
Nanoparticle modification of microfluidic cell separation for cancer cell detection and isolation†
Yun
Zhou
a,
Ziye
Dong
 b,
Hermella
Andarge
a,
Wei
Li
b and
Dimitri
Pappas
*a
b,
Hermella
Andarge
a,
Wei
Li
b and
Dimitri
Pappas
*a
aDepartment of Chemistry and Biochemistry, Texas Tech University, Lubbock, TX 79409, USA. E-mail: d.pappas@ttu.edu; Fax: +1 806-742-1289; Tel: +1 806-834-1103
bDepartment of Chemical Engineering, Texas Tech University, Lubbock, TX 79409, USA
First published on 24th October 2019
Abstract
Cancer is a major health problem in the United States with extremely high mortality. The detection and isolation of cancer cells are becoming increasingly important for cancer diagnosis. We describe a microfluidic device modified with silica nanoparticles to enhance the isolation of cancer cells using affinity separation. The isolation of seven different cancer cell lines spiked into liquid biopsies was demonstrated and compared with unmodified separation devices. Cancer cells were isolated using CD71 which has already been demonstrated to be a widespread “net” for capturing cancer cells of any phenotype as the affinity target. The capture efficiency of our nanoparticle (NP)-modified HB chip showed significant differences compared with the normal HB chip, exhibiting an average increase of 16%. The cell enrichment increased by a factor of 2 over unmodified chips. Patient-derived ALL cells, COG-LL-332, were spiked into blood with concentrations ranging from 1% to 20% of total leukocytes, and isolated with the purity of 41%–65%. The results of this study demonstrated that the increase of cell–chip interactions after nanoparticle modification improved capture efficiency and capture purity, and can be applied to a wide range of cell separations.
Introduction
Cancer and cancer-related diseases affect more than 10 million patients every year worldwide.1 Many types of cancer spread via free circulating tumor cells (CTCs) detaching from the primary tumor and moving along with the bloodstream toward a new site for subsequent disease growth.2 Therefore, the role of CTCs in cancers is not only as the indicator for disease progression, but also as a diagnostic parameter for cancer identification, since they originate from primary tumors.3 The detection and isolation of CTCs in the so-called liquid biopsies are of great importance for cancer diagnosis. However, especially in early-stage cancers, CTCs are a type of rare cells that are present in the bloodstream only at concentrations as low as 1–100 in 109 blood cells.4 Thus, methods applied for the detection of CTCs need to be very sensitive.5Methodologies for the detection of CTCs are generally composed of two steps: isolation (immunological and morphological techniques) and identification (cytometric and nucleic acid techniques).6 Morphology-based isolation approaches are based on the size discrepancies of CTCs working as microfilters to isolate the desired cells from blood samples. This method requires neither functional modification nor complex enrichment procedures.7 However, compared with other isolation approaches, the sensitivity and purity are relatively low.7 Density gradient separations, which are mostly operated using Ficoll-Hypaque (GE Healthcare) and Oncoquick (Greiner Bio-One), are also commonly used to isolate CTCs from whole blood samples.8 The main drawback of this approach is the cross-contamination between different layers. Unlike these two methods, immunological techniques target on specific markers that are expressed on the CTC surface. Epithelial-specific biomarkers can label tumor epithelial cells by specific antigen–antibody bindings, thus to recognize and isolate the desired cells. Nowadays, a CellSearch™ system (Veridex) is the only FDA approved automated immunological separation system for the clinical detection of CTCs. CTCs are distinguished from leukocytes by coupling with EpCAM coated ferrofluid nanoparticles which can be attracted by passing through a magnetic field.9 However, a prior knowledge of cancers is needed since this system can only be accurately applied in analyzing metastatic breast cancer, colorectal cancer, and prostate cancer.10 Besides, it is expensive to have this system in a normal analytical lab.
In this work, we employed microfluidics for cancer cell detection and isolation. Microfluidic devices are becoming a promising and creative approach for the isolation of cancer cells from bulk cell mixtures. Compared with all techniques mentioned above, microfluidics based on affinity surface cell capture is portable, easier to manufacture, and less complex.11 Furthermore, most of the immunological and morphological techniques require millimeters of blood samples to reach the desired outcome. However, only 100 μl samples were used in our work and excellent capture purity and efficiency were obtained. Moreover, the whole experimental process takes only 2 hours with a per-chip cost of approximately $2 USD and is much faster and cheaper than all commercial methods. Besides, anti-CD71 was used as the affinity ligand in this work. CD71 (human transferrin receptor) is expressed abundantly in some proliferative cells, such as cancer cells, activated lymphocytes and serum-induced fibroblasts.12,13 Our previous work demonstrated that CD71 is an excellent ligand for multiple different cancer cell lines and is not specific to cancer phenotype.14 Thus it can be viewed as a pan-cancer screen when prior knowledge about the cancer is not known. Therefore, the coupling of CD71 with our microfluidic device can be used for a wide range of cancer cell detection. We employed seven different cell lines (CCRF-CEM, Ramos, Jurkat, HL-60, PC-3, MDA-231, and COG-LL-332) to serve as models, mimicking cancers and other diseases. The main purpose of this work is to evaluate the enhanced cell capture and separation performance in nanoparticle-modified microfluidic systems.
Multiple nanostructures, such as nanopillars,15,16 nanofibers,17 nanofractals,18etc., have been studied and incorporated into PDMS-based microchips as modifiers to minimize nonspecific bindings, and thus to enhance cell isolation efficiency.19 Most of these nanostructures have also been used for the detection of CTCs in different manners and showed excellent performance due to their similar size to biomolecules and high surface area-to-volume ratio.20,21 Also, Hyeun et al. reviewed current advances in nanomaterials for the detection of CTCs and showed that nanoscale structures provide a new set of tools that have great potential to overcome current limitations in detecting CTC cells.22 Therefore, it is believed that the integration of nanomaterials with microfluidic devices can provide an improved performance in the detection of CTCs with respect to the adhesion and immobilization of biomolecules.22,23 Wang et al. have successfully modified the poly(dimethylsiloxane)(PDMS)-based microfluidic chips with gold nanoparticles and reported that the separation resolution of dopamine and epinephrine increased from 0.62 on a native PDMS device to 1.14 on a modified PDMS microchip.24 The increased interactions, provided by nanostructures, are the main reason for improved separation performance. Thus, this approach can also be used for the isolation of rare cells from blood samples using affinity separations which is based on antigen–antibody interactions.
