
Counting bubbles: precision process control of gas–liquid reactions in flow with an optical inline sensor†
Nikolay
Cherkasov
 *ab,
Antonio José
Expósito
ab,
Yang
Bai
a and
Evgeny V.
Rebrov
*abc
*ab,
Antonio José
Expósito
ab,
Yang
Bai
a and
Evgeny V.
Rebrov
*abc
aSchool of Engineering, University of Warwick, Coventry CV4 7AL, UK. E-mail: n.cherkasov@warwick.ac.uk; e.rebrov@warwick.ac.uk
bStoli Catalysts Ltd, Coventry, CV3 4DS, UK
cDepartment of Biotechnology and Chemistry, Tver State Technical University, Nab. A. Nikitina 22, 170026, Russia
First published on 23rd October 2018
Abstract
Quality by Design encouraged by the US Food and Drug Administration (FDA) in continuous flow synthesis requires tight monitoring of all the reaction input and output parameters to improve reproducibility and eliminate the process rejects. Reaction monitoring, however, relies on costly (over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000$) process analytical technology (PAT) – one of the factors that prevents the wider utilisation of continuous processes. In this work, we show that gas–liquid reactions can be monitored using low-cost (10$) hardware – an optical liquid inline sensor – that allows instantaneous analysis of the gas fraction in a moving stream. We discuss the application of the sensor for various gas–liquid reactions. Gas-consuming reactions such as hydrogenation are the easiest to implement because a sensor without calibration provides accurate readings close to complete consumption of the gas. Gas-evolving reactions can be monitored but require sensor calibration to determine the gas fraction accurately. Operation of the sensor was demonstrated for various hydrogenation reactions self-optimised using a proportional–integral (PID) algorithm which adjusted the substrate concentration to provide high (but not full) pre-defined hydrogen consumption. The optimised hydrogen consumption agreed with the product analysis for a range of substrates hydrogenated under various pressures and with different selectivities. The optical sensor was also proven to be an efficient tool in adapting the reaction conditions to catalyst deactivation in the reaction of 2-methyl-3-butyn-2-ol semi-hydrogenation – the autonomous reactor allowed a turn-over number (TON) of 2.7 × 106 to be reached with a value of 1.5 × 107 expected till a twofold decrease in the catalyst activity. The TON values demonstrated are significantly higher than those observed in batch reactors (∼103) even in the case of catalyst re-use (105) demonstrating a substantial improvement of process sustainability operating with the process control.
000$) process analytical technology (PAT) – one of the factors that prevents the wider utilisation of continuous processes. In this work, we show that gas–liquid reactions can be monitored using low-cost (10$) hardware – an optical liquid inline sensor – that allows instantaneous analysis of the gas fraction in a moving stream. We discuss the application of the sensor for various gas–liquid reactions. Gas-consuming reactions such as hydrogenation are the easiest to implement because a sensor without calibration provides accurate readings close to complete consumption of the gas. Gas-evolving reactions can be monitored but require sensor calibration to determine the gas fraction accurately. Operation of the sensor was demonstrated for various hydrogenation reactions self-optimised using a proportional–integral (PID) algorithm which adjusted the substrate concentration to provide high (but not full) pre-defined hydrogen consumption. The optimised hydrogen consumption agreed with the product analysis for a range of substrates hydrogenated under various pressures and with different selectivities. The optical sensor was also proven to be an efficient tool in adapting the reaction conditions to catalyst deactivation in the reaction of 2-methyl-3-butyn-2-ol semi-hydrogenation – the autonomous reactor allowed a turn-over number (TON) of 2.7 × 106 to be reached with a value of 1.5 × 107 expected till a twofold decrease in the catalyst activity. The TON values demonstrated are significantly higher than those observed in batch reactors (∼103) even in the case of catalyst re-use (105) demonstrating a substantial improvement of process sustainability operating with the process control.
1. Introduction
Gas–liquid reactions are widespread in the chemical industry and range from oil desulfurization performed on a gigaton scale to metathesis on a gram scale. Gas–liquid reactions involve numerous reaction classes and functional groups, but hydrogenations are particularly important because of versatility, and often perfect atom efficiency.1–3A majority of hydrogenation reactions in the fine and pharma industries are carried out in stirred-tank batch reactors.4,5 Low production and economic performance shown by the batch reactors comes from mass and heat transfer limitations as well as myriad operations such as substrate loading, heating, cooling, and reactor cleaning – repetitive and non-productive. The performance can be enhanced with continuous flow chemistry.6,7
Reactions in continuous flow improve micro-mixing, heat dissipation, process safety and reaction control.8,9 Sub-microsecond chemistry becomes possible in flash reactions.10 Flow chemistry, as a result, is getting adopted in the research and manufacturing processes.9 However, a majority of the reactions already converted to flow are either exothermic or hazardous liquid–liquid reactions. Gas–liquid reactions attract disproportionally little attention.9,11–13 The likely reason is that gas–liquid reactions, such as hydrogenations, have lower reaction rates and their hydrodynamics is more difficult to control resulting in limited reproducibility.14
The gas–liquid reactions often require a solid catalyst making them three-phase reactions. High pressure drop, limited product selectivity, formation of hot spots and associated quick catalyst deactivation are the usual problems.15–17 The exact control of residence time is another problem because different two-phase flow regimes can co-exist in different parts of the reactor.16,17 Nevertheless, there are ample examples of carrying out gas–liquid reactions with high selectivity in a continuous fashion achieving excellent catalyst utilisation and process intensification.18–21
Another barrier for wider adoption of continuous flow manufacturing for liquid-phase as well as gas–liquid reactions comes from the need for precise process monitoring, automation and control. Control of all the input operational parameters (temperature, pressure and flow rates) as well as the main reaction indicators (conversion, selectivity) is the key for reproducible process operation.
In a manufacturing process, autonomous operation of continuous flow processes decreases labour costs and improves process safety.9,22–24 Product quality obtained under the precise process control is ensured and any abnormalities are detected and eliminated.25 The US regulator, the Food and Drug Administration (FDA), encourages adoption of continuous flow processes because they provide Quality by Design.26,27
In a research environment, autonomous operation of a flow reactor improves efficiency allowing unprecedented testing throughput and improved reproducibility of the processes.28 Self-optimisation of reaction conditions is one of such examples – the process where the software finds the optimum conditions without human interaction.9,23,29–32 Months of experimental work can be performed in days.
Monitoring the input reaction parameters is straightforward and involves standard equipment such as flow meters, temperature and pressure transducers.11 Monitoring the output reaction parameters is, unfortunately, significantly more difficult because it often requires gaining insights into the chemical composition. The literature shows examples of using online chromatography, NMR, mass, IR, Raman and UV-vis spectroscopy.24,28,33–36 Selecting a suitable reaction monitoring tool involves finding a balance between the (i) price of the process monitoring solution, (ii) versatility for various reactions, and (iii) data quality and acquisition speed. The quality of data is, not surprisingly, prioritized and process monitoring tools cost above $10000 a unit – prohibitively expensive for many labs.
High equipment price and uncertainties in process monitoring impose limitations on process automation. In the current work, we show that excellent data quality in gas–liquid reactions can be achieved at high acquisition speed with low-cost equipment. A 10$ inline optical sensor can be used to control the reaction conditions precisely. The sensor can also be employed in self-optimisation of reaction conditions to provide the required gas consumption and maintain the product yield regardless of the course of catalyst deactivation.
