
Modular design of a trap-and-atomizer device with a gold absorber for selenium collection after hydride generation
Ignacio
Machado
 ab,
Dominik
Vaněk
ac,
Stanislav
Musil
a,
Mariela
Pistón
b,
Jiří
Dědina
*a and
Jan
Kratzer
a
ab,
Dominik
Vaněk
ac,
Stanislav
Musil
a,
Mariela
Pistón
b,
Jiří
Dědina
*a and
Jan
Kratzer
a
aInstitute of Analytical Chemistry of the Czech Academy of Sciences, Veveří 97, 602 00 Brno, Czech Republic. E-mail: dedina@biomed.cas.cz
bFaculty of Chemistry, Grupo de Análisis de Elementos Traza y Desarrollo de Estrategias Simples para Preparación de Muestras (GATPREM), Analytical Chemistry, DEC, Universidad de la República, Gral. Flores 2124, Montevideo, Uruguay
cUniversity of Chemistry and Technology, Prague, Faculty of Chemical Engineering, Technická 5, 166 28 Prague 6, Czech Republic
First published on 21st November 2019
Abstract
A novel design of a device serving to trap hydrides and to volatilize the analyte to be detected by atomic absorption spectrometry, a modular trap-and-atomizer device, was invented to allow a simple replacement of the inlet arm which serves as the trap. A device with a gold wire absorber was employed for selenium collection. The relevant experimental conditions (hydrogen flow rate, trapping temperature and volatilization temperature) were optimized by using 75Se radiotracer experiments and atomic absorption spectrometry experiments. A lossless Se collection was achieved. Two arrangements of the trap were tested to maximize the signal/noise ratio. A limit of detection of 7 pg ml−1 was found for the optimum arrangement. Results of analysis of certified reference materials TMRAIN-04 and SRM 1643e provided good accuracy and precision. Interference of several hydride forming elements was characterized. The modular design appears to be promising for collection of other hydride forming elements.
1 Introduction
Liquid phase sampling inductively coupled plasma mass spectrometry (ICPMS) generally serves as a trademark of unparalleled sensitivity for elemental analysis.1 However, when replacing sample nebulization by hydride generation (HG), ICPMS can be replaced by much simpler and cheaper atomic absorption spectrometry (AAS). In addition, the sensitivity of HG AAS can be substantially enhanced by employing the inherent advantage of HG – simple analyte preconcentration either in a special collection device (usually by cryogenic collection2) or, better, directly in the atomizer.In-atomizer collection after HG3,4 is the most convenient way of analyte preconcentration. It is termed “in situ” collection when it takes place on the surface of the atomizer segment which is aligned in the optical path of the instrument. The procedure of in-atomizer collection after HG consists of the following steps: trapping and volatilization/atomization. In the first step, the hydride carried from a generator is trapped in the atomizer until its evolution is completed. In the second step, the trapped analyte is volatilized and atomized. To characterize individual stages of the procedure of in-atomizer collection, it is useful to define their efficiencies in the following way: HG efficiency is the fraction of the analyte transported in the form of hydride to the atomizer; trapping efficiency is the fraction of the generated hydride trapped in the atomizer; volatilization/atomization efficiency is the fraction of the trapped analyte volatilized and atomized. The collection efficiency is the fraction of the generated hydride which is trapped, volatilized and atomized. For the optimum performance of the method it is highly desirable to reach 100% collection efficiency. With incomplete collection, lower sensitivity is achieved and also a small deviation in an experimental parameter may lead to a significant change in collection efficiency resulting in impaired precision and accuracy. The most reliable method to determine the individual efficiencies is to use radiotracers.3,5–7 An alternative way to find the collection efficiency is to compare the peak area sensitivity obtained by in-atomizer collection and on-line atomization under the same atomizer parameters.8–10
