs have poor stability in water, which hampers their use in adsorption of contaminants in water. Milestones of MOFs synthesis and their application in water pollution remediation is shown in Fig. 1. As water-stable MOFs and modifications have been introduced, they have been researched extensively for the use in the removal of PTEs from water.17,16,14 As shown in Fig. 2, a search was conducted in the Scopus database from 1998–2019 which yields a remarkable increase in the published articles with “heavy metal adsorption” and “MOF” as keywords. As such, research in this area has been expanding noticeably in recent years.18 This review aims to offer an overall insight into the interaction between PTEs including heavy metals and MOFs by focusing on the mechanisms of interaction. Further, this review discusses the synthesis, performance, and characteristics of MOFs and the removal of individual contaminants in separate subsections. More interestingly, we calculate and report partition coefficient values with the recorded adsorption capacities. As adsorption capacities are very much dependent on the initial parameters of laboratory experiments, it does not give a fair comparison. Furthermore, an attempt was taken to identify gaps in existing research as a base for future research.
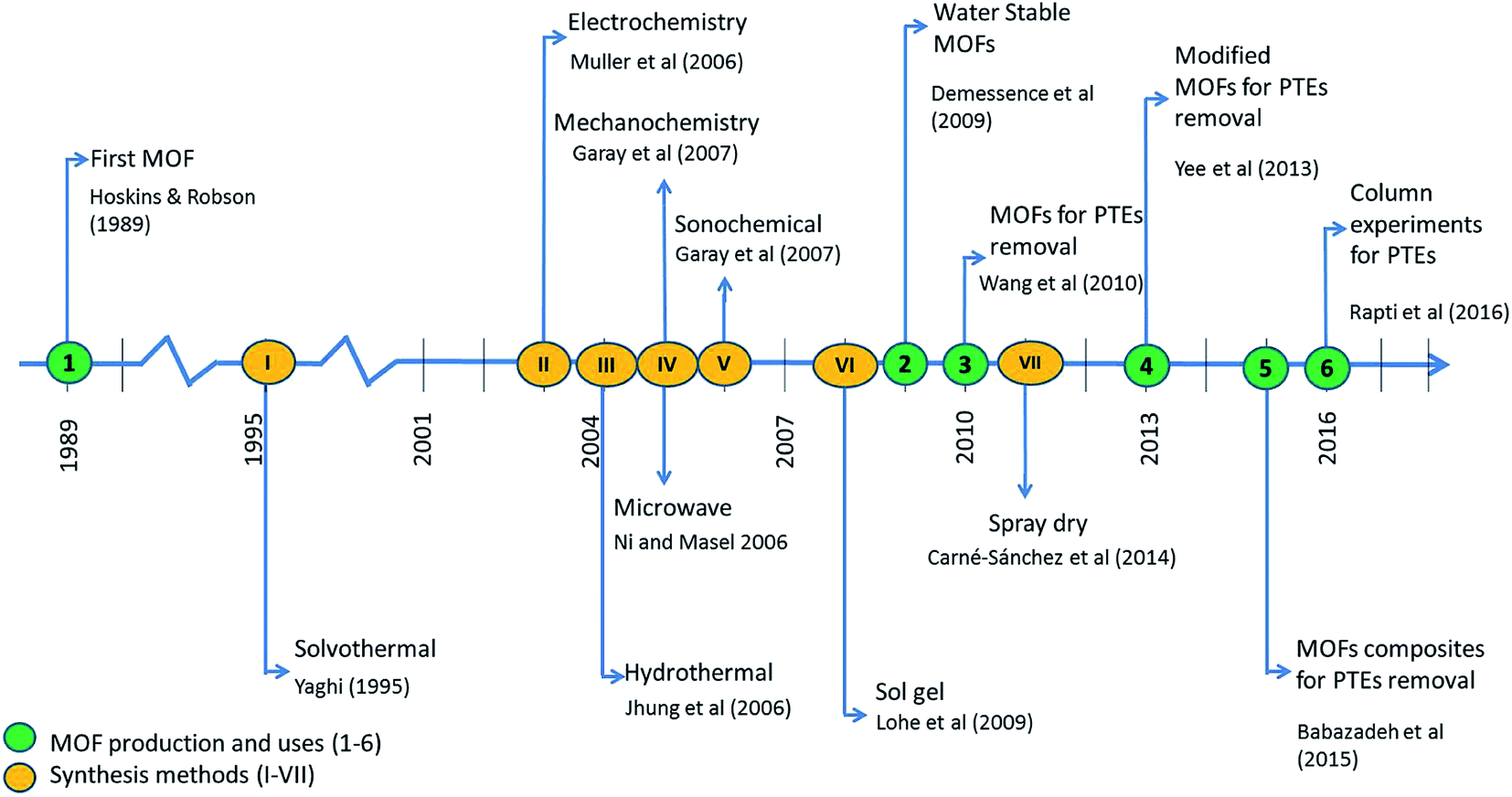 | ||
| Fig. 1 Milestones of MOFs synthesis (I–VII) and their application in water pollution remediation (1-,18 2-,19 3-,20 4-,20 5-,21 6-21/I-,22 II-,23,24 III-,25 IV-,26 V-,27 VI-,28 VII-).29 | ||
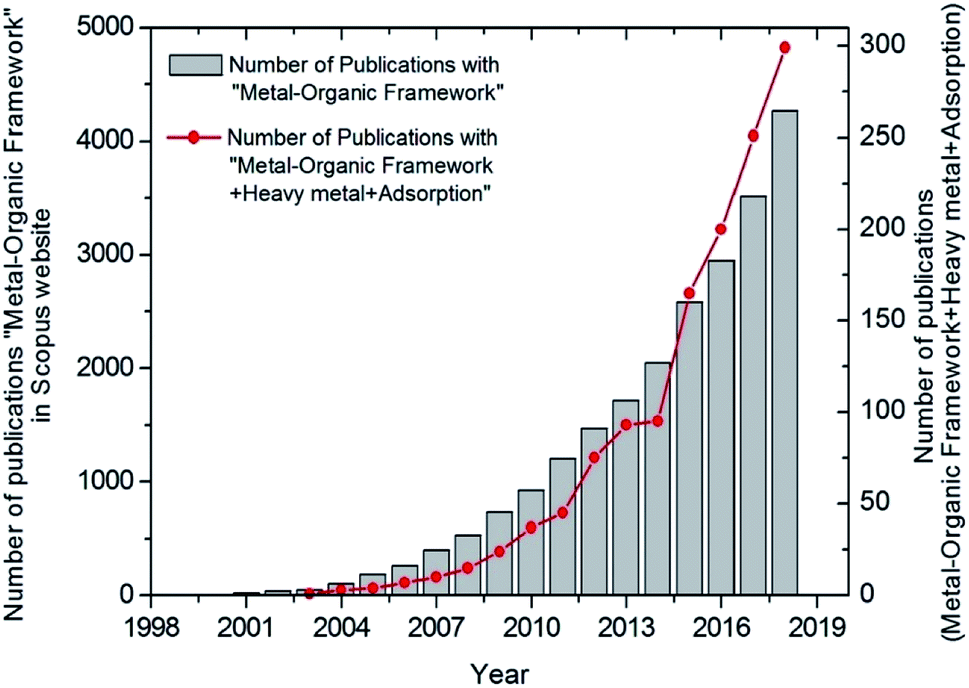 | ||
| Fig. 2 Number of publications on MOFs and metal adsorption based on data in the Scopus database. | ||
2. Metal–organic frameworks as adsorbents in water treatment
2.1 Capacity of the metal–organic framework as adsorbents
Adsorption may happen through physical or chemical interactions of adsorbates and adsorbents. During physisorption, the sorbent and sorbates are interacting through van der Waals forces at the adsorbent surface. Since the attachments are not so secure in physisorption, the sorbents are easily regenerated through a simple change in the system such as solvent exchange, slight pH change, or physical treatment. Chemical adsorption or chemisorption happens through chemical bonding between the adsorbent surface and adsorbate hence the enthalpy is high, which makes regeneration difficult and decreases the reusability of the adsorbent. Among the various physic-chemical properties of MOFs, their large surface area and porosity mark them highly desirable for use as adsorbents as well as advantageous for a variety of applications other than water treatment.30 Adsorption increases as surface area and porosity increase due to higher exchange site accessibility and a greater diffusion rate through the framework. To be viable in practical water treatment applications, an adsorbent must remove contaminants quickly, have a high adsorption capacity, remove a broad range of contaminants, and be reusable.15,18The diverse nature, high surface area, tunable pore size, and high porosity of MOFs make them more attractive for water treatment than other materials, for instance activated carbon, biochar, zeolites, nanomaterials, and chitosan beads.15 Table 3 shows the results of comparative studies indicating that MOFs have the best adsorption capacity for selected emerging contaminants (ECs). Compared to the conventional adsorbents, i.e., carbon nanotubes and zeolite, MOFs have exhibited significant partitioning coefficients indicating high capacity of removal independent of the initial conditions. It is evident from the literature that the maximum adsorption capacity does not provide a good base to select the best adsorbent due to the different initial conditions applied in various experiments, and hence, partition coefficient (PC) is calculated.31,32 As an example, maximum adsorption capacity of diclofenac for AC was 29 and 23% less than 18% SO3H-UiO-66 (sulfonated metal–organic framework, University of Oslo-66) and PCDM-1000 (porous carbons derived from MOF, prepared at 1000 °C) MOFs, whereas PC values showed the same pattern (Table 3).33,34 Although high maximum adsorption capacities have been observed for ciprofloxacin and triclosan for AC, the PC values are less compared to MOFs, which indicates high removal potential of MOFs independent of the initial conditions. The crystalline structure, pore structure, order, size, and shape of MOFs can be adjusted to increase adsorption performance by changing the linkers used in the synthesis process.35 A distinctive feature of MOFs is their ability to retain their structural integrity at the course of synthesis. Moreover, the relationship between the characteristics and properties of MOFs and their diverse composition offers them a status above traditional adsorbents in terms of performance.15 These properties indicate that MOFs can play a significant role as an adsorbent in water treatment. However, concerns over the water stability of MOFs have limited their use in practical water treatment applications. More attention is now being focused towards synthesizing MOFs with stability in water that can be used in remediation of contaminants in aqueous media.
The properties and structure of MOFs, especially their high adsorptive capacity, give them potential as gas storage, sensing, and separation media in clean energy applications, notably for hydrogen and methane gas storage.32 The viability of MOFs for adsorption and diffusion, also make them applicable in membranes, thin-film devices, catalysis, and biomedical imaging. The ultra-high surface area, high porosity, and diverse functionalities of MOFs make them highly efficient at removing contaminants such as PTEs from water. In addition to having adsorption applications, MOFs, which contain metal ions with fluorescence and luminescence properties, can be used as tracers to detect metal ions in water.62,63
One reason that MOFs are promising materials for adsorptive removal and separation of contaminants is their multiple pore sizes, which vary from micro to meso-range.64,65 In contrast to other porous materials such as activated carbon and zeolites, a broad range of pore cavities and functionalities can be seen in MOFs.66 The diversity of pore size allows MOFs to accept a broader range of compounds, which makes them applicable in multiple research areas, including drug delivery,67 adsorption/storage,68 catalysis,69 sensing and detection,70 and luminescence.70
2.2 Modification of metal–organic frameworks
Metal–organic frameworks are further investigated for various modifications to improve the adsorption capacity. Modifications can be introduced through inner and outer surfaces of MOFs by solvent molecules, which can easily penetrate to the interior channels of a MOF due to its high porosity.17,71 Table 2 shows different types of modifiers that can be introduced and the contaminants that can be adsorbed after modification.| MOFs | Modifiers | Contaminants adsorbed | Reference |
|---|---|---|---|
| BUC-17 | Graphene | Anionic dyes | 72 |
| MIL-88(Fe) | Graphene oxide | Methylene blue; rhodamine B | 73 |
| UiO-66 | –NH2 | Doxycycline | 74 |
| MIL-101(Cr) | –SO3Ag | Iodide | 75 |
| ZJU-24 | –COOH | Methylene blue | 76 |
| UiO-66- | –2COOH | Cu(II) | 77 |
| UiO-66 | –OH | Thorium ions | 78 |
| UiO-66 | NHC(S)NHMe | Pb(II), Hg(II), Cd(II) | 18 |
| MIL-101 | –SO3H | Rocephin | 79 |
| MOF-5 | Thiol | Pb(II), Hg(II), Cd(II) | 80 |
| MOF-5 | Fe3O4 | Pb(II), Cd(II) | 18 |
| HKUST-1 | Na2S | Hg(II) | 16 |
| MIL-68 | Na2S | Pb(II), Hg(II), Cd(II) | 18 |
| MIL-53-NH2 | Na2S | Pb(II), Hg(II), Cd(II) | 18 |
Modification of MOFs may be pre-synthetic or post-synthetic. Pre-synthetic functionalization of MOFs may result in the decomposition of functional groups at the high temperatures used during synthesis, while post-synthetic modification does not have this disadvantage.81,82
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–NCRR′, and quinine functional groups, is carried out during PSM.18 Coupling agents, metal joints, pore characteristics, and surface environment can be used to adjust the structure of a MOF to upgrade its structural stability and performance.85 The drawbacks to post-synthetic modification are that it can be time-consuming (1–5 days), and the production rate depends on the pore diameter under standard conditions. Liquid phase PSM (LP-PSM) and vapor phase PSM (VP-PSM) are the two pathways that are commonly practiced for post-synthetic modification. VP-PSM achieves a uniform distribution and high yield within a short period and provides more flexibility in applications.86 Hence, VP-PSM has received more attention than LP-PSM.
