
A cascade approach to 3D cyclic carbamates via an ionic decarboxylative functionalization of olefinic oxamic acids†
Huaqiang
Fan‡
a,
Yi
Wan‡
a,
Peng
Pan
a,
Wenbin
Cai
a,
Shihui
Liu
a,
Chuanxu
Liu
*b and
Yongqiang
Zhang
 *a
*a
aState Key Laboratory of Bioengineering Reactor, Shanghai Key Laboratory of New Drug Design and School of Pharmacy, East China University of Science and Technology, Shanghai 200237, P. R. China. E-mail: yongqiangzhang@ecust.edu.cn
bDepartment of Hematology, Xinhua Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, 200092, P. R. China. E-mail: lioucx@gmail.com
First published on 22nd November 2019
Abstract
An m-CPBA-mediated intramolecular epoxidation-decarboxylative alkoxylation cascade reaction of olefinic oxamic acids has been developed. The distinct ionic decarboxylative mechanism was preliminarily revealed. The protocol features mild reaction conditions and operational simplicity, allowing the construction of diverse medicinally valuable 5–7 membered 3D cyclic carbamate architectures in moderate to high yields.
One of the central goals of organic synthesis is to develop new reactions for the synthesis of complex and biologically important targets using readily accessible starting materials. Realizing this challenging goal relies on uncovering new reactivity, which can transform into a powerful strategy for distinct bond connection and functionalization.
Oxamic acids are conveniently prepared with great structural diversity by the direct coupling of various amines with the feedstock oxalic acid or oxalyl chloride. Intensive research has been focused on the utilization of carbamoyl radicals in situ generated via the oxidative decarboxylation of oxamic acids, which enables the construction of amide, carbamate and urea functionalities through C–C, C–O or C–N bond formation.1–7 In our previous work, the hypervalent iodine(III)-mediated intramolecular decarboxylative Heck-type reaction of 2-vinyl-phenyl oxamic acids was also realized by following a radical pathway, enabling the synthesis of diverse 2-quinolinone structures (Scheme 1a).8 Herein we wish to report an unprecedented intramolecular epoxidation-decarboxylative alkoxylation cascade reaction of olefinic oxamic acids using m-CPBA9 as the sole promoter (Scheme 1b). Distinct from the well-documented radical-engaged decarboxylation of oxamic acids, an ionic decarboxylation approach enabled by imine epoxidation was preliminarily disclosed for C–O bond formation. This protocol features mild and metal-free reaction conditions and operational simplicity, serving as a powerful tool for the synthesis of structurally diverse and medicinally valuable three-dimensional (3D) cyclic carbamates bearing a quaternary carbon centre ortho to the oxygen atom (Fig. S1, ESI†).10–15 Interestingly, in addition to the 5/6-membered 3D cyclic carbamates with better stability, the synthesis of challenging 7-membered structures is also achieved.
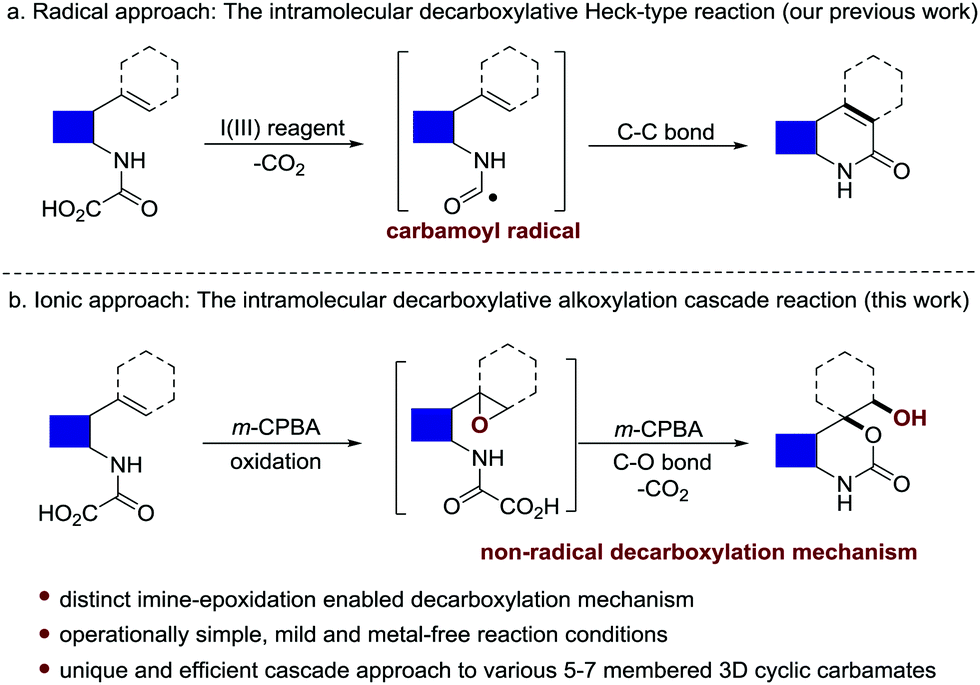 | ||
| Scheme 1 The intramolecular decarboxylative funtionalization of olefinic oxamic acids. | ||
