
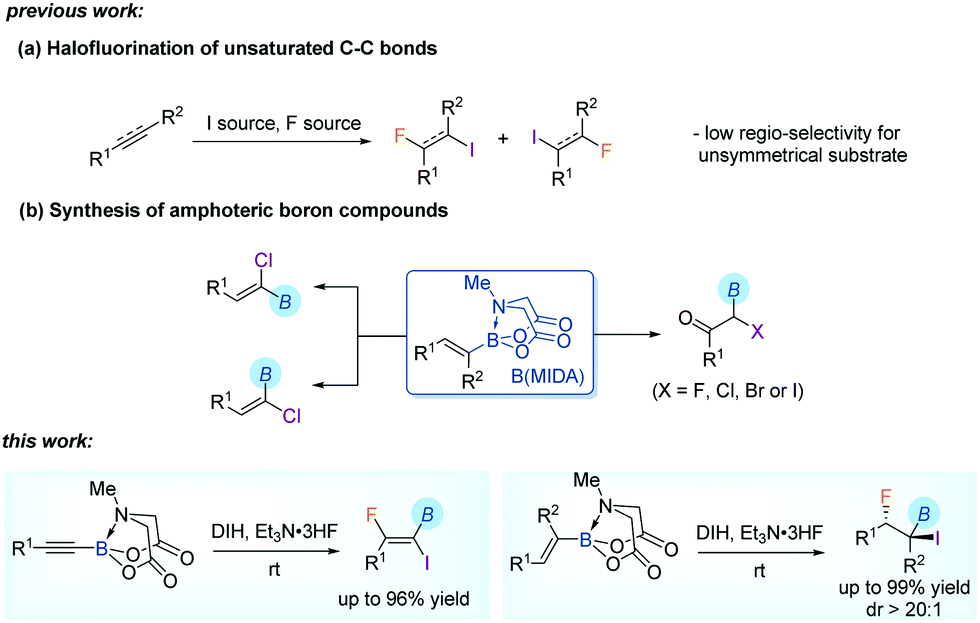
Synthesis of fluorinated amphoteric organoborons via iodofluorination of alkynyl and alkenyl MIDA boronates†
Wen-Xin
Fan
a,
Ji-Lin
Li
a,
Wen-Xin
Lv
a,
Ling
Yang
a,
Qingjiang
Li
 a and
Honggen
Wang
*ab
a and
Honggen
Wang
*ab
aGuangdong Provincial Key Laboratory of Chiral Molecule and Drug Discovery, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China. E-mail: wanghg3@mail.sysu.edu.cn
bState Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources, School of Chemistry and Pharmaceutical Sciences of Guangxi Normal University, Guilin 541004, China
First published on 26th November 2019
Abstract
The iodofluorination of alkynyl and alkenyl MIDA (N-methyliminodiacetyl) boronates led to the synthesis of two types of fluorinated organoborons bearing a valuable C–I bond. The B(MIDA) moiety confers exclusive regioselectivity to the reaction, and the products were formed in generally good yields. Preliminary utility of the products was demonstrated.
There has been considerable interest in the synthesis and chemistry of organofluorines due to their often desirable pharmacokinetic and metabolic properties in pharmaceuticals and agrochemicals.1 One useful approach for the introduction of fluorine to organic molecules is the halofluorination of unsaturated C–C bonds, leading to vicinal halofluorides.2,3 By related methods not only a fluorine atom but also a halogen (chlorine, bromine or iodine) functionality could be incorporated simultaneously. The latter could then serve as a valuable handle for downstream transformations. An important feature of a typical halofluorination reaction is the high stereoselectivity, arising from the initial formation of a three-membered halonium cation intermediate3d and thereafter anti SN2 nucleophilic substitution by a fluoride anion in the mechanism. Nevertheless, the regioselectivity may be problematic when unsymmetrical alkenes or alkynes are used as substrates, although Markovnikov's rule is generally applicable.
