
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpatially resolved NMR spectroscopy of heterogeneous gas phase hydrogenation of 1,3-butadiene with parahydrogen†
Alexandra
Svyatova
ab,
Elizaveta S.
Kononenko
ab,
Kirill V.
Kovtunov
 *ab,
Dmitry
Lebedev
c,
Evgeniy Yu.
Gerasimov
bd,
Andrey V.
Bukhtiyarov
bd,
Igor P.
Prosvirin
bd,
Valerii I.
Bukhtiyarov
d,
Christoph R.
Müller
e,
Alexey
Fedorov
*e and
Igor V.
Koptyug
abd
*ab,
Dmitry
Lebedev
c,
Evgeniy Yu.
Gerasimov
bd,
Andrey V.
Bukhtiyarov
bd,
Igor P.
Prosvirin
bd,
Valerii I.
Bukhtiyarov
d,
Christoph R.
Müller
e,
Alexey
Fedorov
*e and
Igor V.
Koptyug
abd
aInternational Tomography Center, SB RAS, 3 A Institutskaya St., Novosibirsk 630090, Russia. E-mail: kovtunov@tomo.nsc.ru
bNovosibirsk State University, 2 Pirogova St., Novosibirsk 630090, Russia
cDepartment of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog-Weg 1–5, Zürich, CH 8093, Switzerland
dBoreskov Institute of Catalysis SB RAS, 5 Lavrenteva Pr, Novosibirsk 630090, Russia
eDepartment of Mechanical and Process Engineering, ETH Zürich, Leonhardstrasse 21, Zürich, CH 8092, Switzerland. E-mail: fedoroal@ethz.ch
First published on 21st November 2019
Abstract
Magnetic resonance-based methods such as nuclear magnetic resonance (NMR) and magnetic resonance imaging (MRI) are widely used to provide in situ/operando information of chemical reactions. However, the low spin density and magnetic field inhomogeneities associated with heterogeneous catalytic systems containing gaseous reactants complicate such studies. Hyperpolarization techniques, in particular parahydrogen-induced polarization (PHIP), increase significantly the NMR signal intensity. In this study, we test 16 glass tube reactors containing Pd, Pt, Rh or Ir nanoparticles dispersed on a thin layer of TiO2, CeO2, SiO2 or Al2O3 for the hydrogenation of 1,3-butadiene using parahydrogen. The catalytic coatings of Ir and Rh gave hydrogenation products with the highest nuclear spin polarization while the coatings of Pd are the most selective ones for the semihydrogenation of 1,3-butadiene to 1- and 2-butenes. Spatially resolved NMR spectroscopy of the reagent and the product distribution along the reactor axis provided further mechanistic insight into the catalytic function of these reactive coatings under operando conditions.
Introduction
Development of efficient and selective heterogeneous catalysts for hydrogenation of alkynes and dienes is an active research field, motivated by diverse industrial applications ranging from the removal of acetylene from ethylene streams for the synthesis of polyethylene to the stereoselective synthesis of Z-olefins in the fine chemicals sector.1–4 Advances in the understanding of heterogeneous catalysts with spatial visualization of reagents and reaction products inside a working reactor can be made using magnetic resonance imaging (MRI).5–13 However, harnessing the full power of MRI to study heterogeneous catalysts at work has a number of challenges. In particular, in the case of gas–solid reactions the spin density of gaseous media is ca. 3 orders of magnitude lower than that in the condensed phase, which reduces substantially the signal intensity.14 Therefore, MRI of the gas phase products of heterogeneously catalyzed reactions remains a relatively underdeveloped area despite insightful studies, for instance on mapping the spatial distribution of gases and temperature in ethylene hydrogenation in monolith reactors.12,15–17A major improvement in gas imaging can be obtained by exploiting hyperpolarized gases which can provide an up to several orders of magnitude increase in signal intensity and hence in the signal-to-noise ratio (SNR) in NMR experiments. The hyperpolarization methods include, for instance, spin-exchange optical pumping (SEOP),18–21 dynamic nuclear polarization (DNP),22,23parahydrogen-induced polarization (PHIP)24–27 and signal amplification by reversible exchange (SABRE).28–30 All these techniques are powerful, yet their applicability in catalysis research is limited. For instance, noble gases hyperpolarized by SEOP31–33 are rather unreactive and expensive; DNP has a broad range of applications but requires complex and costly equipment.34,35 The SABRE approach transfers hyperpolarization to a substrate without changing its structure,29 making this method inappropriate for studies of catalytic reactors at work. In contrast, the PHIP method is based on the pairwise addition of parahydrogen (p-H2), which is the singlet spin state isomer of hydrogen, to a substrate in a catalytic hydrogenation reaction, forming hyperpolarized products due to the transfer of the singlet spin order from p-H2. Disruption of the magnetic equivalence of the hydrogen nuclei leads to hyperpolarization of nuclear spins. Thus, PHIP36 is the simplest method to produce hyperpolarized gases.37,38 While hyperpolarization of nuclei using PHIP can be observed in both homogeneous25,39,40 and heterogeneous catalysts,36,41,42 advantages of the latter include easier separation of the reaction products from the catalyst, which is particularly important for biomedical MRI applications.43,44 Heterogeneous PHIP (HET-PHIP) studies have often utilized the aforementioned increased spin density of reagents and reaction products in the condensed phase (aqueous media).37,38,45 In addition to the low spin density of gases, challenges for operando studies of heterogeneous reactions include perturbation of the magnetic field homogeneity by solid catalyst bodies and the fast diffusion of gases; all these factors decrease the intensity of the NMR signal of gaseous species.
