
A bottlebrush-architectured dextran polyprodrug as an acidity-responsive vector for enhanced chemotherapy efficiency†
Tian
Zhang
ab,
Yajun
Wang
ab,
Xianbin
Ma
ab,
Cuilan
Hou
c,
Shuangyu
Lv
d,
Die
Jia
ab,
Yi
Lu
ab,
Peng
Xue
 ab,
Yuejun
Kang
*ab and
Zhigang
Xu
*ab
ab,
Yuejun
Kang
*ab and
Zhigang
Xu
*ab
aKey Laboratory of Luminescent and Real-Time Analytical Chemistry (Southwest University), Ministry of Education, School of Materials and Energy, Southwest University, Chongqing, 400715, P. R. China. E-mail: zgxu@swu.edu.cn; yjkang@swu.edu.cn; Fax: +86-023-68253204; Tel: +86-023-68253792
bChongqing Engineering Research Center for Micro-Nano Biomedical Materials and Devices, Southwest University, Chongqing 400715, P. R. China
cDepartment of Cardiology, Shanghai Children's Hospital, Shanghai Jiaotong University, No. 355 Luding Road, Shanghai, 200062, P.R. China
dSchool of Basic Medical Sciences, Henan University, Kaifeng 475001, China
First published on 14th November 2019
Abstract
Compared to normal tissues, unique conditions in the tumor microenvironment, such as a lower pH, can induce accurate release of a drug into specific lesions. This strategy provides an efficient approach to overcome the issues of unexpected drug leakage and poor circulation stability, thereby reducing the side effects and enhancing the effect of cancer treatment. In this study, we designed a class of acid activatable supramolecular nano-prodrugs (DOM@DOX) with a bottlebrush architecture based on the dextran (DEX) polysaccharide, which connects with a hydrophilic polyethylene glycol chain by atom transfer radical polymerization and further conjugates with an anticancer drug doxorubicin (DOX) at the backbone of the copolymer via an acidity-responsive hydrazine bond. Furthermore, the DOM@DOX prodrug has a high drug loading up to 48 wt% for DOX, and the prodrug can maintain a stable nano-sized spherical shape in aqueous solution by a self-assembly strategy. In an acidic environment inside tumor cells, the hydrazine bond of the prodrug breaks, leading to the release of DOX from parental micelles. Owing to the small size of the carrier, the prodrug exhibits good intratumoral permeability, good circulation stability and significant tumor suppression efficiency in tumor-bearing mouse models, which is beneficial for the development of new generation nanomedicine for enhanced chemotherapy.
Introduction
Cancer has become one of the most leading threats to human health worldwide with 18.1 million new cases and 9.6 million cancer-related deaths in 2018.1 Effective control and treatment of cancer are among the top priorities in the healthcare and pharmaceutical industries. In the past few decades, nanotherapeutics have emerged rapidly, which utilize nanoparticles to deliver drugs into specific tumor sites. Compared to traditional chemotherapy, nanocarriers can overcome some common issues in drug delivery, such as rapid clearance of drugs from circulating blood, poor cellular uptake, limited bioavailability of the drug at target tissues and severe side effects.2–6 According to previous reports, various nanocarriers including liposomes,7–9 drug amphiphiles,10–13 inorganic nanoparticles14–16 and polymeric drugs17–24 have been developed to address the challenges of tumor treatment. The abilities to evade blood elution, penetrate and accumulate into deep tumoral tissues, and initiate selective drug release are the most desirable features of novel nanotherapeutic platforms,25,26 and are still urgently needed to overcome the inevitable biological barriers.It is known that polymer-based nanotherapeutics have stable or convertible chemical structures and excellent biocompatibility, and may respond to the tumor microenvironment and produce a remarkable therapeutic effect. Polymeric drug delivery systems typically including linear,27–30 star31–34 and other polymeric nanostructures35–38 have been reported. Linear amphiphilic polymeric prodrugs can self-assemble into micelles in an aqueous solution and enter the tumors due to an enhanced permeability and retention (EPR) effect.39–42 Wang et al. prepared amphiphilic block copolymers (PEOMP-b-PBYOMP) by ring-opening polymerization (ROP) and loaded camptothecin (CPT) by physical embedding to obtain a well-dispersed nanotherapeutic, which showed considerably enhanced toxicity to cancer cells.43 However, the existing polymer-based prodrug delivery systems still face huge challenges owing to complex physiological barriers.44–46 For example, the micellar nanostructure could be easily dissociated depending on the concentration of the polymer and other chemical conditions. Additionally, it is difficult for many drug delivery systems to meet high drug loading and low drug leakage simultaneously.47 Dextran (DEX) is a polysaccharide with good biocompatibility, and is water-soluble, nondigestible and inert for chemotherapeutics. There are abundant hydroxyl groups on DEX molecules, making it possible to realize a wide range of chemical modifications on DEX.48,49 We have previously reported a class of reduction-responsive unimolecular micelles by conjugating a dextran-camptothecin drug through disulfide bonds and they exhibited enhanced antitumor activity against multiple tumor cells,48 serving as an advantageous candidate of DEX for the construction of a non-linear polymeric prodrug for effective cancer treatment.
Herein, we have reported a bottlebrush-architectured DEX prodrug of DEX-POEGMA-b-PDOX (denoted as DOM@DOX) prepared through a two-step polymerization. The anticancer drug DOX was conjugated via pH-responsive hydrazone bonds to the DEX framework as the hydrophobic block to realize controlled release of the drug in the acidic tumor environment. The poly(ethylene glycol) methyl ether methacrylate (OEGMA) monomer was conjugated to the backbone of the copolymer as the hydrophilic block, resulting in an amphiphilic property of the prodrug to achieve self-assembly in aqueous solution (Scheme 1). Compared with a nanomedicine based line-architectured polymer, the bottlebrush-architectured polymer shows a densely spaced polymerization chain (e.g. hydrophilic POEGMA),50 which can provide steric shielding for the loading of small drug molecules, producing minimal side-effects, prolonged blood circulation and enhanced therapeutics. Specifically, the DOM@DOX prodrug delivery system may be beneficial in cancer treatment in the following aspects: (i) the bottlebrush-architecture endows DOM@DOX with the shielding OEGMA layer, which can enhance the solubility and stability of prodrugs in blood; (ii) the pH-sensitive hydrazone bond linking with DOX can be hydrolyzed shedding DOX in the acidic endo/lysosomal microenvironment (pH ∼5.0),51 leading to the real-time release of DOX. Therefore, this acidity-responsive DOM@DOX delivery system is expected to show good biocompatibility and permeability into solid tumors, and thereby enhance tumor therapeutic efficacy with reduced side effects.