2. Experimental
The hydrogenation experiments were performed in an automated system described in our previous work.37 Briefly, the system included two HPLC pumps (Knauer P4.1S) to control the liquid flow rates (Fig. 1). The gas flow rates were set with a set of mass flow controllers (Bronkhorst) and pneumo-actuated valves (Swagelok) placed afterwards.37 The gas and liquid flows were combined in a T-mixer (IDEX). A reactor was placed in a convection oven followed by a back-pressure controller (Equilibar). The reaction was performed in catalyst-coated tube reactors (1.27 mm inner diameter, 1.6 mm outer diameter) provided by Stoli Catalysts Ltd. The catalyst-coated tubes were used to avoid problems associated with liquid channelling and residence time distribution inherent to packed-bed reactors.14 A filter (IDEX A-410) was placed after the catalyst-coated tube to protect the pressure controller from the possible catalyst particles. The liquid products were placed into vials with septa using a Zang Autosam 360 automated sample collector. The samples were analysed with an offline gas chromatograph (Shimadzu GC-2010) equipped with a flame ionisation detector and a Stabilwax column 10 m × 0.15 mm × 0.15 μm. Product conversion and selectivity were calculated using equations provided in the ESI,† S1.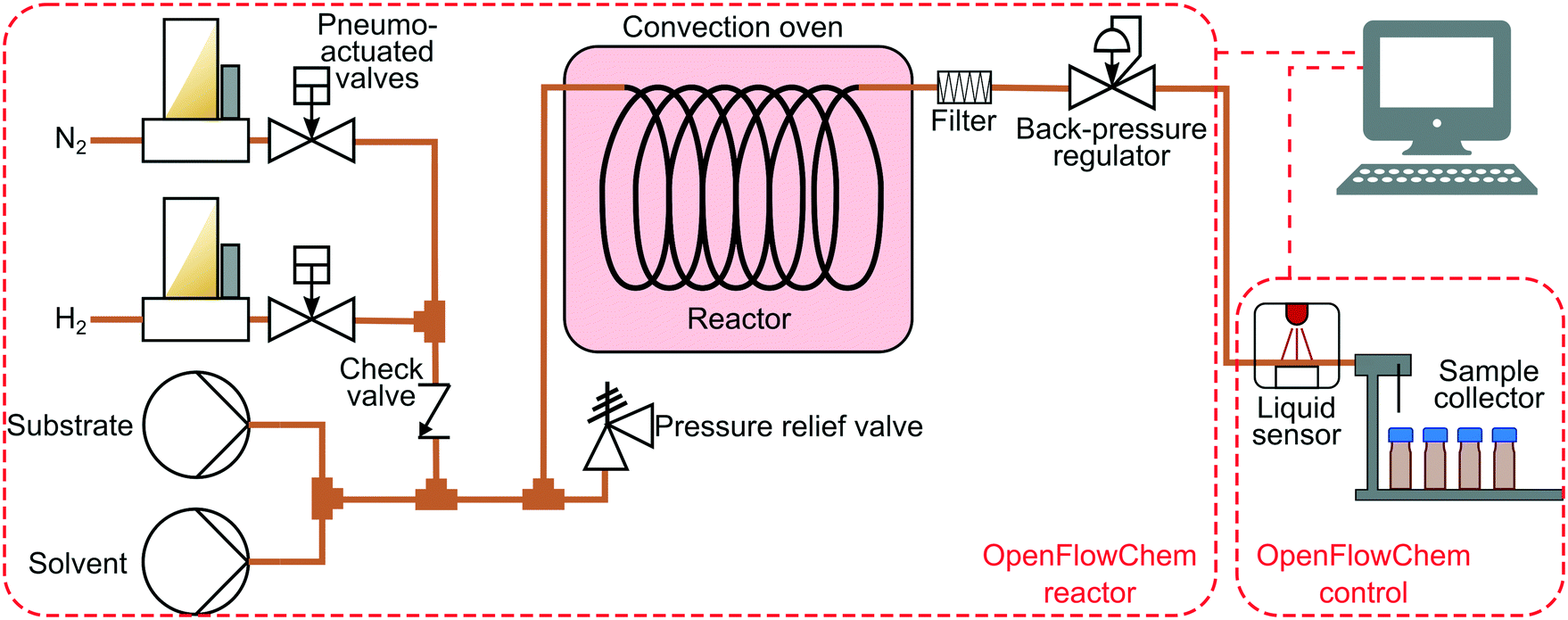 | ||
| Fig. 1 Scheme of the hydrogenation reactor with feedback control and process monitoring with an optical sensor. | ||
The reactor was controlled with OpenFlowChem software37 and contained two systems: (i) the flow reactor until the back-pressure regulator, (ii) and a control system containing the optical sensor and the sample collector. The flow reactor implemented the pre-defined reaction conditions (flow rates, temperature, pressure). The reactions were performed by mixing a 0.5 M substrate solution with isopropanol solvent to maintain a constant liquid flow rate of 1.5 mL min−1 at a reaction temperature of 50–90 °C and a pressure of 1.3–6.0 bar (absolute). A simplified system used for the validation of the optical sensor is described in the ESI,† S2.
3. Results and discussion
3.1. Head-on approach: combine equimolar quantities and let them react
First of all, we would like to discuss why semi-hydrogenation may benefit from complex process control. Indeed, the best control measures are often passive. In the case of temperature control, for example, overheating can be prevented in some cases without any process control because the heat dissipation rate increases with temperature.38 The next step in the process control complexity might be a bimetallic strip that bends at known temperatures or a phase-change material that consumes heat – both are simple and reliable for maintaining the process temperature.39,40 In the case of hydrogenation, it is possible to avoid complex process monitoring and rely on a performance margin. The reaction can be carried out for a contact time longer than required so that deactivation and external disturbances do not affect the full conversion.We took alkyne (MBY) semi-hydrogenation to alkene (2-methyl-3-buten-2-ol, MBE) shown in Fig. 2 as a model and the compound was used for vitamin synthesis.3 A straightforward approach to control the reaction could be feeding an equimolar combination of MBY and H2 with sufficient contact time for full H2 consumption – the performance margin.
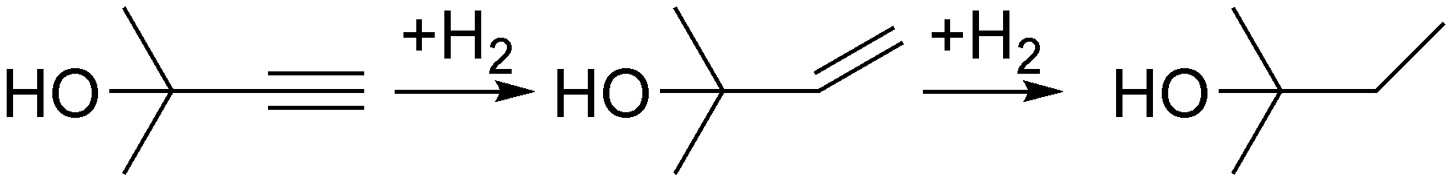 | ||
| Fig. 2 Scheme of the 2-methyl-3-butyn-2-ol (MBY) semi-hydrogenation with alkene as the desired product. | ||
Using the catalysts under hydrogen-lean conditions, unfortunately, brings a lot of problems outlined in Table 1. First of all, the performance margin introduced provides a sub-optimal reactor throughput – the reactor throughput has to be artificially decreased. Secondly, it is difficult to predict when the performance margin “wears out” – the conversion may remain complete for days followed by a sudden drop resulting in contamination of the product with the feedstock. Therefore, a detailed catalyst stability study is required before using the approach. Thirdly, the catalysts tend to leach in the absence of hydrogen.41 Leaching brings contamination of the product stream which might require additional purification. Leaching also alters and removes the catalyst resulting in faster deactivation with more frequent reactor maintenance cycles.41–43 Lastly, any contact time beyond the minimum facilitates side reactions such as isomerisation, oligomerisation, or decomposition.44 Therefore, simplicity brought with the performance margin is fraught with problems.
| Advantages | Disadvantages |
|---|---|
| Simplicity | Performance artificiality limited |
| Quick to set-up | Stability optimisation required |
| No need for process control equipment | Accelerated catalyst deactivation by leaching |
| Product contamination by catalyst leaching | |
| Side-reactions |
Simplicity, nevertheless, might be a substantial incentive to try this simple process control approach. We performed an experimental study of MBY semi-hydrogenation in a 1 m tube reactor wall-coated with a 2.3 wt% Pd/C catalyst. Because the extent of the required performance margin cannot be predicted in advance, we gradually increased the reaction pressure and collected the products. The initial H2 to MBY molar ratio was kept at 105% to ensure high MBE yield despite unavoidable over-hydrogenation to MBA (Fig. 2) that consumes twice as much H2 as MBE.