In our exploration of a new method for the synthesis of 2-quinolinone 2a by using a hypervalent iodine(III)-mediated intramolecular decarboxylative Heck-type reaction of 2-(prop-1-en-2-yl) phenyloxamic acid 1a,8 unexpectedly, a six-membered cyclic carbamate bearing a quaternary carbon center ortho to the oxygen atom 3a (confirmed by single X-ray crystal structure analysis, see Scheme S1 in the ESI† for details) was produced in a low yield (21%, entry 2, Table 1) when 4-FC6H4I was used as the catalyst and m-CPBA (85%) as the oxidant.
|
|
||||
|---|---|---|---|---|
| Entry | Oxidant (equiv.) | Time (h) | 2a | 3a (%) |
| a Reaction conditions: 1a (0.15 mmol, 1.0 equiv.), oxidant (1.5–2.5 equiv.), CHCl3 (3.0 mL), rt, otherwise noted. b Yields were determined by 1H NMR using CH2Br2 as an internal standard. c 0.2 equiv. of 4-FC6H4I was employed. d Isolated yields were reported. e 2.2 equiv. of NaHCO3 was employed. f 2.2 equiv. of KF was employed. g 2.2 equiv. of TFA was employed. h Performed at 50 °C. i Performed on 1.5 g scale. | ||||
| 1 | 4-FPhI(OAc)2 (1.5) | 24 | 80% | — |
| 2 | m-CPBA (1.5) | 24 | — | 21c |
| 3 | m-CPBA (1.5) | 24 | — | 39 |
| 4 | m-CPBA (2.0) | 24 | — | 74 |
| 5 | m-CPBA (2.2) | 24 | — | 88 (79)d |
| 6 | m-CPBA (2.5) | 24 | — | 79 |
| 7 | m-CPBA (2.2) | 24 | — | <10e |
| 8 | m-CPBA (2.2) | 24 | — | <10f |
| 9 | m-CPBA (2.2) | 24 | — | 42g |
| 10 | m-CPBA (2.2) | 36 | — | 84 |
| 11 | m-CPBA (2.2) | 24 | — | 56h |
| 12 | m-CPBA (2.2) | 24 | — | 69d,i |
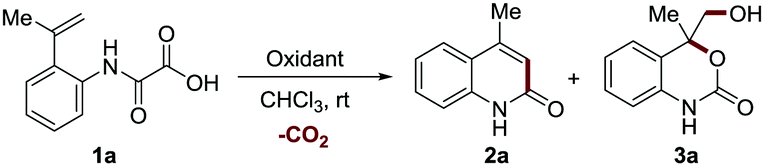
This unexpected outcome inspired us to systematically investigate the interesting process. The use of m-CPBA (85%) as the sole promoter also led to the generation of 3a in a higher yield (39% yield, entry 3) and 2.2 equivalents proved to be the optimal (39–88% yields, entries 3–6). The other oxidants such as hydrogen peroxide (30% H2O2) and oxone (2KHSO5·KHSO4·K2SO4) were also able to promote the reaction albeit with inferior reaction efficiency (Table S1, ESI†). These results, as well as the control experimental studies (Table S1, ESI†), suggest that epoxidation plays a crucial role in this transformation. The addition of base such as NaHCO3 and KF, which were reported to facilitate m-CPBA-mediated epoxidation,17,18 was detrimental to the process (entries 7 and 8). This result indicates that an epoxide ring-opening reaction promoted by the in situ generated 3-chlorobenzoic acid might be involved in this process. However, the use of strong acid TFA as an additive led to a dramatic decrease of the yield (entry 9, 42% yield). Screening of solvents revealed that CHCl3 was optimal for this reaction (Table S2, ESI†). Furthermore, neither running the reaction at a higher temperature (50 °C) nor extending the reaction time to 36 h further improved the reaction efficiency (56–84% yields, entries 10 and 11). Accordingly, the optimal reaction conditions are established as shown in entry 5 (m-CPBA 2.2 equiv., CHCl3, rt, 24 h). Under the optimal reaction conditions, the reaction can be scaled up to 1.5 g and provide the product with 69% isolated yield (entry 12).