N–NCRR′, and quinine functional groups, is carried out during PSM.18 Coupling agents, metal joints, pore characteristics, and surface environment can be used to adjust the structure of a MOF to upgrade its structural stability and performance.85 The drawbacks to post-synthetic modification are that it can be time-consuming (1–5 days), and the production rate depends on the pore diameter under standard conditions. Liquid phase PSM (LP-PSM) and vapor phase PSM (VP-PSM) are the two pathways that are commonly practiced for post-synthetic modification. VP-PSM achieves a uniform distribution and high yield within a short period and provides more flexibility in applications.86 Hence, VP-PSM has received more attention than LP-PSM.2.3 Performances of MOFs against adsorption capacity
In general, adsorption capacity demonstrates the removal potential of contaminants by the adsorbents. As a whole, performance is generally assessed by the equilibrium (or maximum) adsorption capacity. However, the initial loading of the target pollutant (or, more specifically equilibrium concentration or amount) influences the maximum adsorption capacity. If sorbent is exposed to higher concentration of target contaminants, it is supposed to exhibit higher adsorption capacity. This particular weakness elucidates the incapability of adsorption capacity for assessing the actual performance of sorbents. Therefore, in order to compare the capacity of different materials tested in various conditions, partition coefficient (PC) has been proposed as a way to leverage the influences for a fair comparison of each material.31,87 In a solid–liquid system, PC represents the ratio of adsorbate, sorbed in, and on the adsorbent to its equilibrium concentration.88 Partition coefficient values were then obtained by dividing the maximum adsorption capacity value by the equilibrium concentration of the media (eqn (1)). Thus, it has been identified as the true performance of the adsorbent.89 The calculated PC of each various adsorbents including, MOFs, in the removal of emerging contaminants and PTEs, are tabulated in Tables 3 and 4. The PC data reveals that MOFs outperformed other adsorbents such as activated carbon, zeolites etc. supporting the advantages of MOFs depending on the sorbate (Table 3).
 | (1) |
| Emerging contaminants | Adsorbent | Maximum removal Qmax (mg g−1) | Partition coefficient (PC) | References |
|---|---|---|---|---|
| a *Different types of MOFs. | ||||
| Pharmaceuticals and Personal Care Products (PPCPs) | ||||
| Diclofenac | Activated carbon | 76 | 0.76 | 34 |
| Carbon nanotubes | 33.88 | 1.32 | 36 | |
| Zeolite modified with cetylpyridinium chloride (ZCPC-30) | 50.77 | 125.64 | 37 | |
| 18% SO3H-UiO-66* | 263 | 3.57 | 34 | |
| PCDM-1000* | 320 | 6.48 | 33 | |
| Ciprofloxacin | Activated carbon | 231 | 12.2 | 38 |
| Carbon nanotubes | 135 | 5.6 | 38 | |
| NPC-700 derived from ZIF-8* | 416.7 | 0.87 | 39 | |
| Triclosan | Activated carbon | 68 | 2.61 | 40 |
| Graphene | — | 527.25 | 40 | |
| UiO-66-NH–CO–COOH* | 189 | 5.39 | 41 | |
| CDIL@AIPCP* | 212 | 17.0 | 42 | |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| Industrial ECs | ||||
| Dimethyl phthalate | Single wall carbon nanotubes | 2.17 | 0.82 | 43 |
| α-Cyclodextrin | 190 | 39.04 | 44 | |
| β-Cyclodextrin | 206 | 0.01 | 45 | |
| MIL-53BM* | 190 | 17.26 | 46 | |
| MlL-53AlO* | 206.2 | 21.26 | 46 | |
| Phenol | Activated carbon | 398 | 1773.5 | 47 |
| Activated carbon fiber (ACF) | 378 | 1.24 | 48 | |
| MIL-53(Cr)* | 267 | 1.31 | 49 | |
| ZIF-67* | 378 | 50 | ||
| Nitrobenzene | Faujasite | 267 | 1.71 | 51 |
| Nanocrystalline hydroxyapatite | 8.9 | 0.45 | 52 | |
| CAU-1* | 1171 | 1.28 | 53 | |
| MIL-68(Al)* | 1188 | 1.46 | 53 | |
|
||||
| Pesticides | ||||
| Glyphosate | Ni2Al LDH | 172.4 | 2.61 | 54 |
| UiO-67(Zr)* | 537 | 9.58 | 55 | |
| UiO-67(Zr)/GO* | 482 | 7.11 | 56 | |
| 2,4-Dichlorophenoxy-acetic acid | Activated carbon | 286 | 2.16 | 57 |
| USY | 256 | 1.02 | 57 | |
| MIL-53(Cr)* | 556 | 5.28 | 57 | |
| Carbon derived from IL@ZIF-8* | 448 | 3.46 | 58 | |
|
||||
| Dyes | ||||
| Methylene blue | Activated carbon | 26 | 0.12 | 59 |
| MOF-235* | 187 | 0.98 | 59 | |
| HKUST/GO* | 183.49 | 36.69 | 60 | |
| Methyl orange | Activated carbon | 11.2 | 0.03 | 59 |
| MOF-235* | 477 | 2.83 | 59 | |
| EDMIL-101(Cr)* | 160 | 0.79 | 61 | |
| Metal | MOF | Adsorption capacity (mg g−1) unless otherwise indicated | Partition coefficient (PC) (L g−1) | Adsorption equilibrium time (min) | Optimal pH | Reference |
|---|---|---|---|---|---|---|
| Hg | MOF-74-Zn | 63 | 1.32 | 90 | 6 | 119 |
| LMOF-263 | 380 | — | 30 | 4–10 | 120 | |
| MIL-101-Thymine | 52 | 0.26 | 200 | 6 | 121 | |
| UiO-66-NHNHC(S)NHMe | 769 | 137.88 | 5–240 | — | 115 | |
| Pb | MOF-5 | 290 | 7.0 | 360 | 5 | 122 |
| Ln(BTC)(H2O)(DMF)1.1 Ln = Dy | 5.07 | 1.45 | 5–240 | — | 123 | |
| MIL-53(Al) | 492.4 | 0.65 | 120 | — | 124 | |
| UiO-66-NHC(S)NHMe | 233 | 4.92 | 240 | — | 115 | |
| HKUST-1-MW@H3PW12O40 | 98.18 | 26.97 | 120 | 7 | 125 | |
| Cd | Cu3(BTC)2-SO3H | 88.7 | 0.89 | 10 | 6 | 126 |
| UiO-66-NCH(S)NHMe | 49 | 0.28 | 240 | — | 115 | |
| TMU-16-NH2 | 126.6 | 17.34 | 30 | 6 | 127 | |
| HS-mSi@MOF-5 | 98 | 2.62 | 4 | 7 | 115 | |
| Cr | TMU-30 | 145 Cr(VI) | 2.05 | 8 | 2–9 | 128 |
| Cu-BTC | 48 Cr(VI) | 0.98 | — | 7 | 129 | |
| ZJU-101 | 245 Cr(VI) | 0.88 | 15 | — | 130 | |
| UiO-66-NHC(S)NHMe | 118 Cr(III) | 0.88 | 5–240 | — | 115 | |
| Cu | Ln(BTC)(H2O)(DMF)1.1 Ln = Dy | 3.87 | 2.16 | 20 | 3.5 | 123 |
| ZIF-8 | 800 | 1912.05 | 30 | 4 | 131 | |
| UiO-66(Zr)–2COOH | 11 | 183.33 | 60 | 6 | 132 | |
| MOF-5 | 290 | 649 | 180 | 5.2 | 133 |
3. Removal of inorganic contaminants
It has been estimated that more than 20000 MOFs have been synthesized, characterized, and used in various applications such as gas storage, energy generation, sensing, biomedical imaging, chemical catalysis, luminescence, environmental remediation, etc.90 In the case of adsorption, the fundamental aspects of MOFs have been studied and reviewed in numerous reports.14,90,91 Much of this existing research has focused on the remediation of heavy metals ECs. However, the application of MOFs in water treatment has been a growing area of research in recent years. This review separately summarizes the capacity of MOFs to remove inorganic PTEs based on the category of contaminants. In this review, inorganic contaminants are classified into three main categories – metalloids, cationic contaminants and anionic trace elements which have fundamentally different removal mechanisms.
3.1 Removal of metalloids
The six elements, boron, silicon, germanium, arsenic, antimony, and tellurium commonly recognized as metalloids.92 Not many MOF studies have investigated the removal of metalloids, but of those that have, arsenic has been a significant focus. The limited studies available have focused on the simultaneous removal of multivalent metalloids.Since As(V) is the thermodynamically stable and predominant form of arsenic in surface water, MOFs have been synthesized with As(V) adsorption capabilities. The MOF, Fe-BTC, contains iron as the metal node and 1,3,5-benzene tricarboxylic acid as the organic linker. Its adsorption performance has been analyzed under different pH conditions using NaOH and HCl. Fe-BTC has an As(V) removal efficiency higher than 96% for an initial concentration of As(V) 5 mg L−1 at pH of 4. This MOF is 37 times more effective than Fe2O3 nanoparticles for As(V) removal and demonstrates a PC value of 0.17 L g−1. Adsorption of arsenic into the MOF was confirmed using transmission electron microscopy (TEM) and FTIR to show an IR band at 824 cm−1 and interior sites that corresponded to Fe–O–As groups.92,95
Zeolitic imidazolate framework-8 (ZIF-8) for arsenic uptake, has achieved an adsorption capacity of 49.5 mg g−1 of As(V) and 60 mg g−1 of As(III) at neutral pH signifying high removal potential is for As(III). Obviously, As(III) achieves the highest adsorption capacity compared to As(V) due to the high loading concentration of As(III) relative to As(V). Although the same amount of As(III) and As(V) were used for the study, it does not depict the same concentration. Therefore, As(III) depicts a lower PC value of 0.85 L g−1, while As(V) exhibits 1.88 L g−1 value of PC, which specifies that the ZIF-8 shows more than twice higher performance for As(III) than that of the As(V). This comparison itself elucidates the demerit of the adsorption capacity to compare the actual performance of sorbents, specially in the case of studies uses pseudo units such as parts per million or billion etc. Arsenic was adsorbed on to the framework material surface as the arsenate and arsenite ions were more substantial than the ZIF-8 pore spaces. ZIF-8 has different morphologies, however it was found that the surface area does not correlate with the adsorption capacity of the MOF. The replacement of surface hydroxyl groups in Zn–OH of ZIF-8 with arsenic ions is the adsorption mechanism, which was confirmed by FT-IR and X-ray Photoelectron Spectroscopy (XPS).16
MOF-808, which is synthesized by microwave irradiation, was also used to remove As(V) in aqueous media. Rapid adsorption occurred with a removal efficiency of 95% for As(V) was observed within the first 30 min at pH four where the initial As(V) was 5 mg L−1. Weak van der Waal interactions of arsenate and the surface sites of the Zr metal nodes in MOF-808 has been suggested as the adsorption mechanism.96 The arsenic uptake by MOF was independent of other anions in the aqueous media than phosphate as expected.97 The MOF UiO-66 showed a promising arsenic adsorption capacity at a wide pH range, from pH 1–10. However, details of Ce or qe were not reported in order to calculate the PC for MOF-808 and UiO-66, and therefore, PC values were not elucidated. The coordination between hydroxyl groups of the metal node and/or the substitution of 1,4-benzenedicarboxylic acid ligands97 is suggested as the sorption mechanism which was confirmed by PXRD and FT-IR. UiO-66-(SH)2, a thiolated derivative of UiO-66 shows a dual capture mechanism for As(III) and As(V). Arsenate species interacted with the MOF node while thiolated ligands were bound to arsenite species. The uptake of arsenite and arsenate from UiO-66-(SH)2 after 6 h was 10 and 40 mg g−1, respectively.97,98 However, no information was given regarding the possibility of forming thiolated arsenic species during the study, which is a cytotoxic to biological systems.