In order to isolate cancer cells from bulk cell mixtures or blood samples, many affinity based microfluidic cell-capture devices have been reported.25,26 One particular design, the herringbone (HB) chip, has proven to exhibit good performance in both separation efficiency and isolation purity. Unlike the flat microchannel surfaces, herringbone mixers of the HB chip are able to increase the mixing of the flow passing through the channel, and thus to increase collisions between the cells and antibodies coated on channel surfaces.26
In our work, we modified the PDMS-based HB chip with nanoparticles to achieve a better cell isolation and purification compared with the normal HB chip. A human T lymphoblast cell line, CCRF-CEM, was employed for chip reproducibility and stability, capture efficiency, and capture purity investigations. Silica nanoparticles allowed for specific affinity coating and cell capture. They have strong native negative charges and high density of hydroxyl groups on their surfaces allowing to be easily attached on the surface with opposite charges through electrostatic forces, and also be coated with biomolecules.27 Furthermore, their transparent nature enables the clear visualization of microsystems via a fluorescence microscope. Dong et al. reported that nanoparticle-enhanced hollow glass microspheres (HGMS) can be successfully used to capture different cell lines involved in different cancer phenotypes.28 Therefore, we selected the silica nanoparticles as the modifier for our HB chip (Fig. 1A).
 | ||
| Fig. 1 SEM images of nanoparticles (NPs) coated on the herringbone chip surface. Glass nanoparticles were successfully loaded into the channel to increase the surface area-to-volume ratio, as well as to increase interactions between the cells and antibodies. The average size of nanoparticles is 202.5 ± 6.7 nm. The surface coverage is 90.1 ± 4.2%. (B) Schematic of NP-modified HB microchips. Cancer cells (yellow) were spiked into blood cells (red) to simulate liquid biopsies. Target cells are captured by anti-CD71 modified surfaces (Y-shape structure). More target cells can be effectively captured due to the increased surface area-to-volume ratio, as well as the increased antigen–antibody interactions resulting from nanoparticles (gray circle). The inset represents the change in flow pathways (white arrow) after surface modification. (C) Photograph of a NP-modified HB chip. The dimension of the channel is 5 cm × 1 mm × 40 μm (L × W × H). The dimension of each herringbone structure is 150 μm × 1 mm × 40 μm (L × W × H). The angle of each turn is 90°. (D) Representative fluorescence image of captured target cells. Bright dots in red circles represent captured CCRF-CEM cells. Scale bar = 50 μm. | ||
In this work, the nanoparticle (NP)-modified HB chip showed great improvement in capture efficiency and has the ability of maintaining the efficiency at a high level. For all cell lines involved in this study, the NP-modified HB chip showed all positive capture efficiency enhancements compared with normal HB chips (Fig. S1†). Significant differences were observed regarding all cell lines (p < 0.05, 95% confidence interval). Besides, the capture purity of our NP-modified HB chip was also elevated at all spike percentages ranging from 1% to 20%. Furthermore, a patient-derived ALL cell line, COG-LL-332, was isolated from blood biopsies in concentrations as low as 1% with a high capture purity.
Results and discussion
Antibody selection
Iron (Fe2+) is required during cell proliferation for DNA synthesis and other mechanisms.29 However, the storage of intracelluar iron is tightly controlled by cells due to its highly toxic nature.30 CD71, known as the transferrin receptor (TfR), allows for iron uptake via transferrin from blood plasma.31 Thus when available intracellular irons are exhausted during the cell replication process, the cell membrane expression of TfR will increase to acquire more iron molecules.29 Therefore, this CD71 expression enhancement can be used as a good marker for proliferating cells.Furthermore, unlike other commonly used antigens for cancer cells, such as EpCAM, CD71 does not require a priori knowledge of cancer types.32,33 CD71 binds to all proliferating cells with high CD71 expression especially cancer cells which express CD71 at higher levels than any other surrounding somatic cells, and thus can be viewed as the “pan-cancer” screen.14,34 In our previous work, the number of CD71 + CCRF-CEM cells captured in the microchip was reported as 5.6-fold higher than using anti-CD7 and anti-CD10.14 Besides, the binding of anti-CD71 to CCRF-CEM cells was minorly affected by antibody dilution and the capture efficiency can be maintained at a high level.35 Therefore, in this study, we employed anti-CD71 as the capture ligand in the HB chip and NP-modified HB chip to detect different cancer cell lines, and thus to evaluate the performance of our upgraded HB chips for the rare cell detection and isolation.
Chip reproducibility and stability
NP-modified HB chips are shown in Fig. 1. The mean particle size was 202.5 ± 6.7 nm. The size distribution is listed in Fig. S2.† The particles adhere on the channel surface via electrostatic interactions with the surface coverage of 90.1 ± 4.2%. This noncovalent attachment forms the foundation for the subsequent Layer-by-Layer (LbL) deposition of the capture chemistry. The stability of surface coating is an important factor in cell separation. When the surface concentration of antibodies exceeds the antigen expression on the cells, the cell capture is proportional to the cell surface expression. CD71 expression is transient as the cells enter and exit different phases of the cell cycle. We have found in previous work that CD71 expression changes after the cells are subcultured. In order to minimize the expression effects on our measurements, we obtained cells for both NP-HB and HB chips at the same time from the same flask. There was one-hour gap between each pair of chip separations. But based on flowcytometry analysis (Fig. 2), changes in the CD71 expression during this one-hour gap were not significant.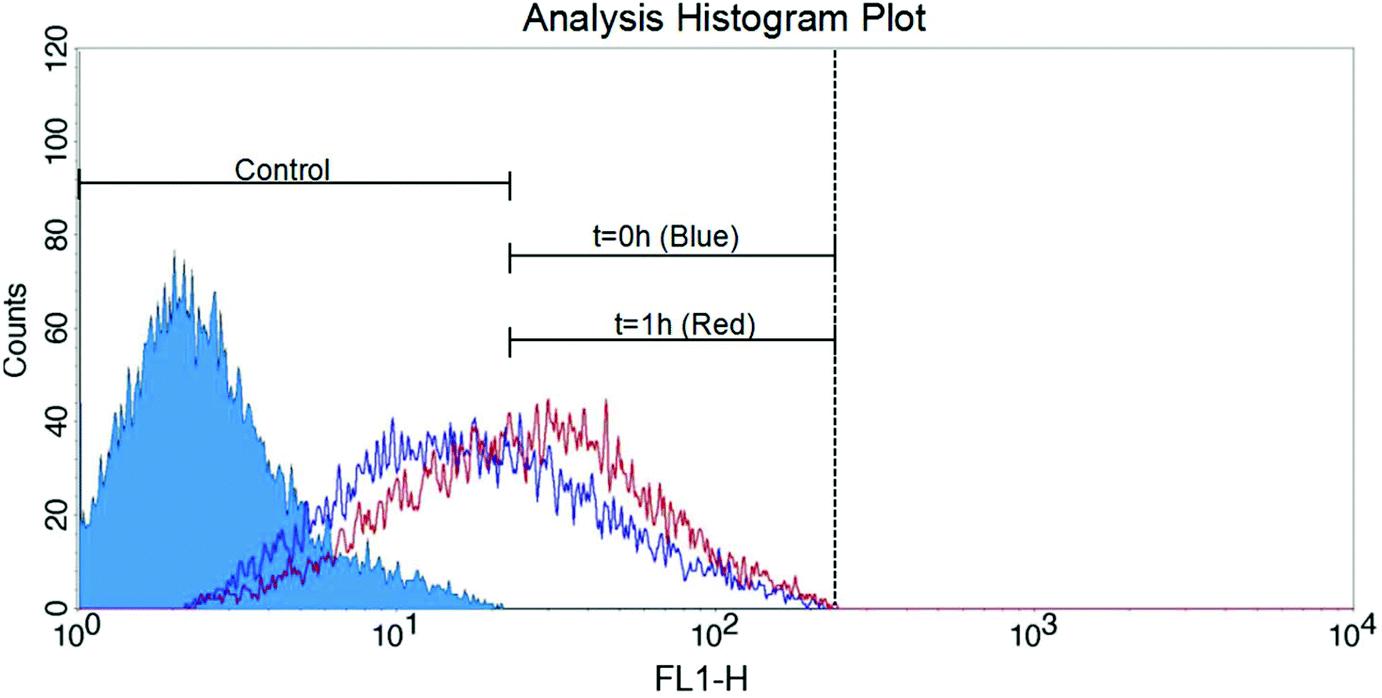 | ||
| Fig. 2 Flow cytometry histogram plot of CCRF-CEM CD71 expression at different times (t = 0 h and t = 1 h). No significant CD71 surface expression changes in CCRF-CEM cells were observed between normal HB and NP-modified HB measurements during this one-hour gap. | ||
Both NP-modified HB chips and normal HB chips maintained a stable capture efficiency for four consecutive separations (Fig. 3A). However, the ratio of NP-modified HB capture efficiency was constant over 5 measurements (1.05–1.14). Each measurement pair was conducted on a different day, resulting in changes of antigen expressions, as well as total cell concentrations. Based on our previous work, the CD71 surface expression of a single cell reached the highest peak on the second day and decreased gradually on every day after.36 Also, as we observed from experiments, the cell concentration for the last trial was 679 cells per μl, while it was 4197 cells per μl for the second trial which had the highest capture efficiency. Hence, the fifth trial capture efficiency may have been influenced by both relatively lower cell population and surface expression after five continuous days of subculture. However, even with the low cell population, the capture efficiency of the NP-modified chip was still maintained at a fair level (76.12%), and higher than that of the normal HB chip (72.81%). Based on our measurements, NP-modified HB chips showed a very stable capture performance, and maintained the capture efficiency at a high level for the first four trails (95 ± 13%) with an 11 ± 2% enhancement compared with normal HB chips. Therefore, the silica nanoparticle coating is stable and can be used for multiple separations with excellent performance.
