
Regioselective SNAr reaction of the phenoxathiin-based thiacalixarene as a route to a novel macrocyclic skeleton†
Tomáš
Landovský
a,
Václav
Eigner
 b,
Martin
Babor
b,
Markéta
Tichotová
c,
Hana
Dvořáková
c and
Pavel
Lhoták
*a
b,
Martin
Babor
b,
Markéta
Tichotová
c,
Hana
Dvořáková
c and
Pavel
Lhoták
*a
aDepartment of Organic Chemistry, University of Chemistry and Technology, Prague (UCTP), Technická 5, 166 28 Prague 6, Czech Republic. E-mail: lhotakp@vscht.cz
bDepartment of Solid State Chemistry, UCTP, 166 28 Prague 6, Czech Republic
cLaboratory of NMR Spectroscopy, UCTP, 166 28 Prague 6, Czech Republic
First published on 22nd November 2019
Abstract
The sulfonyl analogue of phenoxathiin-based thiacalix[4]arene, easily accessible from the parent thiacalix[4]arene, reacts with sodium alkoxides to yield a cleaved product representing a novel type of macrocyclic skeleton with a quasi-calixarene structure. As shown by comparison with other derivatives, the internal strain imposed by the heterocyclic moiety is a driving force of this SNAr reaction.
Thiacalixarenes1 appeared as part of the calixarene family2 more than two decades ago. The presence of sulfur atoms in place of the original CH2 bridges makes these compounds very attractive for supramolecular chemists. Albeit seemingly very similar, considering the macrocycle's topology, thiacalix[4]arenes exhibit surprisingly different chemical behaviour, conformational preferences and complexation properties compared to their parent CH2 counterparts.1
Introduction of sulfur bridges brings about novel types of derivatization reactions, which are impossible for parent calixarenes, such as sulfoxide or sulfone formation, or even S-alkylation3 to form sulfonium ions. In this context, very distinct behavior of the corresponding spirodienone derivatives was demonstrated (Scheme 1). Classical calixarene spirodienones were found to be useful intermediates in the synthesis of calixarene systems with functionalization patterns hardly attainable by other methods.4 For example, the reaction of mono(spirodienone) 1 with HCl leads to the para-chloro substituted derivative 3.5 On the other hand, in the thiacalixarene series, heating of the compound 26 in aqueous HCl invokes the acidic rearrangement of the spirodienone skeleton to form a novel type of thiacalixarene 4 possessing a phenoxathiin moiety.7
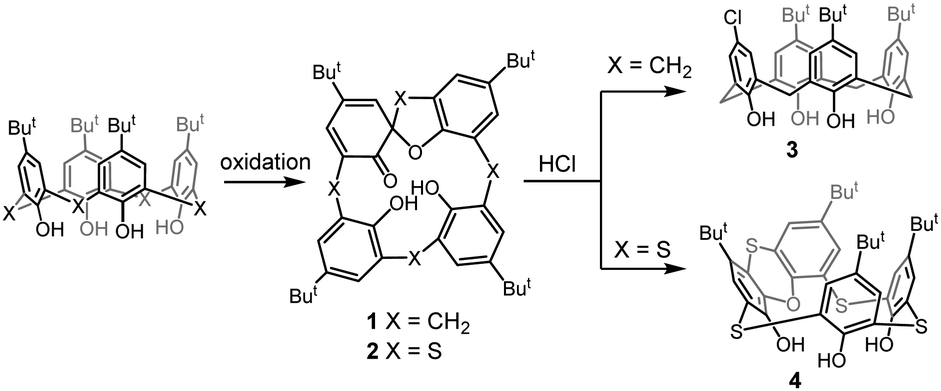 | ||
| Scheme 1 Different reactivity of thiacalixarene vs. calixarene (spirodienone chemistry). | ||
Macrocycle 4, rigidified by the presence of the condensed heterocyclic system, is inherently chiral and potentially useful for the preparation of novel chiral receptors. Moreover, rigid and highly strained calix[4]arenes8 were recently recognized as highly reactive systems in which release of the internal strain is the driving force for subsequent transformations.9
In this paper, we report on the unprecedented regioselective cleavage of the oxidized form of phenoxathiin-based thiacalixarene 4.10 The aromatic nucleophilic substitution reaction (attack of alkoxides) enabled by the presence of a strained heterocyclic moiety together with electron withdrawing functional groups (–SO2–) within the scaffold, has led to a novel type of macrocycle with a quasi-calixarene structure.