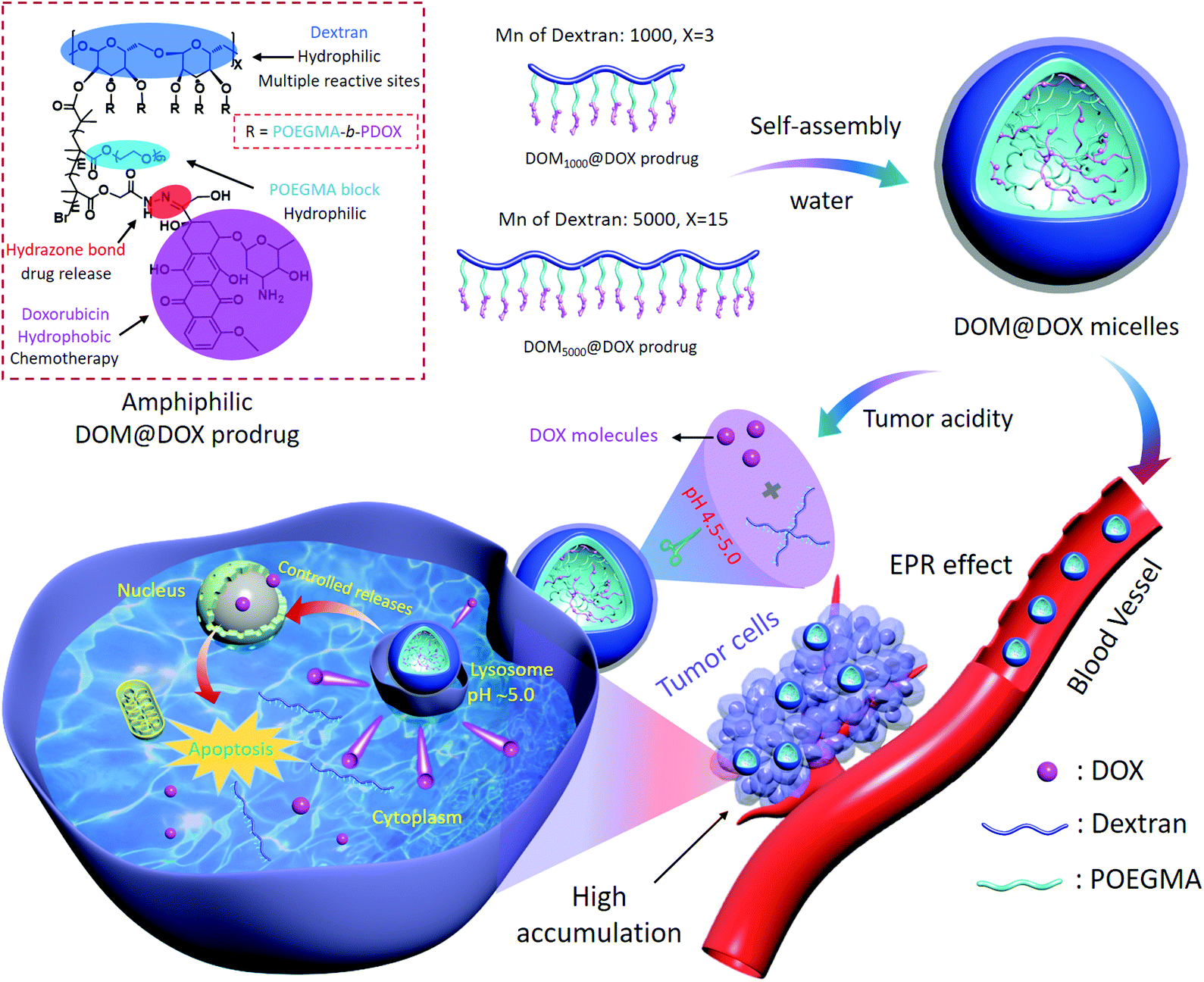 | ||
| Scheme 1 Schematic illustration of the synthetic route of DOM@DOX micelles, drug accumulation via the EPR effect, cell internalization process, and pH-responsive drug release mechanism. | ||
Experimental section
Reagents and materials
Chemical reagents including dextran (DEX, Mw: 1000 Da and 5000 Da, 99.7%), 2-bromoisobutyryl bromide (BIBB, 98%), poly(ethylene glycol) methyl ether methacrylate (OEGMA), tris(2-dimethylaminoethyl)-amine (Me6TREN), trifluoroacetic acid (TFA), hydrazine hydrate and copper(I) bromide (CuBr, 97%) were supplied by Sigma-Aldrich (USA), as well as anhydrous dimethyl sulfoxide (DMSO), N-methyl pyrrolidone (NMP), N,N-dimethylformamide (DMF) and methanol. Doxorubicin (DOX) drugs were supplied by Adamas-beta (China). 3-(4,5 Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), live/dead viability kit, Alexa Fluor® 488 phalloidin (AF-488), Lyso-Tracker Green DND-99 and Cy5-NHS were purchased from Life Technologies (China).Characterization
The chemical structure was characterized by 1H nuclear magnetic resonance (NMR) spectroscopy on a Bruker Avance-600 NMR spectroscope. The hydraulic diameter and zeta potential were detected using a Nano ZS90 Malvern. The morphology of samples was observed using a JEM-1230EX transmission electron microscope (TEM). Fourier-transform infrared spectra (FTIR) were obtained using a Thermo Nicolet 6700 FT-IR spectrophotometer (USA). Gel permeation chromatography (GPC) was performed using an Agilent 1260 which included a 1260 pump, a refractive index detector, and a Styragel HT column. The results of cell viability assay were read with a Tecan SPARK-10 M. Fluorescence imaging was performed with a fluorescence microscope (Olympus IX71), including hematoxylin and eosin (H&E) staining, TdT-mediated dUTP Nick-End Labeling (TUNEL) assay and live/dead staining. The results of cellular uptake were determined by flow cytometry (NovoCyte 2060R, USA). Cell imaging was performed using Confocal Laser Scanning Microscopy (CLSM, Zeiss 800). The near-infrared images in vivo were obtained using a PerkinElmer NIR imaging system (IVIS Lumina Kinetic Series III).Preparation of a DEX–Br polymer
A two-step esterification process was used to prepare a DEX–Br polymer. Firstly, 500 mg of DEX1000 or DEX5000 and 2.5 g of 1-allyl-3-methylimidazolium chloride were added into a 100 mL flask, and the mixture was stirred for 1 h at 80 °C. Then 5 mL of mixed anhydrous NMP and DMF (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were added into the flask. The flask was placed in an ice bath. 7.6 mL of BIBB (61.5 mmol) was added dropwise into the above solution, and the mixture was stirred for 24 h at 25 °C. Then, the mixture was added into water for precipitation. The crude product was dissolved in acetone and precipitated in water, and the purification process was repeated three times. The obtained intermediate was dried for 24 h at 80 °C. Then a second esterification process was carried out to increase the conversion ratio of hydroxyl groups. Briefly, 800 mg of the intermediate was added into a 50 mL flask, and 10 mL of anhydrous NMP was added. The flask was placed in an ice bath. 10 mL of BIBB (40.5 mmol) was added dropwise into the above mixture, which was stirred for 1 h at 0 °C and then for 48 h at 25 °C. After purification, the final product was a white powder of DEX–Br, with a yield of 600 mg.
:1) were added into the flask. The flask was placed in an ice bath. 7.6 mL of BIBB (61.5 mmol) was added dropwise into the above solution, and the mixture was stirred for 24 h at 25 °C. Then, the mixture was added into water for precipitation. The crude product was dissolved in acetone and precipitated in water, and the purification process was repeated three times. The obtained intermediate was dried for 24 h at 80 °C. Then a second esterification process was carried out to increase the conversion ratio of hydroxyl groups. Briefly, 800 mg of the intermediate was added into a 50 mL flask, and 10 mL of anhydrous NMP was added. The flask was placed in an ice bath. 10 mL of BIBB (40.5 mmol) was added dropwise into the above mixture, which was stirred for 1 h at 0 °C and then for 48 h at 25 °C. After purification, the final product was a white powder of DEX–Br, with a yield of 600 mg.
Synthesis of a DEX-POEGMA polymer
DEX-POEGMA was prepared by atom transfer radical polymerization (ATRP). 28.3 mg of DEX–Br (equal to 0.129 mmol Br), 279.3 mg of OEGMA (1.4 mmol), CuBr (15.6 mg, 0.1 mmol), and 2 mL of mixed DMF and DMSO (v/v = 1:1) were added into a 25 mL Schlenk tube under an argon atmosphere. The mixture was subjected to three freeze–pump–thaw cycles with liquid nitrogen and then 46 μL of Me6TREN (0.128 mmol) was added into the mixture solution. The mixture was stirred for 24 h at room temperature. After being dialyzed (MW 3500) against methanol, the solution was dried under vacuum. The product of DEX-POEGMA was obtained.
Preparation of DEX-POEGMA-b-PMGMA (DOM)
DEX-POEGMA-b-PMGMA was prepared by another ATRP reaction. Firstly, 100.05 mg of DEX-POEGMA (0.065 mmol Br), 308.2 mg of MGMA (1.95 mmol), 8 mg of CuBr (0.056 mmol) and DMSO (2.0 mL) were added into a tube under a glovebox. Then 23 μL of Me6TREN (0.064 mmol) was injected into the tube with a micro-syringe. Lastly, the tube was allowed to stand at 25 °C for 24 h. The resulting mixture was dialyzed (MW 3500) against methanol for two days. The product DEX-POEGMA-b-PMGMA was obtained.Synthesis of the DOM@DOX prodrug
Firstly, the DEX-POEGMA-b-PMGMA polymer was treated with hydrazine hydrate for 12 h, producing an intermediate (denoted as DOM@hydrazide). Then the intermediate (100 mg), DOX (35 mg) and trifluoroacetic acid (5 μL) were dispersed in a mixed DMF and methanol solution (v/v = 1:1). After being stirred for two days, the solution was dialyzed using methanol (250 ml × 3) to remove the unreacted DOX. Finally, the solution was dried under vacuum, and the product DOM@DOX was obtained.
Preparation of DOM@DOX micelles
10 mg of DOM@DOX was dissolved in 2 mL DMF and stirred for 1 h. Then, the solution was added dropwise into 6 mL of DI water and stirred for 1 h. Finally, the solution was dialyzed (MW 3500) against DI water (1L × 3) for two days. The DOM@DOX micelles were obtained.DOX release for DOM@DOX
Firstly, 1.0 mL of micelles (500 μg mL−1) were dialyzed against 80 mL of PBS with different pH values (pH = 5.0, 6.8, and 7.4, respectively). The DOX release test was performed at 37 °C for 96 h, under a vibration of 120 rpm. At a designated time-point, 1.0 mL of the external solution was withdrawn from the dialysis bag and the emission wavelength (555 nm) were recorded for calculating the content of DOX using a fluorescence calibration curve.Cell cultures
A human cervical cancer cell line (HeLa), and a human breast cancer cell line (MCF-7) were obtained from the Shanghai Cell Bank of the Chinese Academy of Sciences and were cultivated by using DMEM containing 10% FBS and 1% penicillin/streptomycin (P/S) in an incubator at 37 °C containing 5% CO2.In vitro cytotoxicity
MCF-7 and HeLa cells were selected to measure the cytotoxicity and biocompatibility of DOM@DOX, respectively. Firstly, 200 μl of DMEM containing 1 × 104 cells were added into each well of 96-well plates, and the plates were cultured in the incubator for 12 h. Then fresh DMEM containing free DOX, DOM1000@hydrazide, DOM5000@hydrazide, DOM1000@DOX and DOM5000@DOX was added, respectively. The cells were treated for 72 h. Next, the solution was removed and replaced with 200 μL of an MTT reagent (0.5 mg mL−1). After 4 h, MTT was removed, and 100 μL of DMSO was added and mixed for about 15 min. Finally, the absorbance of the microplates at 570 nm was read with the Tecan Spark-10 plate reader.In vitro live/dead study
HeLa cells were selected for live/dead study to further assess the cell apoptosis caused by the prodrug. Firstly, 1.5 mL DMEM containing 1.3 × 105 HeLa cells was added into each well of a 12-well plate and cultivated for 12 h. Then, free DOX, DOM1000@hydrazide, DOM5000@hydrazide, DOM1000@DOX or DOM5000@DOX (DOX concentration: 20 μg mL−1 and DOM@hydrazide concentration: 30 μg mL−1) in DMEM solution were added into each well and cultured for 24 h. Lastly, the cells in each well were stained with a live/dead kit. After 20 min the images were taken using the fluorescence microscope.In vitro cellular uptake and co-localization studies
In brief, 400 μL of DMEM containing 2.0 × 104 HeLa cells were added into each well of an 8-well plate and cultivated for 12 h. After being incubated with free DOX for 2 h, and DOM1000@DOX or DOM5000@DOX for 0.5 h and 6 h, respectively (DOX concentration: 20 μg mL−1), 400 μL of formalin solution was added into each well and incubated for 0.5 h, followed by the addition of BSA and Triton X-100. After being stained with AF-488, 4′,6-diamidino-2-phenylindole (DAPI), the cells were washed three times and fluorescence images of the cells were recorded using CLSM. Additionally, HeLa cells were cultured as mentioned above. For the co-localization assay, the cells were treated with DOM1000@DOX or DOM5000@DOX for 0.5 h and 6 h. Next, the lysosomes were stained with Lyso Tracker Green DND-99 for another 20 min. Fluorescence images of the cells were obtained by CLSM.The cell uptake process of prodrug micelles was further recorded by flow cytometry. Briefly, 1.5 mL of DMEM containing 1.0 × 105 HeLa cells was added into each well of a 12-well plate, and the plate was cultured for 12 h. After being incubated with free DOX, DOM1000@DOX and DOM5000@DOX in DMEM, the cells were treated for 0.5, 2, 4 and 6 h and the cellular uptake of DOM@DOX was determined by flow cytometry.