We reported recently that model glass reactors with catalytically active Rh/TiO2 coatings can minimize the magnetic field inhomogeneity over the reactor for the catalytic hydrogenation of propene to propane.46 Here, we extend the reactive coating approach to TiO2, CeO2, SiO2, and Al2O3 which are used to support nanoparticles of Rh, Pt, Pd and Ir, giving a total of 16 reactors with a thin layer of reactive coating designed for NMR and MRI investigations of catalytic hydrogenation reactions. We chose 1,3-butadiene as a substrate with the objective of identifying selective semihydrogenation catalysts, which are materials for the formation of 1- or 2-butenes from 1,3-butadiene, as well as identifying catalysts giving the highest polarization of the hydrogenation products. We found that coatings containing Ir and Rh are the most efficient in transferring polarization to the hydrogenation products while coatings with Pd are the most selective for the semihydrogenation of 1,3-butadiene to 1- and 2-butenes. In addition, the distribution of the reagents and the products inside the reactor was mapped. These maps and NMR spectra provide mechanistic insight into the hydrogenation reactions indicating, for instance, that butane is the secondary reaction product in the hydrogenation of 1,3-butadiene with Ir/SiO2 and Rh/CeO2 catalysts, but 2-butene is preferentially formed from 1-butene over Rh/CeO2 or directly from 1,3-butadiene over Pd/CeO2 and Pd/SiO2.
Results and discussion
We have described recently a convenient approach to obtain glass tube reactors with Rh/TiO2 coatings that allowed for MRI visualization of the distribution of reagents and products in the heterogeneous hydrogenation of propene to propane.46 Here, we extend this approach to prepare reactive coatings based on CeO2, SiO2, and Al2O3 oxide supports (Fig. 1a) with the aim of identifying and studying catalysts for the selective semihydrogenation of 1,3-butadiene (Fig. 1b). Towards this end, TiO2, CeO2, SiO2, and Al2O3 were coated via drop-casting on a spinning borosilicate glass tube (10 cm height, 5 mm outer diameter, see the ESI† for details). The coated layers had, after calcination, thicknesses of ca. 10 micrometers for TiO2, SiO2, and Al2O3 and a significantly thinner layer, ca. 1 micrometer, for CeO2 (Fig. 1c–f). These coatings were subsequently decorated with solutions of the respective molecular precursors. For Rh and Ir, we used acetonitrile solutions of chloro-1,5-cyclooctadiene iridium and rhodium dimers and deposited ca. 0.5 μmol of these complexes onto each coating.46 Since tris(dibenzylideneacetone)dipalladium and tris(dibenzylideneacetone)platinum were less soluble in acetonitrile than [(COD)RhCl]2 and [(COD)IrCl]2 complexes, we used a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetonitrile–DCM solvent mixture and deposited ca. 0.4 μmol of Pd and Pt (on the metal basis) on each reactor tube. Overall, 16 reactors were prepared using 4 oxide coatings and 4 metal precursors. Next, the reactors were reduced in situ with H2 (130 °C, 1 h, 1–1.2 mL s−1) yielding metal oxide coatings decorated with catalytically active metallic nanoparticles. According to transmission electron microscopy (TEM) performed for H2-reduced coatings of Rh, Ir, Pd, and Pt metals on silica, chosen as a representative stable support (samples were exposed to ambient air prior to TEM analysis), the formed nanoparticles are small, with an average diameter of ca. 1.5–1.7 nm for Ir, Rh and Pt nanoparticles (NPs), and are slightly larger for Pd NPs (average diameter of 2.3 nm, Fig. 1g–j). Further information about TEM analysis is provided in the ESI.†