Having established the optimal reaction conditions, we next sought to evaluate the substrate scope by varying the substituents on the aromatic ring of 2-(prop-1-en-2-yl)phenyloxamic acid (Scheme 2). The reactions proceeded well for the substrates bearing both electron-withdrawing and weak electron-donating moieties at the 4 or 6 position, providing the products in excellent yields (3b–3g, 80–91% yields). The substrates with these groups at the 5 position displayed slightly inferior reaction efficiency (3i–3o, 70–80% yields). Notably, the oxidation-sensitive alkynyl group was well tolerated (3h, 68% yield). Introducing strong electron-donating groups on the benzene ring was more pronounced detrimental to the reaction (3p–3r, 48–65% yields), which could be attributed to the undesired oxidative side reactions of these highly electron-rich methoxyaniline structures. Both the methyl group at the 3 position and the fused benzene ring at the 3 and 4 positions distinctly reduced the reaction efficiency (3s–3t, 60–64% yields) perhaps owing to the steric hindrance. It should be noted that chloride and cyano groups were well tolerated, providing handles for further functionalization. The N-methylated substrate was also proved to be an effective reactant (3u, 82% yield). Furthermore, this protocol was compatible with the tetrahydroquinoline and indoline derived 2-vinyl phenyloxamic acids, enabling the synthesis of therapeutically valuable tricyclic carbamate structures19 in synthetically useful yields (3v–3w, 56–61% yields).
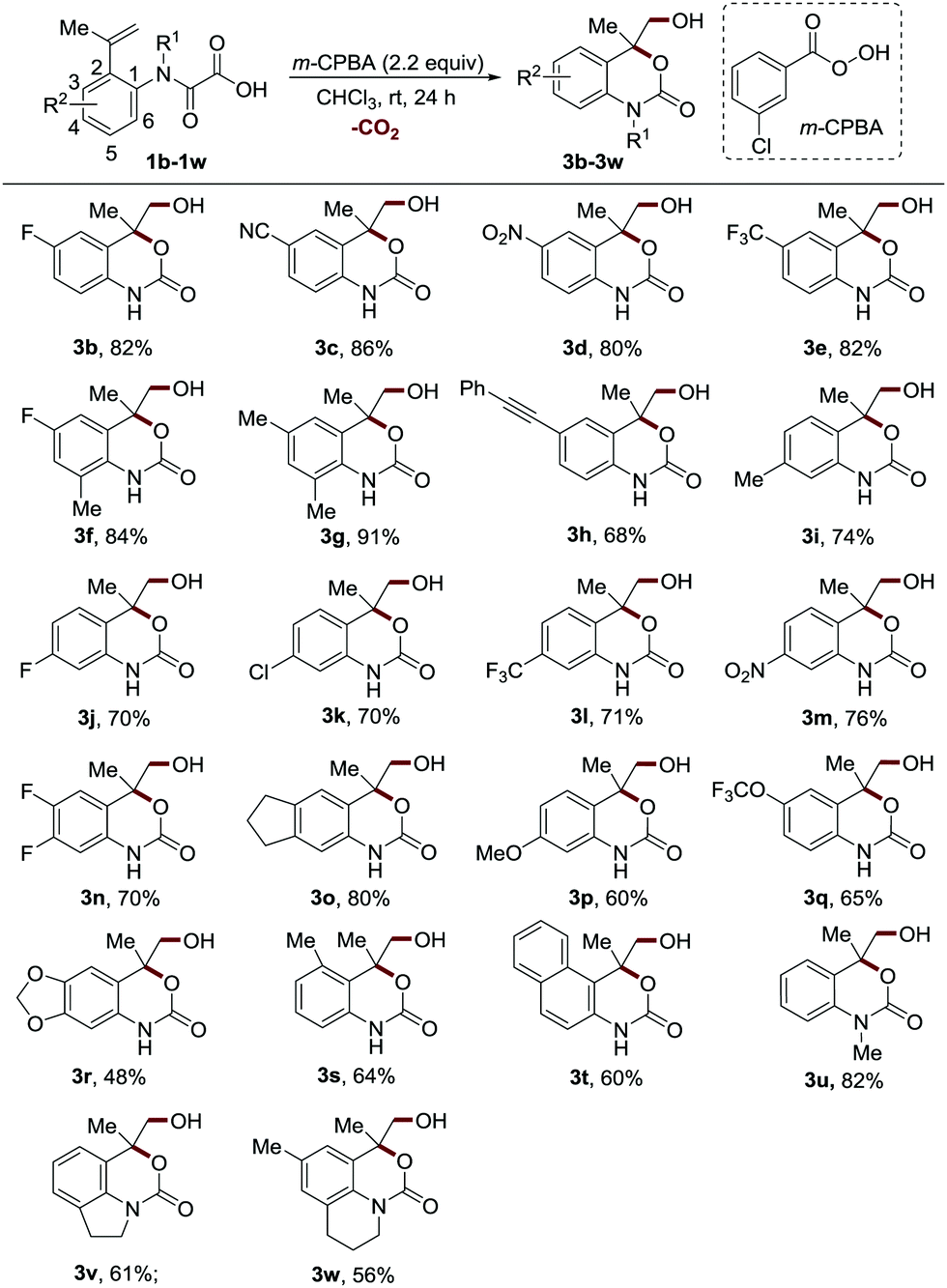 | ||