Deep penetration study in multicellular spheroids (MCS)
Multicellular spheroids (MCS) of MCF-7 cells were cultivated to form a solid tumor in order to assess the deep penetration capability of DOM@DOX. In brief, 100 mg agarose was mixed with 10 mL of 1 × TEA, and heated for 30 s using microwave. Then, 50 μL of 1% agarose was added into each well of the 96-well plate immediately after boiling, followed by exposure to UV-light irradiation for 30 min. 200 μL of DMEM containing 1.0 × 103 cells were added into each well and cultivated for 3–5 days. The solution was refreshed by half every day. Next, the old DMEM was replaced with fresh DMEM (pH = 7.4) containing free DOX and DOM@DOX (DOX concentration: 30 μg mL−1). After 6 h, each well was washed three times using PBS. Then, the fluorescence (red signal of DOX) images of MCS were obtained using CLSM.Animal model
All animal procedures were performed following the guidelines of the Institutional Animal Care and Use Committee (IACUC) of Southwest University and were approved by the Animal Ethics Committee of Southwest University. Female nude mice (6-weeks-old) and KM mice (20–22 g) were provided by Chengdu Da Shuo Experimental Animal Co. (China) and Chongqing Teng Xin Biological Technology Co. (China). Nude mice were injected with 200 μL PBS containing 5 × 106 MCF-7 cells under the armpit. In vivo analyses were carried out after the tumors had grown to about 150 mm3.Blood routine study
KM female mice were used for routine blood tests. Specifically, the KM mice were divided into four groups, and each group was injected with 200 μL saline, free DOX, DOM1000@DOX or DOM5000@DOX (DOX concentration: 500 μg mL−1) through tail veins. Peripheral blood from the orbit of each mouse was collected at day 1 and day 7. Then, the blood indicators including the white blood cell count (WBC), mean corpuscular hemoglobin concentration (MCHC), lymphocyte ratio (Lyphm), red blood cell count (RBC), hemoglobin concentration (HGB), variation coefficient of red blood cell distribution width (RDW), platelets (PLT), hematocrit (HCT) and mean platelet volume (MPV) were analyzed using a hematology analyzer.Pharmacokinetic study
In the plasma pharmacokinetic study, SD mice (180–200 g) were randomly divided into three group (n = 3 per group) and then injected with free DOX, DOM1000@DOX and DOM5000@DOX micelles through tail veins (the DOX concentration: 5 mg kg−1). At predetermined time intervals (i.e., 10 min, 20 min, 0.5, 1, 2, 5, 7, 9, 12 and 24 h), the blood samples were collected from the orbit of each mouse. The plasma samples were obtained by centrifugation (10 min, 3000 rpm) and purified by protein precipitation using three volumes of methanol. The supernatant was collected by centrifugation (10 min, 8000 rpm). Finally, the fluorescence intensity of DOX in the supernatant of each group was measured with the Tecan Spark-10 plate reader.In vivo fluorescence imaging
Firstly, DOM@DOX was labelled with Cy5. Then, free Cy5 or Cy5-labelled DOM@DOX was injected into the nude mice bearing MCF-7 tumors. After 24 h, the optical and fluorescence images of mice and the anatomical tumor, heart, liver, spleen, lung and kidney were obtained by using an NIR imaging system.In vivo therapeutic and histological studies
Firstly, nude mice bearing MCF-7 tumors were divided into four groups (n = 5 per group). Then, each group was injected with saline, free DOX, DOM1000@DOX or DOM5000@DOX (DOX concentration: 500 μg mL−1) through tail veins every third day. The entire treatment last for 11 days. The tumor volumes were calculated as: Tumor volume = width2 × length × 1/2.The tumors and major organs including the kidney, lung, spleen, liver and heart were dissected after treatment, processed in 4% formalin, and excised, sliced, and embedded in paraffin. After that, the collected slices were stained with hematoxylin and eosin (H&E) and TUNEL, respectively, and the optical images were collected using the fluorescence microscope (Olympus IX71).
Statistical analysis
All data are shown as mean ± s.d., and the statistical analysis of results was performed using a method of t-test analysis of variance.Results and discussion
Synthesis and characterization of the DOM@DOX prodrug
The pH-responsive DOM@DOX polymeric prodrug was prepared based on DEX synthesized by atom transfer radical polymerization reaction (ATRP). The route of synthesis is shown in Fig. S1.† Specifically, DEX1000–Br and DEX5000–Br initiators were compounded respectively by the reaction between DEX1000 or DEX5000 and BIBB according to a previous report.48 The 1H NMR results are shown in Fig. S2,† the signals of DEX and –Br correspond to peak a (1.92 ppm) and peak b (5.75 ppm), and about two-thirds of the hydroxyl group could be converted into the –Br group based on the 1H NMR results. A precursor of methyl glycolate methacrylate (MGMA) was synthesized according to the study in ref. 52. Next, DEX–Br reacted with the monomer OEGMA and subsequently with the predesigned monomer MGMA through a two-step ATRP to obtained two kinds of diblock copolymers of DEX1000-POEGMA-b-PMGMA and DEX5000-POEGMA-b-PMGMA, and their 1H NMR profiles are shown in Fig. S3 and S4†, respectively. In Fig. S3,† there are two peaks at 3.38 ppm (peak a) and at 3.64 ppm (peak b), which correspond to the methoxy protons and methylene protons next to the methoxy moiety, respectively. In Fig. S4,† two other characteristic signals at peak e and peak f were related to the methoxy protons and methylene protons close to the ester moiety. These results indicated the successful polymerization of DEX-POEGMA-b-PMGMA (DOM).The obtained DOM was reacted with hydrazine hydrate to prepare DOM@hydrazide. As shown in Fig. S5,† the peak at 3.76 ppm disappeared indicating that the methoxy group was replaced by the hydrazine group. The DOM@hydrazide could react with the ketone group of DOX to produce a DEX-POEGMA-b-PDOX (DOM@DOX) prodrug, as verified by 1H NMR characterization in Fig. S6.† In the spectra, signals at 7.79 ppm (peak c) could be observed clearly, indicating that the DOM@DOX prodrug was synthesized successfully.