:1 acetonitrile–DCM solvent mixture and deposited ca. 0.4 μmol of Pd and Pt (on the metal basis) on each reactor tube. Overall, 16 reactors were prepared using 4 oxide coatings and 4 metal precursors. Next, the reactors were reduced in situ with H2 (130 °C, 1 h, 1–1.2 mL s−1) yielding metal oxide coatings decorated with catalytically active metallic nanoparticles. According to transmission electron microscopy (TEM) performed for H2-reduced coatings of Rh, Ir, Pd, and Pt metals on silica, chosen as a representative stable support (samples were exposed to ambient air prior to TEM analysis), the formed nanoparticles are small, with an average diameter of ca. 1.5–1.7 nm for Ir, Rh and Pt nanoparticles (NPs), and are slightly larger for Pd NPs (average diameter of 2.3 nm, Fig. 1g–j). Further information about TEM analysis is provided in the ESI.†
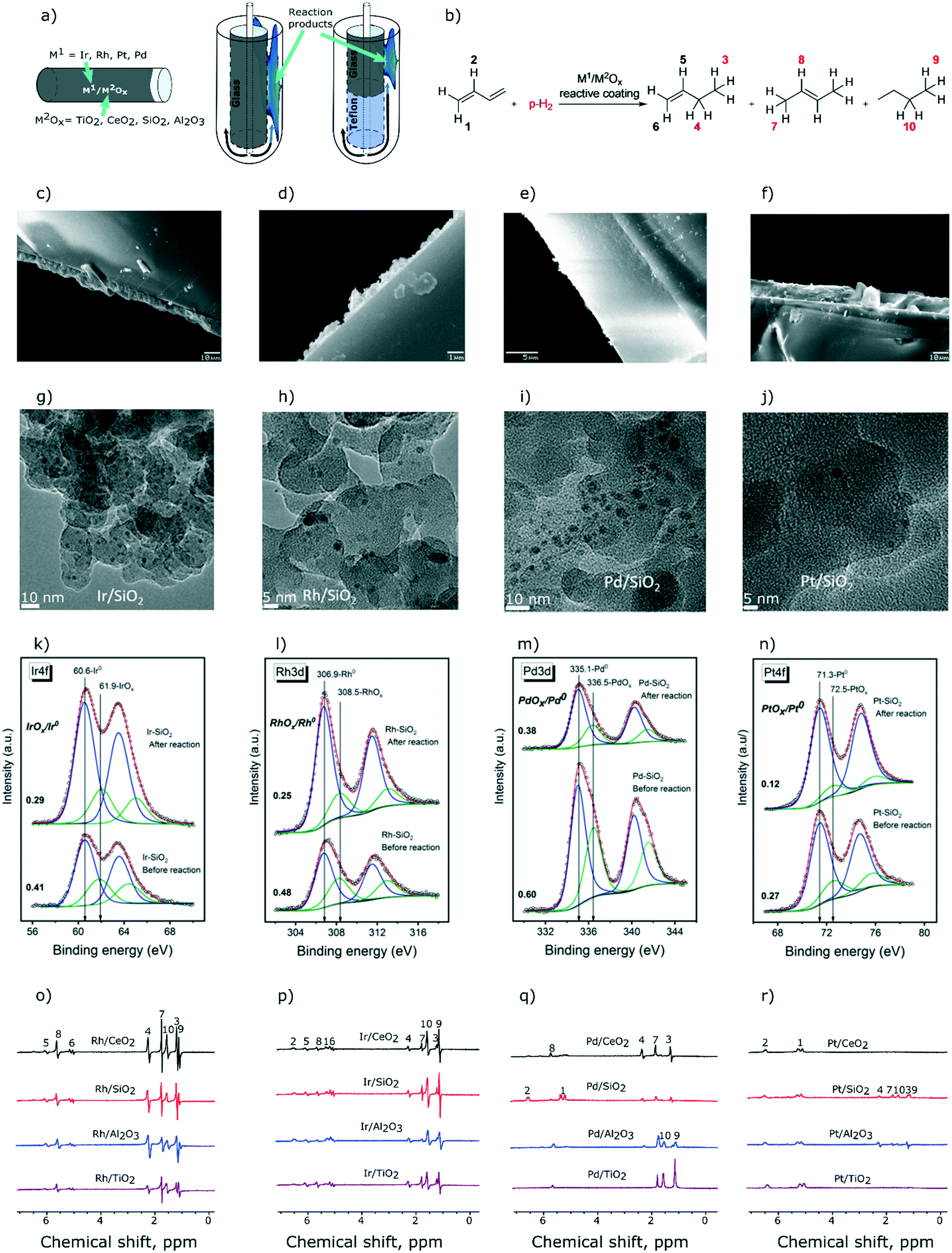 | ||
| Fig. 1 a) Schematic representation of the reactors with catalytic coatings made of TiO2, CeO2, SiO2, and Al2O3 containing Ir, Rh, Pt and Pd active phases (16 compositions in total) investigated in heterogeneous catalytic hydrogenation of b) 1,3-butadiene with p-H2 (1:4 ratio). Photographs of reactive coatings with c) TiO2, d) CeO2, e) SiO2, and f) Al2O3 with scale bars of 10, 1, 5 and 10 microns, respectively. Representative TEM images of the g) Ir/SiO2 h) Rh/SiO2, i) Pd/SiO2, and j) Pt/SiO2 reactive coatings. Deconvolution of k) Ir4f, l) Rh3d, m) Pd3d and n) Pt4f photoelectron spectra measured for silica-supported catalysts. 1H NMR spectra obtained during hydrogenation of 1,3-butadiene at 130 °C using reactive coatings with o) Rh, p) Ir, q) Pd and r) Pt. The gas flow rate was 3.8 mL s−1 except for Rh/TiO2 (5.1 mL s−1), Pd/SiO2 (6.5 mL s−1) and Pd/TiO2 (5.1 mL s−1). | ||
All 16 reactive coatings were investigated by XPS after (i) reduction in H2 and (ii) catalytic hydrogenation of 1,3-butadiene (vide infra). Fig. 1k–n show the deconvolution of the Ir4f, Rh3d, Pd3d and Pt4f spectra of the silica-supported catalysts chosen as representative examples (other supports provide qualitatively similar results). Reduced samples were handled in air, therefore for all four metals, both metallic and oxide states are observed, at lower and higher binding energies, respectively. For all of the samples studied, the contribution of a metallic state increases after the catalytic reaction, probably owing to the lower oxidation tendencies of the active component after exposure to hydrogenation conditions. However, one should note that carburization of the metallic nanoparticles might occur during catalytic testing, yet the XPS signatures of metal carbides of late transition metals are very difficult to distinguish from the signatures of the metal phase, as was recently demonstrated for a Rh/SiO2 catalyst.47 The atomic ratios of the metal to the support as calculated from XPS data are presented in Table S1.† As expected from the quantities of the metal precursors used (discussed above), the metal loadings of Rh and Ir are higher than those of Pd and Pt.