Scheme 2 The substrates bearing different substituents on the aromatic ring.a a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction conditions: see the general procedure B for experimental details, and the isolated yields are reported, unless otherwise noted. Reaction conditions: see the general procedure B for experimental details, and the isolated yields are reported, unless otherwise noted. | ||
The cyclic carbamatation of the phenyloxamic acids with different alkene moieties at the ortho position was then investigated (Scheme 3). The 2-vinyl phenyloxamic acids without any substituents on the alkene group or with the phenyl group at the 1′ position (R3 = Ph, R4 = R5 = H) performed well under the reaction conditions (5a–5h, 66–83% yields). However, strong electron-withdrawing substituents such as the trifluoromethyl group (R3 = CF3) completely shut down the reaction (5i). The reactants with the methyl group at the terminal position of alkene reacted smoothly, giving the products in excellent yields (5j–5k, 84–86% yields). In contrast, the use of phenyl and thienyl groups at this position resulted in a dramatic decrease of the yields (5l–5m, 47–48% yields). The stabilizing effect of aryl on the carbocation might result in a decrease of the eletrophilicity of the epoxide intermediate, which helps to explain this result. However, the steric hindrance effect cannot be totally excluded. It is to be noted that the configuration of alkene (Z or E) has little effect on the reaction efficiency, thus stereoretentively producing the products 5j, 5l and 5m.