We have been interested in the synthetic transformations of alkenyl MIDA (N-methyliminodiacetyl) boronates4 towards the step-economic construction of structurally complex organoborons.5 The reaction of alkenyl MIDA boronates6,7 with different halogen sources allowed us to prepare several halogenated organoborons. These include α-boryl-α-haloketones5a and α-chloroalkenyl boronates,5b both of which are traditionally challenging targets in organic synthesis. Particularly, in line with the importance of fluorine-containing molecules in functional molecules, our attention was also drawn to the synthesis of fluorinated organoborons via a similar late-stage fluorination of a pre-borylated starting material.5c In continuation with these studies, herein, we report an efficient iodofluorination of alkenyl and alkynyl MIDA boronates. The MIDA boron moiety tolerated strong oxidizing reaction conditions and was retained in the product, bringing about additional value to these new fluorinated amphoteric synthons.8 Furthermore, in addition to the high stereoselectivity, an exclusive regioselectivity was observed, thanks to the directing effect of MIDA boron (Scheme 1).
 | ||
| Scheme 1 Iodofluorination and late-stage modification of alkenyl MIDA boronates. | ||
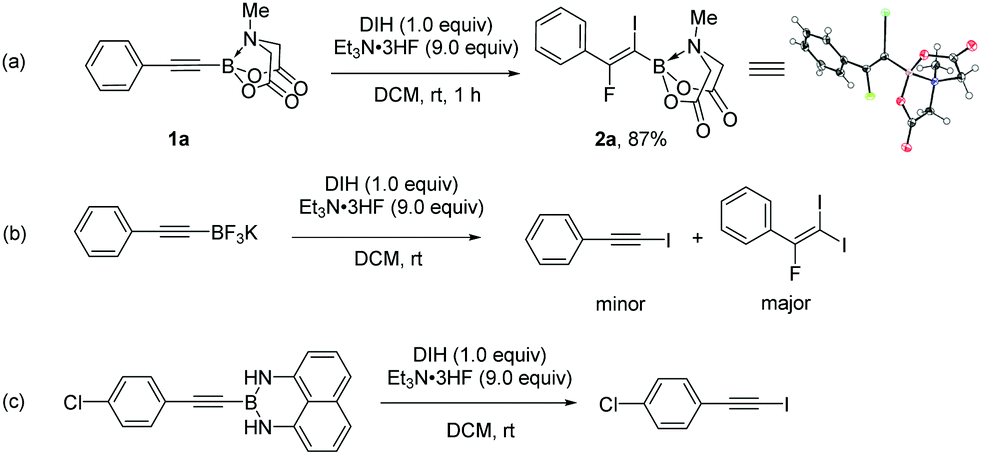
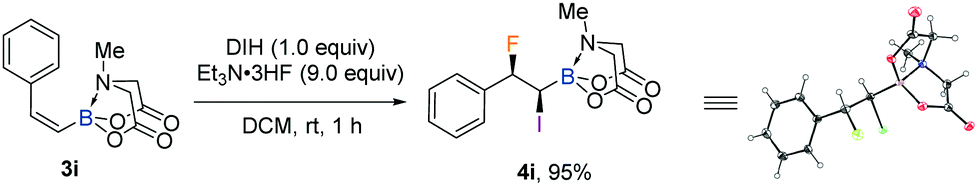
We first explored the iodofluorination of alkynyl MIDA boronates, anticipating to obtain a fluoroalkene bearing a valuable C–B and C–I bond within the same molecule. It should be noted that fluoroalkene has been recognized as an important structural motif in medicinal chemistry.9 Thus, the development of a synthetic method towards multi-functionalized fluoroalkenes is highly desirable. By using analogous conditions of Gouverneur,3k which consists of the use of DIH (1,3-diiodo-5,5,-dimethylhydantoin) as an electrophilic iodo source and Et3N·3HF as a nucleophilic fluorine source in DCM at room temperature, the reaction of phenylethynyl MIDA boronate 1a delivered a trans iodofluorination product 2a in 87% yield (Scheme 2a). The iodo atom was found to be attached α to the C–B bond as clearly defined by X-ray diffraction analysis (CCDC 1942792†).9 The replacement of B(MIDA) moiety with its sp3-B congener BF3K, or sp2-B B(dan), all led to an oxidative deboryliodination reaction, indicating the importance of stability of B(MIDA) in this reaction (Schemes 2b and c).