In addition, the polymerization was traced using gel permeation chromatography (GPC). The number-average molecular weight (Mn) and polymer dispersity index (PDI) of these DOM copolymers were Mn = 30800 g mol−1 (PDI = 1.73) for DEX1000-POEGMA-b-P(MGMA) and Mn = 52300 g mol−1 (PDI = 1.63) for DEX5000-POEGMA-b-PMGMA as shown in Fig. S7.† Compared with the DEX-POEGMA polymer, the GPC peak of DEX-POEGMA-b-PMGMA had a small right shift, which might have been caused by the greater spatial congestion after the grafting of the PMGMA chain.53 Moreover, the degree of polymerization of OEGMA and MGMA on DEX, the DOX loading content (LC), the molecular weight distribution and the size of DOM@DOX in water are summarized in Table S1.†
The characterization of DOM@DOX micelles
The amphiphilic DOM@DOX prodrugs could remain stable in aqueous media, because they can form spherical micelles by self-assembly. The critical micelle concentration (CMC) was determined using a standard pyrene method, and the CMC results were 6.8 μg mL−1 and 12.5 μg mL−1 for DOM1000@DOX and DOM5000@DOX. The results consistently indicated the excellent micellar stability in water medium. The morphology of DOM@DOX micelles was spherical or elliptical (Fig. 1A and D). The size of DOM1000@DOX and DOM5000@DOX was characterized using dynamic light scattering (DLS), and was 37.56 and 49.26 nm in water (Fig. 1B and E), respectively. Furthermore, the DOM@DOX prodrugs could maintain the same over 9 days, indicating the good structural stability (Fig. 1C and F). The zeta potential of DOM@hydrazide was negative and that of free DOX was positive; DOM@DOX was generally neutral, which would be favorable to blood circulation (Fig. 1G). The optical properties including the ultraviolet (UV)-vis and the fluorescence (FL) spectra of DOM@DOX micelles were recorded using a Shimadzu UV-1800 spectrophotometer and a Shimadzu RF-5301PC fluorophotometer (Fig. 1H and I). The absorption peak of DOM@DOX micelles in water was observed at 488 nm, which was similar to the adsorption spectrum of free DOX. In addition, the maximum FL emission intensity of DOM@DOX prodrugs in water was observed at 560 nm, which was also close to that of free DOX. Briefly, the above results indicated the success preparation of DOM@DOX prodrugs.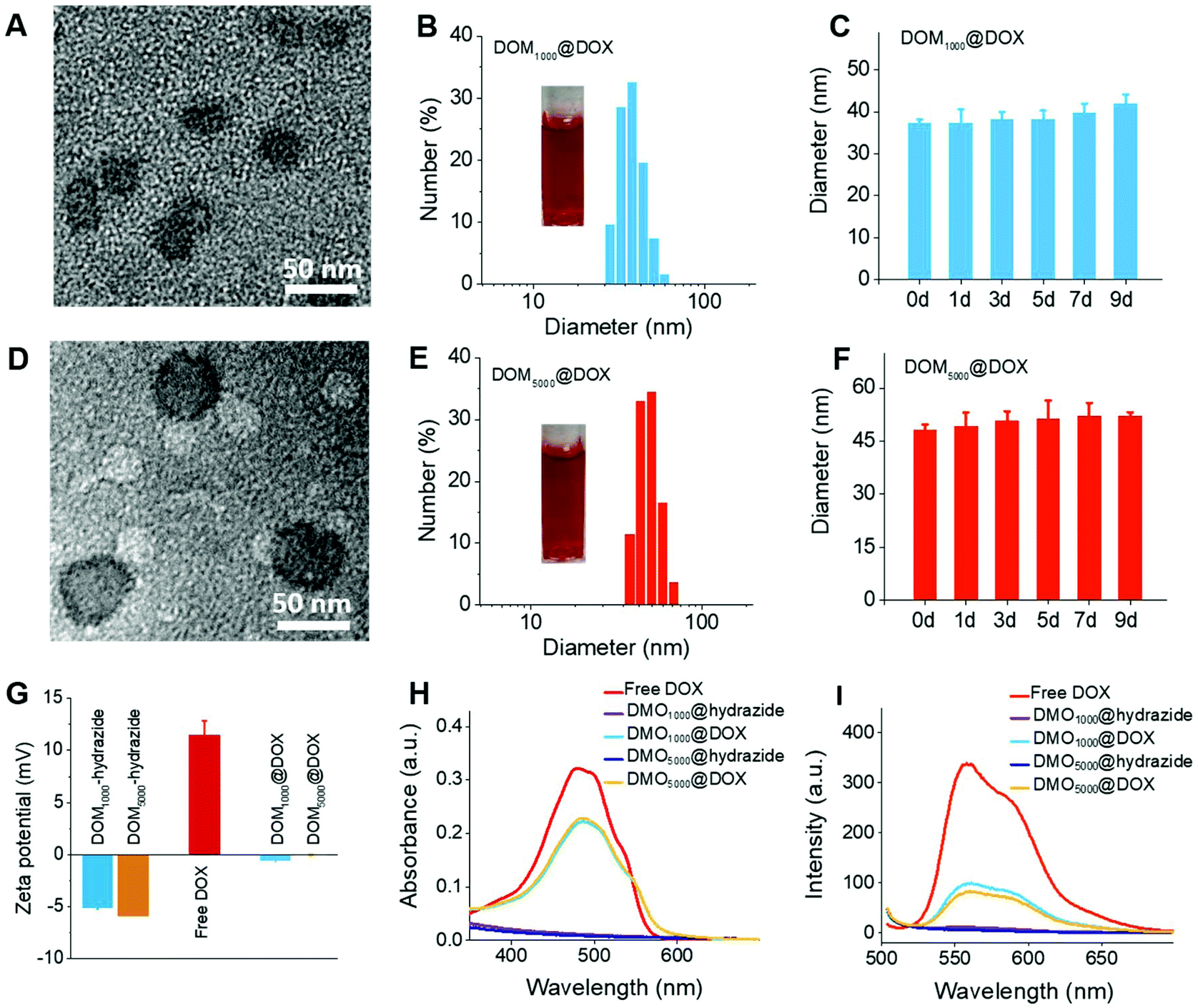 | ||
| Fig. 1 The characterization of DOM@DOX micelles. TEM images, DLS results and micellar stability for DOM1000@DOX (A, B and C) micelles and DOM5000@DOX (D, E and F) micelles, respectively. (G) The zeta potential of DOM@hydrazide, Free DOX and DOM@DOX. UV-vis absorption (H) and fluorescence spectra (I) of DOM@hydrazide, Free DOX and DOM@DOX. Data were expressed as mean ± standard deviation (n = 3). | ||
In vitro DOX release and cytotoxicity studies
Firstly, the drug loading contents (LC%) of these prodrugs were calculated as 42.7 wt% and 48.1 wt% for DOM1000@DOX and DOM5000@DOX using the standard curve of fluorescence intensity against DOX concentration. The drug loading content was larger for DEX with smaller molecular weight, which might have resulted from the weaker steric hindrance. The release of DOX from DOM@DOX was determined by dialysis, and was quantified with the fluorescence intensity at 555 nm. The accumulative release of DOX from DOM1000@DOX and DOM5000@DOX was ∼93% and ∼90%, respectively, at pH 5.0, while it was less than ∼30% for both micelles at pH 6.8 and 7.4 (Fig. 2A).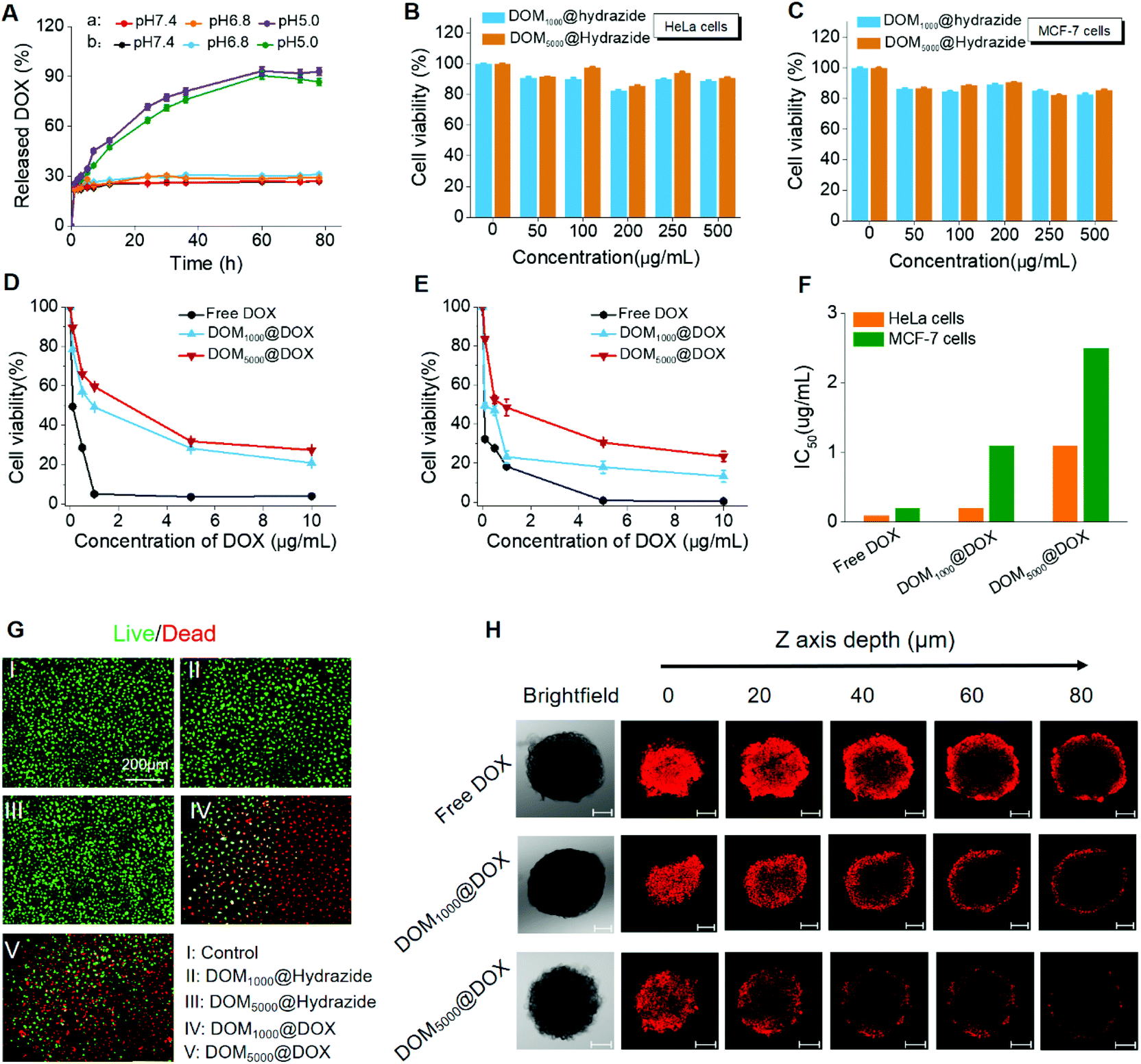 | ||
| Fig. 2 (A) Acidity activated DOX release of DOM1000@DOX (a) and DOM5000@DOX (b). Cell viability of HeLa (B) and MCF-7 (C) cells after treatment with DOM-hydrazide with different concentrations for 72 h. Cell viability of HeLa (D) and MCF-7 cells (E) after treatment with free DOX and DOM@DOX with different concentrations for 72 h. Data were expressed as mean ± standard deviation (n = 3). (F) Half maximal inhibitory concentration (IC50) of free DOX, DOM1000@DOX and DOM5000@DOX. (G) Live/dead fluorescence staining of HeLa cells. The HeLa cells were treated with PBS, DOM1000@hydrazide, DOM5000@hydrazide, DOM1000@DOX and DOM5000@DOX for 24 h. The live and dead cells were stained with FDA (green) and PI (red), respectively. (H) Confocal microscopy images of MCF-7 multicellular spheroids after treatment with free DOX, DOM1000@DOX and DOM5000@DOX (the concentration of DOX was 30 μg mL−1) for 6 h, at pH 7.4. Scale bars: 100 μm. | ||
The possible mechanism of pH-triggered drug release is attributed to the hydrazine bond, which shows a pH-sensitive decomposition in the acidic endo/lysosomal microenvironment.54 This finding demonstrated that the premature drug release could be controlled to a fixed range, which could improve the delivery efficacy of drugs under circulation and promote the therapeutic effect. In addition, the release rate of DOM5000@DOX was less than that of DOM1000@DOX, which might have also resulted from the steric hindrance. Briefly, the DOM@DOX micelle-based delivery system could be activated by the acidity in tumor cells and the drug release and leakage would be decreased under neutral pH in normal tissues.