Fig. 3 shows the effect of reaction pressure on the MBY conversion and MBE selectivity. On increasing the pressure from 1 to 11 bar, the MBY conversion increased because of (i) the higher reaction rate which is proportional to the H2 pressure18,45 and (ii) longer liquid residence time due to compression of H2 bubbles at higher pressure. The MBE selectivity in the 1–11 bar range was 93–95% which is typical for supported Pd catalysts.18,46–49
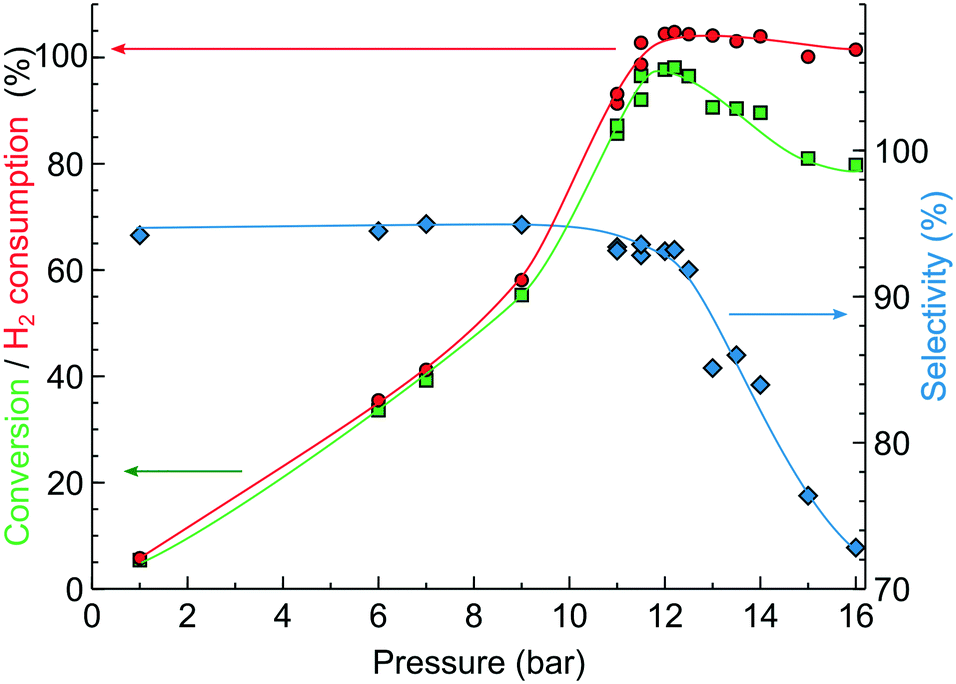 | ||
Fig. 3 Effect of reaction pressure on MBY semi-hydrogenation performance over a 2.3 wt% Pd/C catalyst-coated tube: ( ) MBY conversion, ( ) MBY conversion, ( ) MBE selectivity, and ( ) MBE selectivity, and ( ) H2 consumption calculated from product composition. Reaction conditions: 0.5 mL min−1 5 M MBY, 35 °C, 5% molar excess of H2 to MBY. ) H2 consumption calculated from product composition. Reaction conditions: 0.5 mL min−1 5 M MBY, 35 °C, 5% molar excess of H2 to MBY. | ||
At a pressure of 12 bar, the conversion reached a maximum value of 98.1% and the MBE selectivity decreased to 93%. The decrease in selectivity at high conversion is known to appear from the competitive adsorption of the dominant alkene and minor alkyne species onto the Pd sites.19,20,47,48,50 A single phase (liquid) flow was observed at the reactor outlet confirming full H2 consumption. The product composition confirmed that MBY consumed 103% molar equivalent of H2, in agreement with the introduced feed ratio.
Above 12 bar, H2 had already been fully consumed. A further increase in pressure was expected to bring changes neither in the MBE selectivity nor in conversion. In contrast, the experimental data showed a decrease both in the MBE selectivity and the MBY conversion (Fig. 3). The performance margin introduced by the higher reaction pressure did not provide the desired control over the reaction. The study of this effect is beyond the scope of this work, but it was found to be reproducible over a range of catalysts, pressures and concentrations. This simple model reaction clearly demonstrates that the “performance margin” may bring anything but performance and some form of “complex” process control is beneficial.
3.2. Applicability of an optical sensor
We used a commercially-available 10$ Optek liquid optical sensor for process monitoring. The sensor consists of an infrared light-emitting diode and a photo-transistor with a transparent 1/16′′ tube in between (Fig. 4). The sensor detects liquids based on light refraction and allows analysis of optically-transparent liquids such as water. The output is a binary signal – the presence or absence of liquid in the detection volume of below 0.2 μL. The sensor has an excellent response speed typically in the order of 50 μs; therefore, the measurements can be reliably performed on quickly-moving feeds. | ||
| Fig. 4 Photograph of the Optek optical sensor with 1.59 mm OD tubing. | ||
We used the optical sensor to determine the liquid fraction (LF) – the fraction of liquid in the gas–liquid flow by reading the liquid presence every 100 μs and averaging the data over 10 s. The response time of 10 s is considerably faster than that of chromatography and is comparable to that of spectroscopy.
The accurate LF measurements are obviously possible only for the Taylor flow regime because the relative abundance of liquid slugs is expected to provide the LF.51,52 The annular or slug-annular flow regimes, where the liquid film moves along the reactor wall as a film,19,53,54 seem unsuitable for detection. This is because the light refraction properties of the film may depend on the media, film thickness and velocity resulting in false readings.
The accuracy of the sensor was first verified with a model N2-isopropanol flow with the technical details shown in the ESI,† S2. Fig. 5 shows the relationship between the introduced and the measured LF in the model flow with the dashed line corresponding to the expected measurement. All the LF readings consistently overestimated the correct value by 10–25%. The likely reason is that the meniscus at the gas–liquid boundary introduced strong light refraction so the sensor considered the slug boundary as filled only with liquid underestimating the gas content.
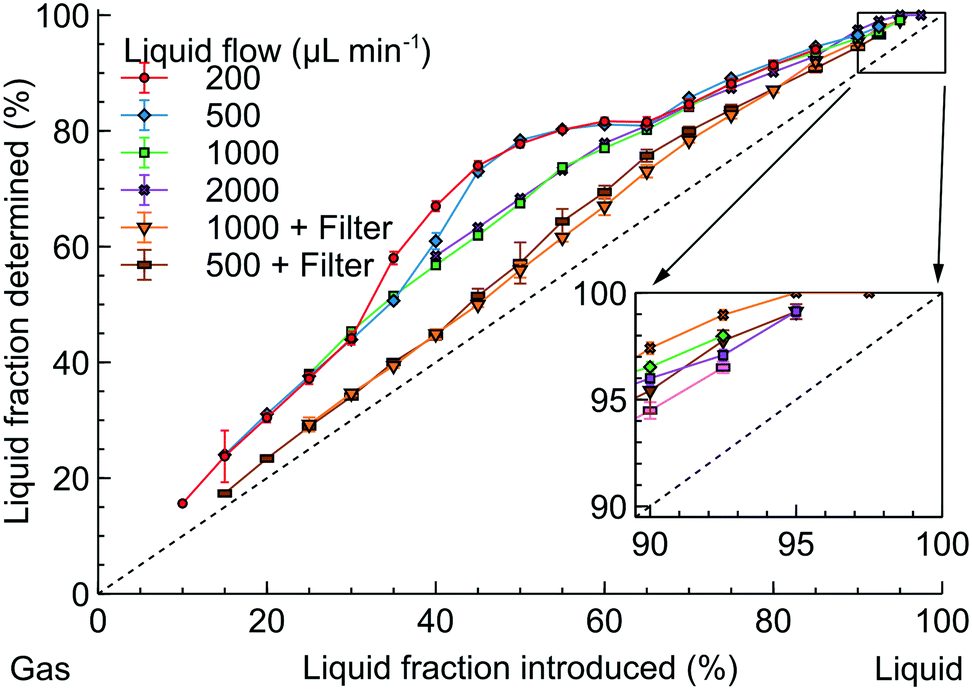 | ||
| Fig. 5 Comparison of the liquid fraction introduced and determined by the optical sensor in the model isopropanol-N2 flow. | ||
When the gas–liquid flow was formed with a T mixer (Fig. 5), a broad “hump” appeared at the 30–60% LF for liquid flow rates of 200 and 500 μL min−1. At higher liquid flow rates, the “hump” disappeared and the LF readings were consistent (within 2%).
The T mixer, however, generated the Taylor flow with constant gas and liquid bubble sizes – not always the flow observed in hydrogenation.15,18,19,47 Therefore, we placed a large-volume filter (300 μL) after the T mixer to introduce flow irregularity. With the filter, no “hump” was observed and the LF readings became considerably closer to the expected LF values likely because the slugs were longer resulting in a smaller number of inter-phase boundaries. However, there was a significant discrepancy between the LF readings observed with and without the filter.
Therefore, the LF readings of the optical sensor depend significantly on the liquid flow rates and slug lengths. The difficulty in controlling the slug lengths after the reactor outlet is a limiting factor in the applicability of process monitoring with the optical sensor. The zoomed-in area in Fig. 5, however, demonstrates that the LF readings above 95% become consistent. Nevertheless, there is also a risk of falsely obtaining 100% LF readings when a small amount of gas is present in the flow – at the introduced LF above 95%. This phenomenon comes, likely, from the sensor inability to detect gas in the bubbly flow – the regime where the gas bubbles are substantially smaller than the channel diameter.54–56
Fig. 6 summarises the findings of the sensor applicability for gas–liquid reactions based on the gas behaviour during the reaction: equimolar substitution, evolution, or consumption. Gas-“neutral” reactions (Fig. 6A) such as carboxybenzyl hydrogenolysis deprotection (where 1 mol of H2 is consumed and replaced with 1 mol of CO2) seem unfeasible to monitor with the optical sensor because the LF readings do not change during the reaction. The nature of the gas, however, may be used to overcome the problem. A possible solution in this example is dissolving CO2 in the liquid media at a high pressure or adding a gas-absorbing alkaline solution. In these cases, the simplicity of the optical sensor may be counterbalanced by complexity introduced by the additional gas-absorption system.