MIL-53(Fe) is another type of MOF that has a variable adsorption capacity for As(V) as the ion in the metal node changes. Lewis acid–base interactions of anionic H2AsO4− and the metal ion of MIL-53 resulted in an adsorption capacity of 21 mg g−1 with a PC value of 1.06 L g−1.99 MIL-53(Al), which is very similar to MOF-53(Fe), with a different ion at the node where its maximum adsorption capacity was reported as 106 mg g−1 for As(V) in the form of HASO42− at a pH of 8. The adsorption mechanism of MOF-53(Al) was confirmed with FT-IR and XPS data. MOF-53(Al) was found to be very effective in removal of arsenic in the presence of other anions, except PO43− which reduced the capacity of adsorption to 14% of its original value.16,100 Chemically stable indium-MOF, AUBM-1 reported a maximum adsorption capacity of 103 mg g−1 for As(V), which is about 5 times higher than that of the MIL-53(Fe) however, more similar PC value of 1 L g−1 was exhibited for both. Zirconium based MOF, NU-1000 depicts a significantly high adsorption capacity of 260 mg g−1 for the removal of As(V), and respective PC value is 21.7 L g−1. However, studies are limited on adsorption of As(III) and As(V) by MOFs, and the data provided are insufficient for evaluating performance. Therefore, thorough studies on adsorption of As by MOFs are essential to be conducted. Among the reported details ZIF-8 demonstrated the best performance for As removal and specifically for As(V). Mostly, the studies on arsenic adsorption by MOFs indicate ion exchange as the prominent mechanism.101 Data on reduction of As(V) to As(III) during adsorption and thermodynamics are lacking. Furthermore, no studies reported the removal of methyl arsenic species by MOFs to the knowledge of the authors.
The most commonly occurring oxidation states of antimony are antimonite and antimonate. Under oxic conditions, Sb(V) is the dominant species that can be found in water above pH 3.104 The MOF, NU-1000, has been used for the removal of Sb(V) from water, due to the thermal, mechanical, and chemical stability it shows, in the pH range of 1–11 and its Zr–OH assemblies on Zr6− nodes, which may be substituted by negatively charged ions.105,106 NU-1000 is an effective Zr-based MOF, which exhibits the highest removal capacity of 260 mg g−1, within 30–48 h. NU-1000 is stable under various aqueous conditions, and its binding mechanism for antimony is revealed by differential pair distribution function analysis.107 NU-1000 shows the highest efficiency for removal of antimony at 136.97 and 287.88 mg g−1 for antimonite and antimonate, respectively.108 NU-1000 MOF demonstrates a better performance for Sb(V) with a PC value of 0.82 L g−1; therefore, NU-1000 is more suitable for the removal of Sb(V), due to the low performance (0.32 L g−1) with Sb(III). At a solution of co-existing with As(III), Sb(III) indicated a higher sorption than As(III) due to stronger Lewis base property and vice versa for Sb(V), where As(V) demonstrate high attraction to NU-1000.
Zr-based MOFs have gained attention in recent research due to their excellent thermal and chemical stability. These properties, have led researchers to introduce amino groups to enhance the adsorption performance of Zr-based MOFs, such as UiO-66.109 The modified MOF, UiO-66(NH2), has shown a high interaction force between Sb and the amino group, which makes it very efficient at adsorbing Sb(III) and Sb(V). UiO-66 has a moderate removal efficiency of 23 mg g−1 for antimonite and 31 mg g−1 for antimonate, while UiO-66(NH2) has a higher efficiency of 37.5 mg g−1 for antimonite and 39 mg g−1 for antimonate.108,110 Both UiO-66 and UiO-66(NH2) MOFs depict an increment of the performance with the increasing temperature. In the removal of Sb(III), UiO-66 exhibits a PC value of 0.12 L g−1, while UiO-66(NH2) shows a PC value of 0.2 L g−1. Therefore, modified MOF is more suitable in the removal of Sb(III) over basic form. However, both basic and modified (0.37 and 0.3 L g−1) MOFs demonstrate similar PC values in the adsorption of Sb(V). Among the different MOFs used in antimony removal, NU-1000 has shown the best performance based on the calculated PC value. Both NU-1000 and UiO-66(NH2) illustrated high performance in removing antimony, with replaceable adsorption sites in their Zr6 nodes suggested as the reason for the speed and efficiency of removal.108
3.2 Removal of cationic trace elements
Thiol-HKUST-1, a MOF that is synthesized solvothermally and modified post-synthetically, exhibited essentially 100% adsorption, removing 714.01 mg g−1 while the initial concentration of Hg(II) is 415.5 mg g−1, within the first 120 min of contact with the contaminated media.113 The same basic framework was magnetically modified to Fe3O4@-SiO2@HKUST-1 using a Fe-containing metal cluster. The maximum Hg(II) uptake reached 264 mg g−1 from a preliminary concentration of 20 mg L−1 of Hg at an optimal pH of 3. However, Thiol-HKUST-1 and Fe3O4@-SiO2@HKUST-1 exhibited comparatively low PC values of 0.5 and 1.29 L g−1 respectively. Magnetic modification has noticeably enhanced the performance of the raw MOF by 44%. In comparison, post-synthetic modification of thiol in the HKUST-1 MOF removes Hg(II) more efficiently than the magnetic modification, but the magnetically modified framework can be regenerated more quickly than the Thiol-HKUST-1. The magnetically-modified HKUST-1 also demonstrated selectivity to Hg(II) in the presence of other metal ions, such as Pb(II) and Cr(III).114
Saleem et al.115 post-synthetically modified the base UiO-66-NH2 to UiO-66-NHC(S)NHMe MOF by covalent modification. This modified framework exhibited a Hg(II) adsorption capacity of 99% after 240 min from a 100 mg L−1 initial concentration. Zr-DMBD is another type of Zr-based MOF that exhibits 100% Hg(II) uptake over 12 h from a solution with an initial concentration of 10 mg L−1 of Hg(II) to a final concentration 0.01 mg L−1.116 Modified MOF UiO-66-NHC(S)NHMe has exhibited an enhanced performance of 137.89 L g−1 in adsorption of Hg(II). However, data reported lacks to calculate PC values for Zr-DMBD, FJI-H12, and [Ni(3-bpd)2(NCS)2]n MOFs. FJI-H12 is a sulfur-modified, novel MOF that was developed by Liang et al.117 and consists of free NCS− groups. The highest removal capacity of FJI-H12 was observed to be 400 mg g−1 in the first hour at an optimal pH of 7. There was no noticeable change in the adsorption capacity with incremental changes to pH in the range from 3 to 6. FJI-H12 recovered 86% of its adsorbent capacity when immersed in a KSCN solution for 24 h.117
The solvothermally-synthesized Zn(hip)(L)(DMF)(H2O) MOF has exhibited a removal capacity of 333 mg g−1 in 1 h. The highest mercury removal capacity was observed at a pH of 5, and this framework performed very well at very low Hg(II) concentrations, such as 5, 10, and 20 μg L−1. Adsorption of Hg(II) is indicated by a color change from green to gray with the MOF [Ni(3-bpd)2(NCS)2]n proposed by Halder et al.118 This MOF is also highly selective for Hg than other ions in solution, such as Pb2+, Cd2+, and As3+. A 94% removal of Hg(II) from a solution at a concentration of 10 μg L−1 was observed within 2 h.118
Considering the high toxicity of Hg, many studies have been conducted to test the performance of various adsorbents for their capacity to remove Hg(II) ions from water. Table 4 summarizes the results of investigations into other types of MOFs, such as MOF-74-Zn,119 LMOF-263,120 and MIL-101-Thymine121 for their Hg(II) removal potential. Although MOF-74-Zn and MIL-101-Thymine exhibited approximately similar adsorption capacities, the PC value of MOF-74-Zn was comparatively high (1.32 L g−1) whereas the calculated PC value for MIL-101-Thymine was less as 0.26 L g−1 (Table 4). This indicates that MOF-74-Zn has a high capacity of removing Hg independent of the initial conditions.
A group of magnetic framework composites was prepared by Ricco et al.124 for the removal of Pb(II), based on a combination of iron oxide nanoparticles and the MIL-53 MOF. Loading the MIL-53(Al@100aBDC) MOF with 50% amino groups led to a significant increase in Pb(II) uptake, with a maximum recorded Pb(II) uptake capacity of 492 mg g−1 within 6 h.124 Even though MIL-53 MOF depicts high adsorption capacity, a low-performance value of 0.64 L g−1, has been exhibited. MnO2-MOF, an extremely effective Pb(II) adsorbent in aqueous media, exhibited an uptake capacity of 917 mg g−1 within 1 h. Due to proton release during the adsorption process, the pH of the solution was reduced to 5.136 At the same time, MnO2-MOF has shown promising removal potential for Cd(II) as well.
Zhang et al.137 introduced HS-mSi@MOF-5, a silica-coated, thiolated MOF-5 derivative, which exhibited a Pb(II) adsorption capacity of 312 mg g−1 within 30 min at an optimal pH of 6.137 In contrast, MOF-5 showed relatively high removal capacities at pH 4 and 6 with a low value at pH 5. It has been suggested that the performance differences with pH variation are due to the presence of both acid and base active sites in the MOF structure. The unmodified version, MOF-5, exhibited a maximum adsorption capacity of 211 mg g−1 for Pb(II) ions.122 However, both MOFs exhibited considerably high performances of 7.03 and 7.63 L g−1 in the removal of Pb(II), respectively. Since both acid and base active sites are present in MOF-5 structure, it is unclear why the uptake of Pb(II) is reduced only at pH 5 compared to pH 4 and 6. For Cd(II) adsorption, HS-mSi@MOF-5 demonstrated a similar equilibrium time (30 min) as for Pb(II) adsorption; however, its adsorption capacity was much less for Cd(II) (98 mg g−1) than for Pb(II).137 In contrast to adsorption behavior of Pb(II), MOF-5 exhibited a lower Cd(II) adsorption capacity (3.6 mg g−1) with a relatively less PC value of 0.01 L g−1, while modified MOF depicts a comparatively high PC value of (2.62 L g−1) in the removal of Cd(II).136,137
The MOF, TMU-5 exhibited a similar equilibrium time and optimal pH for both Pb(II) and Cd(II) adsorption, with a removal capacity of 251 mg g−1 for Pb(II) and 43 mg g−1 for Cd(II).138 HKUST-1 MW@H3PW12O40, which is derived from the MOF HKUST-1, had a maximum Pb(II) uptake capacity of 98 mg g−1 within 10 min. Its Cd(II) adsorption capacity was 32 mg g−1 within 80 min, through the chemisorption mechanism.125 The Cu-terephthalate MOF had an uptake capacity of 80 mg g−1 for Pb(II) and 90 mg g−1 for Cd(II) within 120 min at an optimal pH of 7 with PC values of 2.28 and 2.51 L g−1 respectively.139 Interestingly, the Cu-terephthalate MOF has been compared with graphene oxide and mordenite zeolite to treat high concentrations of Mn, Cu, Zn, Fe, Cd, and Pb in acid drainage from mines, and the Cu-terephthalate MOF exhibited the highest removal capacity.139 Many different types of MOFs, such as AMOF-1,140 3D Co(II) MOF,141 and UiO-66-NHC(S)NHMe,115 have been introduced to remove both Pb(II) and Cd(II) ions from aqueous media. Both PCN-100 (ref. 142) and Dy(BTC)(H2O)(DMF)1.1 (ref. 123) have shown promising results for Cd(II) removal.