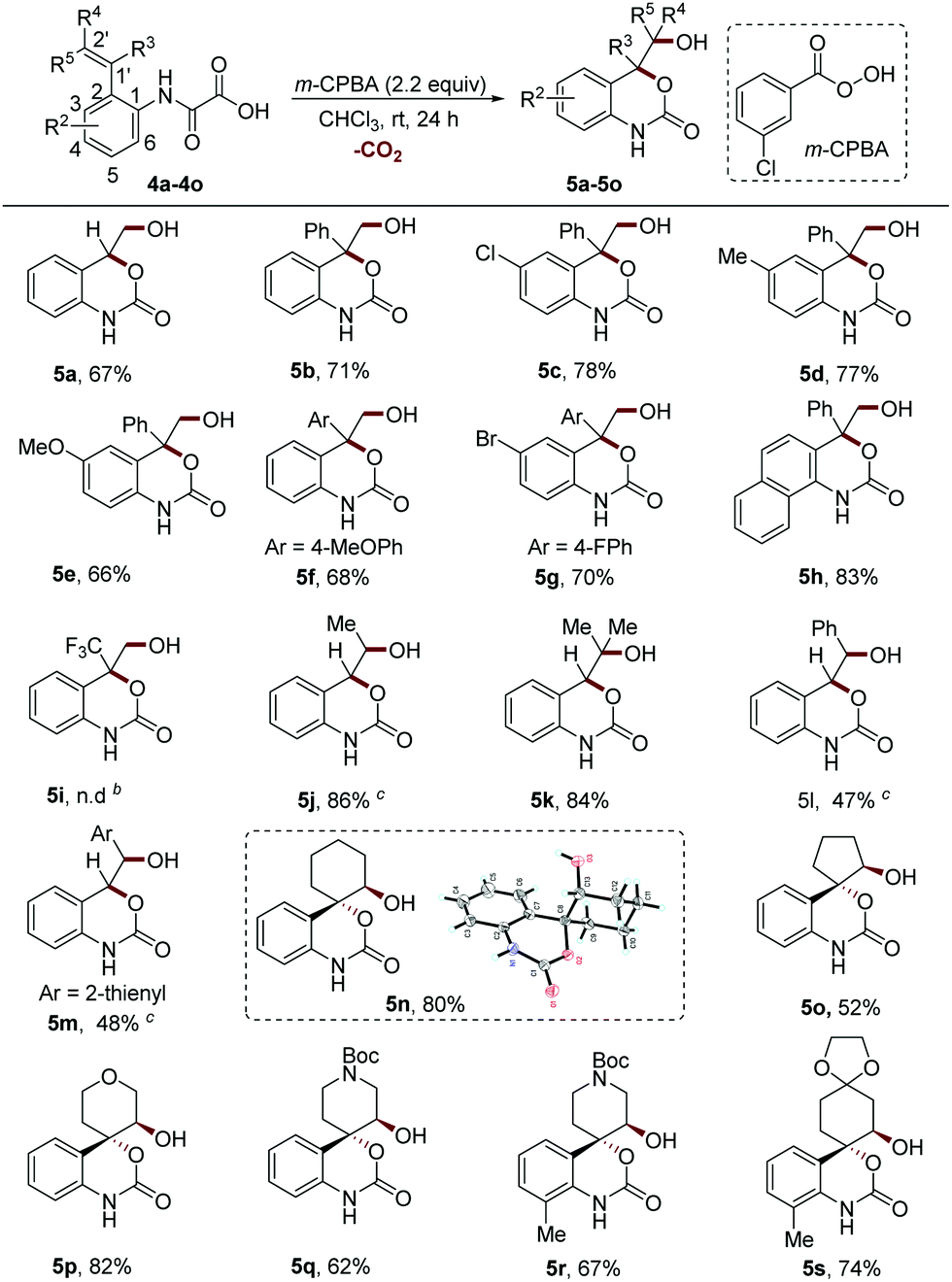 | ||
| Scheme 3 The substrates with structural variation of alkenes.a aReaction conditions: see the general procedure B for experimental details, and the isolated yields are reported, unless otherwise noted. bnot detected. cDiastereomeric ratio (d.r.) determined using 1H NMR spectroscopy: 5j, dr = 3.7:1 (4j, E:Z = 3.2:1); 5l and 5m, dr > 20:1 (4l and 4m, E:Z > 20:1). | ||
In light of the high importance of sp3 and spiro-enriched cyclic carbamates in drug discovery,10,16 we further tested the reactivities of substrates with various cycloalkenyl groups. We found that the reactions of this class of substrates proceeded smoothly to deliver the corresponding sp3-enriched spirocyclic carbamates in moderate to high yields with excellent trans-stereoselectivity (5n–5s, 52–82% yields). This stereospecificity suggests that an SN2 type ring-opening reaction of epoxide might be involved in this process. Notably, the key intermediate for the synthesis of an antihypotensive drug candidate12 can be facilely accessed in a good yield with our method (5q, 62% yield).
To further establish the general application of this transformation, we next sought to examine the feasibility of our method in the synthesis of other ring sized cyclic carbamates (Scheme 4). To our delight, this strategy was also proved to be effective for constructing the 7-membered cyclic carbamate structure (7a, 41% yield), which is difficult to synthesize via the existing methods.20–26 Furthermore, 2-phenylprop-2-en-1-amine derived oxamic acid 8a was also amendable to this protocol, providing 5-membered cyclic carbamate 9a in a synthetically useful yield (32% yield).
