
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, photophysical and electronic properties of tetra-donor- or acceptor-substituted ortho-perylenes displaying four reversible oxidations or reductions†
Julia
Merz
a,
Andreas
Steffen
 ab,
Jörn
Nitsch
a,
Julian
Fink
a,
Claudia B.
Schürger
a,
Alexandra
Friedrich
a,
Ivo
Krummenacher
a,
Holger
Braunschweig
a,
Michael
Moos
c,
David
Mims
c,
Christoph
Lambert
c and
Todd B.
Marder
*a
ab,
Jörn
Nitsch
a,
Julian
Fink
a,
Claudia B.
Schürger
a,
Alexandra
Friedrich
a,
Ivo
Krummenacher
a,
Holger
Braunschweig
a,
Michael
Moos
c,
David
Mims
c,
Christoph
Lambert
c and
Todd B.
Marder
*a
aInstitut für Anorganische Chemie, Institute for Sustainable Chemistry & Catalysis with Boron (ICB), Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: todd.marder@uni-wuerzburg.de
bFaculty of Chemistry and Chemical Biology, TU Dortmund University, Otto-Hahn-Str. 6, 44227 Dortmund, Germany
cInstitut für Organische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
First published on 24th June 2019
Abstract
Via regioselective Ir-catalyzed C–H borylation and subsequent reactions (i.e., viaBr4-Per or (BF3K)4-Per intermediates), we have introduced strong π-donors and acceptors at the 2,5,8,11-positions of perylene leading to unusual properties. Thus, incorporation of four donor diphenylamine (DPA) or four acceptor Bmes2 (mes = 2,4,6-Me3C6H2) moieties yields novel compounds which can be reversibly oxidized or reduced four times, respectively, an unprecedented behavior for monomeric perylene derivatives. Spectroelectrochemical measurements show NIR absorptions up to 3000 nm for the mono-cation radical of (DPA)4-Per and a strong electronic coupling over the perylene bridge was observed indicative of fully delocalized Robin-Day Class III behavior. Both (DPA)4-Per and (Bmes2)4-Per derivatives possess unusually long intrinsic singlet lifetimes (τ0), e.g., 94 ns for the former one. The compounds are emissive in solution, thin films, and the solid state, with apparent Stokes shifts that are exceptionally large for perylene derivatives. Transient absorption measurements on (DPA)4-Per reveal an additional excited state, with a long lifetime of 500 μs, which sensitizes singlet oxygen effectively.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) show very different and unusual properties compared to small aromatic compounds as a result of their extended π-conjugation. Their narrower HOMO–LUMO gaps lead to long-wavelength absorptions and emissions. Furthermore, PAHs such as perylene have lower oxidation and/or reduction potentials, higher mechanical strengths and stronger π–π-interactions.1,2 Perylene diimides (PDIs) have attracted much interest for use as dyes, pigments3,4 and semiconductors5 in diverse applications.6–12 They exhibit high chemical, thermal and photochemical stability, strong absorption and fluorescence and unique self-assembly behavior, which facilitates charge mobility.13–18 In addition, their high electron affinity makes PDIs promising candidates for electronic materials such as organic field effect transistors and solar cells.12,13,19,20The perylene core has 12 positions and can be considered as two naphthalene moieties conjoined at the 1 and 8 positions (Fig. 1).21 Diamagnetic susceptibility studies indicate that the central ring of perylene fails to display fully aromatic character as the naphthalene units dominate the perylene structure.21–23 Furthermore, Nucleus-Independent Chemical Shift (NICS) calculations indicate that the central ring is non-aromatic24 and X-ray structural data demonstrates that the C–C bonds joining the two naphthalene units are relatively long compared to typical aromatic C–C bonds. For the β polymorph of perylene, a C6a–C6b (or C12a–C12b) bond length of 1.474(1) Å was reported at 130 K from single-crystal X-ray diffraction data,25 while a bond length of 1.467(4) Å was reported at 200 K by Botoshansky et al.26 For the α polymorph of perylene, C6a–C6b (or C12a–C12b) bond lengths were reported to be in the range of 1.462–1.480 Å.26–28 These distances are typical of C(sp2)–C(sp2) single bonds.29–31
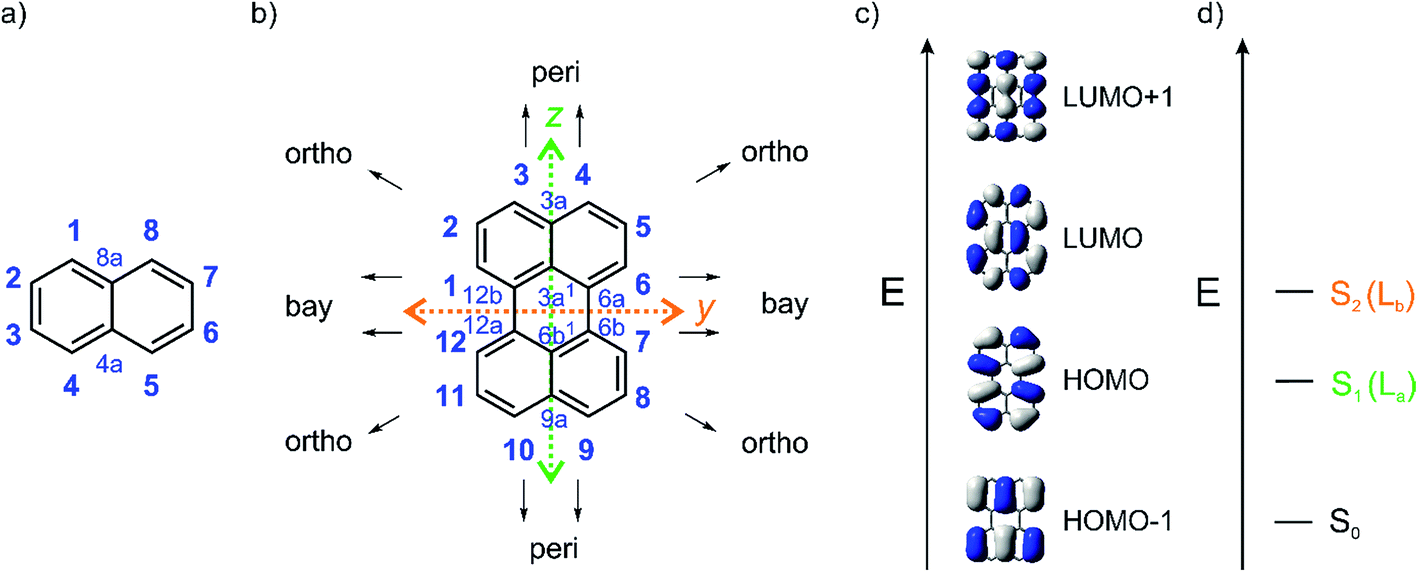 | ||
| Fig. 1 (a) Atom numbering system in naphthalene; (b) atom numbering system with principle Cartesian coordinate system used for perylene; (c) the four frontier orbitals of perylene; and (d) low energy optical transitions of perylene with the Platt nomenclature.32 | ||
This combination of two connected naphthalene units leads to particular photophysical properties. Substituting naphthalene at its 1 and 8 positions leads to a modulation of its La transition in such a way that in perylene the S1 ← S0 transition is polarized along the z-axis (La) and not along the y-axis (Lb) as in naphthalene. Perylene's lowest-lying singlet excited state, observed in the absorption spectrum at 440 nm (ε = 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 in toluene), consists of four vibrational sub-bands with a 1400 cm−1 interval and is assigned to a symmetry allowed HOMO → LUMO transition.33–35 The y-axis-polarized (Lb) S2 ← S0 transition at 253 nm (ε = 52000 M−1 cm−1 in hexane) is energetically well separated from S1 by 16800 cm−1. As the S1 ← S0 transition is allowed, emission from S1 is also (λmax(em) = 444 nm in toluene) allowed and, therefore, the intrinsic lifetime (τ0) of 5.5 ns is quite short, and fluorescence is strong with a quantum yield near unity.35PDI-A's (Fig. 2) lowest energy z-axis-polarized (La) absorption is observed at 526 nm in chloroform, and is stronger than perylene's with ε = 88000 M−1 cm−1.13,36 This compound emits in the yellow-green region at 533 nm in chloroform with τ0 = 4 ns.
000 M−1 cm−1 in toluene), consists of four vibrational sub-bands with a 1400 cm−1 interval and is assigned to a symmetry allowed HOMO → LUMO transition.33–35 The y-axis-polarized (Lb) S2 ← S0 transition at 253 nm (ε = 52000 M−1 cm−1 in hexane) is energetically well separated from S1 by 16800 cm−1. As the S1 ← S0 transition is allowed, emission from S1 is also (λmax(em) = 444 nm in toluene) allowed and, therefore, the intrinsic lifetime (τ0) of 5.5 ns is quite short, and fluorescence is strong with a quantum yield near unity.35PDI-A's (Fig. 2) lowest energy z-axis-polarized (La) absorption is observed at 526 nm in chloroform, and is stronger than perylene's with ε = 88000 M−1 cm−1.13,36 This compound emits in the yellow-green region at 533 nm in chloroform with τ0 = 4 ns.
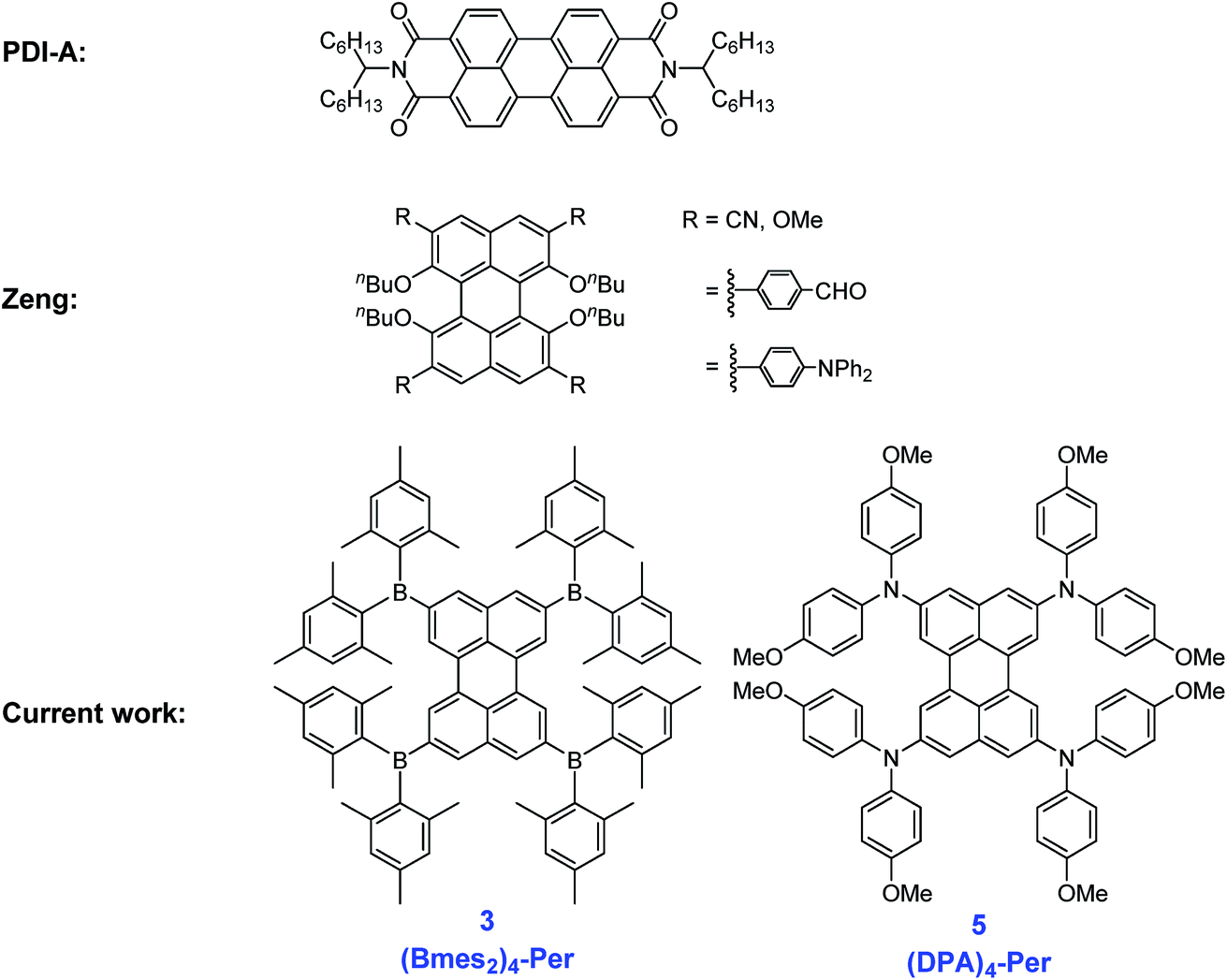 | ||
| Fig. 2 Examples of known perylene derivatives13,44 and our two target perylene derivatives. | ||
As perylenes play an important role in electronic materials, their electronic properties have been studied by several groups.37,38 Cyclic voltammetry measurements show that unsubstituted perylene can be reversibly reduced to its radical anion at −2.07 V and dianion at −2.66 V vs. Fc/Fc+ (DMF/MeCN) and reversibly oxidized at +0.69 V vs. Fc/Fc+ (Table 1).5,38,39 PDIs are a class of compounds that are very easy to reduce, with PDI-A having a reduction potential of −0.98 V vs. Fc/Fc+. Further substitution can give rise to even stronger oxidants.13
| E 1/2[red]1 | E 1/2[red]2 | E 1/2[ox] | |
|---|---|---|---|
| Perylene | −2.07 V vs. Fc/Fc+ (MeCN)5,38,39 | −2.66 V vs. Fc/Fc+ (DMF)5,37 | +1.25 V vs. Fc/Fc+ (MeCN)5,38 |
| −1.67 V vs. SCE (DMF/MeCN)38,39 | −2.21 V vs. SCE (DMF)37 | +1.09 V vs. NHE (DMF or MeCN)38 | |
| −1.63 V vs. SCE (THF)37 | −2.17 V vs. SCE (THF)37 | +0.85 V vs. SCE (MeCN)40,41 | |
| PDI-A | −0.98 V vs. Fc/Fc+ (MeCN)42 | −1.21 V vs. Fc/Fc+ (MeCN)42 | +1.21 V vs. Fc/Fc+ (MeCN)42 |
| −0.58 V vs. SCE5 | −0.81 V vs. SCE5 | +1.61 vs. SCE5 |
However, perylene derivatives without carboxyimide groups at the peri positions are much less well studied due to difficulties in functionalizing the perylene core directly.43–45 One method to functionalize perylene directly is via Ir-catalyzed C–H borylation, which was reported by Marder and co-workers in 2005.45 As a result of the very crowded nature of the active five-coordinate Ir(III)-catalyst, borylation of C–H bonds ortho to a substituent or ring junction is inhibited.46 Therefore, it is possible to borylate the 2,5,8,11-positions of perylene selectively, providing a useful entry point for the synthesis of diverse ortho substituted perylene derivatives.47,48 In 2011, Shinokubo and co-workers49,50 employed a modified Ir-catalyzed borylation reaction with an ortho directing ligand, which enabled the functionalization of the four ortho positions of PDI's with heteroaryl, OMe and OH moieties for the first time. They observed a slight blue-shift in the absorption (from 525 to 512 nm) and emission (from 533 to 516 nm) spectra through the introduction of OH groups, which is caused by an intramolecular hydrogen bonding interaction between the carbonyl and hydroxy groups. The OMe moieties on the other hand, cause a small red shift in the absorption (from 525 to 538 nm) and emission (from 533 to 549 nm) spectra. In 2012, a Suzuki–Miyaura cross-coupling was reported by Ikeda et al. using 2,5,8,11-tetra(Bpin)perylene to give a porphyrin-perylene-porphyrin triad51 and, in 2016, Tran et al. reported Suzuki–Miyaura coupling of our 2,5,8,11-tetra(Bpin)perylene to give microporous coordination polymers.52 However, there are no reports so far of only ortho heteroatom substituted perylenes. Thus, investigations of the effect of substituents on the 2,5,8,11-positions of the perylene core are still lacking. In 2017, Zeng and co-workers44 prepared ortho and bay octa-substituted perylene derivatives by an indirect method, namely oxidative radical–radical coupling of two naphthene units followed by reduction. They synthesized 2,5,8,11-tetrabromo-1,6,7,12-tetra-n-butoxyperylene and showed the versatility of that derivative by performing Suzuki–Miyaura coupling reactions and nucleophilic substitution reactions with copper cyanide or sodium methoxide to place cyano or methoxy moieties at the ortho positions. However, the n-butoxy substituents at the bay positions lead to a twisted core, which influences the properties of these perylene derivatives. In 2017, Hariharan, Shaijumon and co-workers demonstrated, with a twisted PDI, that the energy levels of the reduced species are altered and a single plateau discharge profile can be achieved, which is crucial for rechargeable battery electrodes.53 To investigate the influence of substitution only at the ortho positions on the perylene core, we prepared an ortho perylene derivative with four donors (D) and one with four acceptors (A), due to their strong influence on the frontier orbital levels.