The cytotoxicity of DOM@DOX to cancer cells was determined in HeLa (human cervical cancer cells) and MCF-7 (human breast cancer cells) cells, for evaluating the in vitro tumor inhibiting effects in cancer cells. Firstly, the cytotoxicity of DOM@hydrazide without drug loading was evaluated. Two cells were cultured and treated with DOM1000@hydrazide and DOM5000@hydrazide, in which the concentration ranged from 0 to 500 μg ml−1. No obvious cytotoxicity was observed in cancer cells treated with DOM@hydrazide (Fig. 2B and C). 90% of cells were alive after treatment for 72 h. The above results primarily indicated the good biocompatibility of the DOM@DOX drug delivery system.
As seen in Fig. 2D and E, the cancer cell viability decreased significantly when the effective drug dosage increased from 0 to 10 μg mL−1. Specifically, the viable MCF-7 and HeLa cells at 72 h reduced to only ∼13% and ∼20% after treatment with DOM1000@DOX, or ∼23% and ∼27% after treatment with DOM5000@DOX, respectively. The cytotoxicity was mainly due to the released DOX from DOM@DOX after the breakage of hydrazine bonds in the acidic tumor microenvironment. Moreover, the half maximal inhibitory concentration (IC50) of these two prodrugs showed a notable difference on the two cancer cell lines studied (Fig. 2F). To further assess the cell apoptosis of DOM@DOX, a live/dead cell viability assay was carried out, for which the dead and live cells were stained with PI (red) and FDA (green). As seen in Fig. 2G, both DOM@DOX prodrugs produced remarkable inhibiting effects on tumor cells while DOM@hydrazide groups showed weak cell apoptosis, indicating the outstanding potential of DOM@DOX as a drug delivery system.
In addition, multicellular spheroids (MCSs) of MCF-7 cells were used as a microcosmic model in vitro to investigate the tumor permeation ability of DOM@DOX micelles. The confocal microscopy images of multicellular spheroids were collected by CLSM Z-stack scanning after treatment with free DOX, DOM1000@DOX and DOM5000@DOX at pH = 7.4 for 6 h. As shown in Fig. 2H, when the scanning depth reached 80 μm, the MCSs treated with DOM1000@DOX still showed noticeably stronger fluorescence compared to those treated with free DOX and DOM5000@DOX due to their relatively smaller size, demonstrating a very fine tumor permeation ability by the EPR effect. The above results suggested that the DOM@DOX prodrug has potential for effective enhancement of drug delivery and release.
In vitro cellular uptake evaluated by fluorescence imaging
To trace the cell uptake behavior of DOM@DOX, HeLa cells were incubated with DOM@DOX for 0.5 h and 6 h, followed by fluorescence imaging using CLSM. The lysosomes, nuclei and cytoskeleton of cells were stained with Lyso Tracker Green DND-99, DAPI (blue) and AF-488 (green), respectively. An increase in the fluorescence intensity of DOX (red) was observed in lysosomes (green) from 0.5 h to 6 h (Fig. 3A, B and Fig. S9†). More DOM1000@DOX was swallowed by cells, compared to DOM5000@DOX. With time, the overlap of lysosomes and DOX increased to 0.63 and 0.49 for DOM1000@DOX and DOM5000@DOX, and the higher overlap of DOM1000@DOX might have benefited from its smaller size in comparison with that of DOM5000@DOX. These results indicated that DOM@DOX entered into the cells via an endocytic pathway. The cells were treated with free DOX for 2 h and strong fluorescence of DOX was observed at the nuclei, indicating a rapid internalization process of free DOX (Fig. 3C). Interestingly, the fluorescence intensity of DOX was generally stronger in cells treated with DOM1000@DOX and DOM5000@DOX micelles, compared to that of free DOX. The reason may be the time-dependent drug release behavior of DOM@DOX micelles in the acidic environment. The enlarged images further showed the enhanced drug accumulation over time in the nuclear region.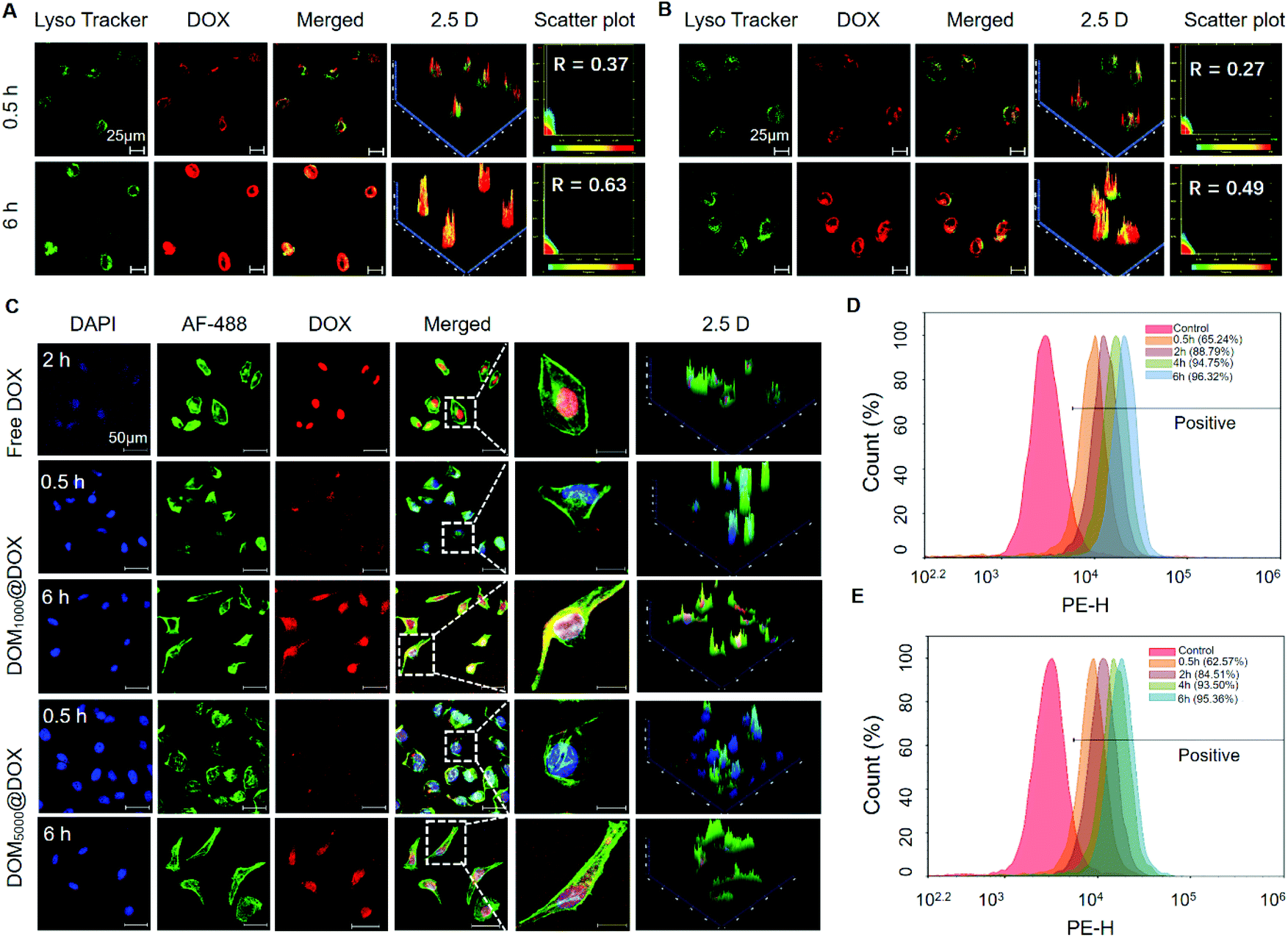 | ||
| Fig. 3 Confocal images showing HeLa cells stained with Lyso-Tracker Green DND-99 after treatment with DOM@DOX (20 μg mL−1 for DOX) for 0.5 h and 6 h (A for DOM1000@DOX and B for DOM5000@DOX). CLSM images of HeLa cells showed the distribution of free DOX after treatment for 2 h and DOM@DOX (equivalent DOX concentration: 20 μg mL−1) after treatment for 0.5 h or 6 h (C). The cellular uptake of DOM@DOX for 0.5 h, 2 h, 4 h and 6 h analyzed using flow cytometry (D for DOM1000@DOX and E for DOM5000@DOX). | ||
Moreover, flow cytometry was used to quantitatively measure the cellular uptake efficiency of DOM@DOX micelles. As shown in Fig. 3D–E and Fig. S10,† the uptake rates of DOM1000@DOX and DOM5000@DOX reached up to 88.79% and 84.51%, respectively, after 2 h of cultivation. Such a difference could be attributed to the size difference between these two micelles, suggesting faster cellular uptake of the smaller micelles. Meanwhile, the cellular uptake of both types of DOM@DOX micelles reached above 90% within 4 h, which demonstrated the remarkable capability of DOM@DOX as an efficient drug transporter into the tumor cells.