Research has shown that media pH, equilibrium time, and media temperature govern both Pb(II) and Cd(II) adsorption by MOFs. Therefore, a comparison of performance (PC values) can be carried out for the adsorption of both Pb(II) and Cd(II) ions. Thiolated MOF-5 derivative (HS-mSi@MOF-5), depicted a PC value of 7.36 L g−1 in the adsorption of Pb(II), while 2.62 L g−1 for Cd(II). With PC values, it can be clearly understand that thiolated MOF-5 is more suitable for the adsorption of Pb(II) over Cd(II). The Cu-terephthalate MOF demonstrated more similar PC values 2.28 L g−1 and 2.51 L g−1 for both Pb(II) and Cd(II), respectively. Therefore, it is more suitable to adsorb both metal ions successfully. Significantly high performance for the adsorption of Pb(II) is exhibited by the MOF HKUST-1-MW@H3PW12O40. A partition coefficient value of 26.97 L g−1 was observed, while 0.24 L g−1 for the adsorption of Cd(II). Another modified MOF, UiO-66-NHC(S)NHMe depicted high performance in the adsorption of Pb(II) (4.92 L g−1) prior to Cd(II) (0.28 L g−1). Among the MOF types, which have the data to calculate PC values, only Cu-terephthalate MOF is identified as a material which performs well in adsorption of both Pb(II) and Cd(II) ions. However, from the obtained PC values, it can clearly understand that most of the MOFs exhibit a better performance to a specific metal ion, therefore, MOFs are highly selective. In the case of Dy(BTC)(H2O)(DMF)1.1, it has been used in removing Cd(II) with a performance of 2.16 L g−1 and noticed as a successful material among the MOFs discussed in this review.
3.3 Removal of anionic trace elements
Hexavalent Cr removal has been examined by a magnetic MOF, Fe3O4@MIL-100Fe, which is recorded a maximum adsorption capacity of 18 mg g−1 within 2 h at an optimal pH of 2, and a PC value of 0.18 L g−1.144 Moreover, azine-functionalized TMU-5 exhibited a maximum uptake capacity of 123 mg g−1 and stopped adsorbing at a pH of 10.138 TMU-30, exhibited an effective removal capacity of 145 mg g−1 within 10 min and comparatively good performance of 2.05 L g−1 to magnetized MOF, Fe3O4@MIL-100Fe.128
Chitosan–MOF(UiO-66) composite, a modified version of UiO-66, showed a high adsorption capacity of 94 mg g−1 for Cr(VI) due to the strong electrostatic attraction between high oxidation state metal ions/oxygen atoms or –NH2 groups and linkers, with an extremely high PC value, 50 L g−1.145 Another UiO-66 based amino-functionalized MOF, MOR-1-HA, was produced by Rapti et al. with a maximum adsorption capacity of 280 mg g−1 and a PC value of 0.31 L g−1.146 However, between two modified versions of UiO-66 MOF, chitosan–MOF composite demonstrates the best adsorption performance compared to MOR-1-HA.
The Zr-based ZJU-101 MOF showed a maximum uptake capacity of 245 mg g−1 within 10 min for a solution of Cr2O72−at an initial concentration of 50 mg L−1. The modified ZJU-101 MOF exhibited 324 times higher adsorption capacity than its precursor, MOF-867. It also showed excellent adsorption selectivity for Cr2O72−.130 In addition to these MOFs, UiO-66-NHC(S)NHMe,115 ZIF-67,147 and Cu-BTC129 have also shown comparatively high adsorption capacities for Cr(VI) from aqueous solutions. Moreover, both ZJU-101 and UiO-66-NHC(S)NHMe MOFs depicted similar PC values, which is 0.88 L g−1. Framework ZIF-67 demonstrated an adsorption performance of 1.05 L g−1.
A study of two new lanthanide-based MOFs, [Ce(L1)0.5(NO3)(H2O)2]·2DMF and Eu3(L2)2(OH)(DMF)0.22(H2O)5.78, showed that [Ce(L1)0.5(NO3)(H2O)2]·2DMF displayed a much higher adsorption capacity (103.95 mg g−1) and faster uptake rate (1.79 g mg−1 min−1) for fluoride than that of its counterpart (57.01 mg g−1).148 With the increase in the temperature, interestingly, MOF demonstrates a reduction in the performance for fluoride. [Ce(L1)0.5(NO3)(H2O)2]·2DMF exhibited PC value of 0.81 L g−1 at the temperatures of 318 K and 0.45 L g−1 at 298 K. Compared to [Ce(L1)0.5(NO3)(H2O)2]·2DMF, Eu3(L2)2(OH)(DMF)0.22(H2O)5.78 depicted a high performance with PC values of 0.26 and 0.17 L g−1 at 318 and 298 K respectively. The MOF MIL-96(Al) has been used in defluoridation, which exhibited a maximum adsorption capacity of 31.69 mg g−1. MIL-96(Al) depicted PC values of 0.95, 1.12, and 1.88 L g−1 at the temperatures of 298, 308, and 318 K, respectively. However, with the increment of the temperature, high performance in adsorption of fluoride is demonstrated.150 Framework UiO-66-NH2 exhibited maximum adsorption capacities of 60, 53, and 42 mg g−1 at 293, 313, and 333 K, respectively. Performance data of MIL-96(Al) and UiO-66-NH2 frameworks also resulted an increasing trend with the increasing temperature.74 Over 80% removal of fluoride (32.13 mg g−1) was achieved by MOF-801, a fumarate-based MOF, within 5 min at room temperature with an adsorption performance of 0.24 L g−1. The maximum fluoride uptake capacity for this nontoxic calcium fumarate (CaFu) MOF was calculated to be as high as 166.11 mg g−1 at 373 K.151 MOF-801 has an adsorption efficiency of 40 mg g−1 at 303 K. This adsorption efficiency remained high and stable in a pH range from 2–10, and was not affected by high ion concentration or the presence of other anions, including Cl−, NO3−, and SO42−. The adsorption performance was recorded as similar to the increasing temperature. At 293 K MOF-801 exhibited a PC value of 0.4 L g−1, while 0.45 L g−1 at 323 K. Mechanism for effective defluoridation by MOF-801 has been suggested to be chemisorption with an exchange of fluoride ions and hydroxyl groups within MOF-801.152
A novel dual ZrLa hydroxide anchored bio-sorbent (ZrLa/PP composites) was studied for fluoride adsorption at temperatures of 20, 30, and 40 °C. The adsorption capacity of ZrLa/PP composites at these temperatures was calculated to be 27.0, 32.5, and 37.2 mg g−1, respectively.153 At pH 3, ZrLa/PP composites showed 0.7 L g−1 of performance value. The ZrLa/PP composites exhibited promising chemical stability, and even after five cycles of column adsorption–desorption, greater than 90% capacity was retained for the adsorbent, indicating a high potential for practical use in water treatment.153
The ZIF-8 and Uio-66 MOFs, with high surface areas (1050 m2 g−1 and 800 m2 g−1 respectively), high total pore volume (0.57 cm3 g−1, 0.45 cm3 g−1 respectively), and average pore diameter (4.5 nm, 3.2 nm respectively) exhibited maximum adsorption capacities of 25 mg g−1 and 20 mg g−1, respectively for fluoride removal.154 Another novel MOF, Ce-1,1′-biphenyl-4,4′-dicarboxylic acid (Ce-bpdc), demonstrated a maximum adsorption capacity of 45.5 mg g−1 for fluoride at 298 K and a pH of 7. This represented a PC value of 1.54 L g−1 and a removal efficiency greater than 80%.155 Furthermore, Ce-bpdc removed fluoride from groundwater samples taken from Yuefang, Jiangji, and Sanyi in China with a removal efficiency of 78, 64, and 50%, respectively, indicating the potential of Ce-bpdc to be an effective adsorbent for this purpose.
The potential of aluminum fumarate (AlFu) MOF, with a surface area of 1156 m2 g−1 and average pore size of 17 Å, was also investigated for the removal of fluoride from groundwater. The results indicated that the AlFu MOF was thermally stable up to 700 °C, with a maximum adsorption capacity for fluoride of 600, 550, 504, and 431 mg g−1, respectively, at 293, 303, 313, and 333 K.156 At 293 and 313 K aluminum fumarate (AlFu), MOF demonstrates PC values of 0.61 and 0.47 L g−1, respectively. The adsorption mechanism was suggested to be substitution of fluoride ions for the hydroxyl ions in the AlFu MOF.156 Similar to the exothermic process in physisorption, a reduction in performance can be clearly observed in most MOFs with the increasing temperature.
3.4 Adsorptive removal of other ions by metal–organic frameworks
The ZrO2 nanoparticles functionalized MIL-101, which is denoted as MIL-101@Zr(DS), provided a medium removal potential for PO43− as 21.28 mg P g−1 with an adsorption performance of 0.23 L g−1.160 Molecular-level dispersion of ZrO2 nanoparticles improved the removal efficiency of phosphate; however, the presence of humic acid has drastically reduced the PO43− adsorption capacity to 2–10 times depending on the varying nanoparticle dispersion in the MOF.160 Solvothermally developed Zr based MOF UiO-66 demonstrates 415 mg g−1 of phosphate uptake capacity, which was significantly more extensive than that of the other Zr-based adsorbents that may possibly be attributed to the strong affinity of PO43− to Zr–OH groups. Amine substituted UiO-66-NH2 was compared with Zr based UiO-66 for PO43− indicated a higher affinity to UiO-66-NH2 via amine–PO43− interaction and the removal capacity doubled at the increment of temperature by two folds.161 Interestingly, once Zr based UiO-66 and UiO-66-NH2 were examined for the removal of PO43− in urine, diluted PO43− in urine (C0 of 14 mg L−1), both MOFs exhibited 100% removal within 10 min, however, at C0 of 500 mg L−1 kinetics were slow however, adsorption capacity remained high 206 and 264 mg g−1 respectively.161 Interestingly, basic MOF UiO-66 depicted 1.6 and 3.6 L g−1 PC values at 293 and 333 K while UiO-66-NH2 MOFs exhibited 1.9 and 4.3 L g−1 at the same temperatures indicating a performance increase with increase in temperature describing chemisorption. However, MOFs for phosphorous adsorption demonstrate an increment in the PC values with the increasing temperature, as same as in a general way. Fe-based MOFs, MIL-101, and NH2-MIL-101 demonstrated a quick reduction of phosphates from the initial 0.60 mg L−1 to 0.045 and 0.032 mg L−1, respectively, in 30 min of exposure with high selectivity over other anions. Adsorption performance of 2.32 L g−1 is observed for Fe-based MOF.162 Similar iron modified MIL-100(Fe) offered a high removal capacity of 93.6 mg g−1 for PO43− with an extremely high performance of 52.12 L g−1 suggested that the electrostatic attractions between positively charged metal sites of MIL-100(Fe) and PO43− to be the primary factor governing the PO43− removal from aqueous media.163
According to the available data, SCU-100 MOF for ReO4− adsorption depicts a PC value of 1.82 L g−1. Single crystal analysis has confirmed that adsorbed ReO4− ions have trapped in the void spaces and framework SCU-100 demonstrates an excellent selectivity for ReO4− due to the formation of Ag–O–Re bonds.165 Both primary and modified frameworks have been tested for adsorption of Th4+, and the best performance of 2.7 L g−1 is exhibited by the modified MOF, UiO-66-(COOH)2. Other MOF types such as UiO-66-COOH, and UiO-66 demonstrate PC values of 0.9 and 0.08 L g−1. The vital role of carboxyl groups is observed with Th4+ ion adsorption performance values of the three framework materials. Binding effects of carboxyl groups and electrostatic interactions enhance the adsorption kinetics. Coordination reaction between carboxyl groups and Th4+ ions has identified as the probable adsorption mechanism, which was then confirmed by FTIR and EXAFS analysis.166 Sulfate/sulfonic acid functionalized MOFs labeled as MOF-808-SO4 and MIL-101-SO3H(Cr) removed >90% radioactive barium (Ba2+) in five minutes and then reached 99% with a maximum removal capacity of 131.1 mg g−1.167 Some radioactive metal ions such as Am(III), Np(IV), and Pu(IV) have not been studied for adsorption by MOFs whereas even the studied conducted already are not in detail as an example, no information exists about the extent of damage to the frameworks of most MOFs by radiation.