Materials with high HOMO energies, such as the compound N,N′-diphenyl-N,N′-bis(3-methylphenyl)-(1,1′-biphenyl)-4,4′-diamine (TPD), are especially useful for hole transport.54–60 Common π-donors that have been used in dyes to boost HOMO energies include amines, with a lone pair on the nitrogen, such as diarylamino, diethylamino, dimethylamino or carbazolyl moieties.61 Diarylamines are among the strongest π-electron donors and have been employed in diverse applications,62–74 due to their outstanding physical, photochemical and electrochemical properties. Furthermore, they are easy to synthesize and handle.75 A methoxy group at the position para to the nitrogen not only increases the electron donating strength of diarylamines, but enables reversible oxidations.75,76 In contrast, materials with low LUMO energies, such as mes2B–(C4H2S)n–Bmes2 (n = 2, 3; mes = 2,4,6-Me3C6H2), are useful for electron transport.77,78 Diarylboryl groups have attracted much interest for use in optoelectronic materials, as the vacant pz-orbital of the three-coordinate boron serves as a strong π-acceptor, interacting with an adjacent π-system.79–91 This conjugation provides the electron-deficient character that gives rise to useful photophysical properties.92–94 For example, attaching a Bmes2 moiety to the 2- and 2,7-positions of pyrene leads to a switch of the energetic order of the LUMO+1 and LUMO, which is a consequence of the mixing of the empty pz-orbital with the pyrene B3u LUMO+1. This mixing leads to a strong stabilization of the LUMO+1 such that it drops below the LUMO in energy.95–97 Kinetic stabilization by bulky groups, such as mesityl or 2,4,6-(CF3)3C6H2 (Fmes), sterically protects the empty pz-orbital from nucleophilic attacks leading to air- and moisture stable materials.79–81,98,99 Incorporation of the boron into a rigid and planar structure is an alternative method to provide increased stability via structural constraints.100
With the above issues in mind, we were motivated to use the Ir-catalyzed C–H borylation reaction45 to prepare new compounds in order to study the effect of strong π-donors and -acceptors at the 2,5,8,11-positions of perylene. We report herein the photophysical and electrochemical properties of the target compounds (Bmes2)4-Per and (DPA)4-Per, which were examined experimentally and theoretically.
Results and discussion
Synthesis and structural characterization
The synthesis of the compounds (Bmes2)4-Per and (DPA)4-Per is summarized in Scheme 1. The starting point of our approach is the high yielding Ir-catalyzed borylation of perylene, which we previously reported in 2005, providing (Bpin)4-Per in 90% yield.45 This synthesis is very convenient, as it can be run on a multigram scale and the tetra-borylated product does not need further purification, such as column chromatography, because the product crystallizes directly from the reaction solution. Suzuki–Miyaura cross-couplings with (Bpin)4-Per have been reported before.51,52 We, on the other hand, transformed this precursor into its potassium trifluoroborate salt (BF3K)4-Per. The synthesis was carried out in air in a THF/water mixture giving a yield of 99%. Potassium trifluoroborate salts are air- and moisture stable, and easy to handle and purify.101,102 Furthermore, they can function as carbon nucleophiles and are important intermediates in diverse synthetic reactions.103–105 However, they can also serve as boron electrophiles. Thus, (BF3K)4-Per reacts with the Grignard reagent mesMgBr to give (Bmes2)4-Per in 86% yield. Marder and co-workers previously demonstrated the utility of this approach to derivatize pyrene with a (Bmes2) moiety.106 Additionally, boroles were prepared from aryl trifluoroborates and aryllithium reagents.99 The utility of a BF3K salt as a boron electrophile for the synthesis of triarylboranes has also been demonstrated by Wagner and co-workers using aryllithium instead of Grignard reagents.107,108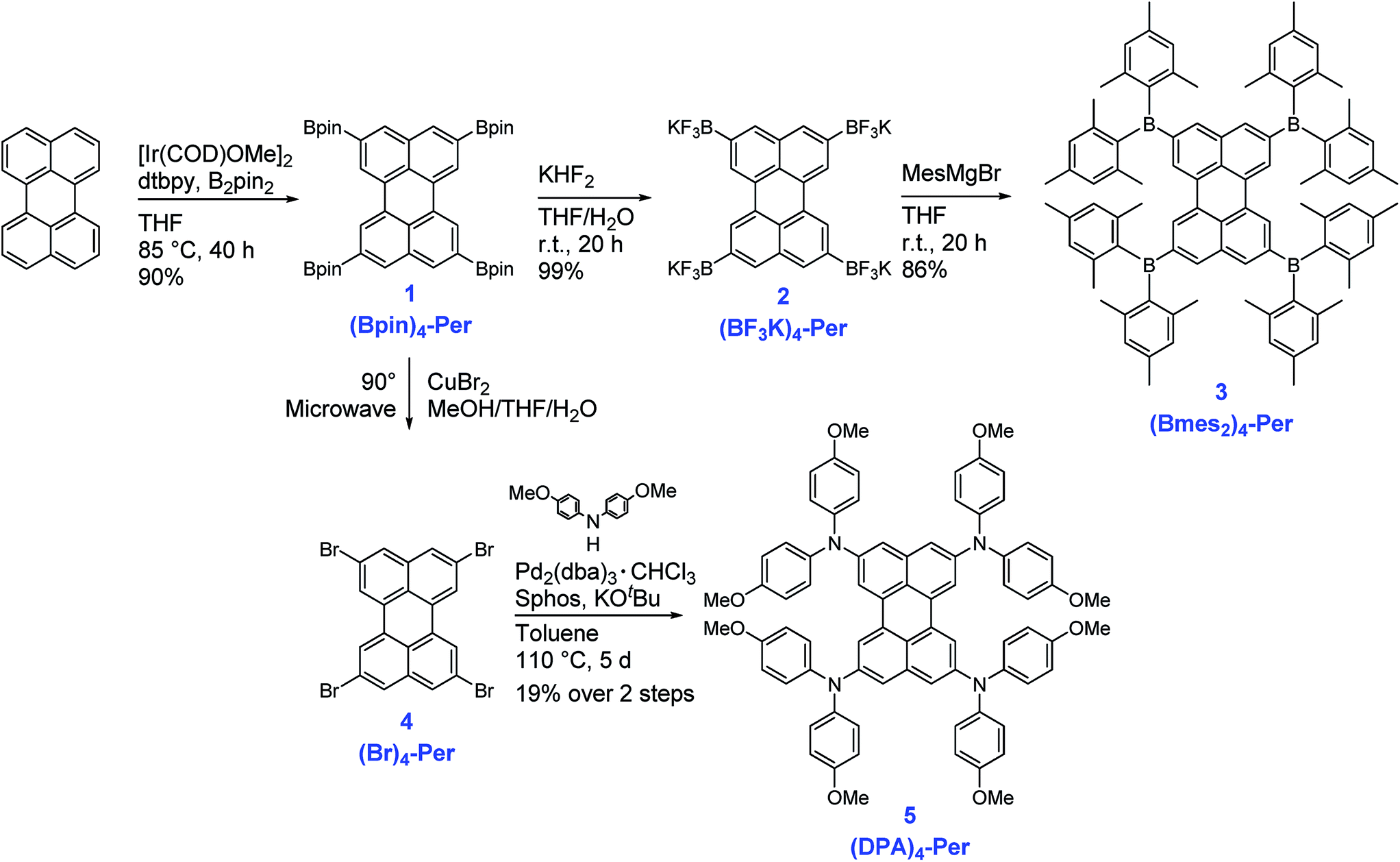 | ||
| Scheme 1 Synthesis of the compounds (Bpin)4-Per, (BF3K)4-Per, (Bmes2)4-Per, (Br)4-Per and (DPA)4-Per. | ||
The precursor (Bpin)4-Per can also be transformed into the corresponding halogenated (Br)4-Per by a halodeboronation.109,110 Thus, (Bpin)4-Per and CuBr2 were suspended in a mixture of THF/MeOH/H2O (1:1:1) and the reaction mixture was irradiated in a microwave reactor at 90 °C for 20 h. We note that performing this reaction in an oil bath instead of a microwave reactor results in a mixture of (Br)3-Per and (Br)4-Per as some protodeborylation occurs. Bromodeborylation of the C–Bpin bond is especially useful as it converts a carbon nucleophile into a carbon electrophile in one easy step. Thus, the (Br)4-Per building block is a further useful intermediate that can serve in numerous types of coupling reactions including a Buchwald–Hartwig amination, which we performed to obtain (DPA)4-Per. The fourfold amination of (Br)4-Per, using Pd2(dba)3·CHCl3 as the catalyst precursor and Sphos as the ligand, was achieved in an overall yield of 19% in two steps starting from (Bpin)4-Per. (DPA)4-Per exhibits very good solubility in common organic solvents. The new products 2–5 were fully characterized by multinuclear NMR spectroscopy, high-resolution mass spectrometry, and elemental analysis.
The solid-state structure of (DPA)4-Per was determined via single-crystal X-ray diffraction. The molecule shows inversion symmetry. In (DPA)4-Per, the individual bond lengths are similar to those of perylene (Table S2†).30 The naphthalene units of the perylene moiety exhibit typical aromatic C–C bond lengths ranging from 1.371(3) to 1.426(3) Å (Fig. 3a and Table S2†). The b and f bonds (1.371(3)–1.380(3) Å) are considerably shorter than the a, c, d, and e bonds (1.412(3)–1.426(3) Å) which is also observed in naphthalene and perylene.31 The length of the C11–C13 bond (Fig. 3a, bond g, Table S2†) connecting the naphthalene units in (DPA)4-Per is 1.470(2) Å at 100 K (Fig. 3). This is in the same range as the related bond distances reported for the two polymorphs of perylene (1.462–1.480 Å)25–28,30,111 and, hence, resembles a C(sp2)–C(sp2) single bond.29–31
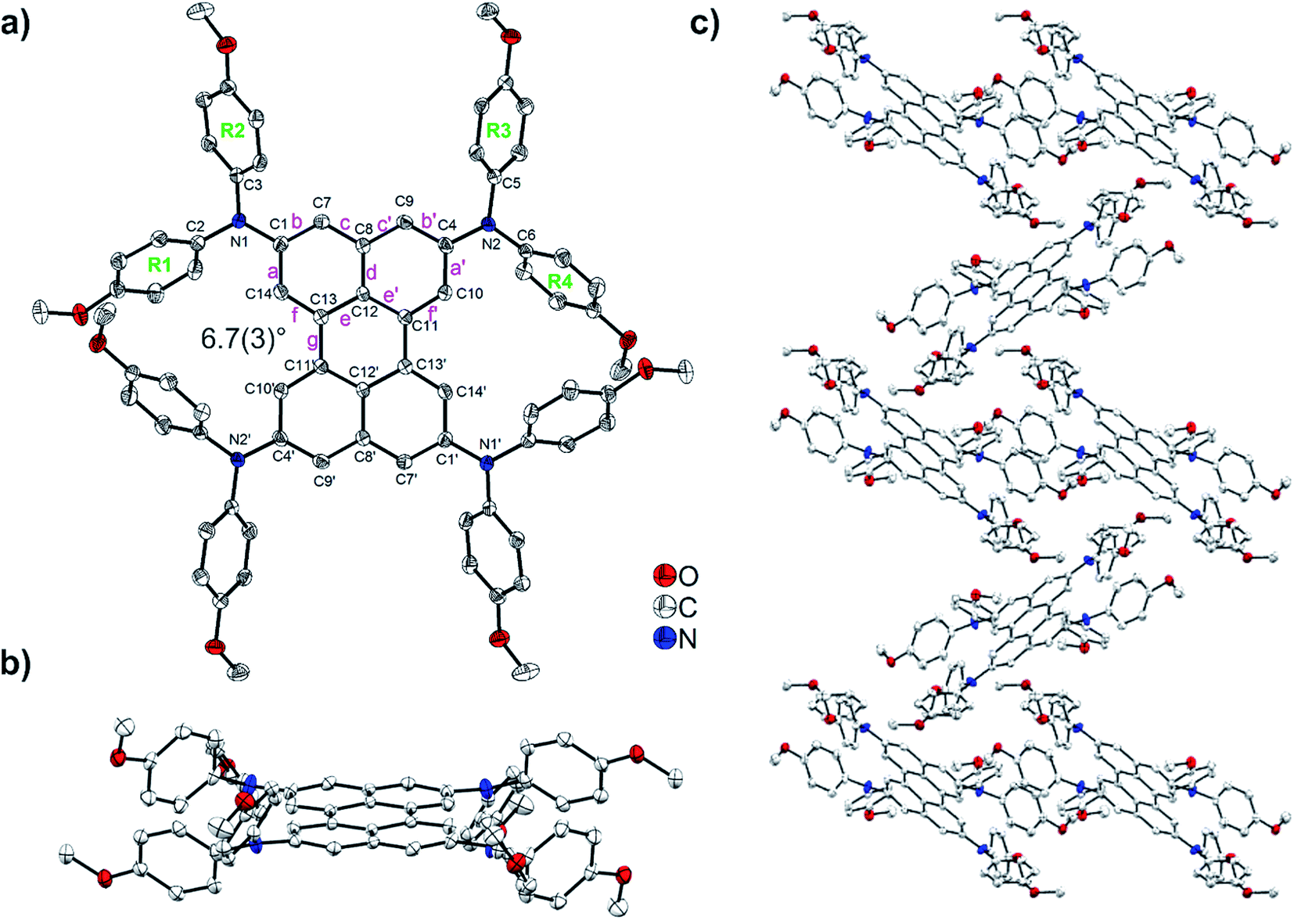 | ||
| Fig. 3 Molecular structure of (DPA)4-Per: (a) top view; (b) side view; and (c) packing of (DPA)4-Per molecules. Hydrogen atoms are omitted for clarity. Element color: carbon (white), nitrogen (blue), oxygen (red). Atomic displacement ellipsoids are shown at the 50% probability level. | ||
The perylene core of (DPA)4-Per is only slightly twisted with a small C10–C11–C13–C14 dihedral angle of 6.7(3)° (Fig. 3a and b). Crystal structures of perylene and peri-substituted perylenes in general show nearly planar perylene cores. For example, dihedral angles in the range 0.1–1.8° were reported for β- and α-perylene.26–28,30,111,112 This shows that the substitution of the sterically demanding DPA moieties at the four ortho positions does not significantly affect the planarity of the perylene core. The nitrogen atoms are virtually trigonal planar with the sum of the angles around N1 being 359.8(2)° and N2 being 359.2(2)°, respectively. The interplanar angles between the terminal phenyl rings bonded to nitrogen and the NC3 planes are 42.9–44.2° for R1, R2, and R3, and 66.0° for R4 (Fig. 3a, Table S2†). In contrast, the crystal structures of most of the perylene compounds containing bay substituents show a strong twist between the naphthalene units of the core due to steric repulsion created by the bay moieties.44,113 For example, dihedral angles of 30.7–34.3° between the two naphthalene units have been reported for bay- and ortho-octasubstituted perylenes by Zeng and co-workers.44 The packing of the (DPA)4-Per molecules in the crystal structure is determined by the large steric demand of the amine moieties. There is no π–π stacking interaction present. Between the methoxy groups of the R1 and R4′ phenyl rings, and their inversion-related groups, intramolecular C–H⋯O interactions exist (Table S3†). Intermolecular C–H⋯C, C–H⋯π, and C–H⋯O interactions are present between methoxy groups, phenyl rings, and the perylene core. They are listed in detail in Table S3.† A Hirshfeld surface analysis was performed in order to quantify the nature and type of intermolecular interactions.114–117 Fingerprint analysis and its breakdown to the individual relative contributions,118–120 shows a major contribution from H⋯H interactions (59%), followed by a significant amount from C⋯H (29%) and O⋯H (10%) interactions, and minor contribution of O⋯C interactions (1%). Only very weak contributions (<1%) from C⋯C, O⋯O, and N⋯H interactions are observed (Fig. S28 and S29†).