In vivo blood compatibility and biodistribution
To study the blood compatibility of DOM@DOX micelles in vivo, we used KM mice and nude mice as models. Firstly, a complete blood test was carried out on KM mice to evaluate the hemocompatibility of DOM@DOX micelles using a Mindray BC-2600Vet hematology analyzer. Comparing with the control group, the routine blood indices including WBC, RDW, HGB, MCHC, RBC, Lyphm, HCT, MPV and PLT were all in the normal ranges as shown in Fig. 4A.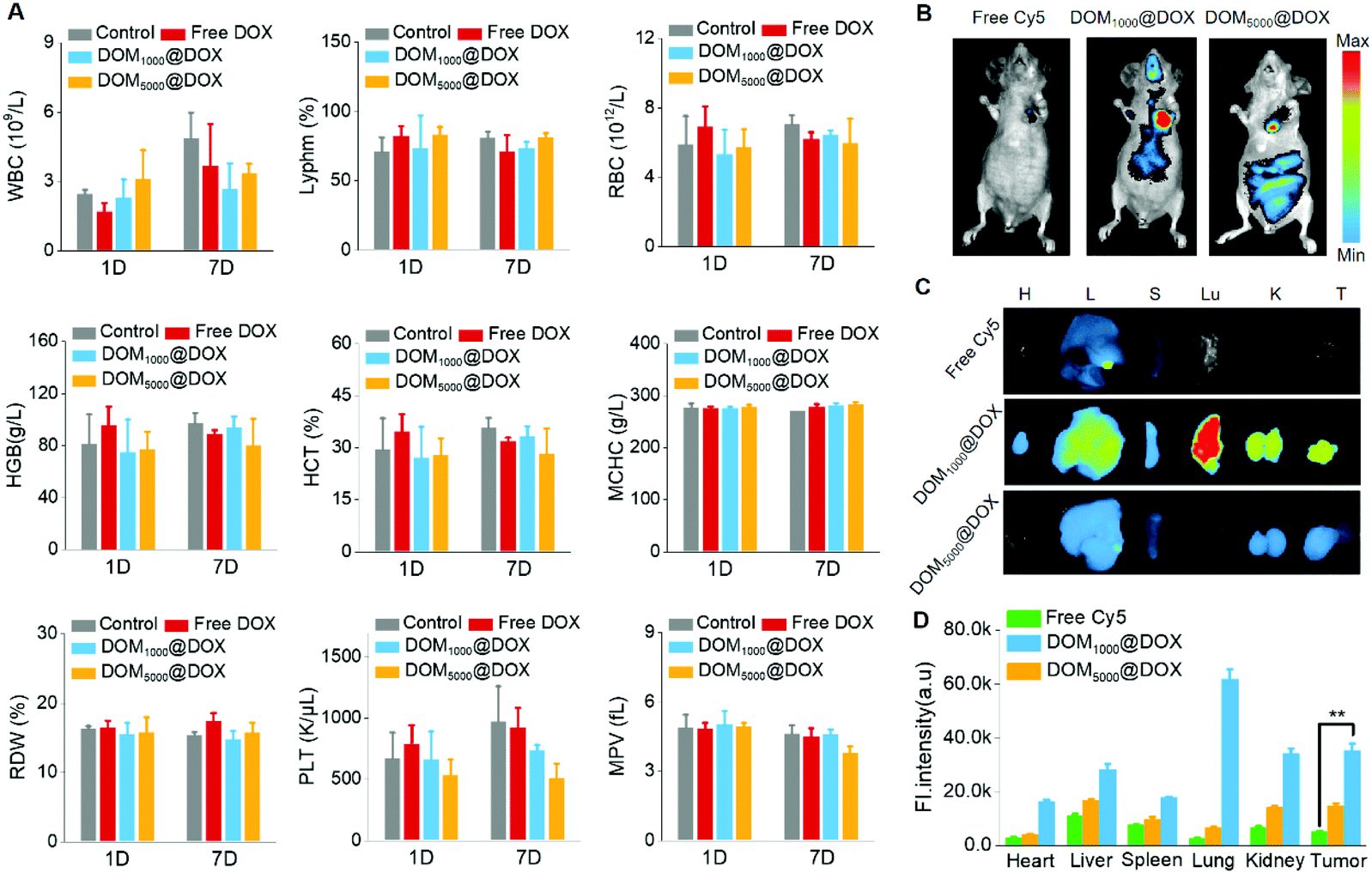 | ||
| Fig. 4 The complete blood indices of KM mice after injection with saline, free DOX and DOM@DOX (DOX dosage: 5 mg kg−1), respectively, at day 1 and day 7 (A). The in vivo fluorescence images of nude mice (B) and ex vivo fluorescence images (C) and quantification (D) of the tumor and major organs, including the heart (H), liver (L), spleen (S), lung (Lu), kidney (K) and tumor (T), showing the in vivo biodistribution of free Cy5 and Cy5-labelled DOM@DOX at 24 h post-treatment. | ||
Furthermore, the pharmacokinetic properties of DOM@DOX micelles were evaluated by determining the concentration of DOX in plasma. SD mice were intravenously injected with free DOX, DOM1000@DOX and DOM5000@DOX micelles, respectively. Fig. S11† indicates that DOM1000@DOX and DOM5000@DOX micelles exhibited similar pharmacokinetic curves, and both significantly prolonged the blood circulation in comparison with the free DOX group. The longer blood circulation of DOM@DOX groups might be caused by the EPR effect of DOM@DOX micelles, leading to increased tumor accumulation.
Cy5-NHS, as a hydrophobic NIR dye, was labelled with DOM@DOX by chemical bonding and used to trace the biodistribution of DOM@DOX micelles in vivo. Specifically, MCF-7 tumor bearing nude mice were administered with Cy5-labelled DOM@DOX or free Cy5. As shown in Fig. 4B, strong fluorescence could be seen at the tumor site after 24 h. Among all three experimental groups, the fluorescence of the group injected with DOM1000@DOX exhibited the strongest intensity, while the free Cy5 group showed the weakest fluorescence because free Cy5 could be easily cleared from blood. These results demonstrated effective drug accumulation of DOM@DOX at the tumor site owing to the enhanced permeability or the EPR effect. The ex vivo fluorescence images of tumors and major organs are shown in Fig. 4C, and their corresponding fluorescence intensity is quantified in Fig. 4D. Generally, the tumors showed relatively stronger fluorescence, which suggested the potential EPR effect of the DOM@DOX prodrug. The strong fluorescence signals in the lungs for DOM1000@DOX might be attributed to the opsonization mechanism.55 Moreover, the fluorescence intensity at the tumor sites for Cy5-labelled DOM@DOX micelles showed an obvious increase compared to that of the free Cy5 group, indicating the significantly higher permeation capability of DOM@DOX into tumors.