Besides the most popular approach of in-atomizer collection, in situ collection in graphite furnaces,3,9,11–15 also in-atomizer collection in quartz tube atomizers (QTAs)3,5,6,8,10,16–28 as well as in dielectric barrier discharge (DBD) atomizers29–34 have been reported. QTAs are usually T-tubes with the longitudinal arm of the T (optical tube) aligned in the optical path of the AAS spectrometer. The central arm of the T-tube (inlet arm) serves for delivery of hydrides carried by a flow of gas from a hydride generator. It should be highlighted that several approaches other than in-atomizer collection after HG were discussed in a review3 and in more recent papers,35–54 namely in situ collection in tube atomizers either of nickel46 or tungsten,47in situ collection in tungsten coils,38,39,54 in-atomizer collection in a tungsten coil placed in the inlet arm of a T-shaped unheated quartz tube either unheated,35 or heated,41,44,45,49 in-atomizer collection in a molybdenum foil strip placed upstream of a miniature diffusion flame3 and, finally, collection in a quartz tube trap followed by atomization in an acetylene–air flame.37,40,42,43,48,50–53 However, neither of these approaches can compete in sensitivity with in-atomizer collection in QTAs. In-atomizer collection in QTAs after HG performs satisfactorily, i.e. 100% collection efficiency can be achieved for Pb,6,16,24,27 Cd,18 Sb,5,17,19,21–23 Bi5,20,22,26 and Sn.28 However, in spite of thorough efforts to optimize both steps of the collection procedure, this approach worked much less successfully for As and Se8 with a collection efficiency of 50% and 70%, respectively. A rather promising approach was developed by Guo and Guo10 who reported SeH2 trapping on a gold wire heated to 200 °C situated in a quartz tube trap. The analyte trapped was volatilized at 900 °C and atomized either in a QTA with AAS detection or in a diffusion flame with atomic fluorescence spectrometric (AFS) detection. This work was focused on AFS detection rather than AAS.
The aim of this work was to explore gold modified QTAs (for AAS detection) to achieve an optimum performance of Se in-atomizer collection including the collection efficiency as close as possible to the ideal 100%. For this investigation, a novel design of a device serving to trap and volatilize analytes was invented to allow a simple replacement of the inlet arm which serves as the trap. The 75Se radiotracer was employed to determine the trapping efficiency.
2 Experimental
2.1 Reagents
Deionized water (<0.1 μS cm−1, Ultrapure, Watrex, USA) was used to prepare all the solutions. The concentrated acids employed were: HCl (p.a., Merck, Germany), HNO3 (p.a., Lach-Ner, Czech Republic) and HF (p.a., Spolchemie, Czech Republic). The blank was 1.0 mol l−1 HCl. Working solutions of Se(IV) were prepared from 1000 μg ml−1 stock solution (Fluka, Germany) by dilution with the blank. If not explicitly stated otherwise, the Se concentration in working solutions was 5 ng ml−1. For interference studies, 1000 μg ml−1 stock solutions of As(III), Sb(III), Bi(III) (Fluka, Germany) and Hg(II) (Merck, Germany) were used to prepare the working solutions in the blank containing constant Se concentration as the analyte. The reductant was a 0.5% (m/v) solution of NaBH4 (Sigma-Aldrich, Germany) in 0.4% (m/v) KOH (p.a., Lach-Ner, Czech Republic) filtered after preparation and stored frozen. The certified reference materials TMRAIN-04 (fortified rainwater) purchased from Environment Canada (Canada) and SRM 1643e (NIST, USA, trace elements in water) with defined Se content were employed to assess the accuracy and precision of the results. Ar (99.996%, SIAD Ltd., Czech Republic) and H2 (99.95%, SIAD Ltd., Czech Republic) were used as carrier gases, while O2 (99.5%, SIAD Ltd., Czech Republic) was introduced into the atomizer.An aqueous solution of radioactive 75Se (half-life 119.8 day), declared as sodium selenite, was purchased from Lacomed Ltd., Czech Republic. To ensure Se(IV) stability, the solution was made to contain 6 mol l−1 HCl.