Photophysical properties
The absorption spectra of (Br)4-Per and (Bpin)4-Per are generally very similar to that of perylene. Their S1 ← S0 transition, which is z-axis polarized (La) and basically attributed to a HOMO → LUMO transition, is allowed with extinction coefficients of 28000–32000 M−1 cm−1 (perylene: 34000 M−1 cm−1). Furthermore, this band has a well-defined vibronic fine structure with an interval of 1400 cm−1 between the sub-bands as in perylene. However, it is slightly bathochromically shifted (310 cm−1 for (Bpin)4-Per and 360 cm−1(Br)4-Per) with respect to the parent perylene. Thus, the S1 ← S0 transition in these derivatives is a pure π → π* transition. As observed previously on pyrene,106 the Bpin moieties barely influence the photophysical properties of perylene, as oxygen substituents at the boron atom decreases its π-acceptor properties.121 Bromo substituents are only weak π-donors; therefore, these two derivatives show hardly any CT character. This is consistent with the observed absorption properties reported for bromo-substituted PDI's.122 It is interesting to observe that, in comparison with the 2,5,8,11-tetrabromo-1,6,7,12-tetra-n-butoxyperylene reported by Zeng and co-workers,44 the S1 ← S0 transition of (Br)4-Per is narrower, structurally better defined and, furthermore, slightly more allowed.123 This indicates some influence of the donating bay substituents on the absorption properties. However, there are no previous reports on perylene derivatives substituted only at the ortho positions for comparison. Stronger π-accepting moieties, such as Bmes2, at the ortho positions leads to a pronounced influence on the S1 ← S0 transition. Hence, the lowest energy band in (Bmes2)4-Per is much broader and is strongly bathochromically shifted (1220 cm−1) with respect to the La band of perylene which indicates a stabilization of the unoccupied orbitals of this derivative in comparison to those of perylene (vide infra).
Furthermore, this transition is much less allowed with an extinction coefficient of 20000 M−1 cm−1. However, this band still possesses a vibronic fine structure, but the modes have rather similar intensity. In comparison to the acceptor version reported by Zeng and co-workers44 (2,5,8,11-tetracyano-1,6,7,12-tetra-n-butoxyperylene) (λmax(abs) = 454 nm) the absorption of (Bmes2)4-Per is further bathochromically shifted (λmax(abs) = 465 nm), which indicates a stronger stabilization of the unoccupied orbitals in our derivative. We have previously shown48,95–97 that CN is a less effective π-acceptor than Bmes2 when attached to pyrene. The influence on the S1 ← S0 transition is even more pronounced in (DPA)4-Per as it does not have any vibrational progression. Its La band is broader and more strongly bathochromically shifted with respect to the La band of perylene (2690 cm−1) than for (Bmes2)4-Per. Hence, the donor DPA substituents exert a larger destabilizing effect on the occupied frontier orbitals than the acceptor Bmes2 stabilizes the empty ones. This is similar to what we observed for 2,7-pyrene derivatives.106 Both target derivatives, (Bmes2)4-Per and (DPA)4-Per have the lowest energy absorptions reported so far for ortho substituted perylene derivatives.
All derivatives show intense fluorescence in the blue to orange region of the electromagnetic spectrum with quantum yields Φ of up to 0.58 (Fig. 4, Table 2). The emission spectra of (Br)4-Per and (Bpin)4-Per are very similar to that of perylene, in that the apparent Stokes shifts124 are very small (300–350 cm−1) and the band shape is a mirror image of its respective S1 ← S0 absorption. The radiative rate constants of both (Br)4-Per and (Bpin)4-Per are the same order of magnitude (kr = 12 × 107 s−1) as that of perylene. Thus, their excited state structures are presumably very similar to those in the ground state. It is interesting to note that 2,5,8,11-tetrabromo-1,6,7,12-tetra-n-butoxyperylene displays a broad emission without vibronic fine structure and a significantly larger apparent Stokes shift of 2120 cm−1, indicating modest geometry changes in its excited state. Furthermore, its emission quantum yield of Φ = 0.30 is only half that of (Br)4-Per (Φ = 0.58) but, as Zeng and co-workers did not report lifetimes or radiative decay rates, a full comparison is not possible. The differences observed must be a result of the bay substituents that lead to a twisted core.126 The significant influence of bay substituents on the photophysical properties of PDI's such as enhanced nonradiative decay was already reported by several groups.13,113,122,127,128 Nevertheless, for PAHs with heavy atoms such as Br, the emissions we measured are intense, and no phosphorescence was detected at 77 K. However, Dreeskamp and Koch already reported129,130 that the bromination of perylene at its peri position (i.e. 3-bromoperylene) does not lead to a quenching of the fluorescence by intersystem crossing (ISC) from S1via spin–orbit coupling. They found that the energy gap between the S1 and T1 of 3-bromoperylene is not small enough for ISC to compete with fluorescence. Furthermore, Hariharan and co-workers demonstrated that the bromination of PDIs also fails to promote ISC and, even in dibromoethane solutions, no effect of the external heavy atoms on ISC was observed.131
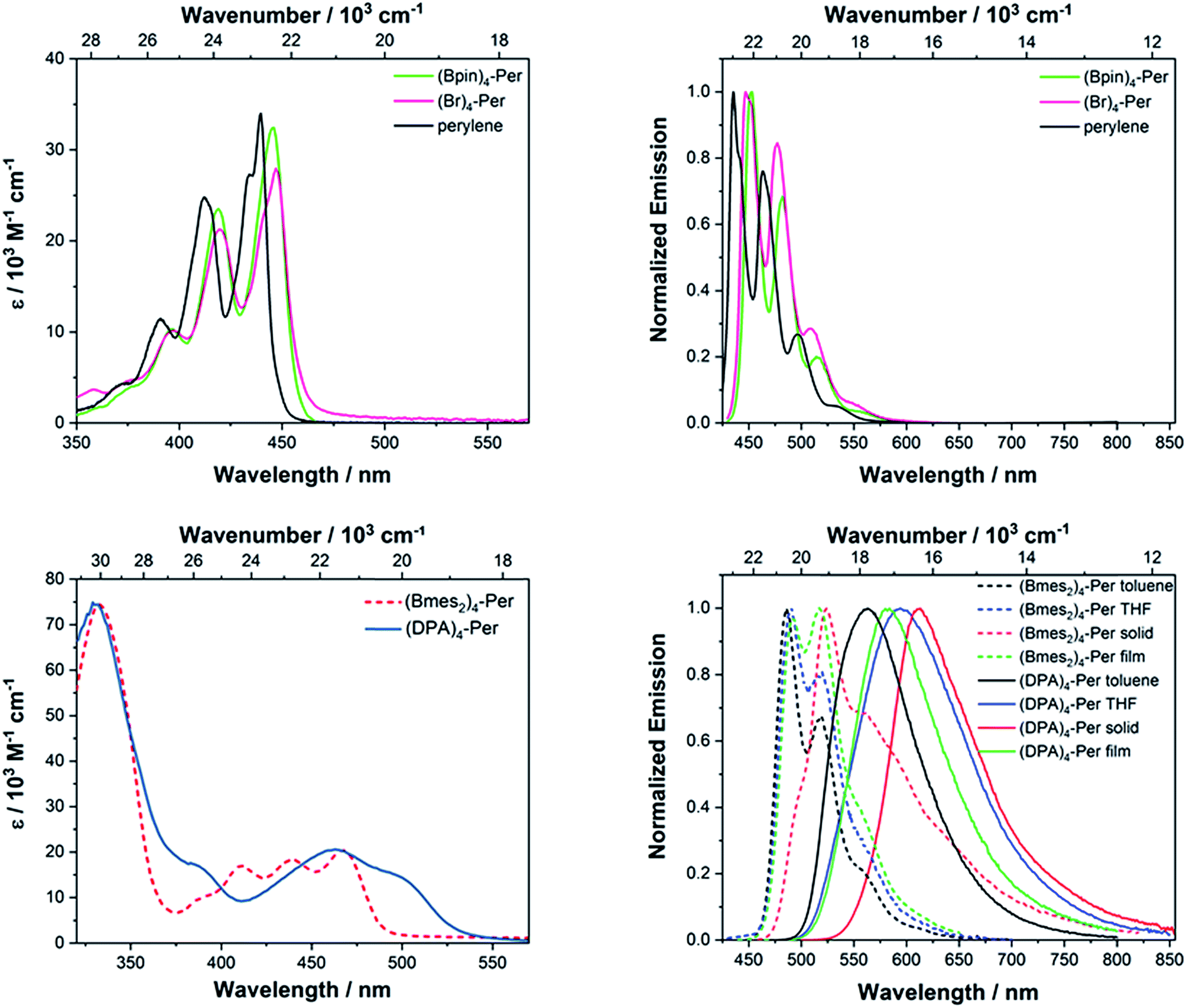 | ||
| Fig. 4 Absorption (left) and emission (right) spectra of (Br)4-Per and (Bpin)4-Per (top) and (Bmes2)4-Per and (DPA)4-Per (bottom) recorded in toluene if not otherwise noted. | ||
| Cpd | Medium | λ abs/nm (ε/103 M−1 cm−1) | λ em /nm | Apparent Stokes124 shift/cm−1 | τ /ns | τ avg /ns | Φ | τ 0/ns | k nr/107 s−1 | k r/107 s−1 |
|---|---|---|---|---|---|---|---|---|---|---|
| a Excited at the respective λabs (max) of S1 ← S0. b Pre-exponential factors Bn scaled to 100 and given in parentheses. c For multi-exponential decays the pure radiative lifetime τ0 = τ/Φ has been approximated by using the experimental average lifetime τ = ∑τnBn/∑Bn with Bn being the pre-exponential factors of the respective lifetime component τn. d From Brites and co-workers.125 e In an O2-saturated solution. f PMMA film doped with 1% of the respective perylene derivative. | ||||||||||
| Perylened | Toluene | 440 (34) | 444 | 4 | 4.0 | — | 0.95 | 4.0 | 1.3 | 24 |
| (Bpin)4-Per | Toluene | 446 (32), 419 (24), 397 (10) | 453 | 350 | 4.9 | — | 0.58 | 8.0 | 8.6 | 12 |
| (Br)4-Per | Toluene | 447 (28), 420 (21), 397 (10) | 453 | 300 | 4.7 | — | 0.55 | 9.0 | 9.6 | 12 |
| (Bmes2)4-Per | Toluene | 465 (20), 440 (18), 412 (17), 333 (74) | 489 | 1060 | 6.6 | — | 0.43 | 15 | 8.6 | 6.5 |
| THF | 466, 441, 412, 335 | 489 | 1010 | 8.6 | — | 0.41 | 21 | 6.7 | 4.8 | |
| Solid | — | 525 | — | 0.9 (87) | 1.2 | 0.04 | 31 | 79 | 3.2 | |
| 3.6 (13) | ||||||||||
| Filmf | — | 490, 517 | — | 7.3 (87) | 8.0 | 0.36 | 22 | 8.0 | 4.5 | |
| 13 (13) | ||||||||||
| (DPA)4-Per | Toluene | 499 (15), 463 (20), 386 (17), 331 (74) | 569 | 2470 | 12 | — | 0.26 | 46 | 6.2 | 2.2 |
| Toluenee | — | 569 | — | 4.6 | — | 0.09 | 51 | 20 | 2.0 | |
| THF | 501, 459, 383, 327 | 593 | 3100 | 8.5 | — | 0.09 | 94 | 11 | 1.1 | |
| Solid | — | 612 | 2.5 (67) | 3.8 | 0.07 | 54 | 26 | 1.9 | ||
| 6.4 (33) | ||||||||||
| Film | — | 580 | — | 12 (86) | 13 | 0.14 | 92 | 6.7 | 1.1 | |
| 20 (14) | ||||||||||
The emission of (Bmes2)4-Per, with λmax(em) = 489 nm, is bathochromically shifted by 2073 cm−1 in comparison to that of perylene and thus has a larger apparent Stokes shift of 1055 cm−1. Its intense emission (quantum yield Φ = 0.43) shows vibronic fine structure in toluene, which does not vanish in THF. Furthermore, this derivative shows no solvatochromism which indicates the absence of charge transfer (CT) character. In the solid state, a green emission with λmax(em) = 525 nm is observed, but the non-radiative decay rate knr is greatly enhanced by more than one order of magnitude (knr = 79 × 107 s−1), which leads to a decreased quantum yield. However, emission in the solid state is rather uncommon for perylene derivatives. The fluorescence of PDIs, for instance, is quenched in the solid state on account of H-type aggregation due to π–π interactions.132 The emission of the donor-substituted derivative (DPA)4-Per is further bathochromically shifted (λmax(em) = 569 nm), shows no vibronic fine structure and is very broad. Furthermore, it exhibits significant solvatochromism, confirming a pronounced CT character. The large apparent Stokes shift (3 100 cm−1 in THF) is untypical for perylenes, as PDIs usually have very small apparent Stokes shifts. However, a large apparent Stokes shift helps to reduce self-quenching and thus to avoid measurement errors.35 The excited state of (DPA)4-Per has an intrinsic lifetime of τ0 = 46 ns and is thus significantly longer lived than perylene (τ0 = 4 ns) and is highly stabilized in a polar environment, as in THF τ0 = 94 ns. The radiative decay rates kr of the target compounds (DPA)4-Per (kr = 2.2 × 107 s−1) and (Bmes2)4-Per (kr = 6.5 × 107 s−1) are one order of magnitude slower than of perylene (kr = 24 × 107 s−1) which is in full agreement with the Strickler–Berg relationship133 as the S1 ← S0 transition of these derivatives is also less allowed. The (DPA)4-Per derivative has the slowest radiative decay which is also in full agreement with the Strickler–Berg relationship133 as its emission is further bathochromically shifted. Interestingly, substituting one DPA moiety at a peri position of perylene results in two emissions at λmax(em) = 530 nm and 630 nm, respectively, while further studies showed125 that the dual emission results from two excited state rotamers with different angular distribution. However, emission from the peri-substituted derivative proceeds significantly faster with radiative decay rates of kr = 8.8 × 107 s−1.125 Hence, substituents at the ortho positions have a distinctly different influence on the excited state properties than substituents at the peri positions.