The attempt to alkylate the lower rim of the sulfonyl derivative 5 with methyl iodide in the presence of sodium hydride in DMF/THF at 65 °C resulted into a rather complicated mixture of partly alkylated products after reacting for two days. Subsequent alkylation of this crude mixture (MeI/Cs2CO3, acetone, reflux) surprisingly led to an almost equimolar mixture of the desired product 6 together with a so far unknown type of macrocycle 7a (Scheme 2) possessing meta- and para-bridging motifs within its skeleton.
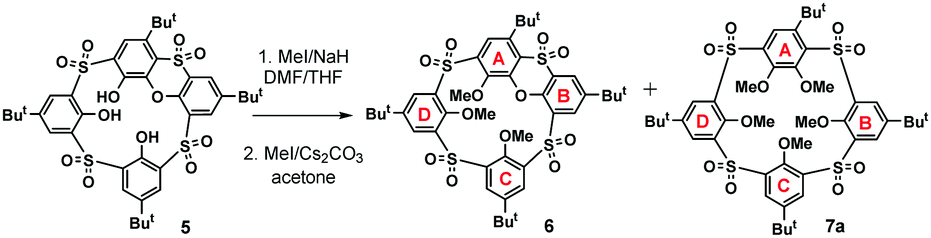 | ||
| Scheme 2 Unprecedented cleavage of the phenoxathiin motif. | ||
Formation of this unexpected macrocycle comprising sulfonylcalixarene and pillararene-like structural elements was confirmed by HRMS. Thus, the HRMS ESI+ analysis of 7a showed a signal at m/z = 957.26582, which is in good agreement with [M + Na]+ (957.26525) cations predicted for this molecule. Moreover, the structure of the macrocycle was confirmed by single crystal X-ray analysis.
Based on the above-mentioned evidence, we propose that compound 7a was formed by the SNAr reaction between the (partly alkylated) phenoxathiin-bearing macrocycle 5 and the in situ generated methoxide anion, followed by further methylation of the remaining phenolate groups in the subsequent alkylation step. The presence of the methoxide anion in the reaction mixture could be ascribed to the reaction of the MeI/NaH system with traces of water present in solvents and/or air humidity. To confirm this hypothesis, compound 6 was treated directly with sodium methoxide in THF under reflux for 2 h, followed by the alkylation with MeI/Cs2CO3, which resulted in the isolation of a single product 7a in 74% cumulative yield (Scheme 3).
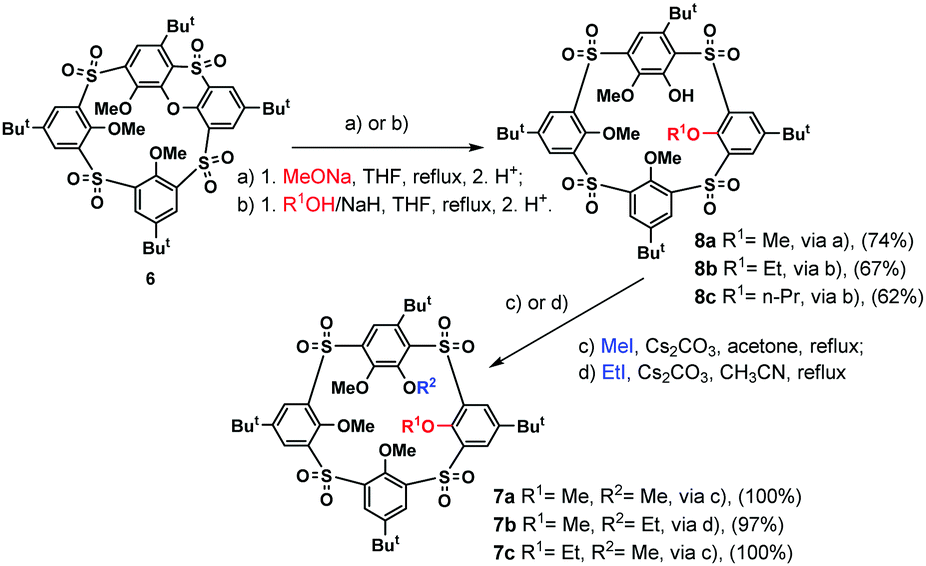 | ||
| Scheme 3 Regioselective cleavage of the phenoxathiin macrocycle 6. | ||
The key step of the whole SNAr mechanism, the attack of the phenoxathiin moiety by the methoxide nucleophile, could proceed in two different ways leading to two distinct intermediates, as shown in Scheme 4. The nucleophile could attack the phenolic moieties A or B, leading to intermediates A or B (route a/b). To shed more light on the regioselectivity of the cleavage reaction, the intermediate 8a formed by the methoxide attack on 6 was isolated (74% yield) after acidic quenching of the reaction mixture.