4. Mechanisms for the removal of potentially toxic elements
Adsorption of PTEs onto MOFs, modified or unmodified, occurs due to their high surface areas and microporosity.18 The effectiveness of removal increases as the surface area and development of micropores increases. Surface polarity and aromaticity are essential characteristics of MOFs, as they influence the adsorption of aqueous organic contaminants, but not inorganic PTEs. The mechanisms for adsorption of PTEs by MOFs can be classified as either chemisorption or physisorption. As shown in Fig. 3, chemisorption includes chemical bonding, coordination, and acid–base interactions. Physisorption includes electrostatic attraction, van der Waals forces, and diffusion. The most common mechanisms for PTE removal via MOFs are suggested as acid–base interaction, π–π interaction, ion exchange, and coordination.16 Other possible applications for MOFs in PTE removal are based on hydrogen bonding.66 Research has shown that MOFs have a unique potential to adsorb relatively inert molecules via hydrogen bonding, which cannot usually be adsorbed by other chemical means, such as acid–base interaction, π–π interaction, and coordination.66 Published data also suggests that unmodified MOFs, in general, have lower adsorption capacities for PTEs than modified MOFs.18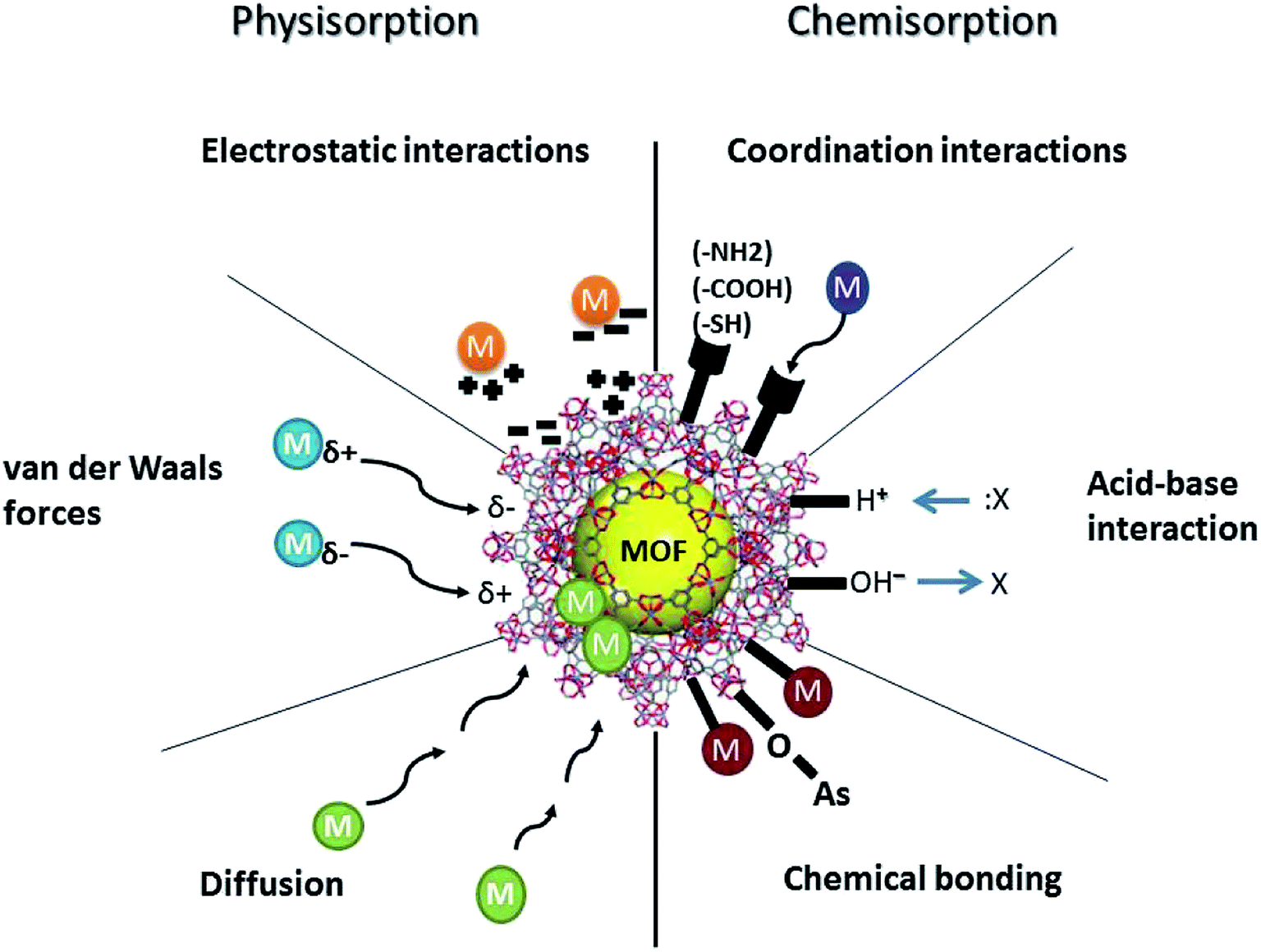 | ||
| Fig. 3 Different physisorption and chemisorption interactions of MOFs with PTEs in aqueous media. | ||
The surface area of a MOF is generally considered to play a crucial role in determining its adsorption capacity. However, that is not always the case. The functional groups in a MOF may override the intrinsic adsorption properties of MOFs where post-synthetic modifications are key to PTE removal. FT-IR analysis before and after contact with PTEs suggests an apparent coordination interaction between the PTEs and the –NH2 and/or other functional groups.141 In some cases, PTEs are adsorbed by the O-containing moieties of the MOFs, while some PTEs, such as Cr, interact with functional groups such as –NH2.16 Coordination interactions may occur between PTEs and such functional groups as amino groups (–NH2), carboxyl groups (–COOH), and thiol (–SH) in modified MOFs.18 Therefore, the functionality of the MOFs critically influences the adsorption capacity.
Electrostatic attraction/repulsion between inorganic contaminants and MOFs is another possible adsorption mechanism. Negatively charged MOF surfaces can facilitate the electrostatic attraction of positively charged cationic PTEs. Fig. 4 illustrates this phenomenon. This electrostatic attraction was reported by various studies related to the adsorption of fluoride and arsenic mainly via surface hydroxyl groups and other functional groups, such as thiols and amines. Both electron-rich and electron-poor functional groups are present in MOFs, hence they can play a role in the adsorption of both electron donors and electron acceptors. Hence, the functionalization of MOFs at the design and synthesis stage, which aims to create a strong affinity to contaminants, is an important factor governing the mechanism of removal for PTEs. Physisorption is mainly controlled by the solution pH, where the surface of the MOFs protonate or deprotonate, allowing PTEs to bind to the MOFs. When the solution pH < pzc, large amounts of hydrated hydrogen ions (H3O+) are present, which will attract negatively charged PTEs. In contrast, when the pH > pzc, the MOF is negatively charged, and there is electrostatic attraction between the MOF and positively charged PTEs.18 This has been well documented for Pb(II), Cd(II), and Hg(II), as well as for Cr(VI) and As(V).137,168,144 Diffusion is a physical mechanism that governs the adsorption of PTEs by MOFs where data modeling shows the process to reflect the intra-particle diffusion equation.137
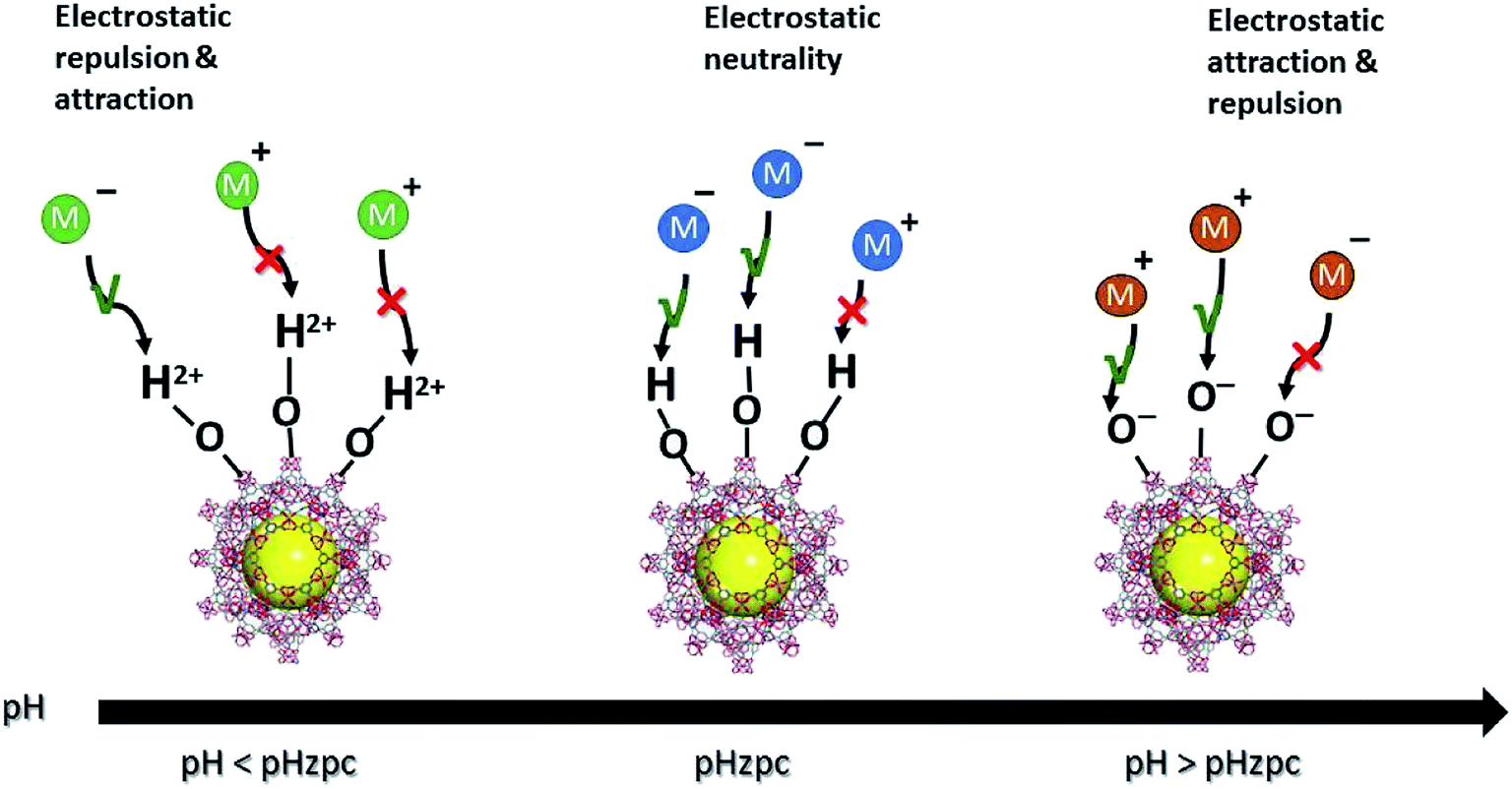 | ||
| Fig. 4 Schematic diagram of the electrostatic attraction/repulsion of charged PTEs based on the zero-point charge (pHzpc) of the MOFs. | ||
It should be noted that the adsorption capacity of a MOF is influenced by many factors of both the adsorbent and adsorbate. These factors include the relatively high surface area, the porosity, the organic linkers, functionalization, and zeta potential of MOFs, and the characteristics of target contaminants, solution pH, and coexisting substances in the adsorbate media. The dominant interaction of the MOF and contaminant may also have a significant influence on the adsorption process and can vary under different conditions. Hence, the real mechanism for adsorption is relatively complex and molecular level investigations are essential for exact prediction.