Reactivity with oxygen
In 1974, Dreeskamp, Koch and co-workers showed that the rate of ISC from the fluorescent singlet to a triplet state can be increased in perylene by an intermolecular energy transfer to halogenated-naphthalene.129,130,134 This precedence motivated us to take a closer look at our systems as the fluorescence quantum yield of (DPA)4-Per is particularly low (φ = 0.26) compared to perylene and triplet sensitizers are of great interest with many applications in phosphorescent materials,135–137 phosphorescent bioimaging,135,137,138 chemosensors,135,137,139 photoinitiated polymerization,137,140 photocatalysis,137,141–144 triplet–triplet annihilation based upconversion,137,145,146 and oncological or antibacterial photodynamic therapy.137,147,148 There are a few reports on triplet states of PDIs and these make use of bimolecular triplet sensitization,149 incorporation of sulfur150 or heavy metals such as Ir,131,151,152 Pt,153 Pd,152,154 or Ru.152 Flamigni and co-workers demonstrated155,156 that unsymmetrical substitution of PDIs is an alternative method to access triplet states. Furthermore, in 2016, Hariharan and co-workers reported131 that, through heavy atom substitution combined with a twisted-core structure of PDIs, triplet states of PDIs become accessible. One indication for an enhanced ISC rate is energy transfer from the triplet state of a compound formed upon photoexcitation to ground state oxygen (3∑g−). This leads to the generation of singlet oxygen (1Δg), which can be detected by its luminescence at 1272 nm. Accordingly, upon excitation of an O2-saturated toluene solution of (DPA)4-Per, an emission at 1272 nm was detected (Fig. 5).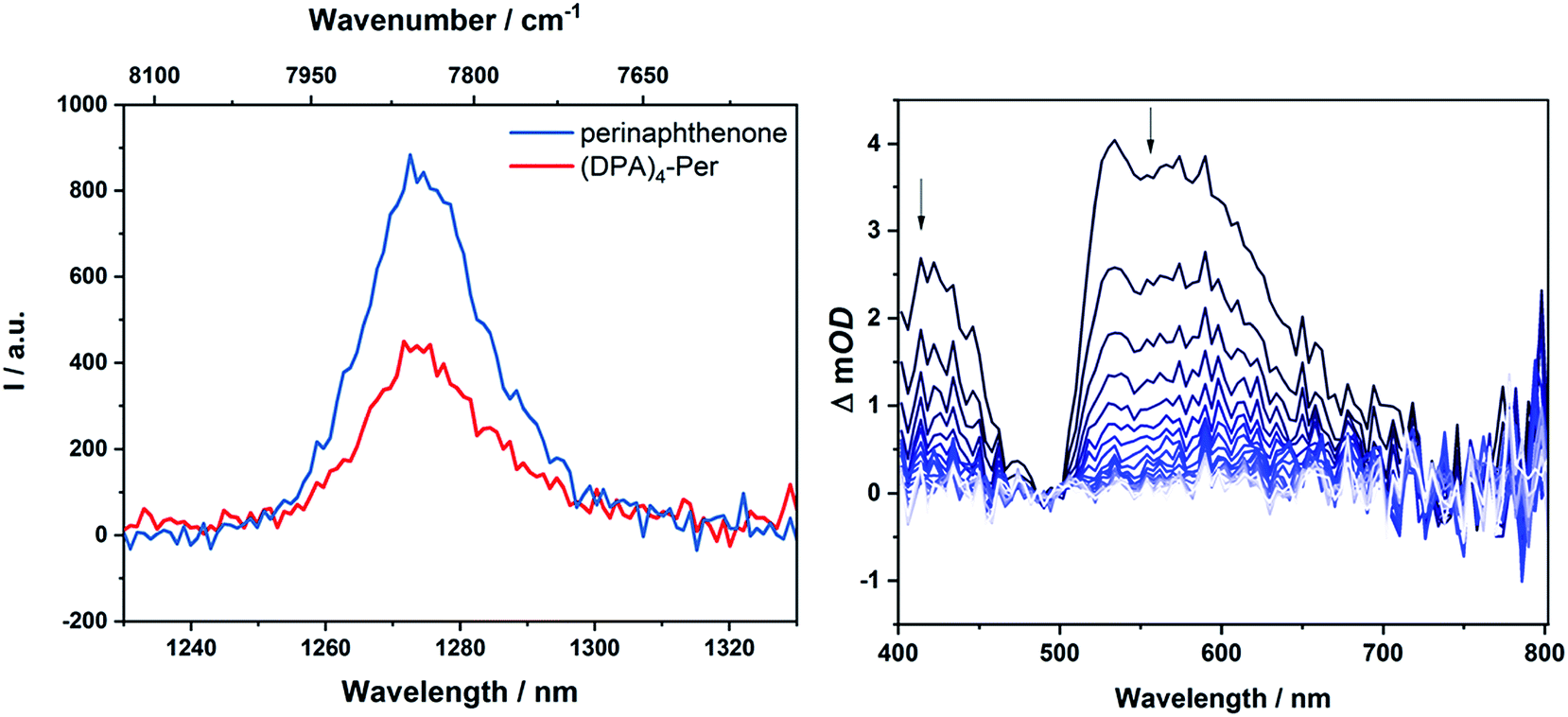 | ||
| Fig. 5 Singlet oxygen luminescence from optically matched toluene solutions of (DPA)4-Per and the standard perinaphthenone; excitation at 340 nm (left); nanosecond transient absorption spectrum of (DPA)4-Per in a degassed DMF solution (right) at intervals of 169.9 μs. | ||
Compared to the standard, perinaphthenone, for which the quantum yield of 1Δg production is close to unity,157(DPA)4-Per sensitizes 1Δg with a quantum yield of 0.60. On the other hand, the derivative (Bmes2)4-Per does not sensitize 1Δg to any measurable extent. However, as the singlet excited state of (DPA)4-Per is long-lived (τ = 12 ns, φ = 0.26) and quenched in an O2-saturated solution (τ = 4.6 ns, φ = 0.09), it is reasonable that the formation of 1Δg is not only a product of its triplet state but also of the singlet excited state.158,159 McLean and co-workers reported that perylene sensitizes 1Δg with a quantum yield of 0.65 in an oxygen-saturated benzene solution even though formation of its triplet state has a vanishing quantum yield, thus, implying 1Δg sensitization from its excited singlet state only.159,160 However, as for (DPA)4-Per the yield of sensitized 1Δg is larger than the fluorescence quantum yield, a significant part must be sensitized from its triplet state. To confirm this, we performed transient absorption measurements on (DPA)4-Per in order to investigate a possible triplet state. Indeed, excited state absorption in the range 400–710 nm was observed revealing a long-lived excited state with a lifetime of 500 μs, which is completely quenched in an O2-equilibrated solution (Fig. S3†).
Electrochemistry
Cyclic voltammetry studies on (Bmes2)4-Per revealed four reversible reductions occurring at −2.04 V, −2.45 V, −2.79 V and −2.98 V with respect to Fc/Fc+ (in THF) to its anion, dianion, trianion and tetraanion, respectively, as shown in Fig. 6 and Table 3. The first reduction is not significantly shifted while the second, third and fourth reductions are cathodically shifted by 0.38 V, 0.72 V and 0.91 V, respectively, compared to the first reduction of perylene.37 The fourth reduction is cathodically shifted by 0.32 V compared to the second reduction of perylene. However, (Bmes2)4-Per shows stability towards very high reduction potentials with the advantage that it has a high electron capacity, as up to four electrons can be stored. This intriguing property is interesting for applications including molecular switches,161,162 receptors,162,163 photoactive dyads164 or photocatalysis.143,165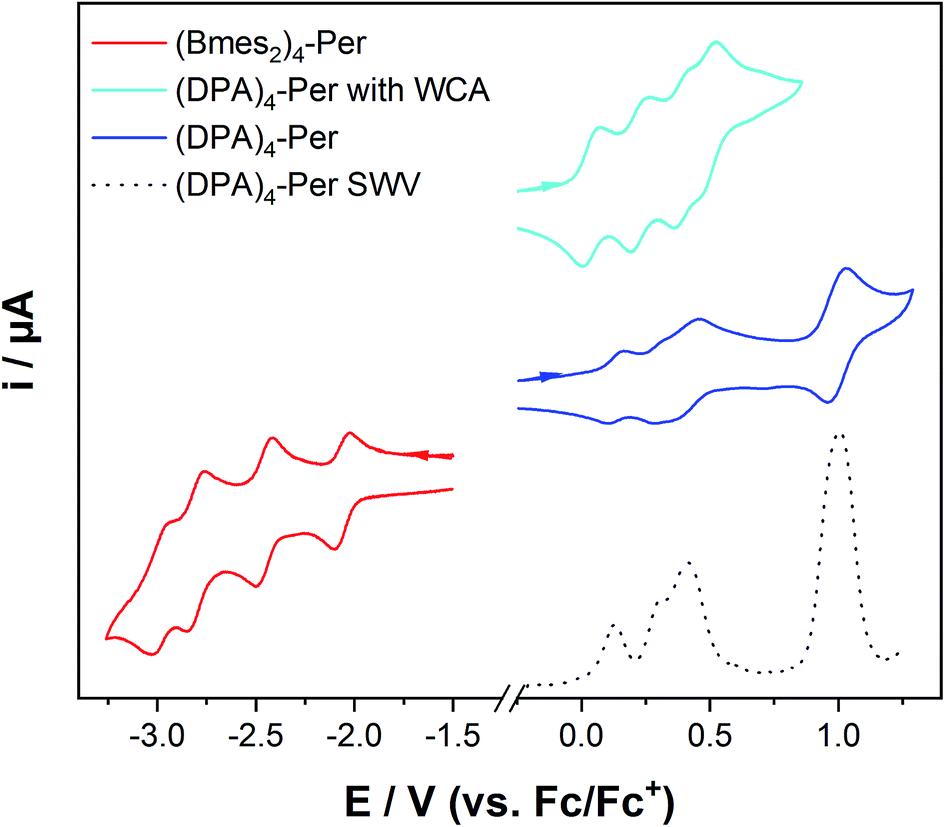 | ||
| Fig. 6 Cyclic voltammograms of (Bmes2)4-Per in THF/0.1 M [n-Bu4N][PF6] at 500 mV s−1 (red) and (DPA)4-Per in CH2Cl2/0.1 M [n-Bu4N][PF6] (blue), and CH2Cl2/0.1 M [Al(O(CF3)3)4][n-Bu4N] (cyan), respectively, at 250 mV s−1, and a square wave voltammogram (dashed line) of (DPA)4-Per in CH2Cl2/0.1 M [n-Bu4N][PF6]. | ||
| E 1/2 [V] [red]1 | E 1/2 [V] [red]2 | E 1/2 [V] [red]3 | E 1/2 [V] [red]4 | E 1/2 [V] [ox]1 | E 1/2 [V] [ox]2 | E 1/2 [V] [ox]3 | E 1/2 [V] [ox]4 | |
|---|---|---|---|---|---|---|---|---|
| a Measured in CH2Cl2/0.1 M [n-Bu4N][Al(OC(CF3)3)4] relative to the Fc/Fc+ couple. | ||||||||
| (Bmes2)4-Per | −2.04 | −2.45 | −2.79 | −2.98 | — | — | — | — |
| (DPA)4-Per | — | — | — | — | 0.13 | 0.31 | 0.41 | 0.98 |
| (DPA)4-Per with WACa | — | — | — | — | 0.040 | 0.24 | 0.41 | 0.51 |
The donor-substituted counterpart, (DPA)4-Per, can be oxidized up to five times according to cyclic voltammetry studies in CH2Cl2/0.1 M [n-Bu4N][PF6]. The first two oxidations are well separated, but the second, third and fourth oxidations are very close to one another and hard to quantify. Therefore, we performed square-wave voltammetry measurements which are known to be more sensitive than cyclic voltammetry.166,167 The first two oxidations are again well separated occurring at 0.13 V and 0.31 V, the third oxidation occurs at 0.41 V and the larger signal intensity of the latter indicates a fourth oxidation within this area. A further oxidation at 0.98 V occurs which is possibly due to the methoxy moieties. We performed a further cyclic voltammetry study using the weakly coordinating anion (WCA)-containing electrolyte [n-Bu4N][Al(OC(CF3)3)4].168 WCAs are known to separate charged species in electrochemical studies better and thus give a larger potential splitting.169 Thus, four clear, reversible oxidations at 0.040 V, 0.24 V, 0.41 V and 0.51 V to the radical cation, dication, radical trication and tetracation, respectively, of (DPA)4-Per were observed, whereas the fifth oxidation is not reversible in this electrolyte.
These numerous reductions and oxidations of our two derivatives are remarkable for perylenes, because PDIs only possess up to two reductions or oxidations and a fourfold reduction has only been reported for bi(PDI)s.170–173 PAHs that can be reduced or oxidized multiple times are rare, with the typical example being fullerenes.174 A recent report by Oki et al. indicated that a pyrrole-fused azacoronene analogue could be reversibly oxidized four times, which was also observed for hexapyrrolohexaazacoronenes.175,176 Nevertheless, the bay and ortho octa-substituted perylenes reported by Zeng and co-workers44 show only a maximum of two oxidations at 0.42 V and 0.72 V in CH2Cl2/0.1 M [n-Bu4N][PF6] for the derivative with four methoxy moieties at the ortho positions (2,5,8,11-tetramethoxy-1,6,7,12-tetra-n-butoxyperylene) vs. Fc/Fc+. These oxidations are significantly shifted to higher potentials compared to (DPA)4-Per. Hence, our donor substituted perylene derivative is among the most electron-rich perylenes reported to date, and the HOMO of (DPA)4-Per must be strongly destabilized, which is unique for perylenes as they usually have very poor electron-donating abilities.177 The compound 2,5,8,11-tetracyano-1,6,7,12-tetra-n-butoxyperylene has only one irreversible reduction at −1.63 V in CH2Cl2/0.1 M [n-Bu4N][PF6]. Thus, Bmes2 as an acceptor at the ortho positions of perylene enables the possibility of multiple reductions, in contrast to the cyano acceptor.44
To investigate the properties of the radical cation, dication, radical trication and tetracation of (DPA)4-Per we performed UV/Vis/NIR spectrochemical measurements in CH2Cl2/0.1 M [n-Bu4N][Al(OC(CF3)3)4]. However, due to the small redox-potential separations, it was not possible to generate exclusively each charged species. Nevertheless, the radical mono-cation can be observed as the “nearly pure” cation without further deconvolution, and is depicted in Fig. 7, while all further spectra are given in the ESI (Fig. S3–S6†). Upon oxidation to the radical mono-cation a very broad and symmetric transition appears between 2400 and 8000 cm−1 with a maximum at ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/char_e0e1.gif) IVCTmax = 3813 cm−1 (2 622 nm, ε = 9 900 M−1 cm−1) in the NIR. The transition at ca. 1100 nm appears at higher oxidation potentials and thus belongs to the dication (Fig. S6†). The analysis of this intervalence charge-transfer band (IV-CT) according to the Mulliken–Hush theory is straightforward as DFT calculations (see below) reveal a vanishing dipole moment difference between ground and excited state upon excitation.178,179 Thus, the radical cation is a delocalized Robin-Day-class-III mixed valence (MV) compound and the electronic coupling between the two diabatic redox states can be evaluated as one half of the energy of the absorption maximum.180–184
IVCTmax = 3813 cm−1 (2 622 nm, ε = 9 900 M−1 cm−1) in the NIR. The transition at ca. 1100 nm appears at higher oxidation potentials and thus belongs to the dication (Fig. S6†). The analysis of this intervalence charge-transfer band (IV-CT) according to the Mulliken–Hush theory is straightforward as DFT calculations (see below) reveal a vanishing dipole moment difference between ground and excited state upon excitation.178,179 Thus, the radical cation is a delocalized Robin-Day-class-III mixed valence (MV) compound and the electronic coupling between the two diabatic redox states can be evaluated as one half of the energy of the absorption maximum.180–184
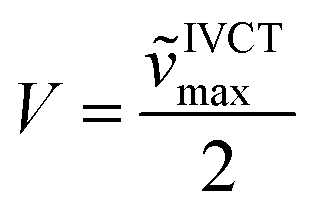 | (1) |
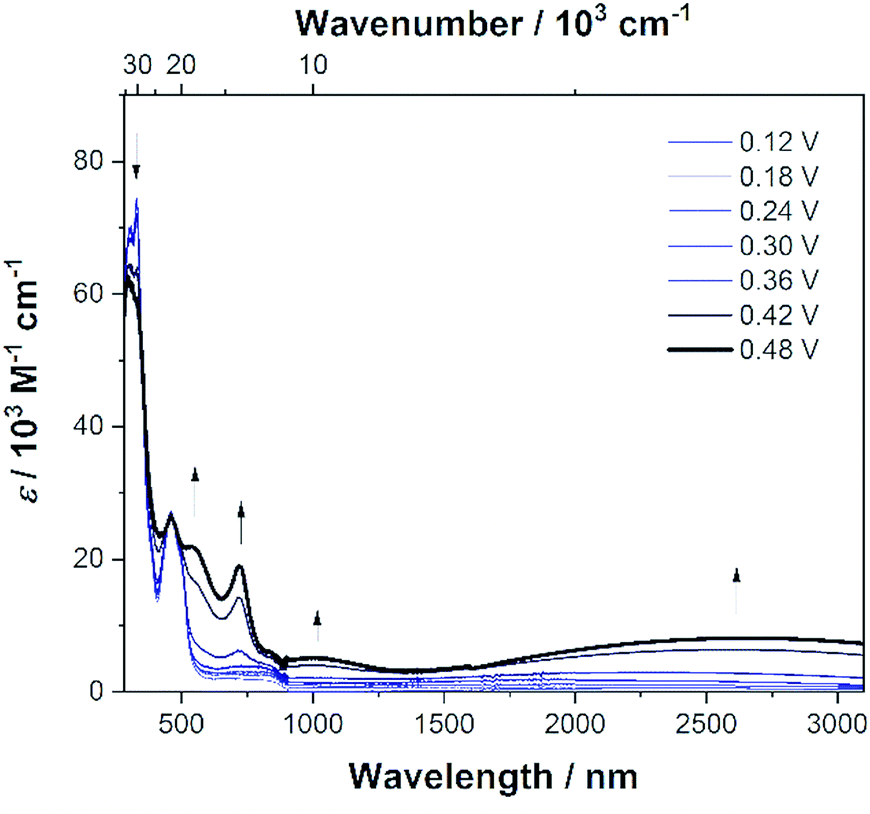 | ||
| Fig. 7 Spectroelectrochemical measurements of the stepwise oxidation process of (DPA)4-Per in CH2Cl2/0.1 M [n-Bu4N][Al(OC(CF3)3)4]. The absorption spectrum of (DPA)4-Per+ is shown as a solid black line. | ||
In this way we evaluated V to be 1807 cm−1 as half of the IV-CT excitation energy (β-HOMO → β-LUMO). This result is fully in line with the TD-DFT computations of the radical mono-cation that we performed as the orbitals involved in the excitation show the expected phase behavior for a Robin-Day-class-III compound (Fig. 8, S36 and Table S4†). Unfortunately, it was not possible to evaluate the electronic coupling within the radical trication, because the redox-potential separations between the dication, radical trication and tetracation are too close to each other. Furthermore, (Bmes2)4-Per could not be studied by spectroelectrochemical measurements, as the absorption spectrum of the neutral species cannot be reproduced in intensity after the reductions (Fig. S8–S12†).