 | ||
| Scheme 2 Exploration of different alkynyl boronates in the iodofluorination reaction. | ||
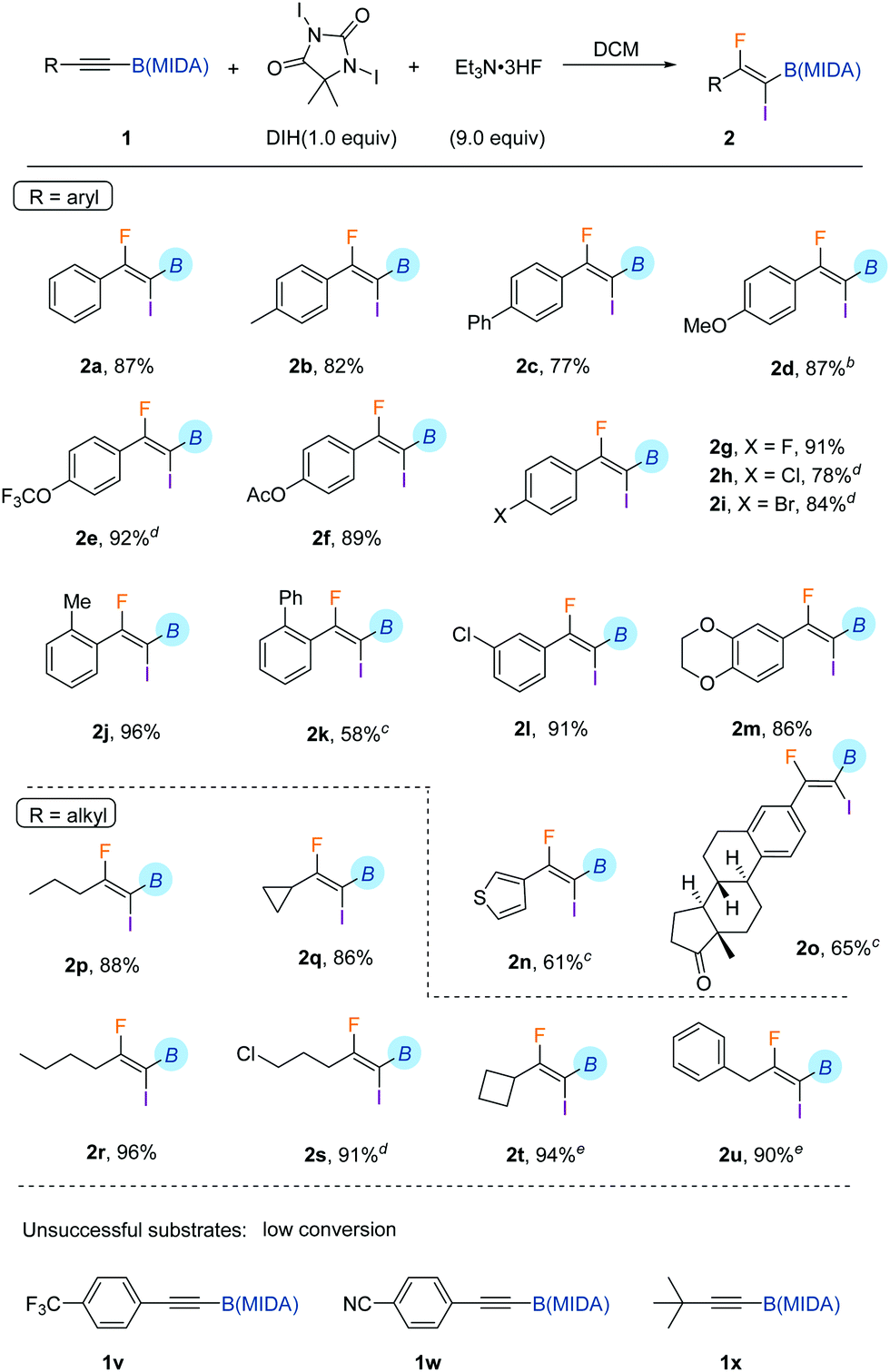
To determine the generality and limitations of this reaction, diverse aryl-substituted alkynyl MIDA boronates were applied. As shown in Table 1, a number of functional groups on the aryl ring were well tolerated under the standard conditions. These include methyl (2b and 2j), phenyl (2c and 2k), methoxy (2d), acetoxyl (2f), trifluoromethoxy (2e), and halogens (2g–2i, 2l). Besides, a thiophenyl substituted substrate was amenable for iodofluorination (2n). And an estrone-derived substrate could also be smoothly converted to the corresponding product 2o in good yield. Strong electron-withdrawing substituents, such as trifluoromethyl (1v), and cyano (1w), retard the reaction, leading to low conversion of the corresponding starting materials. The protocol was applicable to alkyl-substituted alkynyl MIDA boronates as well (2p–2u). And only one regioisomer was detected, indicating the strong directing effect of B(MIDA). Another limitation of the reaction is the incompatibility of tert-butyl groups (1x), probably due to steric reasons. In all these cases, no column chromatography was needed. A simple quenching with aqueous sodium thiosulfate and extraction generally led to a chemically pure product.