5. Production upscale and utilization
Since MOFs were highly promising and attractive to be used in water treatment due to the synthesis of water-stable MOFs, strategies to expand the removal performance of MOFs have attracted focal point in recent years and many different techniques have been examined to improve the adsorption potential of MOFs by varying the additives and procedures during production (Fig. 5). Synthesis of MOFs with large organic linkers may facilitate diffusion of metal ions through the enlarged pores of MOFs increase the adsorption capacity; however, it was limited due to the commercial availability of such linkers.169,106 Similarly, MOFs can be synthesized by incorporating defects via including modulators to the precursor mixture which again will enlarge the pore sizes of the MOFs providing more surface sites for adsorption.170 Inculcating defects into MOFs is considered as simpler as and more economically viable than the MOFs with large organic linkers and furthermore, the produced MOFs are hydrophilic in nature.101 Additionally, functionalization of the organic linker or the metal node improves the capacities or MOFs, and modifications or hybridization such as magnetization strengthens MOFs by including property for easy recovery. The possibility for regeneration is one of the critical parameters of choosing a MOF for water treatment, which satisfies economic viability and various environmental protection guidelines. Regeneration depends on structural and chemical properties of the adsorbent. Regeneration potential of MOFs has been tested by changing temperature/pressure (or vacuum), pH, and modifications.171 One of the effective Cd(II) adsorbents, Cu3(BTC)2-SO3H framework depicted an easy regeneration method which is washing with deionized water and drying.16 The MOF CPO-27, a Ni(II) adsorbent (Ni) depicted regeneration at a temperature of ≥90 °C.172 Furthermore, magnetic modification of MOFs has provided an opportunity for the easily regeneration.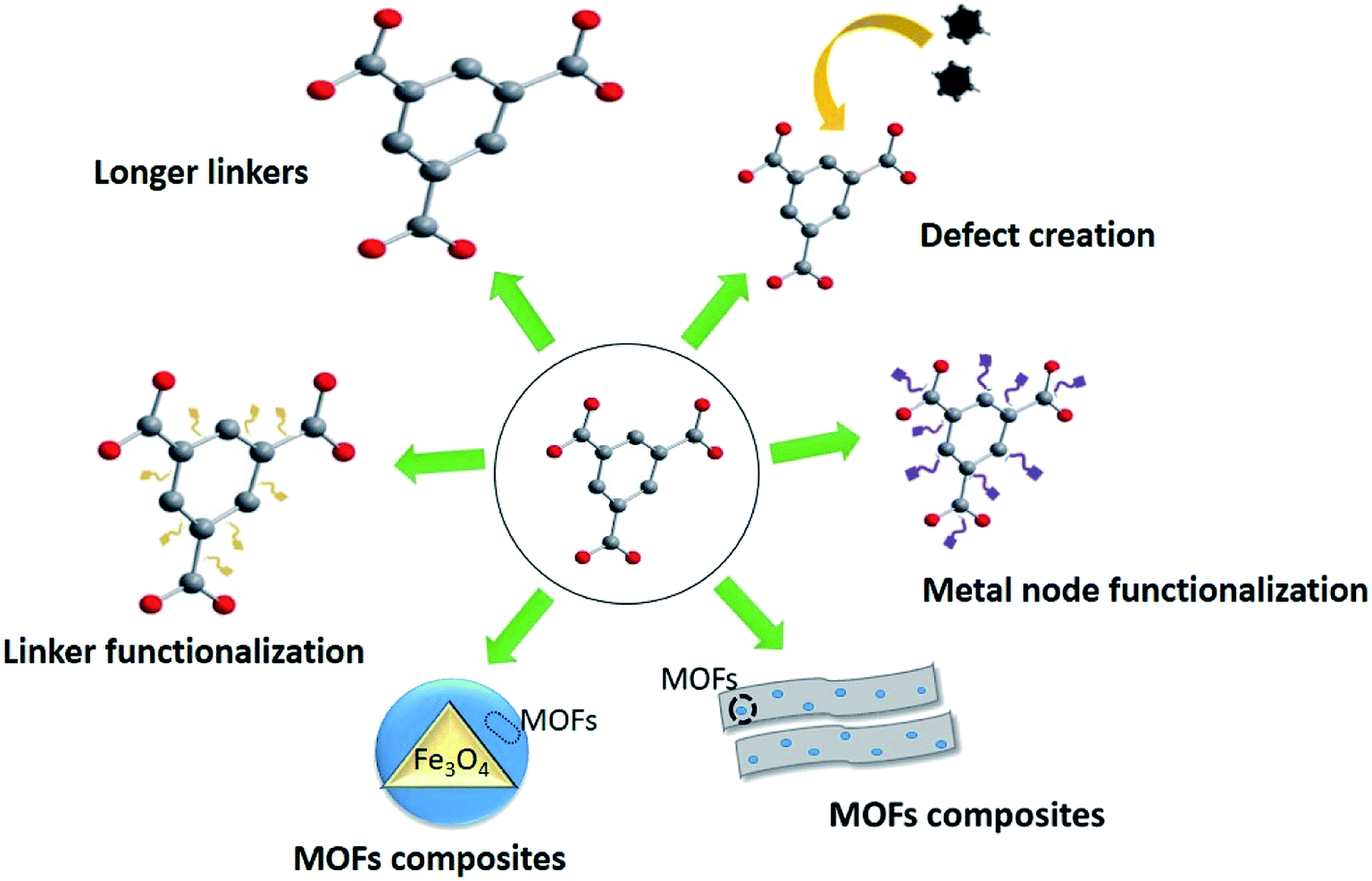 | ||
| Fig. 5 Graphical illustration of strategies for enhancing adsorption performance of MOFs. | ||
Although the above enhancements were promising and tested in laboratory scale, the number of commercial-scale products is limited that indicates a gap between the conversion of laboratory MOFs research to commercialization. It has been observed that the transformation of MOFs from laboratory to commercial level hindered by the scale-up issues such as production capacity (space-time yield), expenditure, properties, purity, and stability. Due to these limitations and challenges, despite the remarkable academic interest which produced thousands of new MOFs with diverse applications, only a few of them are being translated from laboratory to large scale to be used in real-world applications.173 A 3D UiO-66/wood composite membrane has reported, which can be practically applicable in large-scale treatment processes for the removal of organic pollutants with an efficiency of 96%.174 However, not many large-scale applications are not reported for PTEs.
Viable transformation of production of MOFs from laboratory scale to commercial level challenges science and engineering. The challenges in transforming are needed to be identified clearly by the researchers, engineers to explore, improve innovative, and commercially feasible scale-up methods to produce MOFs for more recurrent applications. Some of the challenges of scale-up the production of MOFs are listed as follows.
• The production of stable MOFs for temperature and humidity with maintaining product quality and reproducibility among batches.
• The high volume of organic solvents and linkers for MOFs production necessity mass-scale production of those.
• Scale-up of the production vessels may decrease the surface to volume ratio so that the reaction kinetics will be less which will lead to prolonged reactions hours and low quality of the product.
• The feasibility of production/synthesis technique for commercial application with high space-time yield and reduced usage of heating or external solvents are used.
• Technical capability to produce different MOFs structures from the same piece of equipment.
• Continuous process than batch production which will reduce downtimes, labor costs and constant and consistent production of MOFs.
• Activation and post-modification on a large scale need large volume of chemicals.
6. Future perspectives
PTEs receive significant attention to reduce their presence in water and food sources to protect human health and the environment. Among the many methods available, the removal of PTEs from water using MOFs has become a focal point of research worldwide due to their versatility and high potential for use as adsorbents. Among other characteristics, their wider range of functionalities, chemical activities, higher stability, and their ability to be easily tailored or modified for specific applications make MOFs highly attractive for pollutant removal in the environment. MOFs have consistently shown enhanced adsorption capacity and performance, and the kinetics of removal has been more rapid for MOFs than for conventional adsorbents. Despite their potentially high adsorption capacity for PTEs, a major challenge faced by the majority of MOFs studied to date is poor water stability and poor selectivity. Research has made clear that there is no single, universal MOF, which is an effective adsorbent for many different contaminants simultaneously. Yet, there are opportunities for more detailed molecular level studies, in particular to provide an understanding of the adsorption mechanism of PTEs, to facilitate the design of MOFs that can better address multiple contaminants in variable environments.Several challenges still exist in commercial applications of MOFs for PTE removal in water treatment. The behavior of MOFs in water or aqueous acid/base solutions, their recyclability and reusability (regeneration), and the competitive performance of MOFs in complex wastewater effluents with multiple pollutants are a few of these challenges.15 Competitive sorption should be studied in detail, not only with similar competitive PTEs but also with organic contaminants, dissolved organic compounds, and microbial communities, which have not received enough attention. Development of bi- or heterometallic MOFs to remove metalloids with different valences simultaneously is also a challenge that should be addressed in future research. There is still space in the research arena to develop pioneering technologies for producing water-stable MOFs for small-scale commercial or individual household water treatment systems. Engineered MOFs should be considered for use in either permanent or single-use filters in such systems. Scaling up of MOF production in large scale should receive further attention. A new frontier for adsorptive removal has been opened by the advent of MOFs that function via hydrogen bonding due to their strong adsorptive removal potential.
Exploration of the effects of structural adaptations by ligands in water is another important area of research for the future. At the same time, cost-minimization via introducing simple synthesis methods, easy and cost-effective regeneration/recycling procedures, durability, and sustainable disposal options also need more research. Overall, MOFs will inevitably be considered as an effective alternative to conventional adsorbents provided that their water stability and reusability can be ensured.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research Council of the University of Sri Jayewardenepura is acknowledged for financial support.References
- UN, Transforming our world: the 2030 Agenda for Sustainable Development, 2015 Search PubMed
.
- FAO and IWMI, Water pollution from agriculture: A global review, 2017 Search PubMed
- S. C. Nde and M. Mathuthu, Int. J. Environ. Res. Public Health, 2018, 15, 576 CrossRef
- R. Nieder, D. K. Benbi and F. X. Reichl, in Soil Components and Human Health, Springer, 2018, pp. 375–450 Search PubMed
- R. Milačič, T. Zuliani, J. Vidmar, P. Oprčkal and J. Ščančar, Sci. Total Environ., 2017, 605, 894–905 CrossRef
- A. Shakeri, R. Shakeri and B. Mehrabi, Int. J. Environ. Sci. Technol., 2015, 12, 2201–2212 CrossRef CAS
- M. L. Mitch, J. Environ. Qual., 2002, 31, 109–120 CrossRef
- S. Chatterjee, I. Sivareddy and S. De, J. Environ. Chem. Eng., 2017, 5, 3273–3289 CrossRef CAS
- A. U. Rajapaksha, S. S. Chen, D. C. W. Tsang, M. Zhang, M. Vithanage, S. Mandal, B. Gao, N. S. Bolan and Y. S. Ok, Chemosphere, 2016, 148, 276–291 CrossRef CAS
- M. Hua, S. Zhang, B. Pan, W. Zhang, L. Lv and Q. Zhang, J. Hazard. Mater., 2012, 211, 317–331 CrossRef
- T. Pradeep, Thin Solid Films, 2009, 517, 6441–6478 CrossRef CAS
- C. Santhosh, V. Velmurugan, G. Jacob, S. K. Jeong, A. N. Grace and A. Bhatnagar, Chem. Eng. J., 2016, 306, 1116–1137 CrossRef CAS