In vivo tumor therapeutic assay and infiltration of DOM@DOX into tumors
Nude mice bearing MCF-7 tumors were used to evaluate the tumor suppression effect of DOM@DOX in vivo. Specifically, the mice were treated with saline (control group), free DOX, or DOM@DOX with a drug dosage of 5 mg kg−1 (body weight). During 11 days, the average tumor volume of the control group increased to 1075 mm3 gradually, while those of the groups injected with free DOX, DOM1000@DOX and DOM5000@DOX increased to 441 mm3, 362 mm3 and 494 mm3, respectively, as shown in Fig. 5A and C. The results indicated that the DOM@DOX prodrug produced an obvious tumor suppression effect in comparison with the control group. Compared with the free DOX and DOM5000@DOX groups, the DOM1000@DOX group showed relatively better antitumor efficacy. Meanwhile, using the DOM@DOX prodrug did not induce considerable variation in the mouse body weight as compared with the control group (Fig. 5B), implying minimal harm of the prodrug to the general body health of mice. In contrast, the body weight of the free DOX group decreased at a later stage, which might be attributed to the side effect of DOX molecules. After the final stage of treatment (Fig. 5D and E), the excised tumors from the mice showed an obvious difference and a higher tumor inhibition rate of two DOM@DOX in comparison with the control group.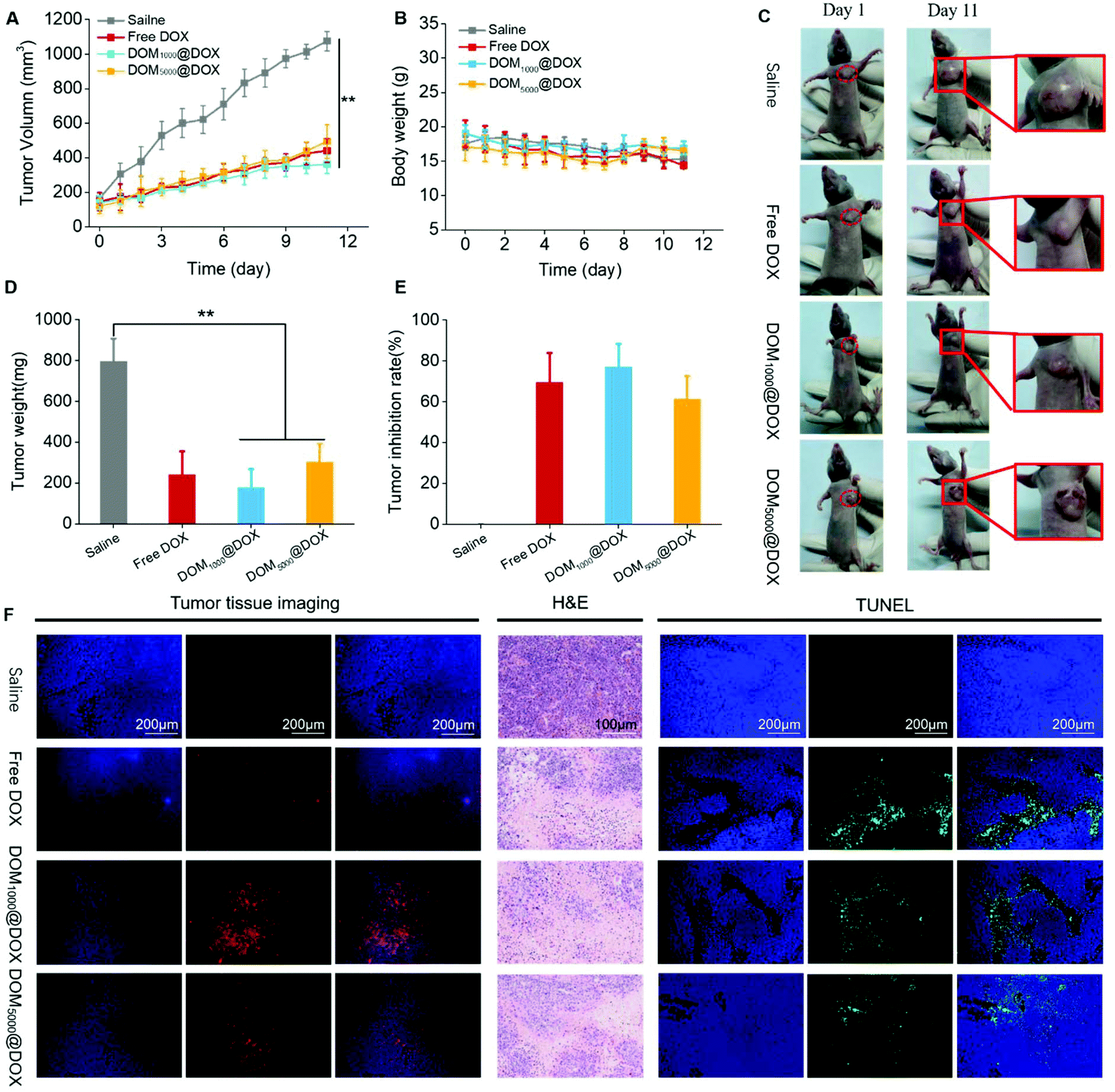 | ||
| Fig. 5 Changes in the tumor volume (A) and body weight (B) of nude mice bearing MCF-7 tumors. The pictures of nude mice bearing MCF-7 tumors at day 1 and day 11 (C). (Data are reported as mean ± s.d., n = 5, *p < 0.05, **p < 0.01.) The weight of excised tumors (D) and their corresponding inhibition rate (E), fluorescence imaging, and H&E and TUNEL staining of the excised tumors (F). | ||
To further verify the infiltration of DOM@DOX into tumors, the slices of sectioned tumors were used to monitor the distribution of the red fluorescence of DOX in entire tumors by fluorescence imaging (Fig. 5F). In Fig. 5F, for the sample from DOM@DOX treated mice, greater fluorescence of DOX was observed in the entire tumor section, indicating the even distribution of the drug across the tumors. In addition, the DOM1000@DOX treated mice presented higher fluorescence intensity than that of DOM5000@DOX. Only a few fluorescence signals of DOX were observed in the samples treated with free DOX, which could have resulted from the rapid clearance of free DOX from the body.
In addition, we carried out TUNEL and H&E staining assays to study the drug-induced apoptosis in tumor tissues (Fig. 5F). The sectioned tumors treated with free DOX or DOM@DOX all showed a significant level of cell apoptosis as compared to the saline-treated control group. H&E staining assay further revealed the predominant necrotic cell stages in the tumors under the chemotherapeutic effect.
The damage by DOM@DOX to the major organs was evaluated (Fig. 6). The mice were treated with saline, free DOX, DOM1000@DOX and DOM5000@DOX. Meanwhile, H&E staining also revealed that application of DOM@DOX induced no obvious damage of the heart, liver, spleen, lung and kidney in tumor tissue, as shown in Fig. 6. These animal experiments indicated that the DOM@DOX delivery system could effectively accumulate in tumor tissues and produce a notable therapeutic effect in vivo. In the meantime, DOM@DOX showed minimal systemic toxicity or side effects to the body health of mice. Hence, DOM@DOX micelles may work as another capable class of nanodrug delivery systems for future cancer therapy.
 | ||
| Fig. 6 Representative microscopy images of H&E stained tumor sections from saline, free DOX, DOM1000@DOX and DOM5000@DOX micelles over 11 days. | ||
Conclusion
In summary, we developed a pH-responsive amphiphilic polymer prodrug DOM@DOX, which could self-assemble to form nanosized micelles in aqueous solution and realize self-controlled drug release in the acidic tumor environment for cancer therapy. The results of in vitro and in vivo characterization demonstrated that the prodrug had good biocompatibility, good circulation stability and tumor permeability attributed to the non-toxic, water-soluble carrier material and the small size of prodrug micelles. This nanosystem remarkably increased the tumor therapeutic efficiency of the drug load, while exhibiting minimal systemic toxicity for normal tissues in vivo due to the pH-response drug release. This work demonstrated a new strategy based on acidic stimuli-responsive micelles as an efficient drug carrier for antitumor applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 51703187 and 31671037); Fundamental Research Funds for Central Universities from Southwest University (No. XDJK2019B005), the Basic and Frontier Research Project of Chongqing (cstc2018jcyjAX0104) and Shanghai Jiaotong University medical technology crossing project (ZH2018QNA31).Notes and references
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jernal, CA-Cancer J. Clin., 2018, 68, 394–424 CrossRef PubMed.
- P. Huang, Y. Gao, J. Lin, H. Hu, H. S. Liao, X. Yan, Y. Tang, A. Jin, J. Song and G. Niu, ACS Nano, 2015, 9, 9517–9527 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- K. Ulbrich, K. Holá, V. Šubr, A. Bakandritsos, J. Tuček and R. Zbořil, Chem. Rev., 2016, 116, 5338–5431 CrossRef CAS PubMed.
- H. S. EI-Sawy, A. M. AI-Abd, T. A. Ahmed, K. M. EI-Say and V. P. Torchilin, ACS Nano, 2018, 12, 10636–10664 CrossRef PubMed.
- J. Q. Jin, Y. Deng, X. Chen and J. Jian, ACS Nano, 2019, 13, 945–977 Search PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS PubMed.
- H. R. Kim, D. G. You, S. J. Park, K.-S. Choi, W. Um, J.-H. Kim, J. H. Park and Y. S. Kim, Mol. Pharm., 2016, 13, 1528–1539 CrossRef CAS PubMed.
- J. J. Shen, H. C. Kim, J. Wolfram, C. F. Mu, W. Zhang, H. R. Liu, Y. Xie, J. H. Mai, H. Zhang, Z. Li, M. Guevara, Z. W. Mao and H. F. Shen, Nano Lett., 2017, 17, 2913–2920 CrossRef CAS PubMed.
- Y. A. Lin, A. G. Cheetham, P. C. Zhang, Y. C. Ou, Y. G. Li, G. S. Liu, D. H. Merino, I. W. Hamley and H. G. Cui, ACS Nano, 2014, 8, 12690–12700 CrossRef CAS PubMed.