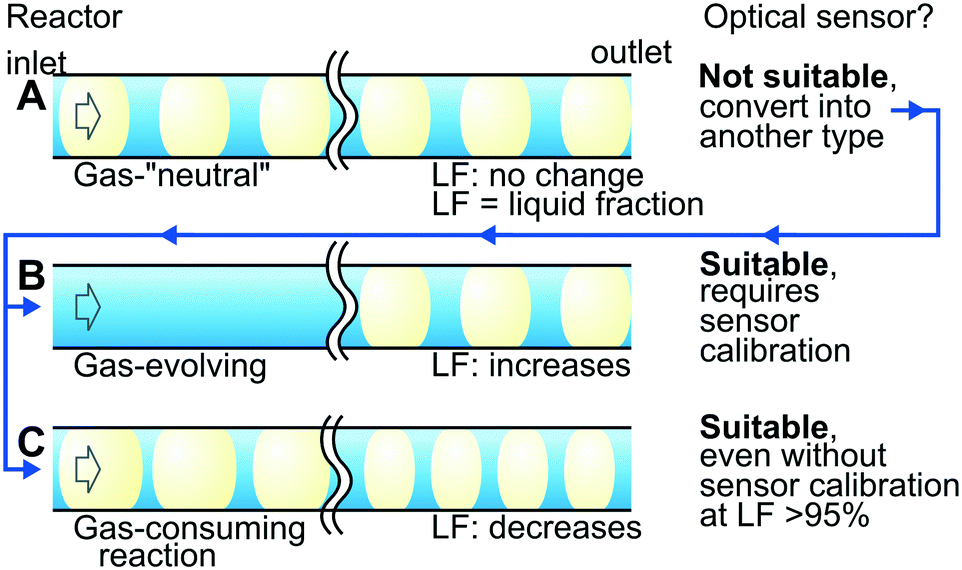 | ||
| Fig. 6 Summary of the optical sensor applicability for monitoring (A) gas-neutral, (B) gas-evolving or (C) gas-consuming reactions. | ||
Gas-evolving reactions (Fig. 6B) such as decarbonylation can be monitored easily with the optical sensor because the LF changes substantially during the reaction. The strong dependence of the LF readings on the liquid flow rate and slug length (Fig. 5), however, requires additional efforts for process monitoring. The optical sensor must be calibrated for the required flow rates with, possibly, slug length control by image analysis.
Gas-consuming reactions (Fig. 6C) such as hydrogenation seem the most suitable for monitoring with the optical sensor. Reliable readings in the LF range of 95–97%, possible without sensor calibration, are beneficial in ensuring almost complete gas consumption. A few remaining hydrogen bubbles saturate the solution and minimise side-reaction and catalyst leaching.41,42 A minor hydrogen content in the product feed maximises the residence time and the reactor throughput. In summary, the negative consequences discussed in Table 1 are eliminated.
3.3. Optimising hydrogenation: adjusting substrate concentration to obtain specified hydrogen consumption
In this work, we used the optical sensor to monitor hydrogenation reactions because these gas-consuming reactions are vital in many fine chemical syntheses.1,3 The experimental system presented in Fig. 1 combined the flows of (i) H2, (ii) a solvent and (iii) a substrate solution. Following the unsuccessful attempts to control the MBY semi-hydrogenation by complete H2 consumption in Fig. 3, we used the optical sensor to monitor and maintain high (but not complete) H2 consumption with the same 2.3 wt% Pd/C catalyst-coated tube.Optimisation was performed using a proportional–integral (PID) algorithm. The LF reading was a process variable (setpoint of 95%), and the MBY concentration was a control variable adjusted to reach the setpoint. Compared to conventional optimisation algorithms such as simplex,23,24,57 the PID algorithm does not assume implacable experimental reproducibility and can handle slow changes in the reaction performance caused by usual phenomena such as catalyst activation/deactivation.18,58 The flow rates of H2 and the solvent were also adjusted by the algorithm, but these were linked to the MBY flow rate by the pre-defined H2 to MBY molar feed ratio and the total liquid flow rate. After reaching the LF setpoint, the system collected the 4 liquid samples for offline GC verification.
Fig. 7A shows the change in the MBY and H2 flow rates over time on stream in the system controlled with the optical sensor. The PID algorithm, using the LF readings, automatically adjusted the MBY flow rate to maintain the LF at 95%. During the experiment, the H2/MBY feed ratio increased stepwise and the LF values dropped momentarily to about 80%. The diminishing LF was compensated by the system with decreasing MBY flow rates allowing for a longer MBY residence time to consume more H2.
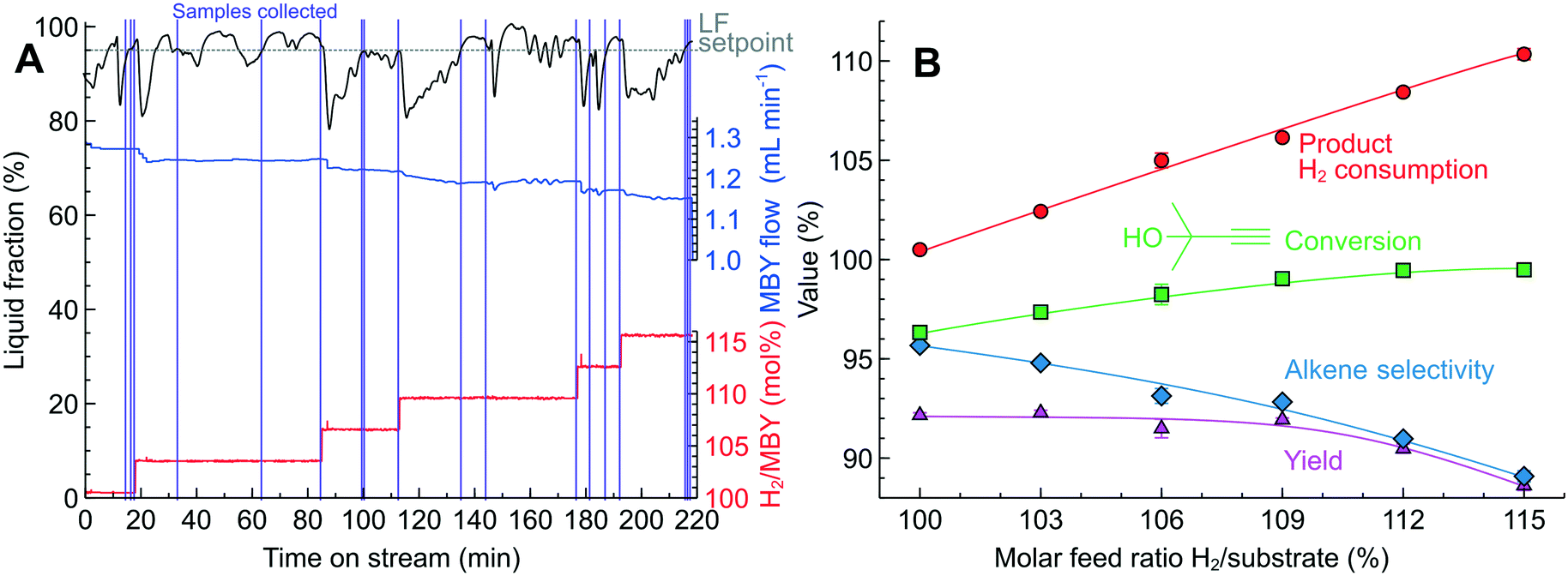 | ||
| Fig. 7 MBY semi-hydrogenation in a 2.3 wt% Pd/C catalyst-coated tube. (A) Performance of the optical sensor and PID reactor control in maintaining the product liquid fraction to 95%. (B) The effect of the H2/MBY feed ratio on the product composition. Concentration 0.39–0.43 M, 70 °C and 2.5 bar. | ||
Once the LF readings stabilised, the system collected 3 liquid samples for each specified H2/MBY feed ratio. The conversion and selectivity for the samples collected was reproducible within ±0.4% which shows excellent process control achieved with the optical sensor. The pre-defined H2/MBY feed ratio agreed with the experimental consumption calculated from the product composition (Fig. 7A). The increasing H2 consumption resulted in a higher MBY conversion but the MBE selectivity decreased due to depletion of MBY species in the solution. Importantly, the MBE yield was constant at around 92% in the H2/MBY feed ratio range from 100 to 109% because the increasing conversion was compensated by decreasing MBE selectivity. The H2/MBY feed ratio above 109% resulted in the declining MBE yield because the conversion could no longer be increased (being close to 100%), but the MBE selectivity decreased due to over-hydrogenation with the available H2.
To validate the process control with the optical sensor, we studied hydrogenation of 3-hexyne-1-ol, a widely used fragrance compound and a model molecule for internal alkyne semi-hydrogenation.59–61 The possible hydrogenation reactions are shown in Fig. 8A.