Turning now to the NMR characterization of all 16 samples, the 1H NMR spectra acquired during 1,3-butadiene hydrogenation with p-H2 under PASADENA conditions25 are presented in Fig. 1o–r. The highest signal intensities were observed for coatings containing Rh and Ir. e.g., SNRs for Rh/CeO2 and Ir/SiO2 are ca. 300 and 220, respectively (Fig. 1o and p; see Table S2† for SNRs of all the catalysts). The lowest signal intensities due to the hydrogenation products were observed for Pt-based catalysts (Fig. 1r, Table S2†), at least partially owing to the lower loading of the Pt metal. Interestingly, while Pd/Al2O3 and Pd/TiO2 did not provide any polarization of the hydrogenation products, Pd/CeO2 and Pd/SiO2 selectively formed hyperpolarized 1-butene without any detectable hyperpolarized 2-butene (Fig. 1q), suggesting that 2-butene is not formed by the isomerization of 1-butene. This means that 1-butene is, at least partially, formed via pairwise hydrogen addition to 1,3-butadiene on Pd/CeO2 and Pd/SiO2 catalysts, in contrast to 2-butene that reveals no pairwise p-H2 addition. From a mechanistic standpoint, this result suggests that 1-butene and 2-butene are either formed on different Pd active sites in Pd/CeO2 and Pd/SiO2 catalysts, or the formation of these products proceeds via different reaction mechanisms. It could be argued that the relaxation processes of the polarized products may play a role leading to a fast decay of hyperpolarization during the isomerization of 1-butene to 2-butene; in this case the relaxation rate should be strongly influenced by the strength of adsorption onto the surface of the oxide support. However, hyperpolarized 2-butene is observed clearly in the case of supported Rh, Ir, and Pt catalysts (Fig. 1o, p and r), which confirms that the relaxation of 1-butene gas during its re-adsorption is not significant. Hyperpolarized 2-butene cannot be formed via pairwise p-H2 addition to 1,3-butadiene in the 1,4-addition step because this would form a symmetrical 2-butene molecule. However, the observation of PHIP effects necessitates the formation of a product molecule with asymmetrically added hydrogen atoms. Therefore, hyperpolarized 2-butene can only be formed via the isomerization of hyperpolarized 1-butene, as observed for supported Rh, Ir, and Pt metal catalysts, in contrast to Pd/CeO2 and Pd/SiO2 catalysts. Clearly, insights provided by PHIP allow conclusions to be drawn on the hydrogenation reaction pathways that are not easily accessible by other experimental methods.
Next, MRI experiments were performed with two selected catalysts, Ir/SiO2 and Rh/CeO2. These catalysts were chosen because they provided the highest intensities and polarization levels of the hydrogenation products during 1,3-butadiene hydrogenation with p-H2. A spin-echo pulse sequence (45–180°-acquisition) was used to visualize the distribution of reagents and products along the length of the reactor. In the experiments using the Ir/SiO2 catalyst, the reaction products were distributed along the whole reactor indicating high reaction rates (Fig. 2a). The concentration of 1-butene and 2-butene increases gradually along the reactor axis while there is a sharp increase in the concentration of butane (x = 1 cm) suggesting that butane is the secondary reaction product.