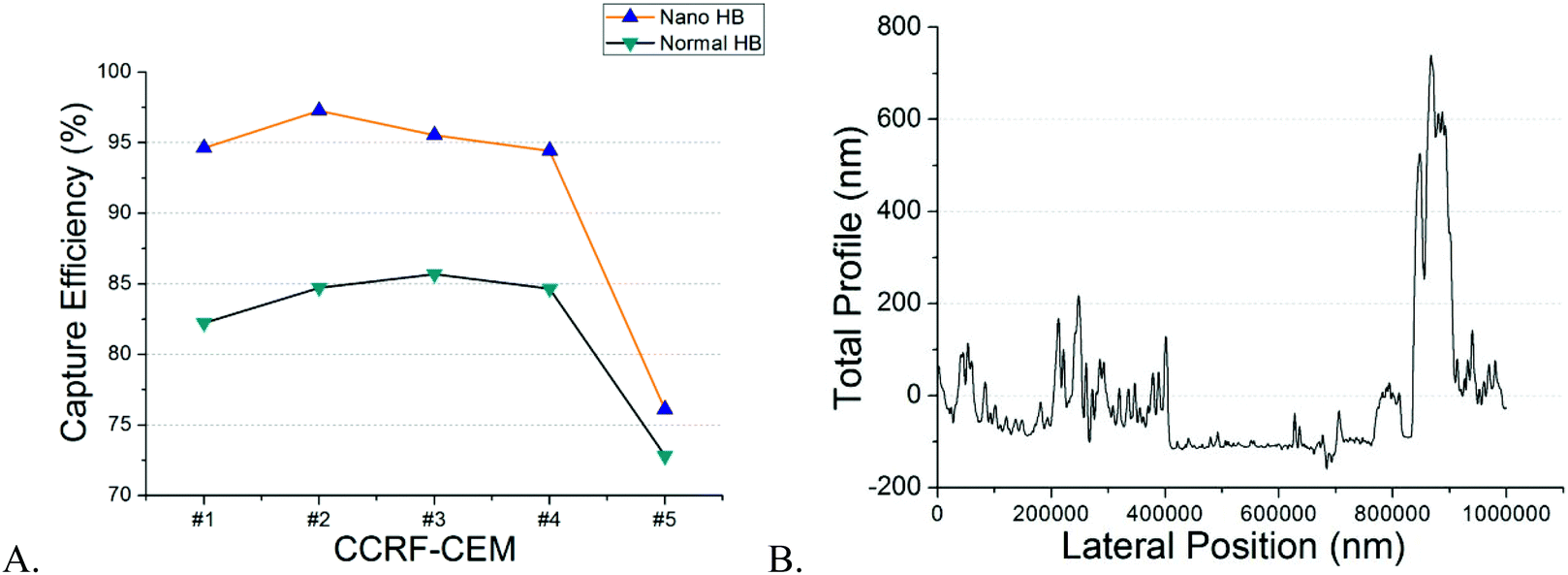 | ||
| Fig. 3 (A) Reproducibility measurements of NP-modified HB chips and normal HB chips (n = 5). The capture efficiency of the NP-modified HB chip was higher than the normal HB chip in separations. The cell concentrations for all measurements were 2508 cells per μL, 4197 cells per μL, 3357 cells per μL, 2006 cells per μL, and 679 cells per μL, respectively. (B) Profilometry analysis of the NP-modified chip surface. Majority of peaks were 50–250 nm higher than the baseline which proved the single layer modification. Same results were observed on both used chip surface and new chip surface. This observation demonstrated the stability of our surface modification. | ||
NP surface distribution may vary from chip to chip, and thus affect the cell capture in different degrees. However, the layer-by-layer (LbL) method we used for surface modification can ensure single layer particle loading, therefore, to maintain the similarity of NP surface distribution at a high level for different microchips. This statement can be proved by the profilometry analysis. We randomly selected 3 new NP-modified chips and 3 used NP-modified chips to prove single layer modification and the stability of nanoparticle loading. All chips demonstrated similar distribution results and an example is shown in Fig. 3B. We used a blade to remove the nanoparticles from a randomly selected region in the channel. The baseline in the figure represents the area without silica nanoparticles. As we can see, the majority peaks on the left side of the baseline are within the height of 50–250 nm which corresponds to the single particle size very well. The highest peak on the right side of the baseline was because of the accumulation of particles caused by blade cutting. Similar results were observed in all new and used NP-modified microchips meaning that the modification is stable and uniform. Also, compared with the channel height (∼40 μm) and cell size (∼15 μm), this single nanoparticle layer is very low and cells will not get stuck in the channel because of this surface modification.
Capture efficiency
Suspended cells can be maintained at a high density in a flask at all times, while adherent cells are limited by the flask surface area.37 Thus, suspended cells typically had higher cell population and cell concentration during measurements than adherent cells. In addition to CCRF-CEM cells, in this part, we employed five more cell lines, including three suspended cell lines and two adherent cell lines, to have a more comprehensive evaluation of our NP-modified chip for the detection of cells in liquid biopsies. The range of cell concentrations for suspended cells was 1040–4450 cells per μL, while it was 46–418 cells per μL for adherent cells. The latter served to mimic the rare CTCs originating from different primary tumors.HL-60, Ramos, and Jurkat are three suspended cell lines having high CD71 expression. The capture efficiency of NP-modified HB chips for CD71 + HL-60 cells reached as high as 94 ± 3%, exhibiting an average increase of 14 ± 6% (Fig. 4A). Similar to HL-60, the capture efficiencies of CD71 + Ramos cells and CD71 + Jurkat cells were 86 ± 3% and 89 ± 9%, exhibiting an increase of 21 ± 8% and 20 ± 4% compared with normal HB chips, respectively. Besides, the T-test was conducted to evaluate the difference between NP-modified HB chip and normal HB chip capture results. Significant increases of capture efficiency were observed for all of these three cell lines with p values of 0.0004, 0.0001, and 0.004, respectively (95% confidence interval).