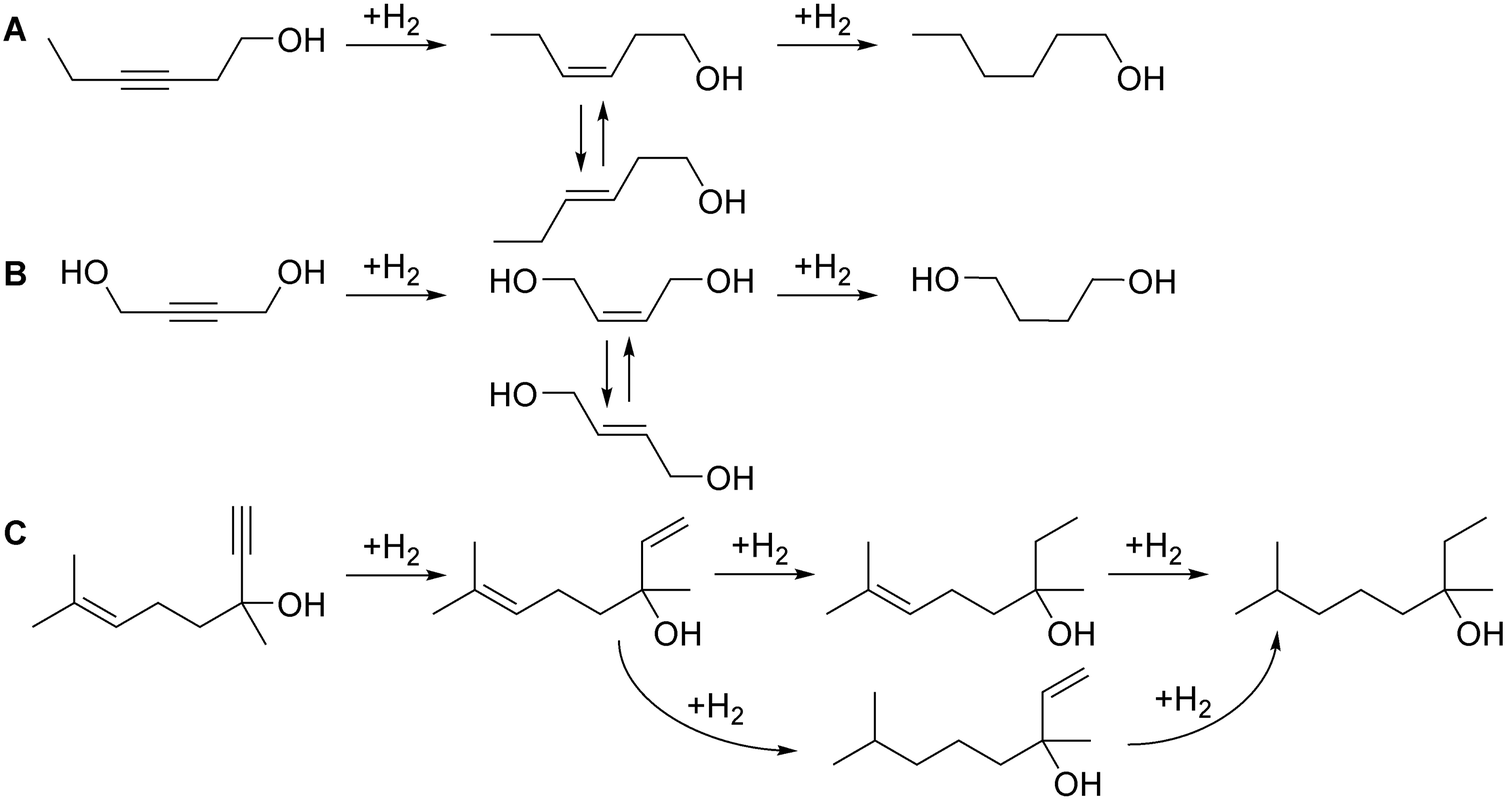 | ||
| Fig. 8 Schemes of (A) 3-hexyne-1-ol, (B) 1,4-butynediol, and (C) dehydrolinalool hydrogenation reactions. | ||
Hydrogenation of 3-hexyne-1-ol was studied in 2.3 wt% Pd/SiO2 and 2.3 wt% Pd/ZnO catalyst-coated tubes. The Pd/ZnO catalyst often provides a higher alkene selectivity in alkyne semi-hydrogenation.19,20,45,46 Compared to MBY (Fig. 7B), the range of 3-hexyne-1-ol hydrogenation products was wider and a significant E-alkene formation was observed caused by Z/E isomerisation.59–61 The increasing H2/substrate feed ratio over the Pd/SiO2 (Fig. 8A) catalyst resulted in a higher alkyne conversion (from 80 to 85%) but a lower Z-alkene selectivity (from 64 to 55%). The highest yield of Z-alkene was 50.5% at a H2/substrate feed ratio of 100%. The results observed over the Pd/ZnO catalyst (Fig. 9B) confirm the expected Z-alkene selectivity increase, which reached 70% with a maximum yield of 62% at a H2/substrate feed ratio of 100%.
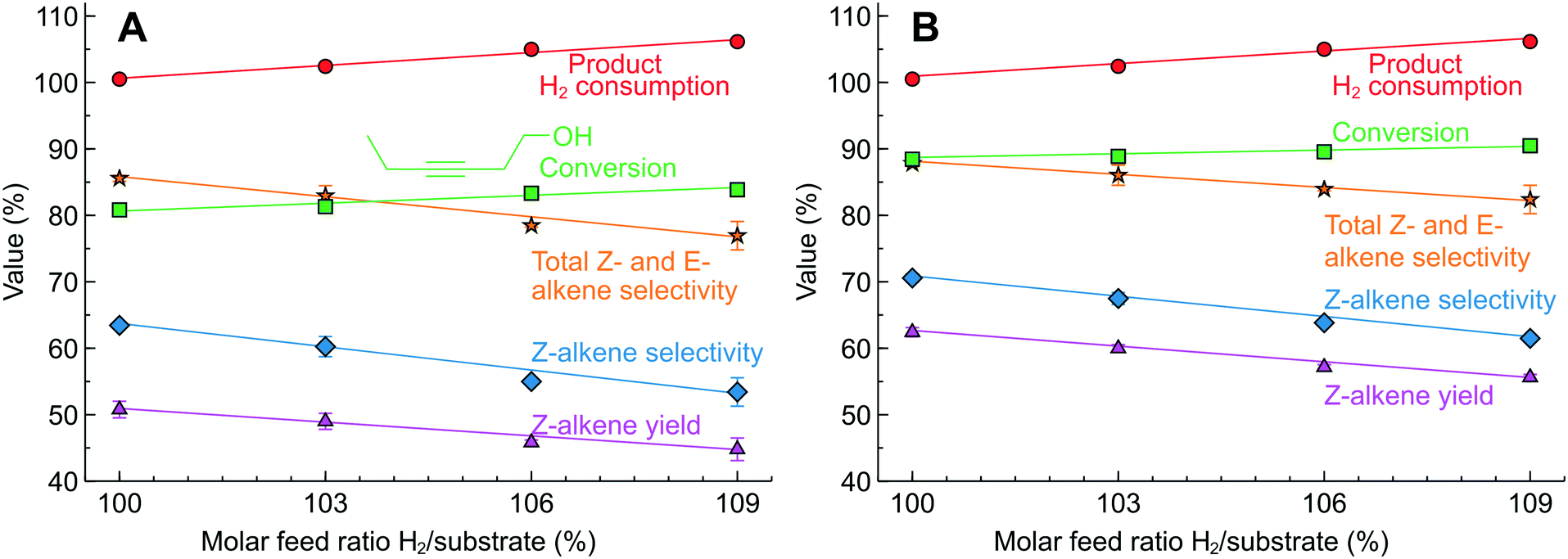 | ||
| Fig. 9 3-Hexyne-1-ol semi-hydrogenation in (A) 2.3 wt% Pd/SiO2 and (B) 2.3 wt% Pd/ZnO catalyst-coated tubes at various H2/substrate feed ratios. Concentration 0.15–0.22 M, 50 °C and 1.3 bar. H2 consumption calculated from product composition. The reactor self-optimised the liquid fraction to 95% for each feed ratio selected. | ||
Therefore, 3-hexyne-1-ol semi-hydrogenation requires further optimisation of the catalyst and reaction conditions to improve the Z-alkene yield. The example presented, however, shows that the precise process control of the H2 consumption is possible even in the case of substantial side-reactions. The precision of process control is evidenced by the excellent agreement between the H2/substrate feed ratio and the H2 consumption calculated from the product chemical composition.
Using the same approach, the study was extended to 1,4-butynediol semi-hydrogenation (Fig. 8B), another model molecule with an internal triple bond and the possibility to form Z- and E-alkenes. In this reaction, we used the more selective 2.3 wt% Pd/ZnO catalyst-coated tube reactor, but the reaction conditions were considerably harsher compared to those studied previously. The reaction pressure was increased to 5 bar and the reaction temperature to 90 °C due to lower hydrogenation activity (in agreement with the literature)62 likely caused by the steric limitation introduced by the OH groups.
Fig. 10 shows the results of 1,4-butynediol semi-hydrogenation experiments. A higher H2/substrate feed ratio increased the conversion from 95 to 97%. The Z-alkene selectivity, meanwhile, decreased from 95 to 90% resulting in a Z-alkene yield around 90% for the H2/substrate feed ratio of 100–106%. The selectivity of 95–90% observed is in line with the literature.63,64 In agreement with the literature, no E-alkene was observed in this reaction compared to 3-hexyne-1-ol (Fig. 9) – the difference highlights the specificity of each compound/catalyst combination.