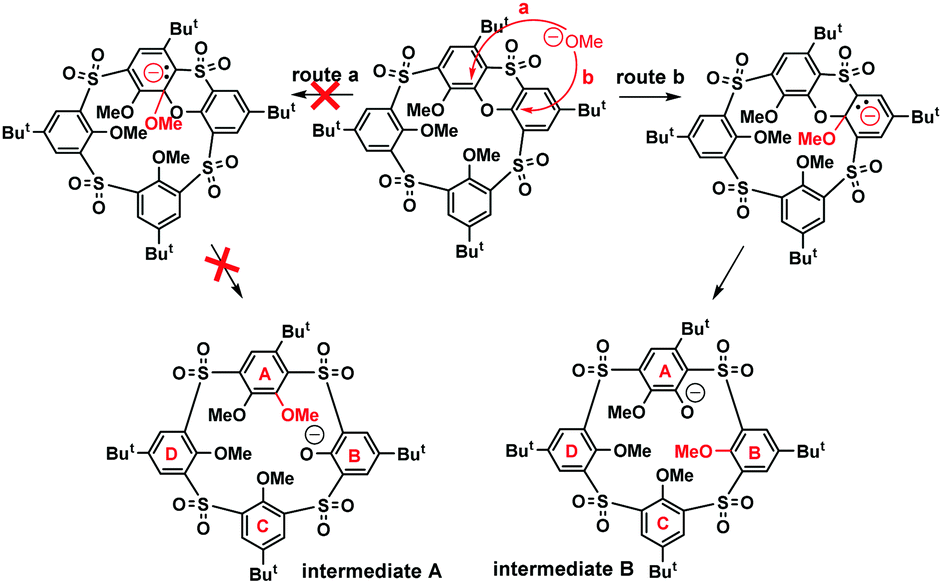 | ||
| Scheme 4 Proposed mechanism/regioselectivity of the phenoxathiin ring cleavage. | ||
Compound 8a, isolated as the sole product, revealed the expected HRMS (ESI+) pattern with m/z = 943.24960 [M + Na]+ and 959.22353 [M + K]+ (compared to the predicted values of m/z = 943.24978 and 959.22317). However, despite our numerous attempts, the exact center of the attack was not assigned unequivocally with the help of just NMR spectroscopy. Fortunately, the final unambiguous proof of the structure was obtained from the single crystal X-ray analysis of the propoxy analogue 8c (Fig. 1) supporting the attack via route b.