2.2 Instrumentation
A GBC model SavantAA atomic absorption spectrometer (GBC, Australia) was employed without background correction. A Se superlamp (Photron, Australia) operated at the 196.0 nm analytical line with a 1.0 nm spectral bandpass and a lamp current of 18 mA (boost current 23 mA). The flow rates of all gases were controlled by means of mass-flow controllers (Cole Parmer, USA).Gamma activity was quantified using an automatic gamma counter (1480 Wizard 3, PerkinElmer) equipped with a NaI(Tl) well-type crystal. A counting time of 60 s was employed. A photoplate (Fuji, Japan) sensitive to gamma radiation was employed for image plate autoradiography. The autoradiograms were obtained with the help of a multi-imager laser scanner system (FX ProPlus Molecular Imager, Bio-Rad, USA).
The apparatus for hydride generation/collection consisted of a hydride generator and a trap-and-atomizer device.
2.3 Hydride generator
The continuous flow hydride generator was made in-house, similar to that described previously.8 Two T-pieces of polyether ether ketone (0.8 mm inner bore) were used to merge sample and reductant flows and, downstream, to merge the reaction mixture flow with the carrier gas flow. A sample (either blank or a working solution) was introduced into the sample channel. The outlet from the second T-piece was connected by a 600 mm length of 1 mm i.d. polytetrafluoroethylene (PTFE) tubing to a 3 ml internal volume gas-liquid separator (GLS) with a forced outlet (see ref. 55 for detailed description of the GLS). A peristaltic pump (Ismatec, Germany) delivered sample and reductant solutions, and removed the waste from the GLS. In all experiments, sample, reductant and waste flow rates were 4.2 ml min−1, 1.2 ml min−1 and 7.4 ml min−1, respectively. The flow rate of H2 evolved from NaBH4 decomposition was calculated, as well as experimentally determined using a manual volumetric flow meter, to be approximately 15 ml min−1.2.4 Modular design of the trap-and-atomizer device
The novel, modular design of a quartz device serving to trap and to volatilize analytes, based on the compact trap-and-atomizer device detailed in ref. 8, was explored. If explicitly stated, the compact device was employed for some comparative measurements.The main feature of the modular trap-and-atomize device (Fig. 1A) was that the inlet arm of the device, serving as the trap, was not sealed to the longitudinal arm but it was easily connectable and demountable.