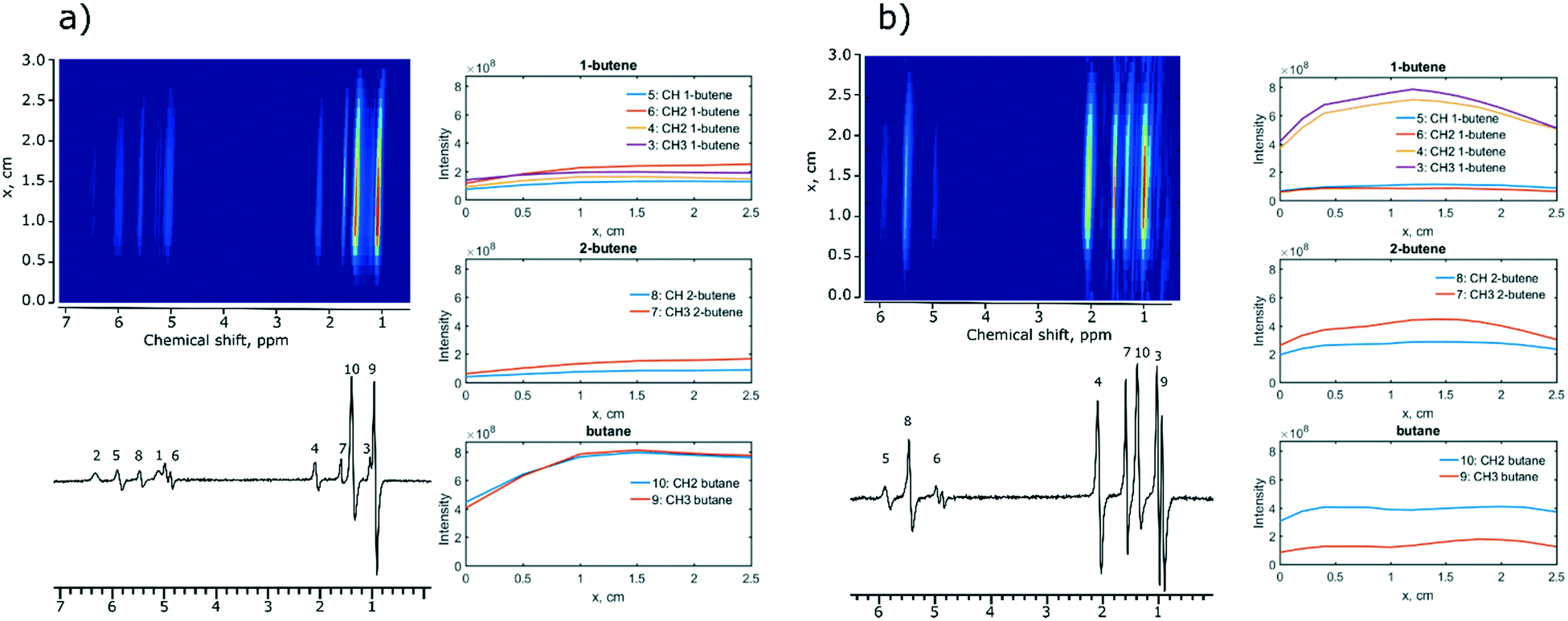 | ||
| Fig. 2 Hydrogenation of 1,3-butadiene over a) Ir/SiO2 and b) Rh/CeO2 catalysts with parahydrogen (1,3-butadiene: p-H2 ratio = 1:4, 5.1 mL s−1 flow rate, 130 °C) as followed by MRI. Top panels: distribution of intensities of MRI signals of the working reactor along its Z-axes. Bottom panels: NMR spectra obtained under PASADENA conditions. Right panel: Plots of the distribution of the reaction products along the length of the reactor. | ||
When the Rh/CeO2 catalyst is subjected to the same conditions, identical hydrogenation products are observed, but the concentration of butane is constant over the entire length of the reactor (Fig. 2b, right), while 1,3-butadiene is not detected by MRI indicating its fast hydrogenation. At the same time, the concentration of 1-butene decreased earlier than the concentration of 2-butene (1.2 cm vs. 1.5 cm, Fig. 2b). This is consistent with the formation rate of butane being slightly faster for the terminal olefin 1-butene than that for the internal 2-butene, in agreement with literature studies.48
For comparison, MRI experiments were also performed using Ir/SiO2 or Rh/CeO2 and normal hydrogen (n-H2). These experiments reproduced the same trends as the ones observed with p-H2 (Fig. S7†). For Rh/CeO2, MR imaging with n-H2 shows 1,3-butadiene at the bottom of the NMR tube and the signal due to 1,3-butadiene decreases along the axis of the reactor, while the signal intensities of 2-butene and butane increase along the reactor axis and stabilize at x = 1.5 cm (Fig. S7b†). Our next control experiment used a Teflon tube placed at the bottom of an NMR tube (Fig. S8†). No MRI signal due to hydrogenation products was observed at the bottom of the NMR tube, however, hydrogenation products formed at the level at which catalytic Ir/SiO2 or Rh/CeO2 coatings were placed, confirming that in the absence of a catalytic coating there is no hydrogenation of 1,3-butadiene (Fig. S8†). This experiment also showed that the constant gas feed flow of 5.1 mL s−1 does not allow for a downward diffusion of the hydrogenation products. Note that the apparent difference in the chemical shift of 1,3-butadiene between the areas with and without the Teflon tube (ca. 0.3 ppm) is due to a different local environment.