- M. L. Hou, Y.-E. Gao, X. X. Shi, S. Bai, X. Q. Ma, B. S. Li, B. Xiao, P. Xue, Y. J. Kang and Z. G. Xu, Acta Biomater., 2018, 77, 228–239 CrossRef CAS PubMed.
- P. Huang, D. L. Wang, Y. Su, W. Huang, Y. F. Zhou, D. X. Cui, X. Y. Zhu and D. Y. Yan, J. Am. Chem. Soc., 2014, 136, 11748–11756 CrossRef CAS PubMed.
- P. P. Liang, X. Y. Huang, Y. Wang, D. P. Chen, C. J. Ou, Q. Zhang, J. J. Shao, W. Huang and X. C. Dong, ACS Nano, 2018, 12, 11446–11457 CrossRef CAS PubMed.
- Z. G. Xu, X. Q. Ma, Y. E. Gao, M. L. Hou, P. Xue, C. M. Li and Y. J. Kang, Mater. Chem. Front., 2017, 1, 1257–1272 RSC.
- Y. H. Li, N. Li, W. Pan, Z. Z. Yu, L. M. Yang and B. Tang, ACS Appl. Mater. Interfaces, 2017, 9, 2123–2129 CrossRef CAS PubMed.
- G. Y. Liu, J. H. Zou, Q. Y. Tang, X. Y. Yang, Y. W. Zhang, Q. Zhang, W. Huang, P. Chen, J. J. Shao and X. C. Dong, ACS Appl. Mater. Interfaces, 2017, 9, 40077–40086 CrossRef CAS PubMed.
- X. L. Hu, S. D. Zhai, G. H. Liu, D. Xing, H. J. Liang and S. Y. Liu, Adv. Mater., 2018, 30, 1706307 CrossRef PubMed.
- X. X. Shi, M. L. Hou, X. Q. Ma, S. Bai, T. Zhang, P. Xue, X. L. Zhang, G. Liu, Y. J. Kang and Z. G. Xu, Biomacromolecules, 2019, 20, 1190–1202 CrossRef CAS PubMed.
- W. J. Zhang, X. L. Hu, Q. Shen and D. Xing, Nat. Commun., 2019, 10, 1704 CrossRef PubMed.
- Z. Su, S. Zhu, A. D. Donkor, C. Tzoganakis and J. F. Honek, J. Am. Chem. Soc., 2011, 133, 6874–6877 CrossRef CAS PubMed.
- T. X. Miao, J. J. Wang, Y. Zeng, G. Liu and X. Y. Chen, Adv. Sci., 2018, 5, 1700513 CrossRef PubMed.
- M. Zhang, C. C. Song, S. Su, F. S. Du and Z. C. Li, ACS Appl. Mater. Interfaces, 2018, 10, 7798–7810 CrossRef CAS.
- H. J. Li, X. J. Du, C. F. Sun, H. X. Wang, Z. T. Cao, X. Z. Yang, Y. H. Zhu, S. Nie and J. Wang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4164–4169 CrossRef CAS PubMed.
- J. Hu, W. H. Zhuang, B. X. Ma, X. Su, T. Yu, G. C. Li, Y. F. Hu and Y. B. Wang, Bioconjugate Chem., 2018, 29, 1897–1910 CrossRef CAS PubMed.
- X. L. Zheng, D. Y. Pan, M. Chen, X. H. Dai, H. Cai, H. Zhang, Q. Y. Gong, Z. W. Gu and K. Luo, Adv. Mater., 2019, 31, 1901586 CrossRef PubMed.
- P. F. Zhang, J. Q. Wang, H. Chen, L. Zhao, B. B. Chen, C. C. Chu, H. Liu, Z. N. Qin, J. Y. Liu, Y. Z. Tan, X. Y. Chen and G. Liu, J. Am. Chem. Soc., 2018, 140, 14980–14989 CrossRef PubMed.
- M. L. Pei, X. Jia, G. P. Li and P. Liu, Mol. Pharm., 2019, 16, 227–237 CrossRef CAS PubMed.
- Y. J. Chen, Z. H. Li, H. B. Wang, Y. Wang, H. J. Han, Q. Jin and J. Ji, ACS Appl. Mater. Interfaces, 2016, 8, 6852–6858 CrossRef CAS PubMed.
- L. Li, X. Pang and G. Liu, ACS Biomater. Sci. Eng., 2018, 4, 1928–1941 CrossRef CAS.
- M. Z. Xu, C. Y. Zhang, J. G. Wu, H. G. Zhou, R. Bai, Z. Y. Shen, F. L. Deng, Y. Liu and J. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 5701–5713 CrossRef CAS.
- Z. G. Xu, S. Y. Liu, H. Liu, C. J. Yang, Y. J. Kang and M. F. Wang, Chem. Commun., 2015, 51, 15768–15771 RSC.
- X. S. Fan, Z. Wang and C. B. He, J. Mater. Chem. B, 2015, 3, 4715–4722 RSC.
- Y. M. Zhao, W. Tian, G. Yang and X. D. Fan, Beilstein J. Org. Chem., 2014, 10, 2696–2703 CrossRef.
- Y. Huang, F. Qiu, D. Chen, L. Y. Shen, S. T. Xu, D. B. Guo, D. Y. Yan and X. Y. Zhu, Small, 2017, 13, 1604062 CrossRef PubMed.
- X. L. Hu, G. H. Liu, Y. Li, X. R. Wang and S. Y. Liu, J. Am. Chem. Soc., 2015, 137, 362–368 CrossRef CAS.
- C. E. Wang, H. Wei, N. Tan, A. J. Boydson and S. H. Pun, Biomacromolecules, 2016, 17, 69–75 CrossRef CAS.
- Q. Chen, Y. Yang, X. Lin, W. Ma, G. Chen, W. Li, X. Wang and Z. Yu, Chem. Commun., 2018, 54, 5369–5372 RSC.
- D. Dheer, D. Arora, S. Jaglan, R. K. Rawal and R. Shanker, J. Drug Targeting, 2017, 25, 1–16 CrossRef CAS.
- Y. Tang, F. Soroush, J. B. Sheffield, B. Wang, B. Prabhakarpandian and M. F. Kiani, Sci. Rep., 2017, 7, 1–14 CrossRef.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS.
- H. Maeda, H. Nakamura and J. Fang, Adv. Drug Delivery Rev., 2013, 65, 71–79 CrossRef CAS PubMed.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed.
- H. Wang, M. Dong, S. Khan, L. Su, R. C. Li, Y. Song, Y. N. Lin, C. H. Komatsu, M. Elsabahy and K. L. Wooley, ACS Macro Lett., 2018, 8, 783–788 CrossRef.
- S. Q. Liu, R. J. Ono, C. Yang, S. J. Gao, J. Y. M. Tan, J. L. Hedrick and Y. Y. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 19355–19364 CrossRef CAS PubMed.
- H. Cabral, K. Miyata, K. Osada and K. Kataoka, Chem. Rev., 2018, 118, 6844–6892 CrossRef CAS PubMed.
- O. Chen, Y. Bobak, O. V. Stasyk and L. A. Kunz-Schughart, Curr. Med. Chem., 2018, 25, 2465–2502 CrossRef CAS PubMed.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed.
- S. Bai, Y. E. Gao, X. Q. Ma, X. X. Shi, M. l. Hou, P. Xue, Y. J. Kang and Z. G. Xu, Carbohydr. Polym., 2018, 182, 235–244 CrossRef CAS.
- Y. Song, Y. Shi, L. P. Zhang, H. Y. Hu, C. Y. Zhang, M. M. Yin, L. X. Chu, X. Y. Yan, M. Y. Zhao, X. M. Zhang, H. J. Mu and K. X. Sun, Mol. Pharm., 2019, 16, 518–532 CrossRef CAS.
- D. Wang, J. Lin, F. Jia, X. Tan, Y. Wang, X. Sun, X. Cao, F. Che, H. Lu, X. Gao, J. Shimkonis, Z. Nyoni, X. Lu and K. Zhang, Sci. Adv., 2019, 5, eaav9322 CrossRef CAS.
- J. Z. Du, H. J. Li and J. Wang, Acc. Chem. Res., 2018, 51, 2848–2856 CrossRef CAS.
- H. Wang, F. Xu, D. Li, X. Liu, Q. Jin and J. Ji, Polym. Chem., 2013, 4, 2004–2010 RSC.
- X. X. Shi, S. Bai, C. Yang, X. Q. Ma, M. L. Hou, J. Chen, P. Xue, C. M. Li, Y. J. Kang and Z. G. Xu, J. Mater. Chem. B, 2018, 6, 5549–5561 RSC.
- M. L. Wang, Y. L. Zhai, H. Ye, Q. Z. Lv, B. J. Sun, C. Luo, Q. K. Jiang, Y. J. Xu, Y. K. Jing, L. Huang, J. Sun and Z. G. He, ACS Nano, 2019, 13, 7010–7023 CrossRef CAS.
- Y. Kuang, K. C. Zhang, Y. Cao, X. Chen, K. W. Wang, M. Liu and R. J. Pei, ACS Appl. Mater. Interfaces, 2017, 9, 12217–12226 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic route and structural characterization including 1H NMR spectra and GPC results of DOM@DOX and their intermediates; additional confocal fluorescence microscopy images and flow cytometry results. See DOI: 10.1039/c9bm01692a |
| This journal is © The Royal Society of Chemistry 2020 |