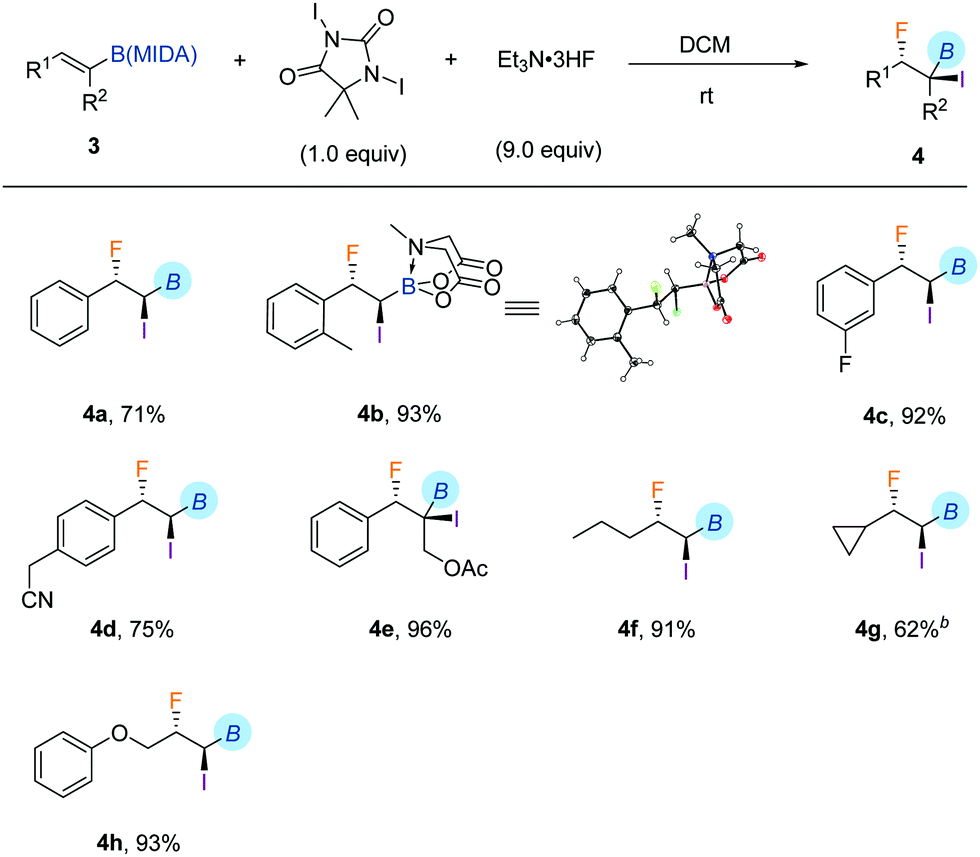
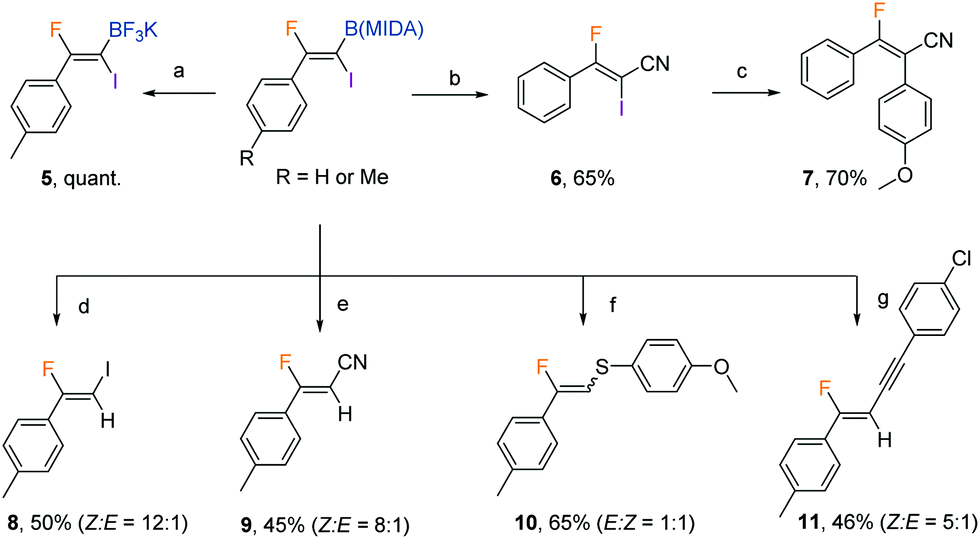
With the iodofluorination of alkynyl MIDA boronates established, we then explored a similar iodofluorination by using alkenyl MIDA boronate as a substrate (Table 2). As expected, by using the identical reaction conditions, phenylvinyl MIDA boronate 3a underwent a reaction smoothly to deliver an anti-iodofluorination adduct 4a. The relative stereochemistry is determined by the X-ray diffraction analysis of 4b (CCDC 1953148†).10 It is worth mentioning that the tri-substituted alkene (4e) was also a good substrate, giving the desired product in 96% yield without erosion of either regio- or stereoselectivity. The alkyl substrates also provided the desired products in good yields and selectivity (4f–4h). The anti-addition nature of the protocol also allowed us to synthesise the cis iodofluorination product (CCDC 1956650†)10 by simply using a Z-type starting material (eqn (1)).
 | (1) |
 | (2) |
 | (3) |
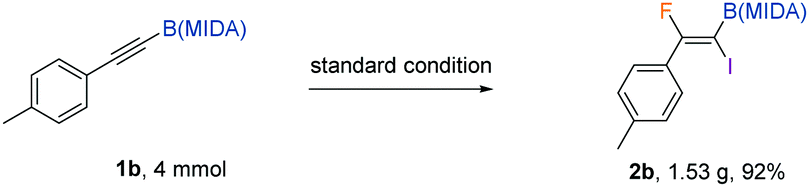
The protocol was amenable to gram-scale synthesis, as evidenced by the excellent yield obtained when multiple millimoles of the alkynyl or alkenyl substrate was employed in the reaction (eqn (2) and (3)). The synthetic utility of the products was preliminarily investigated. The B(MIDA) moiety could be converted to the corresponding