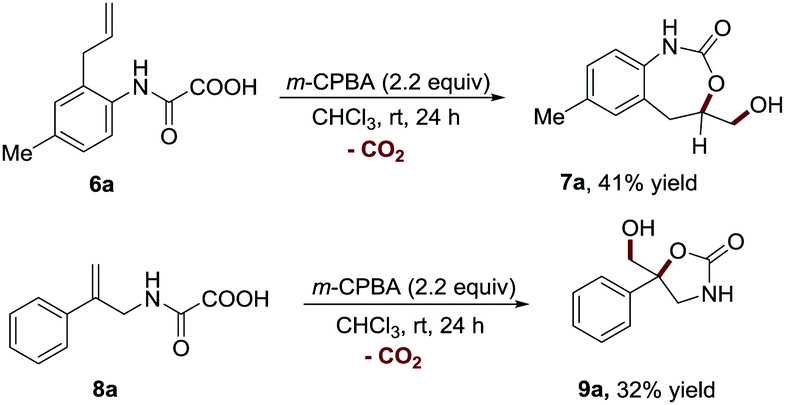 | ||
| Scheme 4 The synthesis of 7- and 5-membered 3D cyclic carbamates. | ||
We sought to elucidate the mechanistic aspects of this interesting reaction. The addition of TEMPO in the reaction has little effect on the outcome (Scheme S2B, ESI†), suggesting that the reaction proceeds with a non-radical decarboxylation mechanism. As far as we are aware, the decarboxylation process with oxamic acids often proceeds via a radical pathway.1–7 The treatment of the methyl esterified 2-vinyl phenyloxamic acid 11a with the reaction conditions provided an oxidative cleavage product of terminal alkene 12a (Scheme S2C, ESI†), which further concludes that epoxidation of the olefin is involved in the process (Scheme S3, ESI†).27,28 Considering the fact that the use of a methylene group in place of the amide carbonyl group of oxamic acid failed to produce the cyclic carbamate structure (Scheme S2D, ESI†); it is believed that the amide group might participate in the intramolecular epoxide ring-opening reaction as a nucleophile to form the benzoxazine intermediate III (Scheme 5).
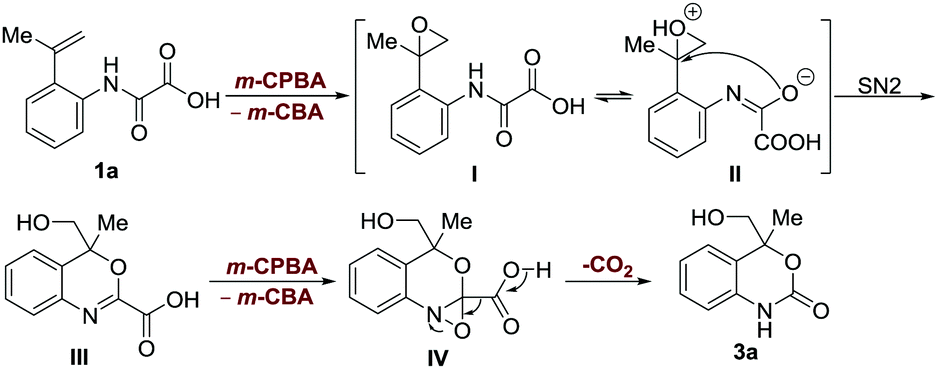 | ||
| Scheme 5 Proposed mechanism. | ||
Based on these studies, a plausible mechanism was proposed (Scheme 5). The reaction is initiated through the epoxidation of alkene to produce the epoxide intermediate I. An SN2 type intramolecular ring-opening reaction of protonated epoxide II enables the generation of a benzoxazine carboxylic acid structure III. Finally, the imine epoxidation-enabled decarboxylation results in the formation of the product 3a. This distinct decarboxylation mechanism was further supported by the control reactions (see the ESI,† Schemes S2E and S4).
The epoxide intermediate-based mechanism inspired us to explore the reaction of 1a using NaClO as the oxidant, where a three-membered cyclic halonium ion intermediate might be in situ generated to facilitate the synthesis of chloro-substituted 3D cyclic carbamates. To our delight, NaClO was also proved to be an effective reaction promoter (Table S3, ESI†), enabling the facile construction of diverse chrolo-substituted 3D cyclic carbamates with inferior reaction efficiency and with a handle for further synthetic elaboration (10a–10d, 38–57% yields, Scheme 6).