We performed additional experiments to obtain the spatial distribution of 1,3-butadiene and its hydrogenation products at high contact times using the Ir/SiO2 catalyst and p-H2; in the case of the more active Rh/CeO2 catalyst, we used n-H2 (Fig. S9†). Both experiments were conducted at a low flow rate of 1.3 mL s−1. An increase in the contact time led, in the case of Ir/SiO2 (Fig. S9a†), to the formation of only the secondary reaction product, butane. The same observation was made for Rh/CeO2 (Fig. S9b†), however, a small amount of 2-butene was still detected implying that Ir/SiO2 has a higher reaction rate compared to that of Rh/CeO2 for the hydrogenation of 1,3-butadiene.
Experimental
NMR and MRI experiments were performed on a Bruker Avance 300 MHz NMR spectrometer. The experiments were conducted using a commercial 10 mm RF probe utilizing a 1H channel. For PHIP experiments, hydrogen was enriched with parahydrogen up to ∼90% using a Bruker parahydrogen generator BPHG 90. The feed gas mixture contained 1,3-butadiene and p-H2 or n-H2 at a 1:4 ratio. Hydrogenation was performed in the 7.1 T magnetic field of the NMR spectrometer at atmospheric pressure and 130 °C. The fabrication of the catalytic reactors was similar to the procedure described previously46 except that drop-casting on a spinning borosilicate glass tube was used instead of dip-coating. One of the produced reactors was placed at the bottom of a 10 mm NMR tube, and the gas mixture was supplied through a 1.6 mm Teflon capillary placed concentrically along the reactor. For control experiments, the Teflon tube was placed at the bottom of the NMR tube, while the reactor was placed above it. The gas flow rate was varied from 1.3 to 6.5 mL s−1 using an Aalborg rotameter. 1H NMR spectra were acquired using a single 45° pulse. 1H MRI was performed using a spin-echo pulse sequence49 with the first tip angle of 45° and using 32 phase encoding gradients. For the analysis, all 16 reactors were cut in half using a diamond broach file (as shown in Fig. S1† for the Ir/SiO2 catalyst).
Conclusions
In conclusion, we demonstrated an operando approach for the visualization of the reagents and the product distribution along the length of a working hydrogenation reactor. We studied 16 different catalyst formulations using nanoparticles of Pt, Pd, Rh, and Ir supported on CeO2, SiO2, Al2O3 or TiO2. This study allowed insights into the reaction mechanism of the 1,3-butadiene hydrogenation reaction on those catalysts to be obtained. It was shown that the catalysts containing Pd nanoparticles are the most selective ones towards butene, while the catalysts containing Ir or Rh nanoparticles demonstrate the highest polarization level in the products. The mechanistic insight into the reaction was obtained by mapping the reagent and product distribution along the length of the reactors. It was shown that 1-butene is formed via pairwise hydrogen addition to 1,3-butadiene on Pd/CeO2 and Pd/SiO2, i.e. 1-butene and 2-butene are formed on different Pd active sites of the catalysts or via different reaction mechanisms. In addition, it was shown that during hydrogenation over the Rh/CeO2 catalyst butane is formed faster from 1-butene than from 2-butene.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AS, ESK, KVK and IVK thank RSF (grant # 19-13-00047) for the support of MRI studies of the hydrogenation reaction. The Novosibirsk team thanks the Russian Ministry of Science and Higher Education (AAAA-A16-116121510087-5) for the access to the NMR/MRI equipment. A. V. B. and I. P. P thank the SB RAS integrated research program (# 0333-2018-0006/II.1.13) for parahydrogen activation studies. AF and CRM thank the Swiss National Science Foundation for support of MRI studies (200020_182692). This work was partially conducted using the equipment of the “National Center of Catalyst Research”.Notes and references
- G. Vile, D. Albani, N. Almora-Barrios, N. López and J. Perez-Ramirez, ChemCatChem, 2016, 8, 21–33 CrossRef CAS.
- S. A. Nikolaev, L. N. Zanaveskin, V. V. Smirnov, V. A. Averyanov and K. L. Zanaveskin, Russ. Chem. Rev., 2009, 78, 231–247 CrossRef CAS.
- L. Delannoy, G. Thrimurthulu, P. S. Reddy and C. Louis, Phys. Chem. Chem. Phys., 2014, 16, 26514–26527 RSC.
- A. Borodziński and G. C. Bond, Catal. Rev.: Sci. Eng., 2008, 4940, 379–469 CrossRef.