potassium trifluoroboronate (5) in quantitative yield upon ligand exchange with KHF2 in MeOH.11 A copper-mediated Chan–Lam-type cyanation5b yielded fluoroalkenyl cyanide (6) in good yield. The C–I bond in 6 could then be used in Suzuki–Miyaura coupling to give a fully substituted fluoroalkene in a stereospecific fashion. Intriguingly, synthetic manipulation on the C–I bond in 2b in the presence of copper all resulted in the formation of deborylation products. The newly introduced functional groups are predominantly located trans to the aryl ring. A metal vinylidene species might be formed as intermediates in these reactions (Table 3).12
The regio- and stereoselectivity is worthy of mention (Scheme 3). Initially, the reaction of DIH with alkynyl MIDA boronates 1 or alkenyl MIDA boronates 3 would form relatively stable three-membered halonium cation intermediates.3d The SN2 nucleophilic ring-opening from the opposite side of iodine accounts for the observed anti stereochemistry of the reaction. The hemilabile nature of the MIDA B–N dative bond makes the boron atom an electron acceptor to some extent.5e,6i For this reason, the development of more cations at the β position is expected, thus leading to a regioselective fluoride substitution. On the other hand, the bulky nature of the B(MIDA) moiety may also dictate the nucleophilic attack at the β position.