 | ||
| Fig. 4 Capture efficiency measurements of different cell lines (n = 6 for each cell line). (A) Suspended cells, including HL-60, Ramos, and Jurkat. (B) Adherent cells, including PC-3 and MDA-231. The capture efficiencies of NP-modified HB chips for HL-60, Ramos, Jurkat, PC-3, and MDA-231 were 94 ± 3%, 85 ± 3%, 89 ± 9%, 74 ± 14%, and 81 ± 6%, exhibiting an improvement of 14 ± 6%, 21 ± 8%, 20 ± 4%, 15 ± 18%, and 14 ± 9%, respectively. The p values for these five cell lines were 0.0004, 0.0001, 0.004, 0.0334, and 0.0008, respectively. These improvements demonstrated a great upgrade of traditional microchips for cancer cell detection. | ||
Even with the relative lower cell concentration compared with suspended cells, the capture efficiency of MDA-231 cells still reached 81 ± 6% in the case of NP-modified HB chips, and showed a significant positive enhancement with an average increase of 14 ± 9% (p = 0.0008, 95% confidence interval) (Fig. 4B). In the case of PC-3, only one of those six trails of NP-modified HB chips showed a decrease of capture efficiency compared with normal HB chips, while others still have an increase of 19 ± 15%. The mean capture efficiency of all six trials was 74 ± 14%, and this high efficiency is sufficient for capturing cells in a concentration as low as 46 cells per μL. The average increase of all six trials was 15 ± 18%, having an overall p value of 0.0334 (95% confidence interval). Based on our previous work, the CD71 expression of PC-3 cells is slightly higher than that of the MDA-231 cells.36 However, in this work, the capture efficiency of PC-3 cells, especially in the first three trials, was lower than that of the MDA-231 cells. The main reason was because of the lower cell population. The concentration for the PC-3 cells was 46–206 cells per μl, while it was 272–418 cells per μl for MDA-231 cells. Similar observations were obtained between adherent cells and suspended cells. As discussed above, the suspended cells had cell concentrations over 3 times higher than the adherent cells, and as a result, all adherent cells showed lower capture efficiency than suspended cells. However, the increased sensitivity using our NP-modified HB chips regarding all different cancer cell lines is particularly important and sufficient for an initial disease diagnosis as well as for minimum residual disease detection when the CTCs are at a very low concentration.
Capture purity
Capture purity is an important indicator of chip performance for rare cell isolation. In this work, we spiked the CCRF-CEM cells in blood samples at the percentage of 20%, 10%, and as low as 1%. As shown in Fig. 5, captured CCRF-CEM cells and non-specific bonded blood cells were observed under different filters and enumerated by image J. Consistently high capture purity was obtained for NP-modified HB chips at all spike percentages (Fig. 6). Based on measurements, a higher spike percentage leads to higher capture purity. In the case of NP-modified chips, the capture purity of 20% cancer cell concentration reached 65 ± 13%, while it was 32 ± 7% for 1% concentration. The driving factor is that the nonspecific binding of leukocytes tends to stay constant, so increasing the number of target cells increases the capture purity.38 Also, the NP-modified chip should have better capture performance than the normal HB chip due to the fact that the nanostructures inside the chip increase the interactions between cells and antibodies. Thus more desired cells should be theoretically captured in NP-modified HB chips than normal HB chips. Compared with the normal HB chip, the NP-modified HB chip had an improvement of capture purity of 9 ± 7%, 10 ± 5%, and 10 ± 7% for 20%, 10%, and 1% spiked cancer cell percentages, respectively. These enhancements illustrated that the NP-modified HB chip was able to isolate cancer cells in different concentrations, and had a satisfactory performance even at low percentage. Based on the T-test results, the p values for 20%, 10%, and 1% measurements were 0.0498, 0.0096, and 0.0114, respectively, meaning that the improvement of our NP-modified chip was extremely effective and significant (95% confidence interval). | ||
| Fig. 5 Representative of images under different filters. White light image (A) showed all captured desired cells and non-specific bonded blood cells. Image (B) showed captured CCRF-CEM cells stained with Mitotracker Green. Image (C) showed all non-specific bonded blood cells stained with propidium iodide. Target cells and non-specific bonded blood cells were observed and counted under different channels of the microscope for capture purity analysis. | ||
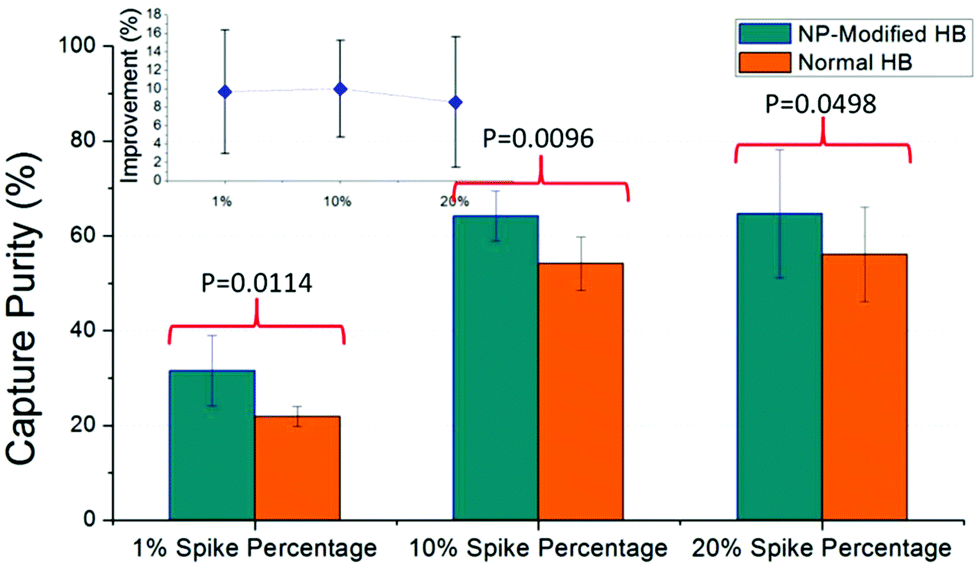 | ||
| Fig. 6 CCRF-CEM cells were spiked into blood samples at 20%, 10%, and 1% spike percentage, and the mixture was measured on both NP-modified HB chips and normal HB chips. Capture purity was calculated to evaluate our chip performance for CD71 + cell capture (n = 6 for each spike percentage). The capture purities of the NP-modified HB chip for 20%, 10%, and 1% spike percentages were 65 ± 13%, 64 ± 5%, and 32 ± 7%, exhibiting an increase of 9 ± 7%, 10 ± 5%, and 10 ± 7%, respectively. The inset represents the improvement of capture purity. | ||
Capture purity of a microfluidic device was mainly decided by the non-specific binding of non-target cells and the specific binding of target cells. Thus in this work, the captured cell numbers of leukocytes and cancer cells are also important parameters to evaluate the effect of NP modification. Using 20% spike percentage as an example, the number of nonspecifically bound leukocytes was similar in NP-HB chips and HB chips (284 ± 67 vs. 213 ± 141, respectively), while the specific capture of CD71 + CCRF-CEM cells was over 2-fold higher in the NP-HB chip (688 ± 288 vs. 297 ± 179, respectively). Similar results were obtained and are listed in Table 1 when the spike percentages were 10% and 1%. This observation showed that the silica nanoparticle surface modification would not lead to the increase of non-specific binding, and the potential factor of the increased number of non-specifically bound blood cells in 20% measurements was due to the higher concentrations of the total cells. This enhanced performance, especially in the low cancer cell percentage, is of great importance and can be further improved by minimizing non-specific bindings.