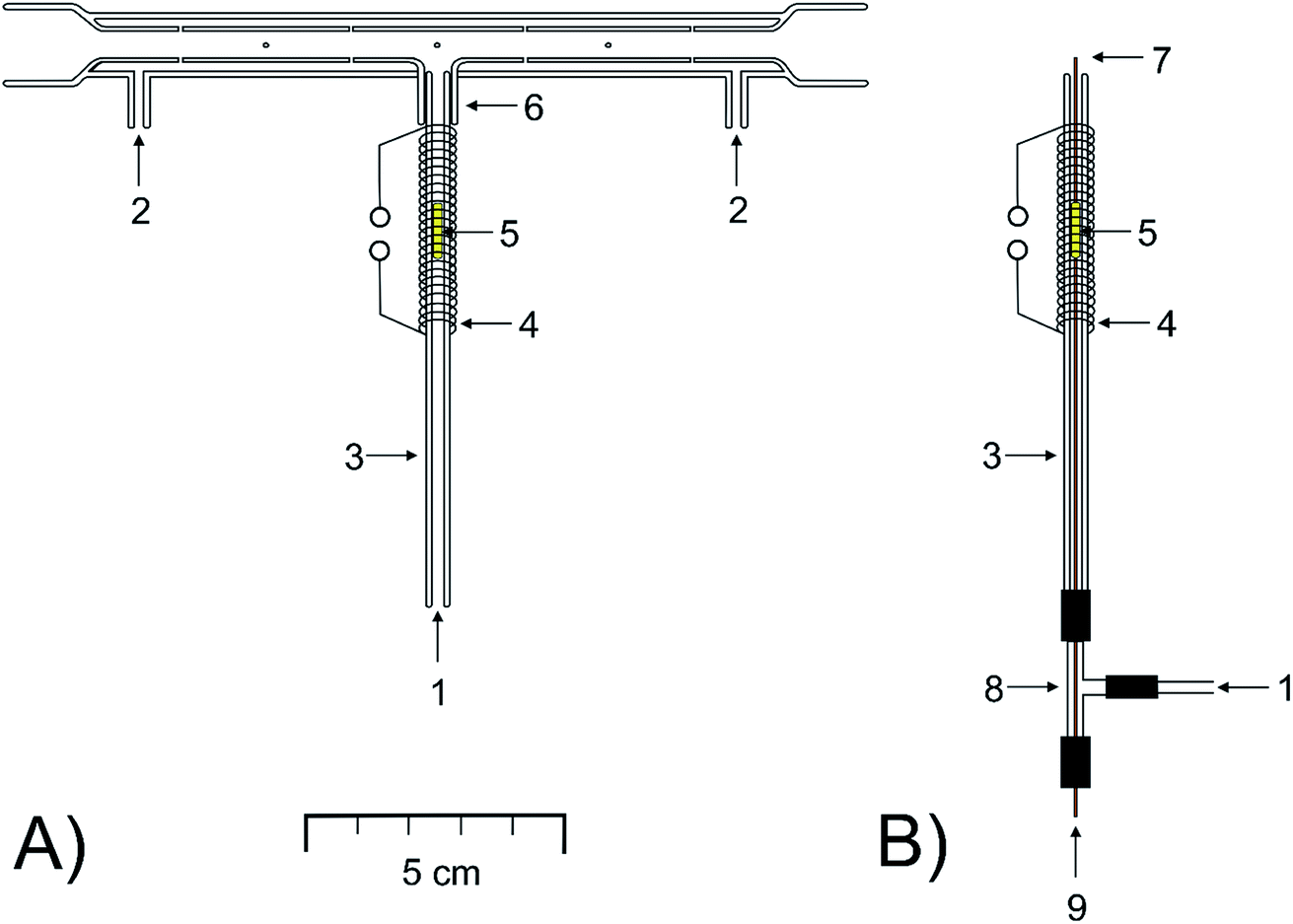 | ||
| Fig. 1 The scheme of the modular design of a trap-and-atomizer device. Two setups of the inlet arm are shown: (A) Au packed inlet arm without a capillary; (B) Au packed inlet arm with a capillary. (1) Gases from the GLS, (2) O2 inlet, (3) inlet arm, (4) heating coil, (5) packed Au wire, (6) 1 cm long short tube, (7) capillary top, (8) fitted T-piece, and (9) H2 flow. | ||
The longitudinal arm (160 mm long), aligned in the optical path of the spectrometer, was equal to that of the multiatomizer described previously (model MM4 in ref. 56). It was made of two concentric tubes. The inner (optical) one (6 mm i.d.) was evenly perforated with 14 orifices.
The inlet arm was a bare quartz tube of 100 mm length (2 mm i.d., 4 mm o.d.). A short tube 1 cm long (4 mm i.d., 6 mm o.d.) was fused to the longitudinal arm in its centre in the right angle (Fig. 1A). The downstream section of the inlet arm 1 cm long could be slid into the sealed short tube tightly to realize its gastight connection with the longitudinal arm.
A flow of oxygen was introduced from the sides (Fig. 1A) into the cavity between both tubes of the longitudinal arm and then passed through the orifices into the optical tube. A commercial heating unit model MHS-20 (PerkinElmer, USA), with temperature control, was used to heat the central part 120 mm long of the longitudinal arm to a temperature of 900 °C. The upper section of the inlet arm 2 cm long, including its 1 cm long section slid into the short tube sealed to the longitudinal arm, was covered by the heating unit. A 4 cm length of heating coil made from 50 cm kanthal wire (4.20 Ω m−1, 0.65 mm diameter, ELCHEMCo) covered the upper part of the inlet arm up to the lower rim of the short tube sealed to the longitudinal arm. The coil was resistively heated by a laboratory power supply source PS 3065-10B (E-A, Elektro-Automatik GmbH, Germany). The gas phase temperature inside the inlet arm heated by the applied current was determined by use of a digital K-type thermocouple thermometer (Omega Engineering, USA). A current between 0 and 4 A resulted in a temperature in the range of 50–1050 °C.