- Y. Taamneh and S. Sharadqah, Appl. Water Sci., 2017, 7, 2021–2028 CrossRef CAS
- P. Kumar, V. Bansal, K.-H. Kim and E. E. Kwon, J. Ind. Eng. Chem., 2018, 62, 130–145 CrossRef CAS
- S. Dhaka, R. Kumar, A. Deep, M. B. Kurade, S. W. Ji and B. H. Jeon, Coord. Chem. Rev., 2019, 380, 330–352 CrossRef CAS
- P. A. Kobielska, A. J. Howarth, O. K. Farha and S. Nayak, Coord. Chem. Rev., 2018, 358, 92–107 CrossRef CAS
- M. Feng, P. Zhang, H.-C. Zhou and V. K. Sharma, Chemosphere, 2018, 209, 783–800 CrossRef CAS
- J. Wen, Y. Fang and G. Zeng, Chemosphere, 2018, 201, 627–643 CrossRef CAS
- A. Demessence, D. M. D. Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784–8786 CrossRef CAS
- S. Wang, E. V Alekseev, J. Diwu, W. H. Casey, B. L. Phillips, W. Depmeier and T. E. Albrecht-Schmitt, Angew. Chem., Int. Ed., 2010, 49, 1057–1060 CrossRef CAS
- M. Babazadeh, R. Hosseinzadeh-Khanmiri, J. Abolhasani, E. Ghorbani-Kalhor and A. Hassanpour, RSC Adv., 2015, 5, 19884–19892 RSC
- M. Y. Omar, Utility patent, 5648508A, 1997
- U. Mueller, M. Schubert, F. Teich, H. Puetter, K. Schierle-Arndt and J. Pastré, J. Mater. Chem., 2006, 16, 626–636 RSC
- U. Müller, H. Puetter, M. Hesse and H. Wessel, WO/2005/049892, 2006
- J. Sung-Hwa, C. Jong-San, H. Young-Kyu, S. Christian and F. Gerard, US Pat., US7855299B2, 2010
- Z. Ni and R. I. Masel, J. Am. Chem. Soc., 2006, 128, 12394–12395 CrossRef CAS
- M. Ulrich, H. Michael, L. Lisa, H. Markus, A. Jan-Dirk and R. Peter, US pat., US7119219B2, 2006
- A. L. Garay, A. Pichon and S. L. James, Chem. Soc. Rev., 2007, 36, 846–855 RSC
- A. Carné-Sánchez, K. C. Stylianou, C. Carbonell, M. Naderi, I. Imaz and D. Maspoch, Adv. Mater., 2015, 27, 869–873 CrossRef
- R. J. Kuppler, D. J. Timmons, Q.-R. Fang, J.-R. Li, T. A. Makal, M. D. Young, D. Yuan, D. Zhao, W. Zhuang and H.-C. Zhou, Coord. Chem. Rev., 2009, 253, 3042–3066 CrossRef CAS
- K. Vikrant and K.-H. Kim, Chem. Eng. J., 2018, 358, 264–282 CrossRef
- K. Vikrant, V. Kumar, Y. S. Ok, K.-H. Kim and A. Deep, TrAC, Trends Anal. Chem., 2018, 105, 263–281 CrossRef CAS
- B. N. Bhadra, I. Ahmed, S. Kim and S. H. Jhung, Chem. Eng. J., 2017, 314, 50–58 CrossRef CAS
- Z. Hasan, N. A. Khan and S. H. Jhung, Chem. Eng. J., 2016, 284, 1406–1413 CrossRef CAS
- O. M. Yaghi, M. O'keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS
- H. Wei, S. Deng, Q. Huang, Y. Nie, B. Wang, J. Huang and G. Yu, Water Res., 2013, 47, 4139–4147 CrossRef CAS
- D. Krajišnik, A. Daković, M. Milojević, A. Malenović, M. Kragović, D. B. Bogdanović, V. Dondur and J. Milić, Colloids Surf., B, 2011, 83, 165–172 CrossRef
- S. A. C. Carabineiro, T. Thavorn-Amornsri, M. F. R. Pereira, P. Serp and J. L. Figueiredo, Catal. Today, 2012, 186, 29–34 CrossRef CAS
- S. Li, X. Zhang and Y. Huang, J. Hazard. Mater., 2017, 321, 711–719 CrossRef CAS
- F. Wang, X. Lu, W. Peng, Y. Deng, T. Zhang, Y. Hu and X. Li, ACS Omega, 2017, 2, 5378–5384 CrossRef CAS
- M. Sarker, J. Y. Song and S. H. Jhung, Chem. Eng. J., 2018, 331, 124–131 CrossRef CAS
- M. Sarker, H. J. An, D. K. Yoo and S. H. Jhung, Chem. Eng. J., 2018, 338, 107–116 CrossRef CAS
- F. Wang, J. Yao, K. Sun and B. Xing, Environ. Sci. Technol., 2010, 44, 6985–6991 CrossRef CAS
- C.-Y. Chen, C.-C. Chen and Y.-C. Chung, Bioresour. Technol., 2007, 98, 2578–2583 CrossRef CAS
- S. Murai, S. Imajo, Y. Takasu, K. Takahashi and K. Hattori, Environ. Sci. Technol., 1998, 32, 782–787 CrossRef CAS
- Z. Li, Y. Wu, J. Li, Y. Zhang, X. Zou and F. Li, Chem.–Eur. J., 2015, 21, 6913–6920 CrossRef CAS
- P. Girods, A. Dufour, V. Fierro, Y. Rogaume, C. Rogaume, A. Zoulalian and A. Celzard, J. Hazard. Mater., 2009, 166, 491–501 CrossRef CAS
- Q.-S. Liu, T. Zheng, P. Wang, J.-P. Jiang and N. Li, Chem. Eng. J., 2010, 157, 348–356 CrossRef CAS
- M. Maes, S. Schouteden, L. Alaerts, D. Depla and D. E. De Vos, Phys. Chem. Chem. Phys., 2011, 13, 5587–5589 RSC
- Y. Pan, Z. Li, Z. Zhang, X.-S. Tong, H. Li, C.-Z. Jia, B. Liu, C.-Y. Sun, L.-Y. Yang and G.-J. Chen, J. Environ. Manage., 2016, 169, 167–173 CrossRef CAS
- J. Reungoat, J.-S. Pic, M.-H. Manero and H. Debellefontaine, Sep. Sci. Technol., 2007, 42, 1447–1463 CrossRef CAS
- W. Wei, R. Sun, J. Cui and Z. Wei, Desalination, 2010, 263, 89–96 CrossRef CAS
- L. Xie, D. Liu, H. Huang, Q. Yang and C. Zhong, Chem. Eng. J., 2014, 246, 142–149 CrossRef CAS
- A. Khenifi, Z. Derriche, C. Mousty, V. Prévot and C. Forano, Appl. Clay Sci., 2010, 47, 362–371 CrossRef CAS
- X. Zhu, B. Li, J. Yang, Y. Li, W. Zhao, J. Shi and J. Gu, ACS Appl. Mater. Interfaces, 2014, 7, 223–231 CrossRef
- Q. Yang, J. Wang, W. Zhang, F. Liu, X. Yue, Y. Liu, M. Yang, Z. Li and J. Wang, Chem. Eng. J., 2017, 313, 19–26 CrossRef CAS
- B. K. Jung, Z. Hasan and S. H. Jhung, Chem. Eng. J., 2013, 234, 99–105 CrossRef CAS
- M. Sarker, I. Ahmed and S. H. Jhung, Chem. Eng. J., 2017, 323, 203–211 CrossRef CAS
- E. Haque, J. W. Jun and S. H. Jhung, J. Hazard. Mater., 2011, 185, 507–511 CrossRef CAS
- L. Li, X. L. Liu, H. Y. Geng, B. Hu, G. W. Song and Z. S. Xu, J. Mater. Chem. A, 2013, 1, 10292–10299 RSC
- E. Haque, J. E. Lee, I. T. Jang, Y. K. Hwang, J.-S. Chang, J. Jegal and S. H. Jhung, J. Hazard. Mater., 2010, 181, 535–542 CrossRef CAS PubMed
- Y. Pan, J. Wang, X. Guo, X. Liu, X. Tang and H. Zhang, J. Colloid Interface Sci., 2018, 513, 418–426 CrossRef CAS
- Q. ZHang, J. Wang, A. M. Kirillov, W. Dou, C. Xu, C.-L. Xu, L. Yang, R. Fang and W.-S. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 23976–23986 CrossRef CAS
- H. Wu, Q. Gong, D. H. Olson and J. Li, Chem. Rev., 2012, 112, 836–868 CrossRef CAS
- B. Van de Voorde, B. Bueken, J. Denayer and D. De Vos, Chem. Soc. Rev., 2014, 43, 5766–5788 RSC
- I. Ahmed and S. H. Jhung, Chem. Eng. J., 2017, 310, 197–215 CrossRef CAS
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Ferey, R. E. Morris and C. Serre, Chem. Rev., 2011, 112, 1232–1268 CrossRef
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2011, 112, 724–781 CrossRef
- V. N. Panchenko, M. N. Timofeeva and S. H. Jhung, Catal. Rev., 2016, 58, 209–307 CrossRef CAS
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC
- B. Liu, K. Vellingiri, S.-H. Jo, P. Kumar, Y. S. Ok and K.-H. Kim, Nano Res., 2018, 11, 4441–4467 CrossRef CAS
- J.-J. Li, C.-C. Wang, H. Fu, J.-R. Cui, P. Xu, J. Guo and J.-R. Li, Dalton Trans., 2017, 46, 10197–10201 RSC
- Y. Wu, H. Luo and H. Wang, RSC Adv., 2014, 4, 40435–40438 RSC
- K.-Y. A. Lin, Y.-T. Liu and S.-Y. Chen, J. Colloid Interface Sci., 2016, 461, 79–87 CrossRef CAS
- X. Zhao, X. Han, Z. Li, H. Huang, D. Liu and C. Zhong, Appl. Surf. Sci., 2015, 351, 760–764 CrossRef CAS
- Q. Zhang, J. Yu, J. Cai, R. Song, Y. Cui, Y. Yang, B. Chen and G. Qian, Chem. Commun., 2014, 50, 14455–14458 RSC
- X. Zhao, D. Liu, H. Huang and C. Zhong, Microporous Mesoporous Mater., 2016, 224, 149–154 CrossRef CAS
- Z. S. Moghaddam, M. Kaykhaii, M. Khajeh and A. R. Oveisi, Spectrochim. Acta, Part A, 2018, 194, 76–82 CrossRef CAS
- X. Zhao, Y. Wei, H. Zhao, Z. Gao, Y. Zhang, L. Zhi, Y. Wang and H. Huang, J. Colloid Interface Sci., 2018, 514, 234–239 CrossRef CAS
- Y. Wu, G. Xu, W. Liu, J. Yang, F. Wei, L. Li, W. Zhang and Q. Hu, Microporous Mesoporous Mater., 2015, 210, 110–115 CrossRef CAS
- C. A. Bauer, T. V Timofeeva, T. B. Settersten, B. D. Patterson, V. H. Liu, B. A. Simmons and M. D. Allendorf, J. Am. Chem. Soc., 2007, 129, 7136–7144 CrossRef CAS
- Y. Bian, N. Xiong and G. Zhu, Technology for the Remediation of Water Pollution: A Review on the Fabrication of Metal Organic Frameworks, 2018, vol. 6 Search PubMed
- H. Deng, C. J. Doonan, H. Furukawa, R. B. Ferreira, J. Towne, C. B. Knobler, B. Wang and O. M. Yaghi, Science, 2010, 327, 846–850 CrossRef CAS
- Z. H. Rada, H. R. Abid, H. Sun and S. Wang, J. Chem. Eng. Data, 2015, 60, 2152–2161 CrossRef CAS
- Z. Yin, S. Wan, J. Yang, M. Kurmoo and M.-H. Zeng, Coord. Chem. Rev., 2017, 378, 500–512 CrossRef
- M. Servalli, M. Ranocchiari and J. A. Van Bokhoven, Chem. Commun., 2012, 48, 1904–1906 RSC
- C.-J. Na, M.-J. Yoo, D. C. W. Tsang, H. W. Kim and K.-H. Kim, J. Hazard. Mater., 2019, 366, 452–465 CrossRef CAS
- H.-C. Hu, X.-S. Chai and D. Barnes, Fluid Phase Equilib., 2014, 380, 76–81 CrossRef CAS
- J. J. Hurst, J. S. Wallace and D. S. Aga, Chemosphere, 2018, 197, 271–279 CrossRef CAS
- D. Song, R. Yang, F. Long and A. Zhu, J. Environ. Sci., 2019, 80, 14–34 CrossRef
- K. Sini, D. Bourgeois, M. Idouhar, M. Carboni and D. Meyer, New J. Chem., 2018, 42, 17889–17894 RSC
- R. E. Vernon, J. Chem. Educ., 2013, 90, 1703–1707 CrossRef CAS
- F. Edition, WHO Chron., 2011, 38, 104–108 Search PubMed
- A. Pell, A. Márquez, J. F. López-Sánchez, R. Rubio, M. Barbero, S. Stegen, F. Queirolo and P. Díaz-Palma, Chemosphere, 2013, 90, 556–564 CrossRef CAS PubMed
- B.-J. Zhu, X.-Y. Yu, Y. Jia, F.-M. Peng, B. Sun, M.-Y. Zhang, T. Luo, J.-H. Liu and X.-J. Huang, J. Phys. Chem. C, 2012, 116, 8601–8607 CrossRef CAS
- Z.-Q. Li, J.-C. Yang, K.-W. Sui and N. Yin, Mater. Lett., 2015, 160, 412–414 CrossRef CAS