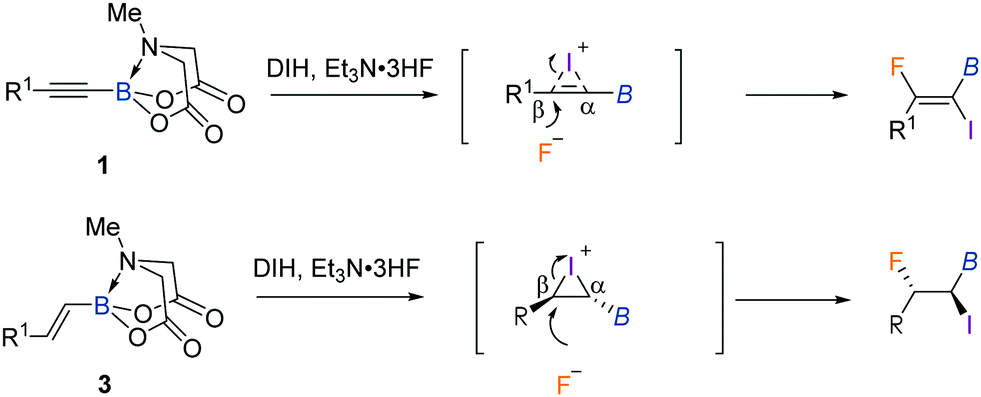 | ||
| Scheme 3 Proposed mechanism. | ||
In summary, by taking advantage of the notable stability of MIDA boronates, we realized an iodofluorination reaction of alkynyl and alkenyl borons. The reaction led to the synthesis of two types of borylated organofluorines bearing a valuable C–I bond. The B(MIDA) confers exclusive regioselectivity to the reaction. Both aryl and alkyl substituents are well compatible and the products were formed in generally good yields. Preliminary utility of the products was demonstrated.
Generous financial support from the Key Project of Chinese National Programs for Fundamental Research and Development (2016YFA0602900), the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01Y093) and Guangdong Provincial Key Laboratory of Chiral Molecule and Drug Discovery (2019B030301005) is gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) A. Choudhary and R. T. Raines, ChemBioChem, 2011, 12, 1801–1807 CrossRef CAS; (b) K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef; (c) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (d) M. Shimizu and T. Hiyama, Angew. Chem., Int. Ed., 2004, 44, 214–231 CrossRef; (e) D. H. Tan, E. Lin, W. W. Ji, Y. F. Zeng, W. X. Fan, Q. J. Li, H. Gao and H. G. Wang, Adv. Synth. Catal., 2018, 360, 1032–1037 CrossRef CAS; (f) J. Wang, M. Sanchez-Rosello, J. L. Acena, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS; (g) K. Yamamoto, J. Li, J. A. O. Garber, J. D. Rolfes, G. B. Boursalian, J. C. Borghs, C. Genicot, J. Jacq, M. van Gastel, F. Neese and T. Ritter, Nature, 2018, 554, 511–514 CrossRef CAS; (h) L. Yang, W. W. Ji, E. Lin, J. L. Li, W. X. Fan, Q. Li and H. Wang, Org. Lett., 2018, 20, 1924–1927 CrossRef CAS.
- For a review: B. Marciniak, J. Walkowiak-Kulikowska and H. Koroniak, J. Fluorine Chem., 2017, 203, 47–61 CrossRef CAS.
- For selected examples, see:
(a) M. Hauptschein and M. Braid, J. Am. Chem. Soc., 1961, 83, 2383–2386 CrossRef CAS;
(b) G. A. Olah, M. Nojima and l. Kerekes, Synthesis, 1973, 780–783 CrossRef CAS;
(c) G. A. Olah, J. T. Welch, Y. D. Vankar, M. Nojima, I. Kerekes and J. A. Olah, J. Org. Chem., 1979, 44, 3872–3881 CrossRef CAS;
(d) P. Esteves, M. de
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Mattos, L. Crespo and R. Ribeiro, Synthesis, 2010, 2379–2382 Search PubMed;
(e) J. Barluenga, P. J. Campos, J. M. Gonzalez, J. L. Suarez, G. Asensio and G. Asensio, J. Org. Chem., 1991, 56, 2234–2237 CrossRef CAS;
(f) M. Kuroboshi and T. Hiyama, Synlett, 1991, 185–186 CrossRef CAS;
(g) D. Dolenc and B. Šket, Synlett, 1995, 327–328 CrossRef CAS;
(h) N. O. Ilchenko, M. A. Cortés and K. J. Szabó, ACS Catal., 2015, 6, 447–450 CrossRef;
(i) Y. Li, X. Liu, D. Ma, B. Liu and H. Jiang, Adv. Synth. Catal., 2012, 354, 2683–2688 CrossRef CAS;
(j) S. Kobayashi, M. Sawaguchi, S. Ayuba, T. Fukuhara and S. Hara, Synlett, 2001, 1938–1940 CrossRef CAS;
(k) L. Pfeifer and V. Gouverneur, Org. Lett., 2018, 20, 1576–1579 CrossRef CAS;
(l) S. Hara, T. Guan and M. Yoshida, Org. Lett., 2006, 8, 2639–2641 CrossRef CAS;
(m) T. Kitamura, S. Mizuno, K. Muta and J. Oyamada, J. Org. Chem., 2018, 83, 2773–2778 CrossRef CAS;
(n) M. Ochiai, M. Hirobe, A. Yoshimura, Y. Nishi, K. Miyamoto and M. Shiro, Org. Lett., 2007, 9, 3335–3338 CrossRef CAS.