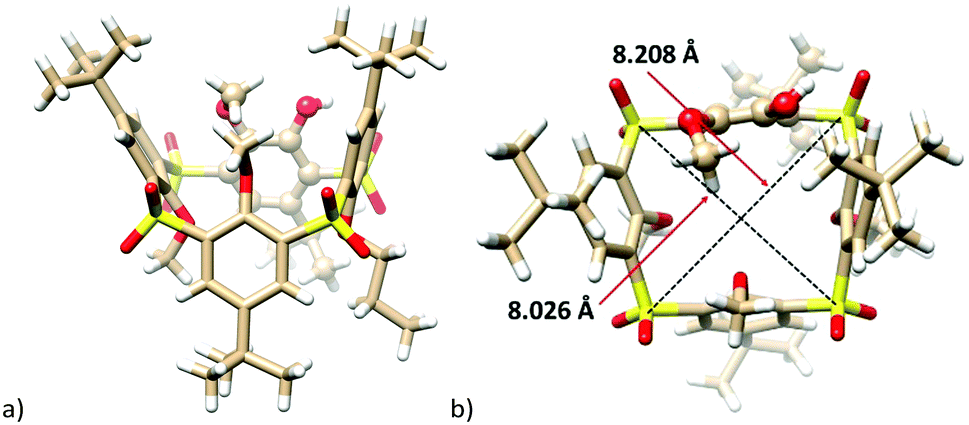 | ||
| Fig. 1 Single crystal X-ray structures of the compound 8c: (a) side-view, (b) top-view (the para-substituted moiety shown as balls for better clarity). | ||
As intuitively expected, the nucleophile regioselectively attacked the B moiety rather than A. Obviously, in this case, the addition of MeO− is stabilized by the presence of two sulfonyl electron withdrawing groups in the ortho-positions, which provides better stabilization for the reaction intermediate B (route b, Scheme 4). Moreover, as shown by similar experiments with common tetrasulfonyl calixarene analogues,11 the presence of the phenoxathiin moiety within the macrocyclic scaffold seems to be the essential prerequisite for the entire transformation, thus indicating that the driving force for SNAr reaction is the minimization of the internal strain imposed by the heterocyclic moiety.
To verify the general applicability of this approach, the SNAr cleavage reaction of the starting sulfonyl derivative 6 was studied with larger alkoxides. The nucleophiles were generated in situ by the addition of sodium hydride to an equimolar amount of an appropriate alcohol (Scheme 3). Cleavage with ethoxide or propoxide resulted in a smooth formation of the desired reaction products 8b and 8c in 67% and 62% yield, respectively. Moreover, the cleavage reaction with sodium tert-butoxide and phenoxide was also tested. While tert-butoxide gave a complicated and inseparable mixture of compounds, no reaction occurred after overnight treatment with sodium phenoxide, probably reflecting its lower nucleophilicity compared to aliphatic alkoxides.
Compounds 8 containing one remaining phenolic –OH group, formed by the opening of the phenoxathiin moiety, represent an excellent option for further functionalization. To verify such a possibility, we performed basic alkylation experiments. In all cases, the alkylation with MeI or EtI in the presence of Cs2CO3 afforded the corresponding product 7 in essentially quantitative yields (Scheme 3).
The final proof of the cleaved structure was obtained by the single crystal X-ray analysis of 8c and 7a. This revealed the crystal of 8c to have the monoclinic system, space group P21/c. As shown in Fig. 1a, the molecule adopts 1,3-alternate conformation with alternating up and down arrangement of the phenolic subunits. These are almost perpendicular to the main plane of the molecule defined by the four sulfur atoms (the corresponding interplanar angles Φ are 74.62°, 77.67°, 78.52°, and 60.39° starting from the para-bridged moiety and continuing clockwise from the top-view (see Fig. 1b)). While the distances between the two neighboring meta-bridging sulfur atoms are 5.52, 5.57 and 5.58 Å, the same distance in the para-substituted moiety is 6.30 Å. Consequently, the cavity of 8c is not square-shaped (like in common thiacalixarenes), but rather trapezoid with the magnitude of the main diagonals of 8.026 Å and 8.208 Å (Fig. 1b). The position of the propyl group in the main skeleton unequivocally confirms that the regio-selectivity/specificity of the SNAr reaction is a consequence of route b (Scheme 4).
The pentamethoxy derivative 7a crystallized in the orthorhombic system, space group Pbcn. The main geometric features are similar to those for 8c, including the 1,3-alternate conformation of the basic skeleton, with the exception of the more pronounced difference between the lengths of the main diagonals: 7.945 and 8.296 Å (see ESI,† Fig. S50).