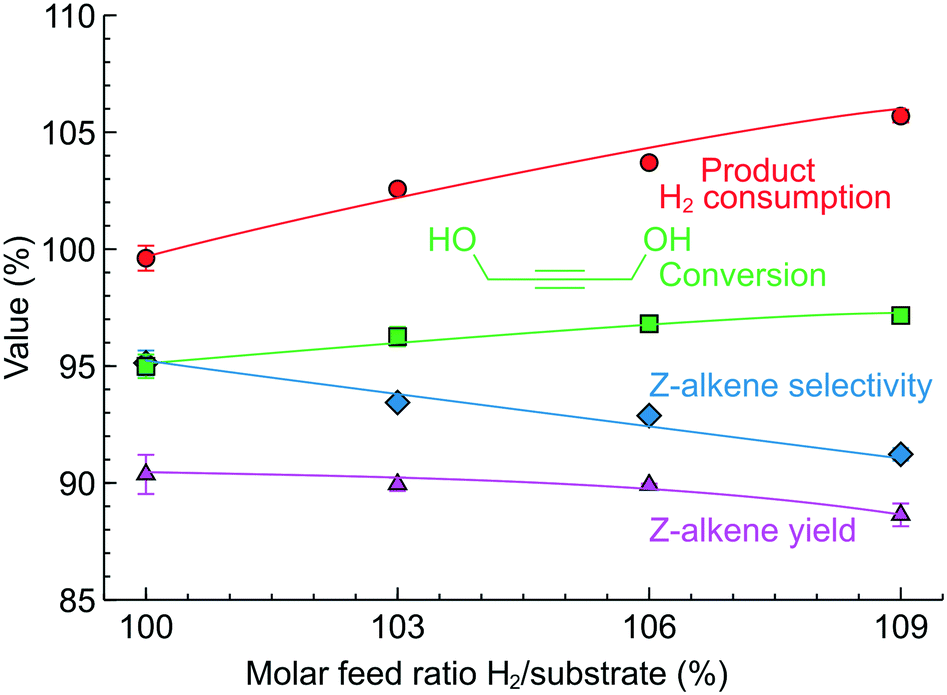 | ||
| Fig. 10 1,4-Butynediol semi-hydrogenation performance over a 2.3 wt% Pd/ZnO catalyst-coated tube at various H2/substrate feed ratios. Concentration 0.10–0.13 M, 90 °C and 5 bar. The reactor self-optimised the liquid fraction to 95% for each feed ratio selected. | ||
The H2 consumption determined from the product analysis showed good agreement with the H2/substrate feed ratio. It is particularly interesting not only as a demonstration of the process control with a different molecule, but also because a substantially higher reaction pressure was used. The agreement between the anticipated and the observed values shows that the H2 gas dissolved in the product feed quickly expands after the back-pressure regulator so the measurement with the optical sensor remains accurate despite the increased reaction pressure.
To explore the possibility of controlling more complex reactions, we studied semi-hydrogenation of dehydrolinalool (DLL), a precursor to linalool fragrance – a molecule that contains a triple as well as a double bond and has a broader range of possible hydrogenation products (Fig. 8C). Fig. 11 shows the effect of the H2/substrate feed ratio on the product distribution observed over the 2.3 wt% Pd/ZnO catalyst-coated tube. The target di-alkene (linalool) selectivity was only 84–79% at a conversion of 88–90%. The resulting linalool yield decreased from 72% to 70% with the addition of the H2 excess highlighting that the higher yield in this reaction requires catalyst optimisation.
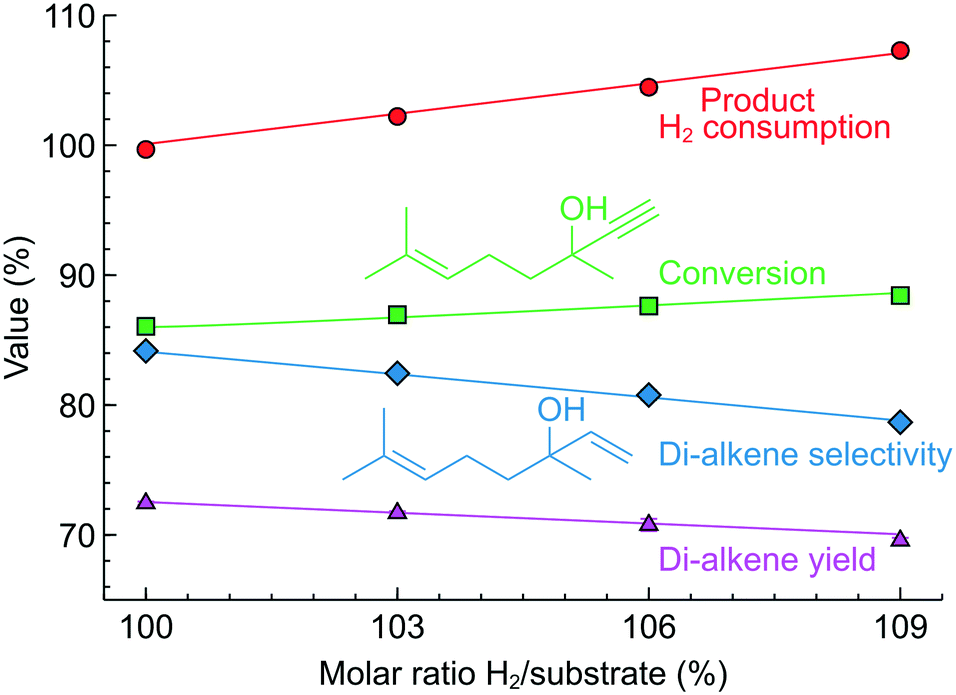 | ||
| Fig. 11 Dehydrolinalool semi-hydrogenation performance in a 2.3 wt% Pd/ZnO catalyst-coated tube at various H2/substrate feed ratios. Concentration 0.10–0.14 M, 50 °C and 1.3 bar. The reactor self-optimised the liquid fraction to 95% for each feed ratio selected. | ||
Nevertheless, the H2 consumption determined from the product composition showed an excellent agreement with the H2/substrate feed ratio.
In summary, the optical sensor was studied for a range of selective hydrogenation reactions under various temperatures and reaction pressures. For all the examples studied, the autonomous reactor controlled with a PID algorithm and the optical sensor was able to self-optimise the substrate concentration to obtain the desired H2 consumption with an accuracy of ±3% and a product yield repeatability better than ±0.4%. The autonomous reactor with LF monitoring provided, in our experience, 3–10 times faster optimisation of the reaction parameters compared to using offline chemical analysis as a reaction monitoring solution.
3.4. Continuous optimisation: maintaining hydrogen consumption in semi-hydrogenation despite deactivation
PID control with the optical sensor may be used not only to find optimal reaction conditions, but to maintain high product conversion despite catalyst deactivation. In the previous work,37 we demonstrated operation of a PID controller in nitrobenzene to aniline hydrogenation over a short period of time. Here, we aimed to maximise catalyst utilisation in a reaction with possible over-hydrogenation. For this task, reaction monitoring and control are critical in maintaining high substrate conversion regardless of inevitable catalyst deactivation.We selected MBY semi-hydrogenation as a model reaction. The reaction poses problems of over-hydrogenation without active process control (Fig. 3). Additionally, the reaction is well studied allowing the application of process intensification to maximise the turn-over number (TON) within a manageable time on stream.18 Under conventional conditions of constant flow rates (ESI,† S3), the MBY conversion declines slowly after a brief increase. The active control of the flow rates provides an opportunity to maintain product yield regardless of changes in catalyst behaviour.
Fig. 12 shows the time on stream experiment carried out in a 2.3 wt% Pd/ZnO catalyst-coated tube in MBY semi-hydrogenation at 100 °C and 9 bar pressure – the conditions chosen to increase the reaction rates. The increasing reaction rates had to be compensated by the higher fluid velocity to ensure adequate mixing and high MBE selectivity by setting the LF setpoint to 70% and optimising the H2/MBY ratio for the H2 consumption of 106%. On starting the reactor, the system adjusted the MBY concentration to maintain the LF readings despite changes in the catalyst behaviour. These changes can be noticed in the MBY flow rates (Fig. 12) which were increasing during the first 5 hours on stream and decreasing afterwards likely caused by catalyst activation and deactivation phenomena. This example highlights that only a feedback control algorithm (in contrast to feed-forward or real-time)65 can be used to maintain constant H2 consumption because the catalyst activation/deactivation phenomena are contingent and are difficult to foresee.