Patient derived cell analysis
The separation of the T-cell leukemia patient-derived cell sample, COG-LL-332, was included in this study to assess the veracity of our work. Using COG-LL-332 cells spiked into blood, we simulated pediatric acute lymphoblastic leukemia (ALL) with lymphoblasts in different concentrations. This disease was traditionally validated when lymphoblasts constituted 30% or more of the nucleated cells in patients’ peripheral blood, while the cutoff value has been adjusted to 20% according to the most recent classification system set by the World Health Organization (WHO).39,40 Therefore in this study, we spiked COG-LL-332 cells in concentrations ranging from 1% to 20% of total leukocytes to evaluate the robustness of our NP-modified chip in capturing the desired cells, as well as to prove the enhanced performance compared with normal herringbone chips in a more practical measurement. After surface modification, the COG-LL-332 capture purity increased from 18%–61% of normal HB chips to 35%–71% of NP-modified HB chips depending on initial concentrations. Especially at the percentage as low as 1%, the increase of capture reached 17 ± 11% with a p value of 0.044. The results demonstrated that the higher spike percentage had higher capture purity in both cases of NP-modified HB chips and normal HB chips (Fig. 7). However, it is important to note that capture purity at the lowest level (41 ± 8%) was still sufficient for cancer cell detection especially in a very low concentration, and can be further improved by mitigating nonspecific bindings. With the surface modification, all spike percentages showed a significant increase (p < 0.05, 95% confidence interval, Fig. 7). Also, this high capture purity at low concentration illustrated that our nanoparticle modified herringbone chip was able to reliably detect ALL lymphoblasts at 1/20 of the WHO limit.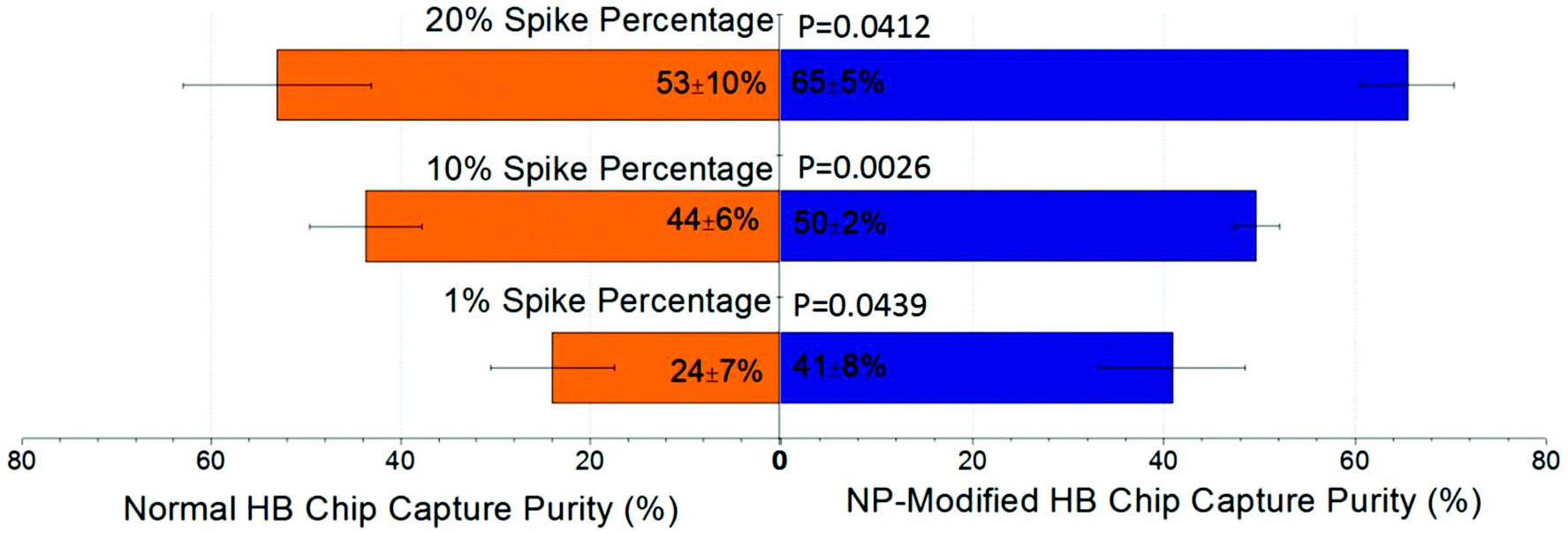 | ||
| Fig. 7 Comparison of capture purity between normal HB chips and NP-modified HB chips (n = 3 for each spike percentage). Higher spike percentage resulted in higher capture purity and the increase of capture purity was observed regarding all COG-LL-332 cell concentrations. Increases of capture purity were 17 ± 11%, 6 ± 8%, and 12 ± 9% for 1%, 10%, and 20%, respectively. P values of all spike percentages were smaller than 0.05 indicating that all capture purity improvements of NP-modified chips were significant. | ||
Experimental
Cell line preparation
Seven cell lines were involved in this study: a human prostate cancer cell line, PC-3 (ATCC CRL-1435); a human breast cancer cell line, MDA-231 (ATCC HTB-26); a human acute myeloid leukemia cell line, HL-60 (ATCC PTS-CCL-240); a human B-cell lymphoma cell line, Ramos (ATCC CRL-1596); a human acute T cell leukemia cell line, Jurkat (ATCC CRL-2900); a human T lymphoblast cell line, CCRF-CEM (ATCC CRM-CCL-119); and a patient-derived pediatric acute lymphoblastic leukemia cell line, COG-LL-332 (http://www.COGcells.org). All cells were cultured in RPMI 1640 medium (Hyclone) supplemented with 10% fetal bovine serum (Hyclone) and penicillin–streptomycin stabilized solution (Sigma-Aldrich). The cells were subcultured every day during the measurements. Cell flasks were stored in an incubator at 37 °C and under a 5% CO2 atmosphere. 6 mL of cell suspension was obtained from the flask for each measurement. The cells were centrifuged at 4500 rpm for 5 min to remove the dead cells and cell debris. The cell pellet was resuspended in phosphatebuffered saline (PBS) to achieve final volume of 1000 μL. 1 μL MitoTracker Green (Invitrogen) (1 mM) was added to stain cells. After 45 min of incubation, cells were washed with PBS solution 3 times, and finally resuspended with 3% bovine serum albumin (BSA) in PBS solution to achieve final volume of 500 μL. The range of final cell concentrations for all measurements was from 46 to 4450 cells per μL.Blood sample preparation
Whole blood samples (BD Multi-Check Control) were purchased from Becton-Dickinson and stored in the refrigerator at 4 °C until use. Upon use, the blood sample was shaken to be homogeneous, and processed at room temperature. The blood lysis process followed our lab procedures.14 During the blood lysis process, 900 μL distilled water was added into each 100 μL blood sample for exactly 30 s. Immediately afterward, 110 μL of concentrated saline buffer (10×) was added to restore osmolarity. The mixture was then centrifuged at 4500 rpm for 5 min. The lysed erythrocytes in the supernatant were discarded, and the white blood cells in the bottom were collected. The pellet of these leukocytes was resuspended with PBS to reach final volume of 1000 μL. 10 μL propidium iodide (1.0 mg mL−1 solution in water, Invitrogen) was added into the diluted sample to stain the leukocytes for 20 min. The mixture was then washed with PBS and centrifuged at 4500 rpm for 5 min 3 times to remove the excess dyes. Finally, the sample was resuspended with 3% BSA in PBS buffer to reach the final volume of 100 μL. The leukocyte concentration ranged from 540 to 1470 cells per μL for all measurements. Blood samples were obtained from a commercial source (Becton Dickinson Multi-Check Control), and are deidentified.Microfluidic device fabrication
Herringbone (HB) chips were fabricated using soft photolithography.41 A herringbone structure design was printed on a high resolution mask (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 dpi, CAD Art Science). Spin coating of negative photoresist SU-8 (Micro Chem) on a 4-inch silicon wafer at 500 rpm for 6 s, followed by 1000 rpm for 30 s, formed a 40 μm thick layer. The wafer was prebaked on a hotplate at 95 °C for 6 min, and then exposed under UV light for 15 s with the high-resolution mask on the top. After the UV light exposure, the wafer was heated again on a hotplate at 95 °C for another 6 min. Then, the wafer was developed with an SU-8 developer (Micro Chem) for 6 min to remove the unexposed photoresist. Finally, the wafer was incubated in a vacuum chamber together with the 1H,1H,2H,2H-perfluorooctyltrichlorosilane (Alfa Aesar) atmosphere overnight to form a hydrophobic surface.