Mattos, L. Crespo and R. Ribeiro, Synthesis, 2010, 2379–2382 Search PubMed;
(e) J. Barluenga, P. J. Campos, J. M. Gonzalez, J. L. Suarez, G. Asensio and G. Asensio, J. Org. Chem., 1991, 56, 2234–2237 CrossRef CAS;
(f) M. Kuroboshi and T. Hiyama, Synlett, 1991, 185–186 CrossRef CAS;
(g) D. Dolenc and B. Šket, Synlett, 1995, 327–328 CrossRef CAS;
(h) N. O. Ilchenko, M. A. Cortés and K. J. Szabó, ACS Catal., 2015, 6, 447–450 CrossRef;
(i) Y. Li, X. Liu, D. Ma, B. Liu and H. Jiang, Adv. Synth. Catal., 2012, 354, 2683–2688 CrossRef CAS;
(j) S. Kobayashi, M. Sawaguchi, S. Ayuba, T. Fukuhara and S. Hara, Synlett, 2001, 1938–1940 CrossRef CAS;
(k) L. Pfeifer and V. Gouverneur, Org. Lett., 2018, 20, 1576–1579 CrossRef CAS;
(l) S. Hara, T. Guan and M. Yoshida, Org. Lett., 2006, 8, 2639–2641 CrossRef CAS;
(m) T. Kitamura, S. Mizuno, K. Muta and J. Oyamada, J. Org. Chem., 2018, 83, 2773–2778 CrossRef CAS;
(n) M. Ochiai, M. Hirobe, A. Yoshimura, Y. Nishi, K. Miyamoto and M. Shiro, Org. Lett., 2007, 9, 3335–3338 CrossRef CAS. - (a) M. Nakatani, Y. Takahashi, A. Ouchi and K. Watanuki, Bull. Chem. Soc. Jpn., 1970, 43, 2072–2075 CrossRef CAS; (b) S. J. Lee, K. C. Gray, J. S. Paek and M. D. Burke, J. Am. Chem. Soc., 2008, 130, 466–468 CrossRef CAS; (c) E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2007, 129, 6716–6717 CrossRef CAS; (d) E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2008, 130, 14084–14085 CrossRef CAS PubMed.
- (a) W. X. Lv, Y. F. Zeng, Q. Li, Y. Chen, D. H. Tan, L. Yang and H. Wang, Angew. Chem., Int. Ed., 2016, 55, 10069–10073 CrossRef CAS; (b) Y. F. Zeng, W. W. Ji, W. X. Lv, Y. Chen, D. H. Tan, Q. Li and H. Wang, Angew. Chem., Int. Ed., 2017, 56, 14707–14711 CrossRef CAS; (c) W. X. Lv, Q. Li, J. L. Li, Z. Li, E. Lin, D. H. Tan, Y. H. Cai, W. X. Fan and H. Wang, Angew. Chem., Int. Ed., 2018, 57, 16544–16548 CrossRef CAS; (d) W.-X. Lv, Z. Li, E. Lin, J.-L. Li, D.-H. Tan, Y.-H. Cai, Q. Li and H. Wang, Chem. – Eur. J., 2019, 25, 4058 CrossRef CAS; (e) E. E. Lin, J.-Q. Wu, F. Schäfers, X.-X. Su, K.-F. Wang, J.-L. Li, Y. Chen, X. Zhao, H. Ti, Q. Li, T.-M. Ou, F. Glorius and H. Wang, Commun. Chem., 2019 DOI:10.1038/s42004-019-0137-0.