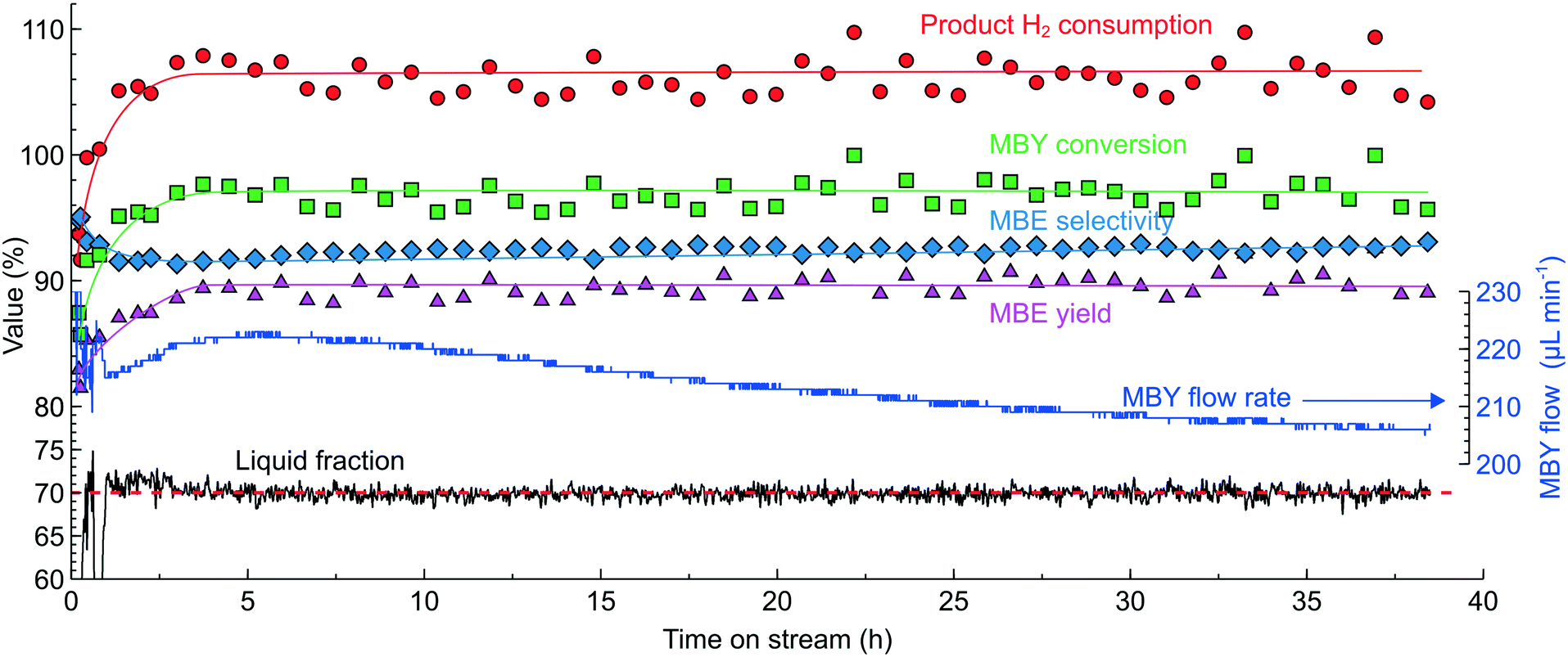 | ||
| Fig. 12 Long-term stability study in MBY semi-hydrogenation in a 0.5 m catalyst-coated tube (2.3 wt% Pd/ZnO) controlled by the optical sensor. Reaction conditions: MBY diluted with isopropanol to 1 mL min−1 total flow rate, 100 °C, 9 bar H2, 6% molar excess of H2 to MBY. The reactor optimised the liquid fraction to 70% continuously. | ||
The samples collected periodically for offline GC analysis show that the system successfully maintained high MBY conversion and MBE yield. The yield of 89.6 ± 1.0% observed is close to the limit possible with a monometallic Pd catalyst. Importantly, the MBE yield was virtually constant regardless of the catalyst behaviour demonstrating the performance of the optical sensor.
In this experiment, the TON value (which shows the number of molecules converted per atom of Pd) exceeded 2.7 × 106. During this experiment, the MBY flow rate decreased only by 6.7% compared to the initial value demonstrating that the catalyst was far from being deactivated. Extrapolating the observed trend considering the exponential decline in the activity, 50% catalyst deactivation will allow a TON value of 1.5 × 107 to be reached in 280 h on stream.
The TON values obtained are drastically higher than those in conventional batch reactors, where the typical values are in the order of 103. Batch hydrogenation with catalyst reuse can provide TON values of 104–105. The notably higher TON in the catalyst-coated tube reactors possible with the optical sensor monitoring demonstrates exceptionally efficient noble metal utilisation. A substantially lower metal requirement, in turn, improves economics of the process and has the potential to introduce more efficient platinum-group catalysts into base-metal hydrogenation.
4. Conclusion
The 10$ refractory liquid optical sensor was shown to be an efficient process monitoring tool in controlling semi-hydrogenation reactions. This approach combines simplicity, robustness, and low cost with high reproducibility required to optimise and maintain a constant hydrogen consumption. High reliability and precision provided by the sensor allow its use in complete as well as selective hydrogenation reactions. A response time of 10 s eliminates analytical delays in process control and excessive material consumption.The autonomous system with process monitoring performed by the optical sensor was demonstrated in several examples of semi-hydrogenation reactions. The proportional–integral–derivative (PID) algorithm allowed for reaction condition optimisation – finding the optimal conditions to ensure the required hydrogen consumption. In the examples, it took 2–15 min to find the conditions and collect a liquid sample for external analysis. Because the system finds and maintains the hydrogen to substrate molar ratio defined by the experimenter, the number of conventional chemical analyses decreases dramatically. With the process control, moreover, all the samples collected have high substrate conversion eliminating the need to analyse many low-conversion samples. In our experience, using process monitoring with an optical sensor provides acceleration in reaction optimisation with a factor of 3–10 compared to using offline chemical analysis as a process monitoring solution.
The utilisation of process monitoring allows not only for optimisation of reaction conditions but maintaining the given hydrogen consumption over a long period of time. The experiment was performed using a model MBY semi-hydrogenation reaction under the intensified reaction conditions for 39 hours on stream at an MBE yield of 89.6 ± 1.0% to obtain a turn-over number of 2.7 × 106. It is more than 3 orders of magnitude above that of the typical batch reactor processes.
Therefore, catalyst-coated tube reactors combined with simple yet robust process monitoring by an optical sensor provide exceptional utilisation of noble metal catalysts.
Conflicts of interest
NC and EVR are founders, directors and shareholders of Stoli Catalysts Ltd.Acknowledgements
The authors are grateful to DSM Nutritional Products for the dehydrolinalool provided and Dr Jonathan Medlock for fruitful discussion of the work. The financial support was provided by the InnovateUK grant (900041).References
- M. Brzozowski, M. O'Brien, S. V. Ley and A. Polyzos, Acc. Chem. Res., 2015, 48, 349–362 CrossRef CAS PubMed.
- J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC.
- W. Bonrath, J. Medlock, J. Schutz, B. Wustenberg, T. Netscher, B. Wüstenberg and T. Netscher, Hydrogenation in the Vitamins and Fine Chemicals Industry – An Overview, in Hydrogenation, ed. I. Karame, InTech, Rijeka, 2012, pp. 69–90 Search PubMed.
- E. H. Stitt, Chem. Eng. J., 2002, 90, 47–60 CrossRef CAS.
- G. S. Calabrese and S. Pissavini, AIChE J., 2011, 57, 828–834 CrossRef CAS.
- V. Hessel, D. Kralisch, N. Kockmann, T. Noël and Q. Wang, ChemSusChem, 2013, 6, 746–789 CrossRef CAS PubMed.
- D. M. Roberge, L. Ducry, N. Bieler, P. Cretton and B. Zimmermann, Chem. Eng. Technol., 2005, 28, 318–323 CrossRef CAS.
- C. Wiles and P. Watts, Green Chem., 2012, 14, 38–54 RSC.
- K. F. Jensen, AIChE J., 2017, 63, 858–869 CrossRef CAS.
- J. Yoshida, Y. Takahashi and A. Nagaki, Chem. Commun., 2013, 49, 9896–9904 RSC.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- S. V. Ley, D. E. Fitzpatrick, R. J. Ingham and R. M. Myers, Angew. Chem., Int. Ed., 2015, 54, 3449–3464 CrossRef CAS PubMed.
- M. Irfan, T. N. Glasnov and C. O. Kappe, ChemSusChem, 2011, 4, 300–316 CrossRef CAS PubMed.
- D. van Herk, P. Castaño, M. Makkee, J. A. Moulijn and M. T. Kreutzer, Appl. Catal., A, 2009, 365, 199–206 CrossRef CAS.
- Y. Bai, N. Cherkasov, S. Huband, D. Walker, R. Walton and E. Rebrov, Catalysts, 2018, 8, 1–18 CrossRef.
- J. Yue, Catal. Today, 2017, 308, 3–19 CrossRef.
- A. Tanimu, S. Jaenicke and K. Alhooshani, Chem. Eng. J., 2017, 327, 792–821 CrossRef CAS.