000 dpi, CAD Art Science). Spin coating of negative photoresist SU-8 (Micro Chem) on a 4-inch silicon wafer at 500 rpm for 6 s, followed by 1000 rpm for 30 s, formed a 40 μm thick layer. The wafer was prebaked on a hotplate at 95 °C for 6 min, and then exposed under UV light for 15 s with the high-resolution mask on the top. After the UV light exposure, the wafer was heated again on a hotplate at 95 °C for another 6 min. Then, the wafer was developed with an SU-8 developer (Micro Chem) for 6 min to remove the unexposed photoresist. Finally, the wafer was incubated in a vacuum chamber together with the 1H,1H,2H,2H-perfluorooctyltrichlorosilane (Alfa Aesar) atmosphere overnight to form a hydrophobic surface.
PDMS was mixed with its curing agent at the weight ratio of 10:1. The mixture was degassed in a vacuum chamber for 30 min, and then poured on the silicon wafer surface, followed by 1 hour of baking at 95 °C. The cured PDMS was peeled off from the silicon wafer, and an 18-gauge blunt needle was used to create an inlet and outlet for the channel. The PDMS piece and a glass slide were processed under oxygen plasma for irreversible bonding. The 30-gauge poly(tetrafluoroethylene) (PTFE) tubes were inserted into the inlet and outlet to introduce samples into the channel.
Nanoparticle modification
Branched polyethylenimine (BPEI) (Mw = 70000, primary, secondary, and tertiary amine groups in approximately 25/50/25 ratio) was purchased from PolyScience. Silica nanoparticles (with an average size of 200 nm) were purchased from General Engineering & Research. The microfluidic chips were modified by the layer-by-layer (LbL) method as described in previous work.42 Briefly, a PDMS replica and a flat PDMS slab were treated with oxygen plasma for 50 s and bonded together to form the microfluidic channels. Afterwards, BPEI solution (2 mg mL−1, pH 9) was immediately injected into the microfluidic channel and allowed to sit for 5 min for absorption, then the solution was removed by vacuum and the channels were rinsed with 1 mL DI-water three times, then silica nanoparticles (NPs) (10 wt% in DI water) were introduced into the channels and allowed a 5 min of absorption time, after which the device was rinsed with DI water. The nanoparticle injection process was repeated 3 times at room temperature.
Affinity surface modification
The bare PDMS-based normal herringbone chip was modified with nanoparticles to form the NP-modified HB chip. And then the same affinity surface modification was applied on both normal HB chips and NP-modified HB chips. The affinity surface modification used a LbL deposition strategy. 8 μL Biotinylated Bovine Serum Albumin (Bio-BSA, Sigma-Aldrich, 5 μL of 1 mg mL−1 in mM Tris-HCl, pH 8.0, 50 mM NaCl) was introduced into the channel for 45 min of incubation. Then, T50 (10 mM Tris-HCl buffer) was infused to rinse the channel and remove the excess Bio-BSA. Then 8 μL neutravidin solution (0.2 mg mL−1 in 10 mM Tris-HCl buffer) was loaded into the channel for 15 min. After 15 min of incubation, the channel was rinsed with T50 solution, followed by distilled water and finally air drying. The chip was then stored in the refrigerator at 4 °C until use. Before the start of each experiment, the neutravidin-modified chip was coated with 6 μL biotin mouse anti-human CD71 (50 μg mL−1, BD Pharmingen) for 20 min of incubation at room temperature.On-chip detection
Before sample loading, the channel was filled with 3% BSA in PBS solution to remove bubbles. All cell concentrations were measured using a hemocytometer to calculate the spike percentage and capture efficiency.For the chip reproducibility and capture efficiency analysis, six cell lines were involved with the concentration range of 46–4450 cells per μL. The sample was introduced into the microfluidic chip at a continuous flow rate of 0.06 mL h−1. The higher flow rate leads to less cell capture, while the lower flow rate can result in more non-specific bindings and worse visualization during cell counting. We tested the flow rate ranging from 0.02 ml h−1 to 0.12 ml h−1 with every 0.02 ml h−1 as a step and finally selected 0.06 ml h−1 by taking all considerations. Capture efficiency was calculated as (eqn (1)):
 | (1) |
For capture purity analysis, CCRF-CEM cells and COG-LL-332 cells were diluted in 3% BSA in PBS solution. The cell sample was spiked into the blood sample at the percentage of 1%, 10%, and 20%. The spike percentage was calculated as (eqn (2)):
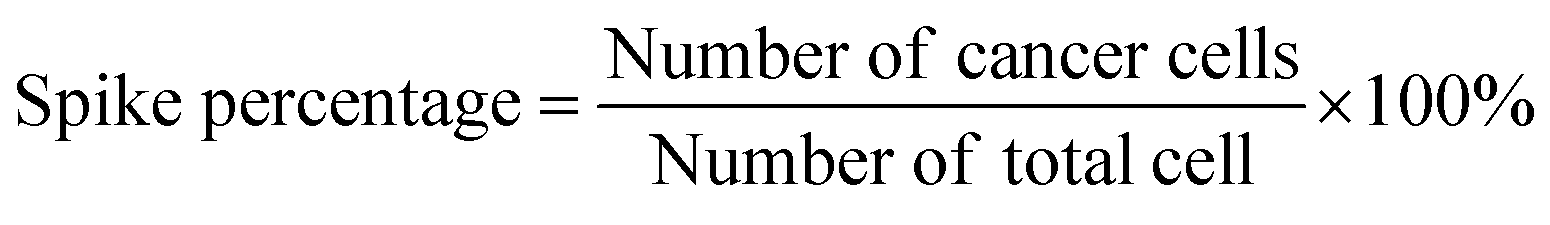 | (2) |
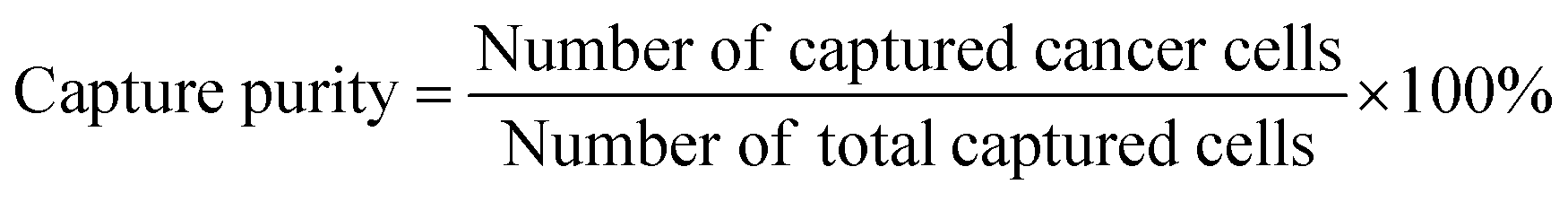 | (3) |
All capture efficiency and purity measurements were repeated 6 times. For the re-use of each chip, the chip was flushed with 70% ethanol and DI water 5 times, respectively, right after each experiment. And then the used chip was kept in an 80 °C oven overnight. Every time before running a new experiment, the used chip would be checked under the microscope to ensure no leftovers in the channel.