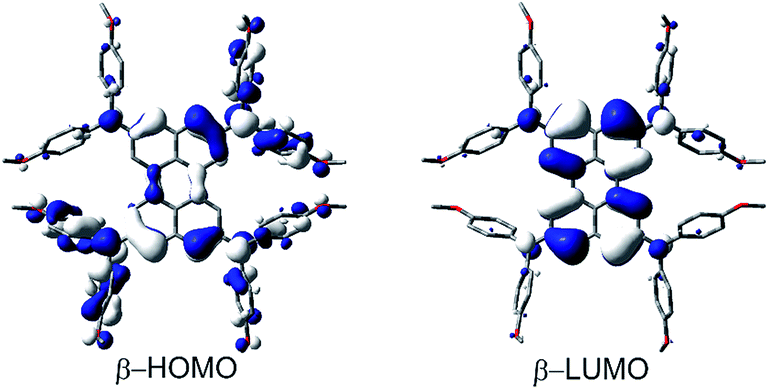 | ||
| Fig. 8 Depiction of the β-HOMO and β-LUMO that are responsible for the lowest energy absorption (IV-CT band) of (DPA)4-Per+. | ||
DFT and TD-DFT calculations
To rationalize the observed trends and properties, we performed DFT and TD-DFT studies on perylene, (DPA)4-Per, and (Bmes2)4-Per. Furthermore, the model derivatives (NH2)4-Per and (BH2)4-Per were calculated and compared with perylene to understand how the frontier molecular orbitals of both compounds are related to the parent perylene system (Fig. S37–S39†). We optimized the ground-states in the gas-phase at the B3LYP/6-31G (d,p) level of theory. Previous studies95,185 have shown that range-separated hybrid functionals are necessary to obtain a reliable picture of the nature and relative energetic ordering of the excited states. Therefore, the CAM-B3LYP functional was employed for the subsequent TD-DFT calculations for the perylene derivatives.Perylene has a large HOMO–LUMO gap (3.05 eV) and its HOMO–1/HOMO and LUMO/LUMO+1 orbitals are well separated. Adding four Bmes2 moieties to the ortho positions of perylene ((Bmes2)4-Per) leads to a 0.20 eV stabilization of the LUMO, which is a combination of the empty pz orbitals of the boron atoms of the Bmes2 fragments and the perylene LUMO resulting in larger orbital coefficients at the ortho positions compared to the parent perylene (Fig. 9).
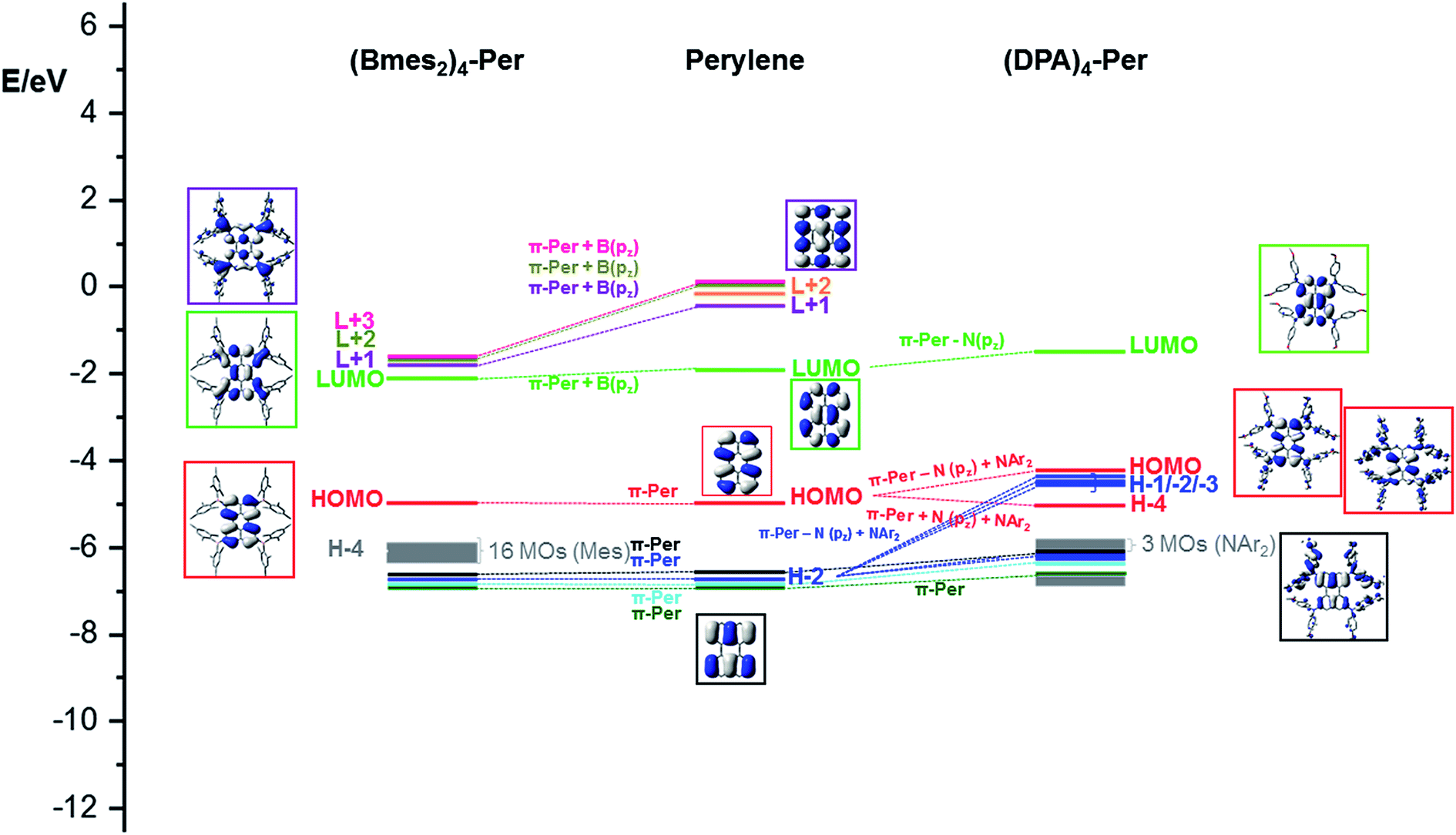 | ||
| Fig. 9 Frontier MO-diagram (B3LYP/6-31+G(d,p)) and depiction of B(mes2)4-Per (left), perylene (middle) and (DPA)4-Per (right) showing the relation between the frontier orbitals of perylene and its acceptor and donor substituted derivatives. The key frontier molecular orbitals are shown in color-coded boxes. | ||
Perylene's LUMO+1, LUMO+3 and LUMO+4 mix even stronger with the Bmes2 fragments, which leads to an increased stabilization of their energies by 1.35 eV, 1.54 eV and 1.58 eV, respectively. Hence, these three orbitals above the LUMO, are all greatly stabilized in (Bmes2)4-Per and are close in energy. This is a plausible explanation for the four electron reduction of this compound that we observed within the cyclic voltammetry studies. The stabilization of the LUMO furthermore explains the observed bathochromic shift of the S1 ← S0 transition, which consists of a pure HOMO → LUMO transition. This contrasts strongly with 2,5,8,11-tetracyano-1,6,7,12-tetra-n-butoxyperylene, which shows that the cyano group does not mix as efficiently with the perylene LUMO and we already made this observation in a closely related pyrene derivative.44,97 This is reflected in the photophysical behavior, as the S1 ← S0 transition of 2,5,8,11-tetracyano-1,6,7,12-tetra-n-butoxyperylene is not as bathochromically shifted as that of (Bmes2)4-Per. Nevertheless, this comparison should be treated with caution as the bay substituents certainly have some influence (vide supra). However, the calculations show that the LUMO is mainly distributed over the four boron centers and the perylene core while the mesityl moieties do not contribute to this orbital (Fig. 9). Even though there is some contribution of the Bmes2 fragments to the LUMO and LUMO+1 to LUMO+3 of (Bmes2)4-Per, these orbitals generally resemble those in unsubstituted perylene. Therefore, the HOMO → LUMO transition of (Bmes2)4-Per does not have much CT character, and is predominantly a local excited state (LE), which agrees with our photophysical studies, as the emission spectrum of (Bmes2)4-Per does not show solvatochromism. The HOMO is not influenced by the Bmes2 fragments and thus does not change much in energy. However, 16 orbitals, which are based on the mesityl fragments only, i.e., are not affected by the perylene core or boron atoms, and form the HOMO−1 to HOMO−16 (Fig. 8, grey) of (Bmes2)4-Per. The former HOMO−1 through HOMO−4 are not altered by the Bmes2 groups and now form the HOMO−17 to HOMO−20 (Fig. 8, black, blue, cyan and green).
In contrast, adding four amine donor moieties to the ortho positions of the perylene core destabilizes the occupied orbitals much more than the virtual ones. The nitrogen pz orbitals of the DPA moieties mix very well with the occupied orbitals of the perylene core, but the π-orbitals of the methoxy phenyl rings also contribute significantly. The mixing of the perylene HOMO with N(pz) and NAr2 orbitals leads to two new perylene-like HOMOs in (DPA)4-Per (HOMO and HOMO−4) (Fig. 8, red). The new HOMO of (DPA)4-Per is strongly destabilized by 0.73 eV while HOMO−4 is slightly stabilized in comparison to the HOMO of perylene. In a similar manner, for HOMO−2 of perylene, two new sets are formed in (DPA)4-Per (Fig. 8, blue). One set (HOMO−1 through HOMO−3) is strongly destabilized by ca. 0.79 eV and the second by ca. 0.59 eV (HOMO–9 and HOMO–10). Perylene's HOMO–1 to HOMO–3 and HOMO−4 (Fig. 9, black, cyan and green), on the other hand, are less destabilized in (DPA)4-Per. These results agree with our observed photophysical properties. The strong destabilizing effect of the HOMO in (DPA)4-Per explains the strong bathochromic shift of the S1 ← S0 transition and as the HOMO of (DPA)4-Per is also distributed over the four amine moieties and displays a pronounced CT character, this is reflected in the broad and structureless absorption and emission spectra as well as the solvatochromic behavior. Thus, the effect of the DPA groups on the occupied orbitals of the perylene is more pronounced than is the case of the Bmes2 moieties, where only the unoccupied orbitals were affected. Furthermore, the observed electrochemical behavior is consistent with our calculations, as the influence on the occupied orbitals results in four orbitals that are very close in energy and around the HOMO of (DPA)4-Per, thus, removing four electrons successively from the system is possible (Fig. 9 and Table 4).
| FC-Sn | E [eV] (E [nm]) | f | Configuration (major contributions > 10%) | |
|---|---|---|---|---|
| Perylene | S1 | 3.21 (387) | 0.446 | H → L (99%) |
| S2 | 4.06 (305) | 0.000 | H → L+1 (58%), H−1 → L (33%) | |
| S3 | 4.34 (285) | 0.004 | H−2 → L (52%), H → L+3 (38%) | |
| S4 | 4.45 (278) | 0.000 | H → L+2 (42%), H−4 → L (24%), H−1 → L (23%) | |
| S5 | 4.88 (254) | 0.000 | H → L+2 (52%), H−4 → L (30%), H → L+1 (13%) | |
| (Bmes2)4-Per | S1 | 3.03 (410) | 0.367 | H → L (96%) |
| S2 | 3.21 (386) | 0.000 | H → L+1 (87%) | |
| S3 | 3.54 (350) | 0.212 | H → L+3 (79%) | |
| S4 | 3.64 (340) | 0.000 | H → L+2 (92%) | |
| S5 | 4.01 (310) | 0.055 | H−4 → L+1 (21%), H−2 → L (20%), H−1 → L+2 (21%), H−3 → L+3 (16%) | |
| (DPA)4-Per | S1 | 3.00 (413) | 0.318 | H → L (91%) |
| S2 | 3.11 (398) | 0.017 | H−1 → L (76%) | |
| S3 | 3.30 (376) | 0.281 | H−2 → L (74%) | |
| S4 | 3.48 (356) | 0.005 | H−3 → L (77%) | |
| S5 | 3.92 (316) | 0.294 | H−4 → L (78%) |
TD-DFT calculations show that the nature of both S1 ← S0 transition in (DPA)4-Per and (Bmes2)4-Per remain HOMO → LUMO in character as in perylene. Thus, these transitions can best be described for the (Bmes2)4-Per derivative as an LE transition with a small CT contribution whereas there is an increased CT contribution for the (DPA)4-Per derivative. This is in accordance with the slightly lower oscillator strengths for the (DPA)4-Per derivative (f = 0.367 for (Bmes2)4-Per; and f = 0.318 for (DPA)4-Per) and lower extinction coefficients (ε = 20000 M−1 cm−1 for (Bmes2)4-Per and ε = 15000 M−1 cm−1 for (DPA)4-Per). However, the red shift of both λmax(abs) and λmax(em) as well as the larger apparent Stokes shift, agrees well with increasing CT admixture for the (DPA)4-Per compound as does the observed solvatochromism, which also results from the CT nature of the lowest excited singlet state, which is not observed for (Bmes2)4-Per.