- I. V. Koptyug, A. A. Lysova, A. V. Matveev, V. N. Parmon and R. Z. Sagdeev, Top. Catal., 2005, 32, 83–91 CrossRef CAS.
- I. V. Koptyug, A. A. Lysova, A. V. Matveev, L. Y. Ilyina, R. Z. Sagdeev and V. N. Parmon, Magn. Reson. Imaging, 2003, 21, 337–343 CrossRef CAS.
- I. V. Koptyug, A. A. Lysova, A. V. Kulikov, V. A. Kirillov, V. N. Parmon and R. Z. Sagdeev, Appl. Catal., A, 2004, 267, 143–148 CrossRef CAS.
- C. R. Müller, D. J. Holland, A. J. Sederman, M. D. Mantle, L. F. Gladden and J. F. Davidson, Powder Technol., 2008, 183, 53–62 CrossRef.
- V. A. Kirillov and I. V. Koptyug, Ind. Eng. Chem. Res., 2005, 44, 9727–9738 CrossRef CAS.
- A. Penn, T. Tsuji, D. O. Brunner, C. M. Boyce, K. P. Pruessmann and C. R. Müller, Sci. Adv., 2017, 3, e1701879 CrossRef.
- L. F. Gladden, Top. Catal., 2003, 24, 19–28 CrossRef CAS.
- L. F. Gladden, F. J. R. Abegão, C. P. Dunckley, D. J. Holland, M. H. Sankey and A. J. Sederman, Catal. Today, 2010, 155, 157–163 CrossRef CAS.
- X. Ren, S. Stapf and B. Blümich, AIChE J., 2005, 51, 392–405 CrossRef CAS.
- V. V. Zhivonitko, A. I. Svyatova, K. V. Kovtunov and I. V. Koptyug, Annu. Rep. NMR Spectrosc., 2018, 95, 83–145 CrossRef CAS.
- J. Ulpts, W. Dreher, M. Klink and J. Thöming, Appl. Catal., A, 2015, 502, 340–349 CrossRef CAS.
- J. Ulpts, L. Kiewidt, W. Dreher and J. Thöming, Catal. Today, 2018, 310, 176–186 CrossRef CAS.
- L. F. Gladden and A. J. Sederman, Annu. Rev. Chem. Biomol. Eng., 2017, 8, 227–247 CrossRef PubMed.
- G. Pavlovskaya, J. Six, T. Meersman, N. Gopinathan and S. P. Rigby, AIChE J., 2015, 61, 4013–4019 CrossRef CAS.
- S. J. Kadlecek, K. Emami, M. C. Fischer, M. Ishii, J. Yu, J. M. Woodburn, M. NikKhah, V. Vahdat, D. A. Lipson, J. E. Baumgardner and R. R. Rizi, Prog. Nucl. Magn. Reson. Spectrosc., 2005, 47, 187–212 CrossRef CAS.
- T. G. Walker, J. Phys.: Conf. Ser., 2011, 294, 012001 CrossRef.
- T. G. Walker and W. Happer, Rev. Mod. Phys., 1997, 69, 629–642 CrossRef CAS.
- A. Abragam and M. Goldman, Rep. Prog. Phys., 1978, 41, 395–467 CrossRef CAS.
- J. Van Bentum, B. Van Meerten, M. Sharma and A. Kentgens, J. Magn. Reson., 2016, 264, 59–67 CrossRef CAS.
- R. A. Green, R. W. Adams, S. B. Duckett, R. E. Mewis, D. C. Williamson and G. G. R. Green, Prog. Nucl. Magn. Reson. Spectrosc., 2012, 67, 1–48 CrossRef CAS.
- C. R. Bowers and D. P. Weitekamp, J. Am. Chem. Soc., 1987, 109, 5541–5542 CrossRef CAS.
- S. Gloggler, S. Wagner and L.-S. Bouchard, Chem. Sci., 2015, 6, 4261–4266 RSC.
- L. E. Olsson, C. Chai, O. Axelsson, M. Karlsson, K. Golman and J. S. Petersson, Magn. Reson. Med., 2006, 737, 731–737 CrossRef.
- A. N. Pravdivtsev, I. V. Skovpin, A. I. Svyatova, N. V. Chukanov, L. M. Kovtunova, V. I. Bukhtiyarov, E. Y. Chekmenev, K. V. Kovtunov, I. V. Koptyug and J.-B. Hovener, J. Phys. Chem. A, 2018, 122, 9107–9114 CrossRef CAS PubMed.
- K. V. Kovtunov, E. V. Pokochueva, O. G. Salnikov, S. F. Cousin, D. Kurzbach, B. Vuichoud, S. Jannin, E. Y. Chekmenev, B. M. Goodson, D. A. Barskiy and I. V. Koptyug, Chem. – Asian J., 2018, 13, 1857–1871 CrossRef CAS.