An inverted epifluorescence microscope (Olympus IX71) was used for the visualization of both fluorescent cells and cells under white light with appropriate filters and a 0.3 NA 10× objective lens. Images were obtained with a cooled charge coupled device (CCD) camera coupled to the microscope. The samples were kept flowing during image acquisition. Moving cells were discriminated from the bound cells by shape. Images were analyzed via Image-J software (National Institutes of Health).
Flow cytometry analysis
Flow cytometry analysis was conducted prior to the on-chip detection. Cells for flow cytometry analysis were obtained together with the cells for the corresponding on-chip detection measurement from the same flask. A 6 mL cell suspension was used for each measurement. The cell suspension was centrifuged at 4500 rpm to remove the dead cells and cell debris and the pellet was resuspended into two vials. One vial was diluted to 500 μL with PBS solution as the unstained control. Another one was diluted to 1000 μL with PBS solution, and stained with 5 μL FITC-anti-CD71 (BD Biosciences) for 20 minutes. The stained cell vial was finally centrifuged at 4500 rpm for 5 min and diluted to 500 μL with PBS.Conclusion
In this work, silica nanoparticles were shown to be able to increase surface interactions and improve cell capture for the isolation of cancer cells from liquid biopsies. The NP-modified HB chip had a better performance over the normal HB chip regarding both capture efficiency and capture purity. The surface treated microchip is capable of isolating cancer cells with high capture efficiency and purity even at a low cell concentration. The T-Test statistically proved that all improvements are effective and significant. In addition, the whole experimental process of our approach takes only 2 hours with a per-chip cost of approximately $2 USD. Thus, compared with other techniques for cancer cell detection, our method: (1) is much cheaper and simple; (2) consumes less sample; (3) sorts CTCs from blood samples directly without any pre-processing of blood which prevents the loss and destruction of CTCs; (4) isolation process is gentle and stable, therefore viable cells can be obtained whereas, for example, the CellSearch™ system can only isolate fixed, non-viable cells; etc. However, there are still some challenges needed to be addressed in our work. First, CTC concentrations range from one to thousands in the patient blood sample. We did not examine the ability of our chip in capturing the desired cell at ultra-low percentage (for example 0.01%). In this case, a longer sample loading time will be needed to allow enough desired cells across the channel. Also, the commercial blood sample we purchased for the lab use has been sterilized and processed. Blood viscosity and blood cell concentration are normally lower than those of the real patient samples. Therefore, the durability of the surface coating and increased non-specific bindings need to be reconsidered if patient samples were applied. However, all these issues can be easily fixed and optimized by changing the channel design and the manner of sample loading. All in all, owing to the portable nature of the microfluidic device, our method has great potential for the future POC detection of a wide range of cancers with high sensitivity and accuracy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from The CH Foundation and CPRIT (RP180862). Z. D. is partially supported by funding from CPRIT (RP170817).References
- A. Jemal, F. Bray, M. M. Center, J. Ferlay, E. Ward and D. Forman, Global cancer statistics, CA-Cancer J. Clin., 2011, 61(2), 69–90 CrossRef.
- M. Cristofanilli, G. T. Budd, M. J. Ellis, A. Stopeck, J. Matera, M. C. Miller, J. M. Reuben, G. V. Doyle, W. J. Allard and L. W. Terstappen, Circulating tumor cells, disease progression, and survival in metastatic breast cancer, N. Engl. J. Med., 2004, 351(8), 781–791 CrossRef CAS.
- Y. Tan, W. Liu, Z. Zhu, L. Lang, J. Wang, M. Huang, M. Zhang and C. Yang, Selection and identification of transferrin receptor-specific peptides as recognition probes for cancer cells, Anal. Bioanal. Chem., 2018, 410(3), 1071–1077 CrossRef CAS.
- H. J. Yoon, T. H. Kim, Z. Zhang, E. Azizi, T. M. Pham, C. Paoletti, J. Lin, N. Ramnath, M. S. Wicha and D. F. Hayes, Sensitive capture of circulating tumour cells by functionalized graphene oxide nanosheets, Nat. Nanotechnol., 2013, 8(10), 735 CrossRef CAS PubMed.
- K. Pantel, R. H. Brakenhoff and B. Brandt, Detection, clinical relevance and specific biological properties of disseminating tumour cells, Nat. Rev. Cancer, 2008, 8(5), 329 CrossRef CAS.
- Y.-F. Sun, X.-R. Yang, J. Zhou, S.-J. Qiu, J. Fan and Y. Xu, Circulating tumor cells: advances in detection methods, biological issues, and clinical relevance, J. Cancer Res. Clin. Oncol., 2011, 137(8), 1151–1173 CrossRef.
- G. Vona, A. Sabile, M. Louha, V. Sitruk, S. Romana, K. Schütze, F. Capron, D. Franco, M. Pazzagli and M. Vekemans, Isolation by size of epithelial tumor cells: a new method for the immunomorphological and molecular characterization of circulating tumor cells, Am. J. Pathol., 2000, 156(1), 57–63 CrossRef CAS.
- G. M. Hayes, R. Busch, J. Voogt, I. M. Siah, T. A. Gee, M. K. Hellerstein, N. Chiorazzi, K. R. Rai and E. J. Murphy, Isolation of malignant B cells from patients with chronic lymphocytic leukemia (CLL) for analysis of cell proliferation: validation of a simplified method suitable for multi-center clinical studies, Leuk. Res., 2010, 34(6), 809–815 CrossRef CAS.
- M. Cristofanilli, The biological information obtainable from circulating tumor cells, Breast, 2009, 18, S38–S40 CrossRef.
- M. Tewes, B. Aktas, A. Welt, S. Mueller, S. Hauch, R. Kimmig and S. Kasimir-Bauer, Molecular profiling and predictive value of circulating tumor cells in patients with metastatic breast cancer: an option for monitoring response to breast cancer related therapies, Breast Cancer Res. Treat., 2009, 115(3), 581 CrossRef.
- T. Fujii, PDMS-based microfluidic devices for biomedical applications, Microelectron. Eng., 2002, 61, 907–914 CrossRef.
- W. A. Jefferies, M. R. Brandon, S. V. Hunt, A. F. Williams, K. C. Gatter and D. Y. Mason, Transferrin receptor on endothelium of brain capillaries, Nature, 1984, 312(5990), 162 CrossRef CAS.
- J. E. Shindelman, A. E. Ortmeyer and H. H. Sussman, Demonstration of the transferrin receptor in human breast cancer tissue. Potential marker for identifying dividing cells, Int. J. Cancer, 1981, 27(3), 329–334 CrossRef CAS.