Conclusion
We have synthesized novel ortho-tetrasubstituted perylenes with a strong DPA donor or a strong Bmes2 acceptor, which represent the first examples of perylenes substituted only at the ortho positions with donors or acceptors. The synthesis of these two systems required two new intermediates (Br)4-Per and (BF3K)-Per, which could potentially be used for the synthesis of many new ortho-perylene derivatives. The donor and the acceptor substituents significantly influence the frontier orbitals which leads to interesting and potentially useful properties. Cyclic voltammetry and square wave voltammetry experiments reveal up to four reversible reductions for the acceptor derivative (Bmes2)4-Per and four reversible oxidations for the donor derivative (DPA)4-Per, which is unprecedented for perylenes and has only been observed previously for bi(PDI)s. Spectroelectrochemical measurements suggest a strong electronic coupling between the amine moieties through the perylene bridge, which can be categorized as a typical Robin-Day-class-III system. (DPA)4-Per reached an unparalleled bathochromic shift of its S1 ← S0 transition (λmax(abs) = 499 nm) and emission (λmax(em) = 593 nm) for ortho-substituted perylenes which do not have carboxyimide moieties at the peri-positions. Both derivatives have unusually long intrinsic singlet lifetimes with that of (DPA)4-Per being 94 ns. Transient absorption measurements reveal an additional excited state with a 500 μs lifetime which effectively sensitizes singlet oxygen. This presumed triplet state could prove useful in organo-photocatalysis.136,140–143 Theoretical studies show that the acceptor Bmes2 couples well with the unoccupied orbitals, which leads to a strong stabilization of the LUMO to LUMO+4 orbitals. However, while the nature of the S1 state is maintained, the HOMO → LUMO transition is less allowed in comparison to the one in perylene. Hence, the radiative decay rates decrease resulting in a longer-lived singlet excited state. The donor DPA also mixes very well with the occupied orbitals, strongly destabilizing the HOMO to HOMO−4 levels. In contrast to the former derivative, its CT character gives rise to a less allowed S1 ← S0 transition which results in the slowest radiative decay reported so far for perylenes. Furthermore, it possesses the strongest bathochromic shift observed for ortho-perylenes and a large apparent Stokes shift.Our synthetic procedure has the potential to produce a wide variety of ortho-perylene derivatives with a broad range of properties as required for diverse applications. As one can anticipate that alternative donors and acceptors could result in typical π-stacking interactions, which are inhibited in our current compounds, the synthesis of such species is under investigation in our laboratory.
Experimental section
General considerations
The catalyst [Ir(OMe)(COD)]2186 and Pd2(dba)3·CHCl3187 were prepared according to literature procedures. All other starting materials were purchased from commercial sources and used as received. Solvents used for the synthesis were HPLC grade, further treated to remove trace water using a commercial solvent purification system and deoxygenated using the freeze–pump–thaw method. Microwave-heating (95 °C) was performed in a Biotage® initiator + reactor. The reactions were set up in standard 20 mL microwave tubes, which were sealed with crimp caps.
The 1H, 13C{1H}, 11B{1H}, 19F, 15N HMBC NMR spectra were recorded in CDCl3, CD2Cl2, or DMSO-d6 solutions either on a Bruker Avance I HD 500 (1H, 500 MHz; 13C, 125 MHz; 11B, 160 MHz, 19F, 470 MHz) or Bruker Avance III HD 300 (1H, 300 MHz; 13C, 75 MHz; 11B, 96 MHz) spectrometer. 1H NMR and 13C spectra are referenced to the residual protiated solvent CHCl3 (1H, δ = 7.26, CDCl3), CD2Cl2 (1H, δ = 5.32, CD2Cl2) and DMSO-d5 (1H, δ = 2.50, DMSO-d6). 13C NMR spectra were broad-band proton decoupled 13C{1H}. 11B{1H} NMR signals were referenced to external BF3·OEt2 and 15N HMBC signals were referenced to MeNO2 + 10% CDCl3. Chemical shifts are listed in parts per million (ppm) and coupling constants in Hertz (Hz). The solid-state magic-angle spinning (MAS) NMR spectra were recorded using a Bruker DSX-400 spectrometer operating at 128 MHz for 11B and 100 MHz for 13C with a 4 mm rotor (o. d.). Chemical shifts were calibrated externally using adamantane (38.48 ppm). Isotropic chemical shifts were estimated by simulating the observed spectrum using the Solid Line Shape Analysis 2.2.4 (SOLA) module in Bruker TopSpin 3.5. The second order quadrupolar powder patterns were simulated to extract the isotropic chemical shifts (δiso), the quadrupolar constants (Cq) and the asymmetry parameters (ηQuad).
HRMS were recorded using a Thermo Scientific Exactive Plus Orbitrap MS system with either an Atmospheric Sample Analysis Probe (ASAP) or by Electrospray Ionization (ESI). Elemental analyses were performed on an Elementar vario MICRO cube elemental analyzer.
Cyclic voltammetry experiments were performed using a Gamry Instruments Reference 600 potentiostat. A standard three-electrode cell configuration was employed using a platinum disk working electrode, a platinum wire counter electrode, and a silver wire, separated by a Vycor tip, serving as the reference electrode. Formal redox potentials are referenced to the ferrocene/ferrocenium ([Cp2Fe]+/0) redox couple by using decamethylferrocene ([Cp2*Fe]; E1/2 = −0.427 V in THF and E1/2 = −0.532 V in CH2Cl2) as an internal standard. Tetra-n-butylammonium hexafluorophosphate ([n-Bu4N][PF6]) or [n-Bu4N][Al(OC(CF3)3)4] were employed as supporting electrolytes. Compensation for resistive losses (iR drop) was employed for all measurements.
A crystal suitable for single-crystal X-ray diffraction was selected, coated in perfluoropolyether oil, and mounted on a MiTeGen sample holder. Diffraction data were collected on a Bruker X8 Apex II 4-circle diffractometer with a CCD area detector using Mo-Kα radiation generated by a Nonius FR591 rotating anode and monochromated by graphite. The crystal was cooled using an Oxford Cryostream low-temperature device. Data were collected at 100 K. The images were processed and corrected for Lorentz-polarization effects and absorption as implemented in the Bruker software packages. The structure was solved using the intrinsic phasing method (SHELXT)188 and Fourier expansion technique. All non-hydrogen atoms were refined in anisotropic approximation, with hydrogen atoms ‘riding’ in idealized positions, by full-matrix least squares against F2 of all data, using SHELXL189 software. Diamond190 software was used for graphical representation. Hirshfeld surfaces were calculated and analyzed using the CrystalExplorer191 program. Other structural information was extracted using Mercury192 and OLEX2193 software. Crystal data and experimental details are listed in Table S1;† full structural information has been deposited with Cambridge Structural Database. CCDC-1881912.
General photophysical measurements
All photophysical measurements were carried out under an argon atmosphere. All solution state measurements were performed in standard quartz cuvettes (1 cm × 1 cm cross section). UV/Vis absorption spectra were recorded using an Agilent 1100 diode array UV/Vis spectrophotometer. Excitation, emission, lifetime and quantum yield measurements were recorded using an Edinburgh Instruments FLSP920 spectrometer equipped with a 450 W Xenon arc lamp, double monochromators for the excitation and emission pathways, and a red-sensitive photomultiplier (PMT-R928P) and a near-IR PMT as detectors. The measurements were made in right-angle geometry mode and all spectra were fully corrected for the spectral response of the instrument. All solutions used in photophysical measurements had a concentration lower than 10−5 M.Fluorescence quantum yield measurements
Fluorescence quantum yields of the samples were measured using a calibrated integrating sphere (150 mm inner diameter) from Edinburgh Instruments combined with the FLSP920 spectrometer described above. For solution-state measurements, the longest wavelength absorption maximum of the compound in the respective solvent was chosen for the excitation. In order to avoid self-absorption, the emission spectra were measured with dilute samples (ca. 0.1 OD at the excitation wavelength).Fluorescence lifetime measurements
Lifetime measurements were conducted using the time-correlated single-photon counting method (TCSPC) on the FLSP920 spectrometer equipped with a high-speed photomultiplier tube positioned after a single emission monochromator. Measurements were made in right-angle geometry mode, and the emission was collected through a polarizer set to the magic angle. Solutions were excited with either a 315 (pulse width 932.5 ps), 376 (pulse width 72.6 ps) or a 472 nm (pulse width 90.6 ps) pulsed diode laser at repetition rates of 1–5 MHz and were recorded at emission maxima. Decays were recorded to 10000 counts in the peak channel with a record length of at least 1000 channels. The band-pass of the monochromator was adjusted to give a signal count rate of <20 KHz. Iterative reconvolution of the IRF with one decay function and nonlinear least-squares analysis were used to analyze the data. The quality of all decay fits was judged to be satisfactory, based on the calculated values of the reduced χ2 and Durbin–Watson parameters and visual inspection of the weighted and autocorrelated residuals.
Transient absorption measurements
Transient absorption spectra were measured with an Edinburgh LP920 laser flash spectrometer equipped with a EKSPLA NT340 Nd:YAG laser with integrated optical parametric oscillator, a 450 W Xe flash lamp, a Hamamatsu R955 photomultiplier and a Tektronix TD3012B oscilloscope for detection of the spectra. The transient maps were obtained by measuring decay profiles in 4 nm steps between ca. 25000 cm−1 (400 nm) and 14085 cm−1 (710 nm). The instrument response (ca. 8 ns) of the set-up was determined by measuring the scattered light using a LUDOX AS-30 colloidal silica suspension in water. Decay curves were fitted with the tailfit function of the spectrometer software. The quality of all decay fits was judged to be satisfactory, based on the calculated values of the reduced χ2 and Durbin–Watson parameters and visual inspection of the weighted and autocorrelated residuals. All solvents were spectroscopic grade and were used without further purification. The sample solutions in the quartz cuvettes were carefully degassed by bubbling argon through the solutions. The samples were excited with ca. 3–6 ns laser pulses at 10 Hz repetition rate. Measurements were performed at pulse energies of 1.2 mJ (excitation at 460 and 550 nm). The stability of the samples was verified by recording the steady-state absorption spectra before and after the time-resolved measurements.
Spectroelectrochemical measurements
Spectroelectrochemical experiments in reflection mode were performed using an Agilent Cary 5000 spectrometer in combination with a designed sample compartment consisting of a cylindrical PTFE cell with an Infrasil® wedge window with an angle of 0.5° and an adjustable three-in-one electrode (6 mm platinum disc working electrode, 1 mm platinum counter electrode and pseudo reference electrode). The potentials were adjusted with a Gamry 600 potentiostat and all experiments were measured at room temperature under an argon atmosphere.Theoretical studies
All calculations (DFT and TD-DFT) were carried out with the program package Gaussian 09 (Rev. E.01)194 and were performed on a parallel cluster system. GaussView 5.0.9 was used to visualize the results, to measure calculated structural parameters, and to plot orbital surfaces (isovalue: ±0.02[ea0−3]1/2). The ground-state geometries were optimized using the B3LYP functional195–197 in combination with the 6-31G(d) basis set.198,199 The optimized geometries were confirmed to be local minima by performing frequency calculations and obtaining only positive (real) frequencies. Based on these optimized structures, the lowest-energy gas-phase vertical transitions were calculated (singlets, 10 states) by TD-DFT, using the Coulomb-attenuated functional CAM-B3LYP200 in combination with the 6-31G+(d,p) basis set. The calculations of the radical mono-cation of (DPA)4-Per were done at the density functional level, using UBLYP with 35% exact-exchange admixture, the SVP basis set and a polarizable continuum model accounting for solvent effects. The time dependent (TD-DFT) calculations were done at the same level of theory.201,202(Bpin)4-Per (1)
In an argon-filled glovebox, perylene (1.00 g, 3.96 mmol, 1.00 eq.), B2pin2 (5.03 g, 19.8 mmol, 5.00 equiv.), [Ir(COD)(μ-OMe)]2 (78.8 mg, 119 μmol, 3.00 mol%), 4,4′-di-tert-butyl-2,2′-bipyridine (dtbpy) (63.8 mg, 238 μmol, 6.00 mol%) and THF (100 mL) were added to a Schlenk flask. The mixture was refluxed at 85 °C in an oil bath for 40 h. After cooling to room temperature, the solvent was removed under reduced pressure. The residue was washed with cyclohexane (2 × 20 mL) and MeOH (2 × 20 mL) to give 1 as a yellow solid (2.69 g, 3.56 mmol, 90%). The spectral data matched those reported previously.451H NMR (300 MHz, CDCl3, r.t.): δ = 8.63 (s, 4H), 8.25 (s, 4H), 1.43 (s, 48H) ppm; 13C{1H} NMR (75 MHz, CDCl3, r.t.): δ = 137.1, 133.4, 132.1, 130.5, 126.8 (br), 126.2, 84.2, 25.1 ppm; 11B{1H} NMR (96 MHz, CDCl3, r.t.): δ = 34.2 ppm; HRMS (ASAP+): m/z calc. for [C44H56B4O8 + H]+ 757.4420; found: [M + H]+ 757.4422 (|Δ| = 0.26 ppm); elemental analysis calc. (%) for C44H56B4O8: C 69.89, H 7.47; found: C 69.69, H 7.35.(BF3K)4-Per (2)
In a round-bottom flask (Bpin)4-Per (1) (378 mg, 500 μmol, 1.00 equiv.) was dissolved in THF (30 mL). To this solution KHF2 (469 mg, 6.00 mmol, 12.0 equiv.) in H2O (3 mL) was added and the mixture was stirred vigorously at room temperature for 20 h. The solvent was removed under reduced pressure and the residue was washed with hot acetone (2 × 40 mL), hot EtOH (2 × 40 mL) and hot MeOH (40 mL) to give the product of 2 as a green solid (335 mg, 496 μmol, 99%). 1H NMR (500 MHz, DMSO-d6, r.t.): δ = 8.11 (s, 4H), 7.40 (s, 4H) ppm; 13C{1H} NMR (125 MHz, DMSO-d6, r.t.): δ = 133.8, 129.0, 128.9, 126.8, 122.2 ppm (one C not observed, likely that attached to B); 11B{1H} NMR (160 MHz, DMSO-d6, RT): δ = 3.9 ppm; 19F{1H} NMR (470 MHz, DMSO-d6, r.t.): δ = −138.7 ppm.(Bmes2)4-Per (3)
Under an argon atmosphere, (BF3K)-Per (2) (1.00 g, 1.48 mmol) and degassed THF (20 mL) were added to a Schlenk flask. The suspension was treated with MesMgBr (0.9 M in THF, 13.6 mmol, 15.1 mL) and the reaction was stirred at room temperature for 72 h. After addition of water (10 mL), the precipitate was collected by filtration. In an ultrasonic bath, the residue was washed successively with H2O, THF, hexane, ethyl acetate, MeOH and CH2Cl2. After each washing process, the solid was separated from the solvent by centrifugation. The product was obtained as a yellow solid (1.58 g, 1.27 mmol, 86%). 1H NMR (500 MHz, CDCl3, r.t.): δ = 8.11 (s, 4H), 7.87 (s, 4H), 6.79 (s, 16H), 2.35 (s, 24H), 2.00 (s, 48H) ppm; 13C{1H} NMR (125 MHz, CDCl3, r.t.): δ = 141.5, 140.9, 140.0, 138.7, 133.8, 132.7, 130.6, 129.4, 128.4, 23.7, 21.4 ppm (one C not observed, likely that attached to B); 11B{1H} SSNMR (128 MHz, r.t.): δ = 72.7 ppm; HRMS (APCI+): m/z calc. for [C92H96B4 + H]+ 1245.7957; found: [M + H]+ 1245.7983 (|Δ| = 2.09 ppm); elemental analysis calcd (%) for C92H96B4: C 88.75, H 7.77; found: C 87.80, H 7.92. As is common for related organo-Bmes2 compounds, the carbon analysis of (Bmes2)4-Per is 0.95% below the calculated value, while the hydrogen analysis is satisfactory. This has been described previously to the formation of boron carbide.89,203,204(Br)4-Per (4)
Under aerobic conditions, CuBr2 (710 mg, 3.17 mmol, 12.0 equiv.), (Bpin)4-Per (1) (200 mg, 0.26 mmol, 1.00 equiv.), MeOH (5 mL), THF (5 mL) and H2O (5 mL) were added to a 20 mL microwave vial. The vial was sealed and heated to 95 °C for 20 h in a microwave reactor. Reaction progress was monitored by HRMS and revealed that the reaction was not finished, hence another portion of CuBr2 (710 mg, 3.17 mmol, 12.0 equiv.) was added and the reaction mixture was heated at 95 °C for another 20 h. The precipitate was collected by centrifugation and washed with H2O, MeOH, EDTA (0.1 M) and THF. The solubility of (Br)4-Per is too low to obtain NMR spectra in solution. However, the solubility was enough to obtain absorption and emission spectra. Hence, this crude product was used (568 mg) without further purification for the following Buchwald–Hartwig amination reaction. A small part was further purified by Kugelrohr sublimation at 250 °C (0.001 mbar) to afford the title compound as an analytically pure red solid. The yield loss during the Kugelrohr sublimation of (Br)4-Per was so large that using the crude product for the following reaction gave higher overall yields. 13C{1H} SSNMR (100 MHz, r.t.) δ = 134.8, 129.6, 127.6, 124.3, 118.0; HRMS (ASAP+): m/z calcd for [C20H8Br4 + H]+: 568.7385; found [M + H]+ 568.7363 (|Δ| = 3.87 ppm); elemental analysis calcd (%) for C20H8Br4: C 42.30, H 1.42; found: C 42.47, H 1.56.(DPA)4-Per (5)
Under an argon atmosphere, crude (Br)4-Per (4) (568 mg, 1.00 mmol), bis(4-methoxyphenyl)amine (1.15 g, 5.00 mmol, 5.00 equiv.), Pd2(dba)3·CHCl3 (20.7 mg, 0.02 mmol, 2.0 mol%), Sphos (16.4 mg, 0.04 mmol, 4.00 mol%), KOtBu (0.56 g, 5.00 mmol, 5.00 equiv.) and toluene were added to a Young's tube, which was then sealed. The reaction mixture was stirred at 110 °C for 5 d, until reaction monitoring indicated that the reaction was complete. After cooling to room temperature, water was added, and the organic layer was extracted with CH2Cl2 (3 × 200 mL). The combined organic extracts were washed with brine and dried over Na2SO4. After removing the solvent under reduced pressure, the crude product was purified by flash chromatography (silica, cyclohexane/CH2Cl2 first 1:4 then gradually increased to 0:1). (DPA)4-Per was obtained as an orange-red solid (220 mg, 189 μmol, 19% yield over 2 steps starting from 1). 1H NMR (500 MHz, CD2Cl2, r.t.): δ = 7.33 (d, J = 2.1 Hz, 4H), 7.01–6.99 (m, 16H), 6.80–6.77 (m, 16H), 6.63 (d, J = 2.1 Hz, 4H), 3.78 ppm (s, 24H); 13C{1H} NMR (125 MHz, CD2Cl2, r.t.): δ = 156.5, 147.7, 140.9, 137.5, 131.6, 127.0, 120.1, 115.1, 115.0, 114.0, 55.8 ppm; 15N NMR (500 MHz, CD2Cl2, RT): δ = −286.3 ppm; HRMS (APCI+): m/z calcd for [C76H64N4O8 + H]+: 1161.4797; found [M + H]+ 1161.4781 (|Δ| = 1.38 ppm).