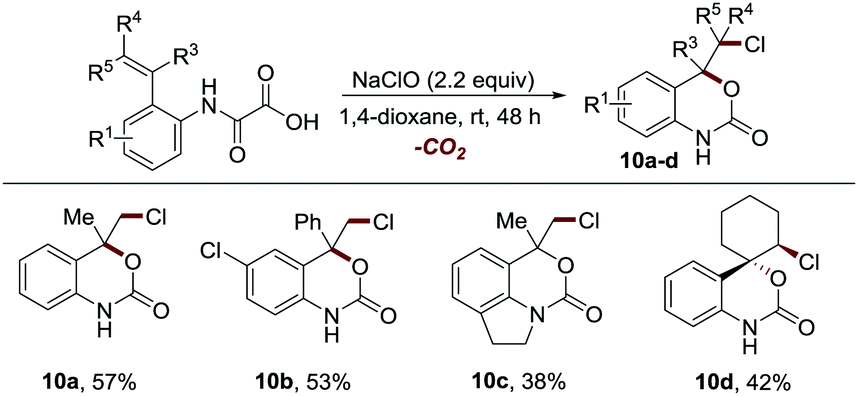 | ||
| Scheme 6 Synthetic application.a aReaction conditions: see the general procedure C for experimental details, and the isolated yields are reported, unless otherwise noted. | ||
In conclusion, we describe here an unprecedented metal-free intramolecular epoxidation-decarboxylative alkoxylation cascade reaction of readily accessible and easily handled olefinic oxamic acids using m-CPBA as the sole promoter. Distinct from the well-established radical-engaged decarboxylation of oxamic acids, the new process follows an ionic pathway in which imine epoxidation enables the decarboxylative C–O bond formation. The new reactivity transforms into an alternative approach for the synthesis of structurally diverse and medicinally valuable 5–7 membered 3D cyclic carbamate architectures. Furthermore, this protocol is mild and operationally simple and displays excellent functional group tolerance and broad substrate scope. Further exploration of the asymmetric version of this transformation and validation of the proposed mechanism are still ongoing in our laboratory.
Financial support from the program of the National Natural Science Foundation of China (21602060, 21871086, Y.-Q. Z.; 21572055, 21738002, W. W.) and the Chinese Fundamental Research Funds for the Central Universities (ECUST: WY1714033) is gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Q.-F. Bai, C. Jin, J.-Y. He and G. Feng, Org. Lett., 2018, 20, 2172 CrossRef CAS.
- H. Huang, G.-J. Zhang and Y.-Y. Chen, Angew. Chem., Int. Ed., 2015, 54, 7872 CrossRef CAS.
- M. Yuan, L. Chen, J.-W. Wang, S.-J. Chen, K.-C. Wang, Y.-B. Xue, G.-M. Yao, Z. W. Luo and Y.-H. Zhang, Org. Lett., 2015, 17, 346 CrossRef CAS.
- H. Wang, L.-N. Guo, S. Wang and X.-H. Duan, Org. Lett., 2015, 17, 3054 CrossRef CAS.
- M.-Z. Li, C. Wang, P. Fang and H.-B. Ge, Chem. Commun., 2011, 47, 6587 RSC.
- G. G. Pawar, F. Robert, E. Grau, H. Cramail and Y. Landais, Chem. Commun., 2018, 54, 9337 RSC.
- A. H. Jatoi, G. G. Pawar, F. Robert and Y. Landais, Chem. Commun., 2019, 55, 466 RSC.
- H.-Q. Fan, P. Pan, Y.-Q. Zhang and W. Wang, Org. Lett., 2018, 20, 7929 CrossRef CAS.
- H. Hussain, A. Al-Harrasi, I. R. Green, I. Ahmed, G. Abbas and N. U. Rehman, RSC Adv., 2014, 4, 12882 RSC.
- A. K. Ghosh and M. Brindisi, J. Med. Chem., 2015, 58, 2895 CrossRef CAS.
- P. M. Cox and N. N. Bumpus, ACS Med. Chem. Lett., 2014, 5, 1156 CrossRef CAS.