- W. Li, Y. Zhang, C. P. Reynolds and D. Pappas, Microfluidic Separation of Lymphoblasts for the Isolation of Acute Lymphoblastic Leukemia Using the Human Transferrin Receptor as a Capture Target, Anal. Chem., 2017, 89(14), 7340–7347 CrossRef CAS.
- S. Wang, Y. Wan and Y. Liu, Effects of nanopillar array diameter and spacing on cancer cell capture and cell behaviors, Nanoscale, 2014, 6(21), 12482–12489 RSC.
- S. Mondésert-Deveraux, D. Aubry and R. Allena, In silico approach to quantify nucleus self-deformation on micropillared substrates, Biomech. Model. Mechanobiol., 2019, 1–15 Search PubMed.
- L. Ma, G. Yang, N. Wang, P. Zhang, F. Guo, J. Meng, F. Zhang, Z. Hu, S. Wang and Y. Zhao, Trap Effect of Three-Dimensional Fibers Network for High Efficient Cancer-Cell Capture, Adv. Healthcare Mater., 2015, 4(6), 838–843 CrossRef CAS.
- F. Zhang, Y. Jiang, X. Liu, J. Meng, P. Zhang, H. Liu, G. Yang, G. Li, L. Jiang and L.-J. Wan, Hierarchical nanowire arrays as three-dimensional fractal nanobiointerfaces for high efficient capture of cancer cells, Nano Lett., 2015, 16(1), 766–772 CrossRef.
- E. Guihen and J. D. Glennon, Nanoparticles in separation science—recent developments, Anal. Lett., 2003, 36(15), 3309–3336 CrossRef CAS.
- X.-R. Xia, N. A. Monteiro-Riviere and J. E. Riviere, An index for characterization of nanomaterials in biological systems, Nat. Nanotechnol., 2010, 5(9), 671 CrossRef CAS.
- M. A. Dobrovolskaia and S. E. McNeil, Immunological properties of engineered nanomaterials, Nat. Nanotechnol., 2007, 2(8), 469 CrossRef CAS.
- H. J. Yoon, M. Kozminsky and S. Nagrath, Emerging role of nanomaterials in circulating tumor cell isolation and analysis, ACS Nano, 2014, 8(3), 1995–2017 CrossRef CAS.
- I. Wong and C.-M. Ho, Surface molecular property modifications for poly (dimethylsiloxane)(PDMS) based microfluidic devices, Microfluid. Nanofluid., 2009, 7(3), 291 CrossRef CAS.
- A.-J. Wang, J.-J. Xu, Q. Zhang and H.-Y. Chen, The use of poly (dimethylsiloxane) surface modification with gold nanoparticles for the microchip electrophoresis, Talanta, 2006, 69(1), 210–215 CrossRef CAS PubMed.
- A. A. S. Bhagat, H. Bow, H. W. Hou, S. J. Tan, J. Han and C. T. Lim, Microfluidics for cell separation, Med. Biol. Eng. Comput., 2010, 48(10), 999–1014 CrossRef.
- S. Wang, A. Thomas, E. Lee, S. Yang, X. Cheng and Y. Liu, Highly efficient and selective isolation of rare tumor cells using a microfluidic chip with wavy-herringbone micro-patterned surfaces, Analyst, 2016, 141(7), 2228–2237 RSC.
- Z. Dong, C. C. Ahrens, D. Yu, Z. Ding, H. Lim and W. Li, Cell isolation and recovery using hollow glass microspheres coated with nanolayered films for applications in resource-limited settings, ACS Appl. Mater. Interfaces, 2017, 9(18), 15265–15273 CrossRef CAS.
- Z. Dong, D. Yu, Q. Liu, Z. Ding, V. J. Lyons, R. K. Bright, D. Pappas, X. Liu and W. Li, Enhanced capture and release of circulating tumor cells using hollow glass microspheres with a nanostructured surface, Nanoscale, 2018, 10, 16795–16804 RSC.
- K. Rittenhouse-Diakun, H. V. Leengoed, J. Morgan, E. Hryhorenko, G. Paszkiewicz, J. Whitaker and A. Oseroff, The role of transferrin receptor (CD71) in photodynamic therapy of activated and malignant lymphocytes using the heme precursor δ-aminolevulinic acid (ALA), Photochem. Photobiol., 1995, 61(5), 523–528 CrossRef CAS.
- F. Lin and A. W. Girotti, Photodynamic action of merocyanine 540 on leukemia cells: iron-stimulated lipid peroxidation and cell killing, Arch. Biochem. Biophys., 1993, 300(2), 714–723 CrossRef CAS.
- R. Newman, C. Schneider, R. Sutherland, L. Vodinelich and M. Greaves, The transferrin receptor, Trends Biochem. Sci., 1982, 7(11), 397–400 CrossRef CAS.
- S. Kim, S.-I. Han, M.-J. Park, C.-W. Jeon, Y.-D. Joo, I.-H. Choi and K.-H. Han, Circulating tumor cell microseparator based on lateral magnetophoresis and immunomagnetic nanobeads, Anal. Chem., 2013, 85(5), 2779–2786 CrossRef CAS.
- P. Chen, Y.-Y. Huang, K. Hoshino and X. Zhang, Multiscale immunomagnetic enrichment of circulating tumor cells: from tubes to microchips, Lab Chip, 2014, 14(3), 446–458 RSC.
- D. K. Marsee, G. S. Pinkus and H. Yu, CD71 (transferrin receptor) an effective marker for erythroid precursors in bone marrow biopsy specimens, Am. J. Clin. Pathol., 2010, 134(3), 429–435 CrossRef.
- K. Kato, M. Toda and H. Iwata, Antibody arrays for quantitative immunophenotyping, Biomaterials, 2007, 28(6), 1289–1297 CrossRef CAS.
- V. J. Lyons, A. Helms and D. Pappas, The effect of protein expression on cancer cell capture using the Human Transferrin Receptor (CD71) as an affinity ligand, Anal. Chim. Acta, 2019, 1076, 154–161 CrossRef CAS PubMed.
- D. Pappas, Practical cell analysis, John Wiley & Sons, 2010 Search PubMed.
- Y. Zhang, V. Lyons and D. Pappas, Fundamentals of affinity cell separations, Electrophoresis, 2018, 39(5–6), 732–741 CrossRef CAS.
- M. Antica, Acute Leukemia-The Scientist's Perspective and Challenge, 2011 Search PubMed.
- R. Weinkauff, E. H. Estey, P. Starostik, K. Hayes, Y. O. Huh, C. Hirsch-Ginsber, M. Andreeff, M. Keating, H. M. Kantarjjan and E. J. Freireich, Use of peripheral blood blasts vs bone marrow blasts for diagnosis of acute leukemia, Am. J. Clin. Pathol., 1999, 111(6), 733–740 CrossRef CAS.
- M. A. Unger, H.-P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Monolithic microfabricated valves and pumps by multilayer soft lithography, Science, 2000, 288(5463), 113–116 CrossRef CAS PubMed.
- Z. Dong, L. Tang, C. C. Ahrens, Z. Ding, V. Cao, S. Castleberry, J. Yan and W. Li, A benchtop capillary flow layer-by-layer (CF-LbL) platform for rapid assembly and screening of biodegradable nanolayered films, Lab Chip, 2016, 16(23), 4601–4611 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Capture efficiency of six different cancer cell lines on both NP-modified HB chips and non-modified HB chips; nanoparticle size distribution. See DOI: 10.1039/c9an01719d |
| This journal is © The Royal Society of Chemistry 2020 |