Variable-temperature (VT) NMR spectroscopy, conveniently used for the conformational behaviour study of calixarenes, has somewhat failed in the case of phenoxathiin-based thiacalix[4]arene analogues 7 and 8 due to their extreme conformational mobility. This feature is the most evident in 1H temperature dependent spectra of pentamethoxy derivative 7a, where no spectral changes have been observed for the whole temperature range (173–403 K). This fact, however, does not confirm the rigidity of this system (although it usually does), as we could find more conformational species after the introduction of larger substituents, such as ethyl and propyl 8b and 8c. The ethyl derivative 8b reveals only broad complex spectra indicating the presence of equilibrium between several conformers. The larger propyl substituent in 8c enabled us to isolate a single conformer, as the sole product of the reaction, which was determined as the 1,3-alternate by X-ray analysis (see Fig. 1). Interestingly, upon dissolution in CD2Cl2, CDCl3 or C2D2Cl4, the pure compound gradually changed into the mixture of two conformers, indicating a transformation of the original 1,3-alternate8c to another conformer. This suggests that the propyl group is bulky enough to induce the formation of two different stereoisomers (atropisomers). Thus, standing of C2D2Cl4 solution of 8c at 298 K for 40 hours led to a mixture of 1,3-alternate with the new conformer in ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio (Fig. 2). On the other hand, the 1H NMR spectra measured in CD3CN or THF-d8 remained virtually unchanged during the time of the experiment (40 h).
:2 ratio (Fig. 2). On the other hand, the 1H NMR spectra measured in CD3CN or THF-d8 remained virtually unchanged during the time of the experiment (40 h).
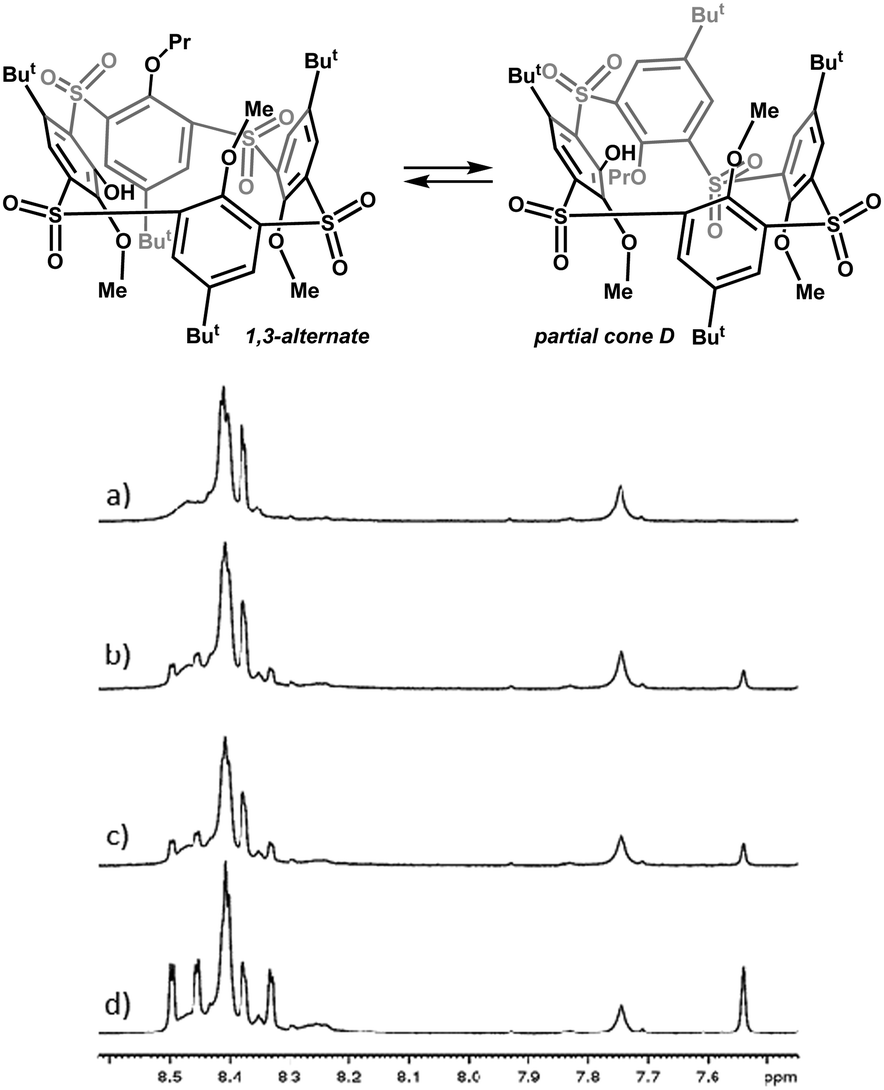 | ||
| Fig. 2 The fragments of 1H NMR spectra of 8c in C2D2Cl4, 298 K. (a) Freshly dissolved 1,3-alternate, (b) after standing for 10 hours, (c) after standing for 20 hours, and (d) after standing for 40 hours. The newly arising conformer was determined as partial cone D (pacoD). | ||
To determine the structure of the newly-emerged conformer, we performed its complete NMR analysis including NOE at 393 K.12 The NOE contacts between C3-B5, B3-tBu(A) and Pr(B)-tBu(D) unambiguously identified the partial cone conformer with ring D inverted (pacoD) as indicated in Fig. 3.