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We are grateful for generous financial support by the Bavarian State Ministry of Science, Research, and the Arts for the Collaborative Research Network “Solar Technologies go Hybrid”, the DFG (GRK 2112) and the University of Würzburg.References
- H. Ito, K. Ozaki and K. Itami, Angew. Chem., Int. Ed., 2017, 56, 11144–11164 CrossRef CAS.
- J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS.
- H. Zollinger, Color Chemistry: Syntheses, Properties, and Applications of Organic Dyes and Pigments, Wiley-VCH, Weinheim, 3rd edn, 2003 Search PubMed.
- W. Herbst, G. Wilker and K. Hunger, Industrial Organic Pigments: Production, Properties, Applications, Wiley-VCH, Weinheim, 3rd edn, 2004 Search PubMed.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS.
- T. Kaehler, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2019 DOI:10.1002/anie.201905823.
- K. Peneva, G. Mihov, F. Nolde, S. Rocha, J.-i. Hotta, K. Braeckmans, J. Hofkens, H. Uji-i, A. Herrmann and K. Müllen, Angew. Chem., Int. Ed., 2008, 47, 3372–3375 CrossRef CAS.
- X. Zhang, S. Rehm, M. M. Safont-Sempere and F. Würthner, Nat. Chem., 2009, 1, 623–629 CrossRef CAS PubMed.
- T. Weil, T. Vosch, J. Hofkens, K. Peneva and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 9068–9093 CrossRef CAS.
- S. Bhosale, A. L. Sisson, P. Talukdar, A. Fürstenberg, N. Banerji, E. Vauthey, G. Bollot, J. Mareda, C. Röger, F. Würthner, N. Sakai and S. Matile, Science, 2006, 313, 84–86 CrossRef CAS.
- D. Görl, X. Zhang and F. Würthner, Angew. Chem., Int. Ed., 2012, 51, 6328–6348 CrossRef.
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS.
- F. Würthner, Chem. Commun., 2004, 14, 1564–1579 RSC.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef.
- H. Langhals, Helv. Chim. Acta, 2005, 88, 1309–1343 CrossRef CAS.
- L. Chen, C. Li and K. Müllen, J. Mater. Chem. C, 2014, 2, 1938–1956 RSC.
- G. Li, Y. Zhao, J. Li, J. Cao, J. Zhu, X. W. Sun and Q. Zhang, J. Org. Chem., 2015, 80, 196–203 CrossRef CAS.
- Z. Chen, U. Baumeister, C. Tschierske and F. Würthner, Chem.–Eur. J., 2007, 13, 450–465 CrossRef CAS.
- C. W. Struijk, A. B. Sieval, J. E. J. Dakhorst, M. van Dijk, P. Kimkes, R. B. M. Koehorst, H. Donker, T. J. Schaafsma, S. J. Picken, A. M. van de Craats, J. M. Warman, H. Zuilhof and E. J. R. Sudhölter, J. Am. Chem. Soc., 2000, 122, 11057–11066 CrossRef CAS.
- C. D. Dimitrakopoulos and P. R. L. Malenfant, Adv. Mater., 2002, 14, 99–117 CrossRef CAS.
- I. B. Berlman, Handbook of Fluorescence Spectra of Aromatic Molecules, Academic Press, New York, 2nd edn, 1971 Search PubMed.
- D. M. Donaldson, J. M. Robertson and J. G. White, Proc. R. Soc. London, Ser. A, 1953, 220, 311–321 CrossRef CAS.
- H. Shiba and G. Hazato, Bull. Chem. Soc. Jpn., 1949, 22, 92–96 CrossRef CAS.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS.
- A. Ranganathan and G. U. Kulkarni, Experimental Crystal Structure Determination, CCDC 215338, 2004 Search PubMed.
- M. Botoshansky, F. H. Herbstein and M. Kapon, Helv. Chim. Acta, 2003, 86, 1113–1128 CrossRef CAS.
- A. Camerman and J. Trotter, Proc. R. Soc. London, Ser. A, 1964, 279, 129–146 CrossRef CAS.
- T. M. Krygowski, A. Ciesielski, B. Swirska and P. Leszczynski, Pol. J. Chem., 1994, 2097–2107 CAS.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- A. Ranganathan and G. U. Kulkarni, Proc. - Indian Acad. Sci., Chem. Sci., 2003, 115, 637–647 CrossRef CAS.
- J. T. Markiewicz and F. Wudl, ACS Appl. Mater. Interfaces, 2015, 7, 28063–28085 CrossRef CAS.
- J. R. Platt, J. Chem. Phys., 1949, 17, 484–495 CrossRef CAS.
- J. Tanaka, Bull. Chem. Soc. Jpn., 1963, 36, 1237–1249 CrossRef CAS.
- Y. Tanizaki, T. Yoshinaga and H. Hiratsuka, Spectrochim. Acta, Part A, 1978, 34, 205–210 CrossRef.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 2010 Search PubMed.
- H. Langhals, J. Karolin and L. B.-Å. Johansson, Faraday Trans., 1998, 94, 2919–2922 RSC.
- B. S. Jensen and V. D. Parker, J. Am. Chem. Soc., 1975, 97, 5211–5217 CrossRef CAS.
- C. E. Crespo-Hernandez, D. M. Close, L. Gorb and J. Leszczynski, J. Phys. Chem. B, 2007, 111, 5386–5395 CrossRef CAS.
- M. Montalti and S. L. Murov, Handbook of Photochemistry, Taylor & Francis, Boca Raton, 3rd edn, 2006 Search PubMed.
- T. C. Werner, J. Chang and D. M. Hercules, J. Am. Chem. Soc., 1970, 92, 5560–5565 CrossRef CAS.
- E. S. Pysh and N. C. Yang, J. Am. Chem. Soc., 1963, 85, 2124–2130 CrossRef CAS.
- J. Salbeck, H. Kunkely, H. Langhals, R. W. Saalfrank and J. Daub, Chimia, 1989, 43, 6–9 CAS.
- Y. Zagranyarski, L. Chen, D. Jänsch, T. Gessner, C. Li and K. Müllen, Org. Lett., 2014, 16, 2814–2817 CrossRef CAS.
- Y. Li, Y. Hong, J. Guo, X. Huang, H. Wei, J. Zhou, T. Qiu, J. Wu and Z. Zeng, Org. Lett., 2017, 19, 5094–5097 CrossRef CAS PubMed.
- D. N. Coventry, A. S. Batsanov, A. E. Goeta, J. A. K. Howard, T. B. Marder and R. N. Perutz, Chem. Commun., 2005, 2172–2174 RSC.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS.
- Boronic acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, ed. D. Hall, Wiley-VCH, Weinheim, 2nd edn, 2011 Search PubMed.
- A. G. Crawford, Z. Liu, I. A. I. Mkhalid, M.-H. Thibault, N. Schwarz, G. Alcaraz, A. Steffen, J. C. Collings, A. S. Batsanov, J. A. K. Howard and T. B. Marder, Chem.–Eur. J., 2012, 18, 5022–5035 CrossRef CAS.
- S. Nakazono, Y. Imazaki, H. Yoo, J. Yang, T. Sasamori, N. Tokitoh, T. Cédric, H. Kageyama, D. Kim, H. Shinokubo and A. Osuka, Chem.–Eur. J., 2009, 15, 7530–7533 CrossRef CAS.
- T. Teraoka, S. Hiroto and H. Shinokubo, Org. Lett., 2011, 13, 2532–2535 CrossRef CAS.
- S. Ikeda, N. Aratani and A. Osuka, Chem. Commun., 2012, 48, 4317–4319 RSC.
- L. D. Tran, J. Ma, A. G. Wong-Foy and A. J. Matzger, Chem.–Eur. J., 2016, 22, 5509–5513 CrossRef CAS.
- H. Banda, D. Damien, K. Nagarajan, A. Raj, M. Hariharan and M. M. Shaijumon, Adv. Energy Mater., 2017, 7, 1701316 CrossRef.
- P. J. Low, M. A. J. Paterson, H. Puschmann, A. E. Goeta, J. A. K. Howard, C. Lambert, J. C. Cherryman, D. R. Tackley, S. Leeming and B. Brown, Chem.–Eur. J., 2004, 10, 83–91 CrossRef CAS.
- V. Bulvic, G. Gu, P. E. Burrows, S. R. Forrest and M. E. Thompson, Nature, 1996, 380, 29 CrossRef.
- N. Tamoto, C. Adachi and K. Nagai, Chem. Mater., 1997, 9, 1077–1085 CrossRef CAS.
- E. Bellmann, S. E. Shaheen, S. Thayumanavan, S. Barlow, R. H. Grubbs, S. R. Marder, B. Kippelen and N. Peyghambarian, Chem. Mater., 1998, 10, 1668–1676 CrossRef CAS.
- M. C. Harris and S. L. Buchwald, J. Org. Chem., 2000, 65, 5327–5333 CrossRef.
- U. Mitschke and P. Bäuerle, J. Mater. Chem., 2000, 10, 1471–1507 RSC.
- P. M. Borsenberger and D. S. Weiss, Organic Photoreceptors for Xerography, Marcel Dekker, New York, 1998 Search PubMed.
- O. Kwon, S. Barlow, S. A. Odom, L. Beverina, N. J. Thompson, E. Zojer, J.-L. Bredas and S. R. Marder, J. Phys. Chem., 2005, 109, 9346–9352 CrossRef CAS.
- M. Stolka, J. F. Yanus and D. M. Pai, J. Phys. Chem., 1984, 88, 4707–4714 CrossRef CAS.
- P. M. Borsenberger and D. S. Weiss, Organic Photoreceptors for Imaging Systems, Dekker, New York, 1993 Search PubMed.
- M. Thelakkat, R. Fink, F. Haubner and H.-W. Schmidt, Macromol. Symp., 1998, 125, 157–164 CrossRef CAS.
- M. Thelakkat, Macromol. Mater. Eng., 2002, 287, 442 CrossRef CAS.
- C. W. Tang, Appl. Phys. Lett., 1986, 48, 183–185 CrossRef CAS.
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- E. S. Kolb, R. A. Gaudiana and P. G. Mehta, Macromolecules, 1996, 29, 2359–2364 CrossRef CAS.
- H. Fujikawa, S. Tokito and Y. Taga, Synth. Met., 1997, 91, 161–162 CrossRef CAS.
- C. Giebeler, H. Antoniadis, D. D. C. Bradley and Y. Shirota, Appl. Phys. Lett., 1998, 72, 2448–2450 CrossRef CAS.
- M. Redecker, D. D. C. Bradley, M. Inbasekaran, W. W. Wu and E. P. Woo, Adv. Mater., 1999, 11, 241–246 CrossRef CAS.
- T. Braig, D. C. Müller, M. Groß, K. Meerholz and O. Nuyken, Macromol. Rapid Commun., 2000, 21, 583–589 CrossRef CAS.
- W. E. Moerner and S. M. Silence, Chem. Rev., 1994, 94, 127–155 CrossRef CAS.
- S. Dapperheld, E. Steckhan, K.-H. G. Brinkhaus and T. Esch, Chem. Ber., 1991, 2557–2567 CrossRef CAS.
- S. Amthor, B. Noller and C. Lambert, Chem. Phys., 2005, 316, 141–152 CrossRef CAS.
- C. Lambert and G. Nöll, J. Am. Chem. Soc., 1999, 121, 8434–8442 CrossRef CAS.
- T. Noda and Y. Shirota, J. Am. Chem. Soc., 1998, 120, 9714–9715 CrossRef CAS.
- Y. S. M. Kinoshita, Chem. Lett., 2001, 30, 614–615 CrossRef.
- C. D. Entwistle and T. B. Marder, Angew. Chem., Int. Ed., 2002, 41, 2927–2931 CrossRef CAS.
- C. D. Entwistle and T. B. Marder, Chem. Mater., 2004, 16, 4574–4585 CrossRef CAS.
- L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846–863 RSC.
- S. Y. A. Wakamiya, Bull. Chem. Soc. Jpn., 2015, 88, 1357 CrossRef.
- Y. Ren and F. Jäkle, Dalton Trans., 2016, 45, 13996–14007 RSC.
- A. W. S. Yamaguchi, Pure Appl. Chem., 2006, 78, 1413–1424 Search PubMed.
- F. Jäkle, Coord. Chem. Rev., 2006, 250, 1107–1121 CrossRef.
- S.-Y. Li, Z.-B. Sun and C.-H. Zhao, Inorg. Chem., 2017, 56, 8705–8717 CrossRef CAS PubMed.
- F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef PubMed.
- S.-B. Zhao, P. Wucher, Z. M. Hudson, T. M. McCormick, X.-Y. Liu, S. Wang, X.-D. Feng and Z.-H. Lu, Organometallics, 2008, 27, 6446–6456 CrossRef CAS.
- S. Griesbeck, Z. Zhang, M. Gutmann, T. Lühmann, R. M. Edkins, G. Clermont, A. N. Lazar, M. Haehnel, K. Edkins, A. Eichhorn, M. Blanchard-Desce, L. Meinel and T. B. Marder, Chem.–Eur. J., 2016, 22, 14701–14706 CrossRef CAS.
- S. Griesbeck, M. Ferger, C. Czernetzi, C. Wang, R. Bertermann, A. Friedrich, M. Haehnel, D. Sieh, M. Taki, S. Yamaguchi and T. B. Marder, Chem.–Eur. J., 2019, 25, 7679–7688 CAS.
- S. Griesbeck, E. Michail, C. Wang, H. Ogasawara, S. Lorenzen, L. Gerstner, T. Zang, J. Nitsch, Y. Sato, R. Bertermann, M. Taki, C. Lambert, S. Yamaguchi and T. B. Marder, Chem. Sci., 2019, 41, 2927 Search PubMed.
- S. Yamaguchi, T. Shirasaka and K. Tamao, Org. Lett., 2000, 2, 4129–4132 CrossRef CAS.
- N. Matsumi, K. Naka and Y. Chujo, J. Am. Chem. Soc., 1998, 120, 5112–5113 CrossRef CAS.