- C. Prum, R. Dolphen and P. Thiravetyan, J. Environ. Manage., 2018, 213, 11–19 CrossRef CAS
- C. O. Audu, H. G. T. Nguyen, C.-Y. Chang, M. J. Katz, L. Mao, O. K. Farha, J. T. Hupp and S. T. Nguyen, Chem. Sci., 2016, 7, 6492–6498 RSC
- T. A. Vu, G. H. Le, C. D. Dao, L. Q. Dang, K. T. Nguyen, Q. K. Nguyen, P. T. Dang, H. T. K. Tran, Q. T. Duong and T. V. Nguyen, RSC Adv., 2015, 5, 5261–5268 RSC
- J. Li, Y. Wu, Z. Li, M. Zhu and F. Li, Water Sci. Technol., 2014, 70, 1391–1397 CrossRef CAS
- J. Li, X. Wang, G. Zhao, C. Chen, Z. Chai, A. Alsaedi, T. Hayat and X. Wang, Chem. Soc. Rev., 2018, 47, 2322–2356 RSC
- M. Filella, N. Belzile and Y.-W. Chen, Earth-Sci. Rev., 2002, 57, 125–176 CrossRef CAS
- T. E. McKone and J. I. Daniels, Regul. Toxicol. Pharmacol., 1991, 13, 36–61 CrossRef CAS
- J. E. Mondloch, W. Bury, D. Fairen-Jimenez, S. Kwon, E. J. DeMarco, M. H. Weston, A. A. Sarjeant, S. T. Nguyen, P. C. Stair and R. Q. Snurr, J. Am. Chem. Soc., 2013, 135, 10294–10297 CrossRef CAS
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS
- A. J. Howarth, M. J. Katz, T. C. Wang, A. E. Platero-Prats, K. W. Chapman, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2015, 137, 7488–7494 CrossRef CAS
- S. Rangwani, A. Howarth, M. R. DeStefano, C. D. Malliakas, A. E. Platero-Prats, K. W. Chapman and O. K. Farha, Polyhedron, 2018, 151, 338–343 CrossRef CAS
- J. Li, X. Li, T. Hayat, A. Alsaedi and C. Chen, ACS Sustainable Chem. Eng., 2017, 5, 11496–11503 CrossRef CAS
- M. Kim and S. M. Cohen, CrystEngComm, 2012, 14, 4096–4104 RSC
- X. He, X. Min and X. Luo, J. Chem. Eng. Data, 2017, 62, 1519–1529 CrossRef CAS
- A. J. Howarth, M. J. Katz, T. C. Wang, A. E. Platero-, K. W. Chapman, J. T. Hupp, O. K. Farha, A. J. Howarth, M. J. Katz, T. C. Wang, A. E. Platero-prats, K. W. Chap-, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2015, 137, 7488–7494 CrossRef CAS
- J. W. Jun, M. Tong, B. K. Jung, Z. Hasan, C. Zhong and S. H. Jhung, Chem.–Eur. J., 2015, 21, 347–354 CrossRef CAS
- F. Ke, L.-G. Qiu, Y.-P. Yuan, F.-M. Peng, X. Jiang, A.-J. Xie, Y.-H. Shen and J.-F. Zhu, J. Hazard. Mater., 2011, 196, 36–43 CrossRef CAS PubMed
- L. Huang, M. He, B. Chen and B. Hu, J. Mater. Chem. A, 2015, 3, 11587–11595 RSC
- H. Saleem, U. Rafique and R. P. Davies, Microporous Mesoporous Mater., 2016, 221, 238–244 CrossRef CAS
- K.-K. Yee, N. Reimer, J. Liu, S.-Y. Cheng, S.-M. Yiu, J. Weber, N. Stock and Z. Xu, J. Am. Chem. Soc., 2013, 135, 7795–7798 CrossRef CAS
- L. Liang, Q. Chen, F. Jiang, D. Yuan, J. Qian, G. Lv, H. Xue, L. Liu, H.-L. Jiang and M. Hong, J. Mater. Chem. A, 2016, 4, 15370–15374 RSC
- S. Halder, J. Mondal, J. Ortega-Castro, A. Frontera and P. Roy, Dalton Trans., 2017, 46, 1943–1950 RSC
- Y. Y. Xiong, J. Q. Li, L. Le Gong, X. F. Feng, L. N. Meng, L. Zhang, P. P. Meng, M. B. Luo and F. Luo, J. Solid State Chem., 2017, 246, 16–22 CrossRef CAS
- N. D. Rudd, H. Wang, E. M. A. Fuentes-Fernandez, S. J. Teat, F. Chen, G. Hall, Y. J. Chabal and J. Li, ACS Appl. Mater. Interfaces, 2016, 8, 30294–30303 CrossRef CAS
- X. Luo, T. Shen, L. Ding, W. Zhong, J. Luo and S. Luo, J. Hazard. Mater., 2016, 306, 313–322 CrossRef CAS
- J. M. Rivera, S. Rincón, C. Ben Youssef and A. Zepeda, J. Nanomater., 2016, 2016, 9 Search PubMed
- A. Jamali, A. A. Tehrani, F. Shemirani and A. Morsali, Dalton Trans., 2016, 45, 9193–9200 RSC
- R. Ricco, K. Konstas, M. J. Styles, J. J. Richardson, R. Babarao, K. Suzuki, P. Scopece and P. Falcaro, J. Mater. Chem. A, 2015, 3, 19822–19831 RSC
- F. Zou, R. Yu, R. Li and W. Li, ChemPhysChem, 2013, 14, 2825–2832 CrossRef CAS
- Y. Wang, G. Ye, H. Chen, X. Hu, Z. Niu and S. Ma, J. Mater. Chem. A, 2015, 3, 15292–15298 RSC
- M. Roushani, Z. Saedi and Y. M. Baghelani, Environmental Nanotechnology, Monitoring & Management, 2017, 7, 89–96 Search PubMed
- L. Aboutorabi, A. Morsali, E. Tahmasebi and O. Buyukgungor, Inorg. Chem., 2016, 55, 5507–5513 CrossRef CAS
- A. Maleki, B. Hayati, M. Naghizadeh and S. W. Joo, J. Ind. Eng. Chem., 2015, 28, 211–216 CrossRef CAS
- Q. Zhang, J. Yu, J. Cai, L. Zhang, Y. Cui, Y. Yang, B. Chen and G. Qian, Chem. Commun., 2015, 51, 14732–14734 RSC
- Y. Zhang, Z. Xie, Z. Wang, X. Feng, Y. Wang and A. Wu, Dalton Trans., 2016, 45, 12653–12660 RSC
- Y. Zhang, X. Zhao, H. Huang, Z. Li, D. Liu and C. Zhong, RSC Adv., 2015, 5, 72107–72112 RSC
- N. Bakhtiari and S. Azizian, J. Mol. Liq., 2015, 206, 114–118 CrossRef CAS
- Y. Manawi, G. McKay, N. Ismail, A. K. Fard, V. Kochkodan and M. A. Atieh, Chem. Eng. J., 2018, 352, 828–836 CrossRef CAS
- M. S. Tizo, L. A. V Blanco, A. C. Q. Cagas, B. R. B. Dela Cruz, J. C. Encoy, J. V Gunting, R. O. Arazo and V. I. F. Mabayo, Sustainable Environ. Res., 2018, 28, 326–332 CrossRef CAS
- Q. Qin, Q. Wang, D. Fu and J. Ma, Chem. Eng. J., 2011, 172, 68–74 CrossRef CAS
- J. Zhang, Z. Xiong, C. Li and C. Wu, J. Mol. Liq., 2016, 221, 43–50 CrossRef CAS
- E. Tahmasebi, M. Y. Masoomi, Y. Yamini and A. Morsali, Inorg. Chem., 2014, 54, 425–433 CrossRef
- E. Rahimi and N. Mohaghegh, Mine Water Environ., 2016, 35, 18–28 CrossRef CAS
- A. Chakraborty, S. Bhattacharyya, A. Hazra, A. C. Ghosh and T. K. Maji, Chem. Commun., 2016, 52, 2831–2834 RSC
- A. Abbasi, T. Moradpour and K. Van Hecke, Inorg. Chim. Acta, 2015, 430, 261–267 CrossRef CAS
- Q.-R. Fang, D.-Q. Yuan, J. Sculley, J.-R. Li, Z.-B. Han and H.-C. Zhou, Inorg. Chem., 2010, 49, 11637–11642 CrossRef CAS
- A. F. C. Campos, H. A. L. de Oliveira, F. N. da Silva, F. G. da Silva, P. Coppola, R. Aquino, A. Mezzi and J. Depeyrot, J. Hazard. Mater., 2019, 362, 82–91 CrossRef CAS
- Q. Yang, Q. Zhao, S. Ren, Q. Lu, X. Guo and Z. Chen, J. Solid State Chem., 2016, 244, 25–30 CrossRef CAS
- K. Wang, X. Tao, J. Xu and N. Yin, Chem. Lett., 2016, 45, 1365–1368 CrossRef CAS
- S. Rapti, A. Pournara, D. Sarma, I. T. Papadas, G. S. Armatas, Y. S. Hassan, M. H. Alkordi, M. G. Kanatzidis and M. J. Manos, Inorg. Chem. Front., 2016, 3, 635–644 RSC
- X. Li, X. Gao, L. Ai and J. Jiang, Chem. Eng. J., 2015, 274, 238–246 CrossRef CAS
- A. Ma, F. Ke, J. Jiang, Q. Yuan, Z. Luo, J. Liu and A. Kumar, CrystEngComm, 2017, 19, 2172–2177 RSC
- M. Vithanage and P. Bhattacharya, Environ. Chem. Lett., 2015, 13(2), 131–147 CrossRef CAS
- N. Zhang, X. Yang, X. Yu, Y. Jia, J. Wang, L. Kong, Z. Jin, B. Sun, T. Luo and J. Liu, Chem. Eng. J., 2014, 252, 220–229 CrossRef CAS
- F. Ke, C. Peng, T. Zhang, M. Zhang, C. Zhou, H. Cai, J. Zhu and X. Wan, Sci. Rep., 2018, 8, 939 CrossRef
- X.-H. Zhu, C.-X. Yang and X.-P. Yan, Microporous Mesoporous Mater., 2018, 259, 163–170 CrossRef CAS
- Y. Shang, X. Xu, B. Gao and Q. Yue, J. Cleaner Prod., 2018, 199, 36–46 CrossRef CAS
- B. Kamarehie, Z. Noraee, A. Jafari, M. Ghaderpoori, M. A. Karami and A. Ghaderpoury, Data in Brief, 2018, 20, 799–804 CrossRef PubMed
- C. Zhao, Y. Cui, F. Fang, S. O. Ryu and J. Huang, Adv. Condens. Matter Phys., 2017, 2017, 8305765 Search PubMed
- J. Dechnik, C. Janiak and S. De, J. Hazard. Mater., 2016, 303, 10–20 CrossRef
- X. Zhang, F. Sun, J. He, H. Xu, F. Cui and W. Wang, Chem. Eng. J., 2017, 326, 1086–1094 CrossRef CAS
- S. Mazloomi, M. Yousefi, H. Nourmoradi and M. Shams, J. Environ. Health Sci. Eng., 2019, 17, 209–218 CrossRef
- H. Qiu, L. Yang, F. Liu, Y. Zhao, L. Liu, J. Zhu and M. Song, Environ. Sci. Pollut. Res., 2017, 24, 23694–23703 CrossRef CAS
- T. Liu, J. Feng, Y. Wan, S. Zheng and L. Yang, Chemosphere, 2018, 210, 907–916 CrossRef CAS
- K.-Y. A. Lin, S.-Y. Chen and A. P. Jochems, Mater. Chem. Phys., 2015, 160, 168–176 CrossRef CAS
- Q. Xie, Y. Li, Z. Lv, H. Zhou, X. Yang, J. Chen and H. Guo, Sci. Rep., 2017, 7, 3316 CrossRef
- M. Nehra, N. Dilbaghi, N. K. Singhal, A. A. Hassan, K.-H. Kim and S. Kumar, Environ. Res., 2019, 169, 229–236 CrossRef CAS
- Y. Feng, H. Jiang, S. Li, J. Wang, X. Jing, Y. Wang and M. Chen, Colloids Surf., A, 2013, 431, 87–92 CrossRef CAS
- D. Sheng, L. Zhu, C. Xu, C. Xiao, Y. Wang, Y. Wang, L. Chen, J. Diwu, J. Chen and Z. Chai, Environ. Sci. Technol., 2017, 51, 3471–3479 CrossRef CAS
- N. Zhang, L.-Y. Yuan, W.-L. Guo, S.-Z. Luo, Z.-F. Chai and W.-Q. Shi, ACS Appl. Mater. Interfaces, 2017, 9, 25216–25224 CrossRef CAS PubMed
- Y. Peng, H. Huang, D. Liu and C. Zhong, ACS Appl. Mater. Interfaces, 2016, 8, 8527–8535 CrossRef CAS PubMed
- L. Huang, M. He, B. Chen and B. Hu, J. Mater. Chem. A, 2016, 4, 5159–5166 RSC
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed
- J. Li, Y. Liu, X. Wang, G. Zhao, Y. Ai, B. Han, T. Wen, T. Hayat, A. Alsaedi and X. Wang, Chem. Eng. J., 2017, 330, 1012–1021 CrossRef CAS
- I. Majchrzak-Kucęba and D. Bukalak-Gaik, J. Therm. Anal. Calorim., 2016, 125, 1461–1466 CrossRef
- M. Kadhom and B. Deng, Applied Materials Today, 2018, 11, 219–230 CrossRef
- M. Rubio-Martinez, C. Avci-Camur, A. W. Thornton, I. Imaz, D. Maspoch and M. R. Hill, Chem. Soc. Rev., 2017, 46, 3453–3480 RSC
- R. Guo, X. Cai, H. Liu, Z. Yang, Y. Meng, F. Chen, Y. Li and B. Wang, Environ. Sci. Technol., 2019, 53, 2705–2712 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2019 |