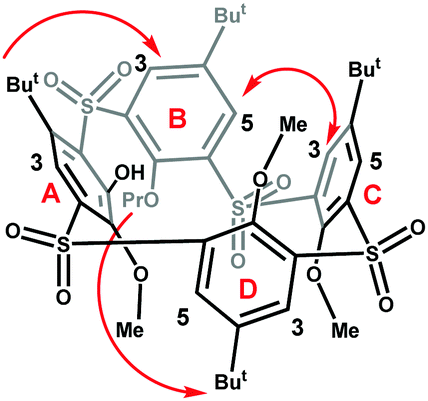 | ||
| Fig. 3 NOE contacts found for 8c at 373 K (600 MHz, C2D2Cl4), (for corresponding NOE spectra see ESI,† Fig. S52). | ||
An intrinsic property of all novel prepared derivatives is the inherent chirality of these systems. To demonstrate this feature, the 1H NMR spectrum of compound 7a was measured in the presence of chiral shift agents. While the presence of Pirkle's alcohol ((R)-1-anthracen-9-yl-2,2,2-trifluoroethanol) did not lead to any changes in the spectrum, the application of europium-based chiral shift agent (europium tris[3-(heptafluoropropylhydroxymethylene)-(+)-camphorate]) led to doubling of some signals (see the behaviour of A3 singlet at 7.96 ppm in Fig. S51a, ESI†) showing thus the presence of a racemic mixture.
In conclusion, the sulfonyl analogue of phenoxathiin-based thiacalix[4]arene 6, easily accessible from the parent thiacalix[4]arene, reacts with sodium alkoxides to yield the cleaved product representing a novel type of macrocyclic scaffold with a quasi-calixarene structure. The presence of the phenoxathiin moiety within the starting macrocycle seems to be essential for the entire transformation, thus indicating that the driving force for the SNAr reaction is the minimization of the internal strain imposed by the heterocyclic moiety. The regioselectivity of phenoxathiin cleavage was confirmed by the single crystal X-ray analysis. The conformational preferences and dynamic behavior of the resulting macrocyclic skeleton were studied using dynamic NMR experiments. All cleaved products represent inherently chiral building blocks potentially useful in the design of novel chiral receptors. Further supramolecular utilization of these compounds as well as the application of other types of nucleophiles for the cleavage reaction is currently under research.
This research was supported by the Czech Science Foundation (Grant 17-18108S). Financial support from specific university research (MSMT No 21/2019) is also acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For recent reviews on thiacalixarenes see: (a) R. Kumar, Y. O. Lee, V. Bhalla, M. Kumar and J. S. Kim, Chem. Soc. Rev., 2014, 43, 4824 RSC; (b) N. Morohashi, F. Narumi, N. Iki, T. Hattori and S. Miyano, Chem. Rev., 2006, 106, 5291 CrossRef CAS; (c) P. Lhotak, Eur. J. Org. Chem., 2004, 1675 CrossRef CAS.