- N. Matsumi, K. Naka and Y. Chujo, J. Am. Chem. Soc., 1998, 120, 10776–10777 CrossRef CAS.
- A. G. Crawford, A. D. Dwyer, Z. Liu, A. Steffen, A. Beeby, L.-O. Pålsson, D. J. Tozer and T. B. Marder, J. Am. Chem. Soc., 2011, 133, 13349–13362 CrossRef CAS.
- L. Ji, R. M. Edkins, A. Lorbach, I. Krummenacher, C. Bruckner, A. Eichhorn, H. Braunschweig, B. Engels, P. J. Low and T. B. Marder, J. Am. Chem. Soc., 2015, 137, 6750–6753 CrossRef CAS.
- L. Ji, A. Lorbach, R. M. Edkins and T. B. Marder, J. Org. Chem., 2015, 80, 5658–5665 CrossRef CAS.
- H. C. Brown and V. H. Dodson, J. Am. Chem. Soc., 1957, 79, 2302 CrossRef CAS.
- Z. Zhang, R. M. Edkins, M. Haehnel, M. Wehner, A. Eichhorn, L. Mailänder, M. Meier, J. Brand, F. Brede, K. Müller-Buschbaum, H. Braunschweig and T. B. Marder, Chem. Sci., 2015, 6, 5922–5927 RSC.
- Z. Zhou, A. Wakamiya, T. Kushida and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 4529–4532 CrossRef CAS.
- G. A. Molander and R. Figueroa, Aldrichimica Acta, 2005, 38, 49 CAS.
- S. Darses and J.-P. Genet, Chem. Rev., 2008, 108, 288–325 CrossRef CAS.
- G. Berionni, B. Maji, P. Knochel and H. Mayr, Chem. Sci., 2012, 3, 878–882 RSC.
- A. J. J. Lennox, Organotrifluoroborate Preparation, Coupling and Hydrolysis, Springer, Heidelberg, 2013 Search PubMed.
- Organic Reactions, ed. S. E. Denmark, John Wiley & Sons, Inc, Hoboken, 79th edn, 2013, vol. 79 Search PubMed.
- J. Merz, J. Fink, A. Friedrich, I. Krummenacher, H. H. Al Mamari, S. Lorenzen, M. Haehnel, A. Eichhorn, M. Moos, M. Holzapfel, H. Braunschweig, C. Lambert, A. Steffen, L. Ji and T. B. Marder, Chem.–Eur. J., 2017, 23, 13164–13180 CrossRef CAS.
- K. Schickedanz, J. Radtke, M. Bolte, H.-W. Lerner and M. Wagner, J. Am. Chem. Soc., 2017, 139, 2842–2851 CrossRef CAS PubMed.
- K. Schickedanz, T. Trageser, M. Bolte, H.-W. Lerner and M. Wagner, Chem. Commun., 2015, 51, 15808–15810 RSC.
- A. D. Ainley and F. Challenger, J. Chem. Soc., 1930, 2171–2180 RSC.
- A. L. S. Thompson, G. W. Kabalka, M. R. Akula and J. W. Huffman, Synthesis, 2005, 36, 547 Search PubMed.
- C. Näther, H. Bock, Z. Havlas and T. Hauck, Organometallics, 1998, 17, 4707–4715 CrossRef.
- M. Bolte, Experimental Crystal Structure Determination, CCDC 1546182, 2017 Search PubMed.
- F. Würthner, Pure Appl. Chem., 2006, 78, 2341–2349 Search PubMed.
- M. A. Spackman and P. G. Byrom, Chem. Phys. Lett., 1997, 267, 215–220 CrossRef CAS.
- J. J. McKinnon, A. S. Mitchell and M. A. Spackman, Chem.–Eur. J., 1998, 4, 2136–2141 CrossRef CAS.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627–668 CrossRef.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- A. Parkin, G. Barr, W. Dong, C. J. Gilmore, D. Jayatilaka, J. J. McKinnon, M. A. Spackman and C. C. Wilson, CrystEngComm, 2007, 9, 648 RSC.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 267, 3814 RSC.
- L. Weber, V. Werner, M. A. Fox, T. B. Marder, S. Schwedler, A. Brockhinke, H.-G. Stammler and B. Neumann, Dalton Trans., 2009, 1339 RSC.
- C. Huang, S. Barlow and S. R. Marder, J. Org. Chem., 2011, 76, 2386–2407 CrossRef CAS.
- We note, that Zeng and co-workers44 do not list their extinction coefficients. Therefore, we estimated them from their Fig. 1.
- The Stokes shift is defined as the energy difference between the 0–0 transitions of the absorption and the emission. However, as the first transition is broad, the band maxima of the absorption and emission have been used to determine the Stokes shift throughout the paper. Consequently, the term “apparent Stokes shift” is used.
- É. Torres, M. N. Berberan-Santos and M. J. Brites, Dyes Pigm., 2015, 112, 298–304 CrossRef.
- We note that we also measured the emission of (Br)4-Per in CH2Cl2 for full comparison with the brominated derivative from Zeng and co-workers as they only give photophysical results in CH2Cl2 (see ESI Fig. S1†).
- M.-J. Lin, Á. J. Jiménez, C. Burschka and F. Würthner, Chem. Commun., 2012, 48, 12050–12052 RSC.
- F. Schlosser, V. Stepanenko and F. Würthner, Chem. Commun., 2010, 46, 8350–8352 RSC.
- M. Zander, H. Dreeskamp and E. Koch, Z. Naturforsch., A: Phys. Sci., 1974, 29, 1518–1519 CAS.
- H. Dreeskamp, E. Koch and M. Zander, Chem. Phys. Lett., 1975, 31, 251–253 CrossRef CAS.
- K. Nagarajan, A. R. Mallia, V. S. Reddy and M. Hariharan, J. Phys. Chem. C, 2016, 120, 8443–8450 CrossRef CAS.
- S. K. M. Nalluri, J. Zhou, T. Cheng, Z. Liu, M. T. Nguyen, T. Chen, H. A. Patel, M. D. Krzyaniak, W. A. Goddard, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2019, 141, 1290–1303 CrossRef.
- S. J. Strickler and R. A. Berg, J. Chem. Phys., 1962, 37, 814–822 CrossRef CAS.
- H. Dreeskamp, A. Läufer and M. Zander, Z. Naturforsch., A: Phys. Sci., 1983, 38, 698–700 Search PubMed.
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- H. Xiang, J. Cheng, X. Ma, X. Zhou and J. J. Chruma, Chem. Soc. Rev., 2013, 42, 6128–6185 RSC.
- A. Gut, Ł. Łapok, D. Drelinkiewicz, T. Pędziński, B. Marciniak and M. Nowakowska, Chem.–Asian J., 2018, 13, 55–65 CrossRef CAS.
- Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007–3030 RSC.
- Y. Feng, J. Cheng, L. Zhou, X. Zhou and H. Xiang, Analyst, 2012, 137, 4885–4901 RSC.
- J.-P. Fouassier, F. Morlet-Savary, J. Lalevée, X. Allonas and C. Ley, Materials, 2010, 3, 5130–5142 CrossRef CAS.
- M. L. Marin, L. Santos-Juanes, A. Arques, A. M. Amat and M. A. Miranda, Chem. Rev., 2012, 112, 1710–1750 CrossRef CAS.
- N. Noto, T. Koike and M. Akita, Chem. Sci., 2017, 8, 6375–6379 RSC.
- N. Noto, Y. Tanaka, T. Koike and M. Akita, ACS Catal., 2018, 8, 9408–9419 CrossRef CAS.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS.
- J. Zhao, S. Ji and H. Guo, RSC Adv., 2011, 1, 937 RSC.
- M. R. Detty, S. L. Gibson and S. J. Wagner, J. Med. Chem., 2004, 47, 3897–3915 CrossRef CAS.
- L. Sobotta, P. Skupin-Mrugalska, J. Mielcarek, T. Goslinski and J. Balzarini, Mini-Rev. Med. Chem., 2015, 15, 503–521 CrossRef CAS.
- W. E. Ford and P. V. Kamat, J. Phys. Chem., 1987, 91, 6373–6380 CrossRef CAS.
- A. J. Tilley, R. D. Pensack, T. S. Lee, B. Djukic, G. D. Scholes and D. S. Seferos, J. Phys. Chem. C, 2014, 118, 9996–10004 CrossRef CAS.
- M. T. Vagnini, A. L. Smeigh, J. D. Blakemore, S. W. Eaton, N. D. Schley, F. D'Souza, R. H. Crabtree, G. W. Brudvig, D. T. Co and M. R. Wasielewski, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15651–15656 CrossRef CAS.
- M. Schulze, A. Steffen and F. Würthner, Angew. Chem., Int. Ed., 2015, 127, 1590–1593 CrossRef.
- A. A. Rachford, S. Goeb and F. N. Castellano, J. Am. Chem. Soc., 2008, 130, 2766–2767 CrossRef CAS.
- H. Weissman, E. Shirman, T. Ben-Moshe, R. Cohen, G. Leitus, L. J. W. Shimon and B. Rybtchinski, Inorg. Chem., 2007, 46, 4790–4792 CrossRef CAS.
- B. Ventura, H. Langhals, B. Böck and L. Flamigni, Chem. Commun., 2012, 48, 4226–4228 RSC.
- L. Flamigni, A. Zanelli, H. Langhals and B. Böck, J. Phys. Chem. A, 2012, 116, 1503–1509 CrossRef CAS.
- E. Oliveros, S. H. Bossmann, S. Nonell, C. Martí, G. Heit, G. Tröscher, A. Neuner, C. Martínez and A. M. Braun, New J. Chem., 1999, 23, 85–93 RSC.
- C. Schweitzer and R. Schmidt, Chem. Rev., 2003, 103, 1685–1757 CrossRef CAS.
- A. A. Abdel-Shafi and F. Wilkinson, J. Phys. Chem. A, 2000, 104, 5747–5757 CrossRef CAS.
- A. J. McLean, D. J. McGarvey and T. G. Truscott, J. Chem. Soc., Faraday Trans., 1990, 86, 3075–3080 RSC.
- J. D. Harris, M. J. Moran and I. Aprahamian, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9414–9422 CrossRef CAS.
- A. B. Buades, V. Sanchez Arderiu, D. Olid-Britos, C. Viñas, R. Sillanpää, M. Haukka, X. Fontrodona, M. Paradinas, C. Ocal and F. Teixidor, J. Am. Chem. Soc., 2018, 140, 2957–2970 CrossRef CAS.
- M. Frasconi, I. R. Fernando, Y. Wu, Z. Liu, W.-G. Liu, S. M. Dyar, G. Barin, M. R. Wasielewski, W. A. Goddard and J. F. Stoddart, J. Am. Chem. Soc., 2015, 137, 11057–11068 CrossRef CAS.
- J. Kübel, R. Schroot, M. Wächtler, U. S. Schubert, B. Dietzek and M. Jäger, J. Phys. Chem. C, 2015, 119, 4742–4751 CrossRef.
- Y.-M. Tian, X.-N. Guo, M. W. Kuntze-Fechner, I. Krummenacher, H. Braunschweig, U. Radius, A. Steffen and T. B. Marder, J. Am. Chem. Soc., 2018, 140, 17612–17623 CrossRef CAS.
- L. Ramaley and M. S. Krause, Anal. Chem., 1969, 1362–1365 CrossRef CAS.
- G. C. Barker and I. L. Jenkins, Analyst, 1952, 77, 685–696 RSC.
- I. Raabe, K. Wagner, K. Guttsche, M. Wang, M. Grätzel, G. Santiso-Quiñones and I. Krossing, Chem.–Eur. J., 2009, 15, 1966–1976 CrossRef CAS.
- F. Barrière and W. E. Geiger, J. Am. Chem. Soc., 2006, 128, 3980–3989 CrossRef.
- Y. Fan, K. Ziabrev, S. Zhang, B. Lin, S. Barlow and S. R. Marder, ACS Omega, 2017, 2, 377–385 CrossRef CAS.
- W. Jiang, C. Xiao, L. Hao, Z. Wang, H. Ceymann, C. Lambert, S. Di Motta and F. Negri, Chem.–Eur. J., 2012, 18, 6764–6775 CrossRef CAS.
- W. Jiang, L. Ye, X. Li, C. Xiao, F. Tan, W. Zhao, J. Hou and Z. Wang, Chem. Commun., 2014, 50, 1024–1026 RSC.
- H. Horinouchi, H. Sakai, Y. Araki, T. Sakanoue, T. Takenobu, T. Wada, N. V. Tkachenko and T. Hasobe, Chem.–Eur. J., 2016, 22, 9631–9641 CrossRef CAS.
- L. Echegoyen and L. E. Echegoyen, Acc. Chem. Res., 1998, 31, 593–601 CrossRef CAS.
- M. Takase, V. Enkelmann, D. Sebastiani, M. Baumgarten and K. Müllen, Angew. Chem., Int. Ed., 2007, 46, 5524–5527 CrossRef CAS.
- K. Oki, M. Takase, S. Mori, A. Shiotari, Y. Sugimoto, K. Ohara, T. Okujima and H. Uno, J. Am. Chem. Soc., 2018, 140, 10430–10434 CrossRef CAS.
- Y. Shibano, T. Umeyama, Y. Matano, N. V. Tkachenko, H. Lemmetyinen and H. Imahori, Org. Lett., 2006, 8, 4425–4428 CrossRef CAS.
- B. Badger and B. Brocklehurst, Trans. Faraday Soc., 1970, 66, 2939–2947 RSC.
- We note that it is a semantic issue as to whether small delocalized systems are called “MV” compound as it is arbitrary or even impossible to identify redox centers. In fact, the term “charge-resonance” compound is often used.178 However, in this paper we use the term “mixed valence” compounds.
- A. Heckmann and C. Lambert, Angew. Chem., Int. Ed., 2012, 51, 326–392 CrossRef CAS.
- B. S. Brunschwig and N. Sutin, Coord. Chem. Rev., 1999, 187, 233–254 CrossRef CAS.
- B. S. Brunschwig, C. Creutz and N. Sutin, Chem. Soc. Rev., 2002, 31, 168–184 RSC.
- R. J. Cave and M. D. Newton, Chem. Phys. Lett., 1996, 249, 15–19 CrossRef CAS.
- R. J. Cave and M. D. Newton, J. Chem. Phys., 1997, 106, 9213 CrossRef CAS.
- M. J. G. Peach, P. Benfield, T. Helgaker and D. J. Tozer, J. Chem. Phys., 2008, 128, 44118 CrossRef.
- R. Uson, L. A. Oro, J. A. Cabeza, H. E. Bryndza and M. P. Stepro, in Inorganic Syntheses, ed. Kirschner (Hg.), 1985, pp. 126–130 Search PubMed.
- S. S. Zalesskiy and V. P. Ananikov, Organometallics, 2012, 31, 2302–2309 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
- K. B. H. Putz, Diamond, Crystal and Molecular Structure Visualization, Crystal Impact, H. Putz & K., Brandenburg GbR, Bonn (Germany), 2017 Search PubMed.
- M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer17, University of Western Australia, 2017 Search PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081 CrossRef CAS.
- G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham and W. A. Shirley, J. Chem. Phys., 1988, 2193–2218 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- M. Renz, K. Theilacker, C. Lambert and M. Kaupp, J. Am. Chem. Soc., 2009, 131, 16292–16302 CrossRef CAS.
- M. Kaupp, M. Renz, M. Parthey, M. Stolte, F. Würthner and C. Lambert, Phys. Chem. Chem. Phys., 2011, 13, 16973–16986 RSC.
- J. C. Collings, S.-Y. Poon, C. Le Droumaguet, M. Charlot, C. Katan, L.-O. Pålsson, A. Beeby, J. A. Mosely, H. M. Kaiser, D. Kaufmann, W.-Y. Wong, M. Blanchard-Desce and T. B. Marder, Chem.–Eur. J., 2009, 15, 198–208 CrossRef CAS PubMed.
- S.-F. Liu, Q. Wu, H. L. Schmider, H. Aziz, N.-X. Hu, Z. Popović and S. Wang, J. Am. Chem. Soc., 2000, 122, 3671–3678 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1881912. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02420d |
| This journal is © The Royal Society of Chemistry 2019 |