- R. W. Adams, J. A. Aguilar, K. D. Atkinson, M. J. Cowley, P. I. P. Elliott, S. B. Duckett, G. G. R. Green, I. G. Khazal, J. Lopez-Serrano and D. C. Williamson, Science, 2009, 323, 1708–1711 CrossRef CAS.
- A. B. Baranga, S. Appelt, M. V. Romalis, C. J. Erickson, A. R. Young, G. D. Cates and W. Happer, Phys. Rev. Lett., 1998, 80, 2801–2804 CrossRef CAS.
- Z. I. Cleveland, G. E. Pavlovskaya, N. D. Elkins, K. F. Stupic, J. E. Repine and T. Meersmann, J. Magn. Reson., 2008, 195, 232–237 CrossRef CAS PubMed.
- K. F. Stupic, Z. I. Cleveland, G. E. Pavlovskaya and T. Meersmann, J. Magn. Reson., 2011, 208, 58–69 CrossRef CAS PubMed.
- M. S. Albert and F. T. Hane, Hyperpolarized and Inert Gas MRI: From Technology to Application in Research and Medicine, Elsevier, 2017 Search PubMed.
- A. Zagdoun, A. Lesage, C. Copéret, L. Emsley, A. J. Rossini and M. Lelli, Acc. Chem. Res., 2013, 46, 1942–1951 CrossRef PubMed.
- K. V. Kovtunov, I. E. Beck, V. I. Bukhtiyarov and I. V. Koptyug, Angew. Chem., Int. Ed., 2008, 47, 1492–1495 CrossRef CAS.
- L. B. Bales, K. V. Kovtunov, D. A. Barskiy, R. V. Shchepin, A. M. Coffey, L. M. Kovtunova, A. V. Bukhtiyarov, M. A. Feldman, V. I. Bukhtiyarov, E. Y. Chekmenev, I. V. Koptyug and B. M. Goodson, J. Phys. Chem. C, 2017, 121, 15304–15309 CrossRef CAS.
- J. McCormick, S. Korchak, S. Mamone, Y. N. Ertas, Z. Liu, L. Verlinsky, S. Wagner, S. Glöggler and L. S. Bouchard, Angew. Chem., Int. Ed., 2018, 57, 10692–10696 CrossRef CAS PubMed.
- S. B. Duckett and N. J. Wood, Coord. Chem. Rev., 2008, 252, 2278–2291 CrossRef CAS.
- D. Blazina, S. B. Duckett, J. P. Dunne and C. Godard, Dalton Trans., 2004, 2601–2609 RSC.
- K. V. Kovtunov, D. A. Barskiy, A. M. Coffey, M. L. Truong, O. G. Salnikov, A. K. Khudorozhkov, E. A. Inozemtseva, I. P. Prosvirin, V. I. Bukhtiyarov, K. W. Waddell, E. Y. Chekmenev and I. V. Koptyug, Chem. – Eur. J., 2014, 20, 11636–11639 CrossRef CAS.
- K. V. Kovtunov, V. V. Zhivonitko, I. V. Skovpin, D. A. Barskiy and I. V. Koptyug, Top. Curr. Chem., 2012, 338, 123–180 CrossRef PubMed.
- R. V. Shchepin, A. M. Coffey, K. W. Waddell and E. Y. Chekmenev, Anal. Chem., 2014, 86, 5601–5605 CrossRef CAS.
- R. V. Shchepin, W. Pham and E. Y. Chekmenev, J. Labelled Compd. Radiopharm., 2014, 54, 517–524 CrossRef.
- K. V. Kovtunov, D. A. Barskiy, R. V. Shchepin, O. G. Salnikov, I. P. Prosvirin, A. V. Bukhtiyarov, L. M. Kovtunova, V. I. Bukhtiyarov, I. V. Koptyug and E. Y. Chekmenev, Chem. – Eur. J., 2016, 22, 16446–16449 CrossRef CAS.
- K. V. Kovtunov, D. Lebedev, A. Svyatova, E. V. Pokochueva, I. P. Prosvirin, E. Y. Gerasimov, V. I. Bukhtiyarov, C. R. Müller, A. Fedorov and I. V. Koptyug, ChemCatChem, 2019, 11, 969–973 CrossRef CAS.
- X. Huang, D. Teschner, M. Dimitrakopoulou, A. Fedorov, B. Frank, R. Kraehnert, F. Rosowski, H. Kaiser, S. Schunk, C. Kuretschka, R. Schlögl, M.-G. Willinger and A. Trunschke, Angew. Chem., Int. Ed., 2019, 58, 8709–8713 CrossRef CAS.
- C. Hu, J. Sun, D. Kang, Q. Zhu and Y. Yang, Catal. Sci. Technol., 2017, 7, 2717–2728 RSC.
- E. L. Hahn, Phys. Rev., 1950, 80, 580–594 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cy02100k |
| This journal is © The Royal Society of Chemistry 2020 |