The modular trap-and-atomizer device was employed in three setups of the inlet arm: either empty, as detailed above, or packed by Au wire, under two arrangements as specified below.
2.5 Procedure
Measurements were performed either in the on-line atomization mode or in the collection mode. If not stated otherwise the flow rate of carrier gas was 75 ml min−1 H2 and oxygen flow rate was 40 ml min−1. Carrier gas was introduced upstream of the GLS in both modes of measurement. In some measurements a mixture of argon and hydrogen, containing a hydrogen fraction between 0 and 100%, was employed as the carrier gas. The oxygen flow rate varied from 4.5 to 40 ml min−1 depending on the hydrogen fraction (see Section 3.1).Throughout the on-line atomization mode measurement performed in the trap-and-atomizer device with an empty inlet arm (Section 2.4), HG was in progress (peristaltic pump was on), the inlet arm was unheated, and the atomic absorption signal from the analyte atomized in the optical tube was monitored continuously. If explicitly stated, the on-line atomization mode measurement performed in a setup of the trap-and-atomizer device with a Au packed inlet arm employed heating of the inlet arm to the temperature of 1020 °C. A typical measurement consisted in establishing the baseline for blank introduction into the sample channel. Then an actual sample (either blank or a working solution) was introduced into the sample channel for 30 s, to be subsequently replaced by the blank for 30 s. The signal obtained in the on-line atomization mode was integrated for 60 s beginning at the start of introduction of the sample.
The collection mode procedure consisted of two steps: (i) trapping, in which the analyte was trapped in the inlet arm and (ii) volatilization, in which the trapped analyte was released and transferred into the optical tube to be atomized there. If not explicitly stated otherwise, hydrogen was employed as the carrier gas in both steps of the collection procedure. The start and length of the integration period are detailed below. The procedure slightly differed for individual arrangements of the inlet arm (Section 2.4).
Volatilization step: heating of the inlet arm to the volatilization temperature (1020 °C if not explicitly stated otherwise) was activated. At the same time, the signal recording was switched on with an integration time of 60 s. After that the inlet arm heating was switched off, the inlet arm was cooled down and the whole procedure could be repeated.
If explicitly stated, hydrogen flow was replaced by Ar as the carrier gas in the trapping step and in the first 20 s of the volatilization step. Then the Ar flow was replaced by H2 flow and simultaneously the signal recording was switched on. The signal was recorded for 15 s instead of 60 s since this time was sufficient to record the complete analyte peak – absorbance safely returned to the baseline. Then the inlet arm heating was switched to trapping temperature to start the collection mode procedure again.
Volatilization step: heating of the inlet arm to the volatilization temperature (1020 °C) was activated and simultaneously the carrier H2 flow bypassed the GLS by means of a three-way valve – it was introduced via the capillary (see Fig. 1B) directly to the optical tube. In the following period of 20 s, before the volatilization temperature was reached, there was no gas flow through the GLS and the trap. Subsequently, H2 flow was redirected again to the generator (upstream of the GLS) so that hydrogen flew through the inlet arm and, at the same time, the signal recording was switched on. 20 s was sufficient for reaching the baseline. Analogously as in the case of the arrangement with Au wire without a capillary, the inlet arm heating was then switched off and the whole procedure could be repeated.
2.6 Measurements with a radiotracer
A 75Se radiotracer was employed to quantify the trapping efficiency in the Au packed inlet arm with a capillary (Section 2.4.1). The working solution was prepared from the commercial 75Se radiotracer solution stored in 6 mol l−1 HCl by appropriate dilution with 1 mol l−1 HCl. Se isotopes in addition to the radioactive 75Se yielded an estimated total Se concentration of approximately 2.5 ng ml−1 (based on the data provided by the manufacturer and taking into account dilution during preparation). If explicitly stated, working solutions with the 75Se radiotracer were fortified by the addition of the non-radioactive Se (21 or 210 ng ml−1). The typical activity taken for each experiment was 2.8 kBq corresponding to 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cpm in the automatic gamma counter.