- H. Takai, H. Obase, M. Teranishi, A. Karasawa, K. Kubo, K. Shuto, Y. Kasuya, M. Hashikami, N. Karashima and K. Shigenobu, Chem. Pharm. Bull., 1985, 33, 1129 CrossRef CAS.
- C. P. Leslie, J. Bentley, M. Biagetti, S. Contini, R. D. Fabio, D. Donati, T. Genski, S. Guery, A. Mazzali, G. Merlo, D. A. Pizzi, F. Sacco, C. Seri, M. Tessari, L. Zonzini and L. Caberlotto, Bioorg. Med. Chem. Lett., 2010, 20, 6103 CrossRef CAS.
- L.-H. Zhuang, C. M. Tice, Z. Xu, W. Zhao, S. Cacatian, Y.-J. Ye, S. B. Singh, P. Lindblom, B. M. McKeever, P. M. Krosky, Y. Zhao, D. Lala, B. A. Kruk, S. Meng, L. Howard, J. A. Johnson, Y. Bukhtiyarov, R. Panemangalore, J. Guo, R. Guo, F. Himmelsbach, B. Hamilton, A. Schuler-Metz, H. Schauerte, R. Gregg, G. M. McGeehan, K. Leftheris and D. A. Claremon, Bioorg. Med. Chem., 2017, 25, 3649 CrossRef CAS.
- Y.-D. Zhang, J.-P. Wu, G.-H. Li, K. R. Fandrick, J. Gao, Z. Tan, J. Johnson, W. Li, S. Sanyal, J. Wang, X.-F. Sun, J. C. Lorenz, S. Rodriguez, J. T. Reeves, N. Grinberg, H. Lee, N. Yee, B.-Z. Lu and C. H. Senanayake, J. Org. Chem., 2016, 81, 2665 CrossRef CAS.
- N. C. Tran, H. Dhondt, M. Flipo, B. Deprez and N. Willand, Tetrahedron Lett., 2015, 56, 4119 CrossRef CAS.
- J. L. G. Ruano, J. Alemán, C. Fajardo and A. Parra, Org. Lett., 2005, 7, 5493 CrossRef CAS.
- F. A. Davis, J. Lamendola, U. Nadir, E. W. Kluger, T. C. Sedergran, T. W. Panunto, R. Billmers, R. Jenkins and I. J. Turchi, J. Am. Chem. Soc., 1980, 102, 2000 CrossRef CAS.
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845 CrossRef CAS.
- A. Garzan, A. Jaganathan, M. N. Salehi, R. Yousefi, D. C. Whitehead, J. E. Jackson and B. Borhan, Chem. – Eur. J., 2013, 19, 9015 CrossRef CAS.
- N. Salehi Marzijarani, R. Yousefi, A. Jaganathan, K. D. Ashtekar, J. E. Jackson and B. Borhan, Chem. Sci., 2018, 9, 2898 RSC.
- Y.-M. Yu, Y.-N. Huang and J. Deng, Org. Lett., 2017, 19, 1224 CrossRef CAS.
- K. Kobayashi, S. Fukamachi, D. Nakamura, O. Morikawa and H. Konishi, Heterocycles, 2008, 75, 95 CrossRef CAS.
- L. Sun, J.-H. Ye, W.-J. Zhou, X. Zeng and D.-G. Yu, Org. Lett., 2018, 20, 3049 CrossRef CAS.
- J.-H. Ye, L. Song, W.-J. Zhou, T. Ju, Z.-B. Yin, S.-S. Yan, Z. Zhang, J. Li and D.-G. Yu, Angew. Chem., Int. Ed., 2016, 55, 10022 CrossRef CAS.
- Y. Takeda, S. Okumura, S. Tone, I. Sasaki and S. Minakata, Org. Lett., 2012, 14, 4874 CrossRef CAS.
- M. O. F. Goulart, A. G. Cioletti, J. D. de Souza Filho, C. A. De Simone, E. E. Castellano, F. S. Emery, K. C. G. De Moura, M. C. F. R. Pinto and A. V. Pinto, Tetrahedron Lett., 2003, 44, 3581 CrossRef CAS.
- F. V. Singh, H. M. S. Milagre, M. N. Eberlin and H. A. Stefani, Tetrahedron Lett., 2009, 50, 2312 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details and NMR spectra. CCDC 1902836 and 1902837. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc07709j |
| ‡ These two authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2020 |