- N. Cherkasov, Y. Bai and E. Rebrov, Catalysts, 2017, 7, 1–16 CrossRef.
- N. Cherkasov, A. O. Ibhadon and E. V. Rebrov, Appl. Catal., A, 2016, 515, 108–115 CrossRef CAS.
- L. B. Okhlopkova, S. V. Cherepanova, I. P. Prosvirin, M. A. Kerzhentsev and Z. R. Ismagilov, Appl. Catal., A, 2018, 549, 245–253 CrossRef CAS.
- Z. Wu, E. Calcio Gaudino, M. Manzoli, K. Martina, M. Drobot, U. Krtschil and G. Cravotto, Catal. Sci. Technol., 2017, 7, 4780–4791 RSC.
- J. C. Pastre, D. L. Browne and S. V. Ley, Chem. Soc. Rev., 2013, 42, 8849–8869 RSC.
- V. Sans and L. Cronin, Chem. Soc. Rev., 2016, 45, 2032–2043 RSC.
- B. J. Reizman and K. F. Jensen, Acc. Chem. Res., 2016, 49, 1786–1796 CrossRef CAS PubMed.
- S. Mascia, P. L. Heider, H. Zhang, R. Lakerveld, B. Benyahia, P. I. Barton, R. D. Braatz, C. L. Cooney, J. M. B. Evans, T. F. Jamison, K. F. Jensen, A. S. Myerson and B. L. Trout, Angew. Chem., Int. Ed., 2013, 52, 12359–12363 CrossRef CAS PubMed.
- L. Yu, Continuous Manufacturing Has a Strong Impact on Drug Quality|FDA Voice, https://blogs.fda.gov/fdavoice/index.php/2016/04/continuous-manufacturing-has-a-strong-impact-on-drug-quality/, (accessed 6 July 2017).
- S. L. Lee, T. F. O'Connor, X. Yang, C. N. Cruz, S. Chatterjee, R. D. Madurawe, C. M. V. Moore, L. X. Yu and J. Woodcock, J. Pharm. Innov., 2015, 10, 191–199 CrossRef.
- D. Perera, J. W. Tucker, S. Brahmbhatt, C. J. Helal, A. Chong, W. Farrell, P. Richardson and N. W. Sach, Science, 2018, 359, 429–434 CrossRef CAS PubMed.
- N. Holmes, G. R. Akien, A. J. Blacker, R. L. Woodward, R. E. Meadows and R. A. Bourne, React. Chem. Eng., 2016, 1, 366–371 RSC.
- V. Sans, L. Porwol, V. Dragone and L. Cronin, Chem. Sci., 2014, 6, 1258–1264 RSC.
- A. Echtermeyer, Y. Amar, J. Zakrzewski and A. Lapkin, Beilstein J. Org. Chem., 2017, 13, 150–163 CrossRef CAS PubMed.
- D. E. Fitzpatrick and S. V. Ley, Tetrahedron, 2017, 74, 3087–3100 CrossRef.
- A. J. Parrott, R. A. Bourne, G. R. Akien, D. J. Irvine and M. Poliakoff, Angew. Chem., Int. Ed., 2011, 50, 3788–3792 CrossRef CAS PubMed.
- D. E. Fitzpatrick, C. Battilocchio and S. V. Ley, Org. Process Res. Dev., 2016, 20, 386–394 CrossRef CAS.
- Z. Amara, E. S. Streng, R. A. Skilton, J. Jin, M. W. George and M. Poliakoff, Eur. J. Org. Chem., 2015, 2015, 6141–6145 CrossRef CAS.
- E. Danieli, J. Perlo, A. L. L. Duchateau, G. K. M. Verzijl, V. M. Litvinov, B. Blümich and F. Casanova, ChemPhysChem, 2014, 15, 3060–3066 CrossRef CAS PubMed.
- N. Cherkasov, Y. Bai, A. J. Exposito and E. V. Rebrov, React. Chem. Eng., 2018, 3, 769–780 RSC.
- L. P. Pagani, G. E. Apostolakis, P. Hejzlar, L. P. Pagani, G. E. Apostolakis and P. Hejzlar, Nucl. Technol., 2005, 149, 129–140 CrossRef CAS.
- T. Narushima, N. T. Kinahan and J. J. Boland, Rev. Sci. Instrum., 2005, 76, 095113 CrossRef.
- R. Kizilel, A. Lateef, R. Sabbah, M. M. Farid, J. R. Selman and S. Al-hallaj, J. Power Sources, 2008, 183, 370–375 CrossRef CAS.
- P. Albers, J. Pietsch and S. F. Parker, J. Mol. Catal. A: Chem., 2001, 173, 275–286 CrossRef CAS.
- M. Argyle and C. Bartholomew, Catalysts, 2015, 5, 145–269 CrossRef CAS.
- J. A. Moulijn, A. E. Van Diepen and F. Kapteijn, Appl. Catal., A, 2001, 212, 3–16 CrossRef CAS.
- S. Kataoka, Y. Takeuchi, A. Harada, T. Takagi, Y. Takenaka, N. Fukaya, H. Yasuda, T. Ohmori and A. Endo, Appl. Catal., A, 2012, 427–428, 119–124 CrossRef CAS.
- E. V. Rebrov, E. A. Klinger, A. Berenguer-Murcia, E. M. Sulman and J. C. Schouten, Org. Process Res. Dev., 2009, 13, 991–998 CrossRef CAS.
- S. K. Johnston, N. Cherkasov, E. Pérez-Barrado, A. Aho, D. Y. Murzin, A. O. Ibhadon and M. G. Francesconi, Appl. Catal., A, 2017, 544, 40–45 CrossRef CAS.
- N. Cherkasov, A. O. Ibhadon and E. V. Rebrov, Lab Chip, 2015, 15, 1952–1960 RSC.
- N. Cherkasov, A. O. Ibhadon, A. McCue, J. A. Anderson and S. K. Johnston, Appl. Catal., A, 2015, 497, 22–30 CrossRef CAS.
- Z. Wu, N. Cherkasov, G. Cravotto, E. Borretto, A. O. Ibhadon, J. Medlock and W. Bonrath, ChemCatChem, 2015, 7, 952–959 CrossRef CAS.
- N. Cherkasov, M. Al-Rawashdeh, A. O. Ibhadon and E. V. Rebrov, Catal. Today, 2016, 273, 205–212 CrossRef CAS.
- N. Harries, J. R. Burns, D. A. Barrow and C. Ramshaw, Int. J. Heat Mass Transfer, 2003, 46, 3313–3322 CrossRef CAS.
- A. Günther, S. A. Khan, M. Thalmann, F. Trachel and K. F. Jensen, Lab Chip, 2004, 4, 278–286 RSC.
- A.-K. Liedtke, F. Bornette, R. Philippe and C. de Bellefon, Chem. Eng. J., 2013, 227, 174–181 CrossRef CAS.
- C. X. Zhao and A. P. J. Middelberg, Chem. Eng. Sci., 2011, 66, 1394–1411 CrossRef CAS.
- A. Günther, M. Jhunjhunwala, M. Thalmann, M. A. Schmidt and K. F. Jensen, Langmuir, 2005, 1547–1555 CrossRef PubMed.
- A. N. Tsoligkas, M. J. H. Simmons and J. Wood, Chem. Eng. Sci., 2007, 62, 4365–4378 CrossRef CAS.
- R. J. Ingham, C. Battilocchio, D. E. Fitzpatrick, E. Sliwinski, J. M. Hawkins and S. V. Ley, Angew. Chem., Int. Ed., 2015, 54, 144–148 CrossRef CAS PubMed.
- Y. Liu, P. Gao, N. Cherkasov and E. V. E. V. Rebrov, RSC Adv., 2016, 6, 100997–101007 RSC.
- C. Moreno-Marrodan, F. Liguori and P. Barbaro, Beilstein J. Org. Chem., 2017, 13, 734–754 CrossRef CAS PubMed.
- P. T. Witte, P. H. Berben, S. Boland, E. H. Boymans, D. Vogt, J. W. Geus and J. G. Donkervoort, Top. Catal., 2012, 55, 505–511 CrossRef CAS.
- L. A. Saudan, Acc. Chem. Res., 2007, 40, 1309–1319 CrossRef CAS PubMed.
- F. Liguori and P. Barbaro, J. Catal., 2014, 311, 212–220 CrossRef CAS.
- J. Wood, L. Bodenes, J. A. Bennett, K. Deplanche and L. E. Macaskie, Ind. Eng. Chem. Res., 2010, 49, 980–988 CrossRef CAS.
- C. Berguerand, I. Yuranov, F. Cárdenas-Lizana, T. Yuranova and L. Kiwi-Minsker, J. Phys. Chem. C, 2014, 118, 12250–12259 CrossRef CAS.
- F. Zhao, D. Cambié, V. Hessel, M. G. Debije and T. Noël, Green Chem., 2018, 20, 2459–2464 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8re00186c |
| This journal is © The Royal Society of Chemistry 2019 |