- (a) M. L. Lepage, S. Lai, N. Peressin, R. Hadjerci, B. O. Patrick and M. Perrin, Angew. Chem., Int. Ed., 2017, 56, 15257–15261 CrossRef CAS; (b) R. N. Khanizeman, E. Barde, R. W. Bates, A. Guérinot and J. Cossy, Org. Lett., 2017, 19, 5046–5049 CrossRef CAS PubMed; (c) J. Taguchi, T. Ikeda, R. Takahashi, I. Sasaki, Y. Ogasawara, T. Dairi, N. Kato, Y. Yamamoto, J. W. Bode and H. Ito, Angew. Chem., Int. Ed., 2017, 56, 13847–13851 CrossRef CAS; (d) B. E. Uno, E. P. Gillis and M. D. Burke, Tetrahedron, 2009, 65, 3130–3138 CrossRef CAS; (e) M. Lüthy and R. J. K. Taylor, Tetrahedron Lett., 2012, 53, 3444–3447 CrossRef; (f) J. Cornil, P.-G. Echeverria, P. Phansavath, V. Ratovelomanana-Vidal, A. Guérinot and J. Cossy, Org. Lett., 2015, 17, 948–951 CrossRef CAS; (g) M. G. McLaughlin, C. A. McAdam and M. J. Cook, Org. Lett., 2015, 17, 10–13 CrossRef CAS; (h) B. Quiclet-Sire and S. Z. Zard, J. Am. Chem. Soc., 2015, 137, 6762–6765 CrossRef CAS PubMed; (i) V. B. Corless, A. Holownia, H. Foy, R. Mendoza-Sanchez, S. Adachi, T. Dudding and A. K. Yudin, Org. Lett., 2018, 20, 5300–5303 CrossRef CAS PubMed.
- (a) M. Kischkewitz, K. Okamoto, C. Muck-Lichtenfeld and A. Studer, Science, 2017, 355, 936–938 CrossRef CAS PubMed; (b) L. Zhang, G. J. Lovinger, E. K. Edelstein, A. A. Szymaniak, M. P. Chierchia and J. P. Morken, Science, 2016, 351, 70–74 CrossRef CAS PubMed; (c) A. Bonet, M. Odachowski, D. Leonori, S. Essafi and V. K. Aggarwal, Nat. Chem., 2014, 6, 584–589 CrossRef CAS PubMed.
- (a) Z. He and A. K. Yudin, J. Am. Chem. Soc., 2011, 133, 13770–13773 CrossRef CAS PubMed; (b) Z. He, A. Zajdlik and A. K. Yudin, Acc. Chem. Res., 2014, 47, 1029–1040 CrossRef CAS PubMed; (c) J. D. St. Denis, Z. He and A. K. Yudin, ACS Catal., 2015, 5, 5373–5379 CrossRef CAS; (d) P. Trinchera, V. B. Corless and A. K. Yudin, Angew. Chem., Int. Ed., 2015, 54, 9038–9041 CrossRef CAS PubMed; (e) D. B. Diaz, C. C. Scully, S. K. Liew, S. Adachi, P. Trinchera, J. D. St Denis and A. K. Yudin, Angew. Chem., Int. Ed., 2016, 55, 12659–12663 CrossRef CAS PubMed.
- (a) H. Yanai and T. Taguchi, Eur. J. Org. Chem., 2011, 5939–5954 CrossRef CAS; (b) B. Malo-Forest, G. Landelle, J. A. Roy, J. Lacroix, R. C. Gaudreault and J. F. Paquin, Bioorg. Med. Chem. Lett., 2013, 23, 1712–1715 CrossRef CAS PubMed; (c) B. Metayer, G. Compain, K. Jouvin, A. Martin-Mingot, C. Bachmann, J. Marrot, G. Evano and S. Thibaudeau, J. Org. Chem., 2015, 80, 3397–3410 CrossRef CAS PubMed.
- See the ESI† for details.
- Q. I. Churches, J. F. Hooper and C. A. Hutton, J. Org. Chem., 2015, 80, 5428–5435 CrossRef CAS PubMed.
- (a) C. Bruneau and P. H. Dixneuf, Acc. Chem. Res., 1999, 32, 311–323 CrossRef CAS; (b) B. M. Trost, Chem. Ber., 1996, 129, 1313–1322 CrossRef CAS; (c) B. M. Trost, F. D. Toste and A. B. Pinkerton, Chem. Rev., 2001, 101, 2067–2096 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, analytical data, NMR spectra of products and crystallographic data. CCDC 1942792, 1953148 and 1956650. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc08386c |
| This journal is © The Royal Society of Chemistry 2020 |