- For selected books on calixarenes and their applications, see: (a) Calixarenes and Beyond, ed. P. Neri, J. L. Sessler and M. X. Wang, Springer Int. Publishing, Switzerland, 2016 Search PubMed; (b) C. D. Gutsche, Calixarenes An introduction, The Royal Society of Chemistry, Thomas Graham House, Cambridge, 2nd edn, 2008 Search PubMed; (c) Calixarenes in the Nanoworld, ed. J. Vicens, J. Harrowfield and L. Backlouti, Springer, Dordrecht, 2007 Search PubMed; (d) Calixarenes 2001, ed. Z. Asfari, V. Böhmer, J. Harrowfield and J. Vicens, Kluwer Academic Publishers, Dordrecht, 2001 Search PubMed.
- (a) O. Kundrat, V. Eigner, H. Dvorakova and P. Lhotak, Org. Lett., 2011, 13, 4032 CrossRef CAS; (b) O. Kundrat, H. Dvorakova, S. Böhm, V. Eigner and P. Lhotak, J. Org. Chem., 2012, 77, 2272 CrossRef CAS.
- For selected examples of the spirodienone route to derivatization of calixarenes, see e.g.: (a) C. Gatea, F. Troisi, C. Talotta, T. Pierro and P. Neri, J. Org. Chem., 2012, 77, 3634 CrossRef; (b) F. Troisi, T. Pierro, C. Gaeta and P. Neri, Tetrahedron Lett., 2009, 50, 4416 CrossRef CAS; (c) S. Simaan and S. E. Biali, J. Org. Chem., 2004, 69, 95 CrossRef CAS; (d) S. Simaan, K. Agbaria and S. E. Biali, J. Org. Chem., 2002, 67, 6136 CrossRef CAS; (e) K. Agbaria and S. E. Biali, J. Am. Chem. Soc., 2001, 123, 12495 CrossRef CAS; (f) K. Agbaria, J. Wöhnert and S. E. Biali, J. Org. Chem., 2001, 66, 7059 CrossRef CAS.
- A. M. Litwak, F. Grynszpan, O. Aleksiuk, S. Cohen and S. E. Biali, J. Org. Chem., 1993, 58, 393 CrossRef CAS.
- N. Morohashi, M. Kojima, A. Suzuki and Y. Ohba, Heterocycl. Commun., 2005, 11, 249 CAS.
- K. Polivkova, M. Simanova, J. Budka, P. Curinova, I. Cisarova and P. Lhotak, Tetrahedron Lett., 2009, 50, 6347 CrossRef CAS.
- (a) K. Flidrova, P. Slavik, V. Eigner, H. Dvorakova and P. Lhotak, Chem. Commun., 2013, 49, 6749 RSC; (b) P. Slavik, V. Eigner and P. Lhotak, CrystEngComm, 2016, 18, 4964 RSC.
- (a) P. Slavik, H. Dvorakova, M. Krupicka and P. Lhotak, Org. Biomol. Chem., 2018, 16, 838 RSC; (b) P. Slavik, M. Krupicka, V. Eigner, L. Vrzal, H. Dvorakova and P. Lhotak, J. Org. Chem., 2019, 84, 4229 CrossRef CAS.
- T. Landovsky, H. Dvorakova, V. Eigner, M. Babor, M. Krupicka, M. Kohout and P. Lhotak, New J. Chem., 2018, 42, 20074 RSC.
- The reaction of tetramethoxy tetrasulfonyl thiacalix[4]arene (calixarene having four –SO2– bridges without additional phenoxathiin moiety) with alkoxides under identical conditions did not give any substitution products of the basic skeleton, showing thus the key role of the phenoxathiin moiety in the SNAr reaction.
- Measuring at elevated temperature provided better spectral resolution, thus it was possible to irradiate selectively almost all proton resonances in 1D NOE experiments.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, full characterization of compounds 7 and 8, X-ray structure of 7a, 1H NMR spectrum of 7a with chiral shifting agent, NOE experiments, standing of 8c1,3-alt solution experiments and VT NMR spectra. CCDC 1943887 and 1955160. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc08335a |
| This journal is © The Royal Society of Chemistry 2020 |