000 cpm in the automatic gamma counter.
All the experiments were carried out under an exhaust fume hood. If not explicitly stated otherwise radiotracer experiments were performed with the same experimental conditions as used with AAS measurements. Pure hydrogen was always used as the carrier gas. The only difference to the apparatus employed in AAS was that absorbing columns packed with granulated activated carbon were appended either downstream of the inlet arm (radio_setup IA) or to both ends of the longitudinal arm (radio_setup HA) (see ref. 57 for the column description). As discussed recently57 the columns can quantitatively trap the generated 75SeH2 so that they also prevent 75Se radiotracer release to the atmosphere.
The procedure used for the trapping step was performed (see Section 2.5.2). After finishing the procedure, the appended device (i.e. either the inlet arm with the column or the modular trap-and-atomizer device with columns) was left to cool down and it was dismantled into individual parts. The radioactivity of parts which were small enough to be placed into sample vials of the automatic gamma counter was counted directly. The larger parts which could not be placed into the well-type crystal detector were leached using HNO3 mixed with deionized water (1:2) and afterwards using a mixture of HF and HNO3 (3:7), with subsequent quantification of the activity in the leachate. This made it possible to quantify the analyte fraction retained in individual parts of the appended device as the ratio of the radioactivity of a given part to the total radioactivity of the appended device. The geometric effect was corrected for.
The spatial distribution of 75Se in the trap-and-atomizer device was assessed using image plate autoradiography. To obtain the radiograms, the device was fixed on a sheet of paper and placed onto a photoplate sensitive to gamma radiation.
2.7 Conventions
Peak areas of AAS signals are invariably employed as the analytical quantity. If sensitivity is mentioned it always stands for the peak area related to the analyte atom mass introduced into the generator. Averages of at least 3 replicates of peak area values are presented in figures and in the text. Uncertainties are derived from standard deviations. If not explicitly given otherwise the precision of measurements was better than 4% (relative standard deviation).The complexity of radioactive measurements did not make it possible to perform a sufficient number of replicate determinations to calculate average values and corresponding standard deviations in most cases. Results of all individual experiments are therefore given. Their corresponding uncertainties, given by the counting statistics are typically lower than 1% of given values.
3 Results and discussion
3.1 Performance of the modular design of the trap-and-atomizer device in the on-line atomization mode
The on-line atomization mode with the compact design of the trap-and-atomizer device was used as a reference. In accordance with the previous findings,8 carrier Ar at a flow rate of 75 ml min−1 was chosen for optimal repeatability and sensitivity. The optimum oxygen flow rate was 4.5 ± 0.4 ml min−1. The sensitivity obtained during several months was 0.52 ± 0.03 s ng−1. The optimum oxygen flow rate as well as the sensitivity correspond reasonably well to the previous measurements.8,56 The long-term monitored sensitivity of analogous measurements (on-line atomization mode) with the “empty inlet arm” setup of the modular trap-and-atomizer device (see Section 2.4) yielded 0.54 ± 0.02 s ng−1. The same optimum oxygen flow rate as for the compact design was found. It can be concluded that the performance, in terms of sensitivity as well as repeatability, of the modular-trap-and atomizer device is equal to that of its compact design. Gas-tight connection of the inlet and longitudinal arms of the modular design has been proven.As discussed below, a high fraction of hydrogen in the atomizer atmosphere is required in the volatilization step of the collection mode. In order to find the collection efficiency the sensitivity in the on-line atomization mode must be determined under the corresponding fraction of hydrogen in the atomizer atmosphere. Therefore, the influence of hydrogen fraction in the carrier gas between 0 and 100% on sensitivity in the on-line atomization mode was investigated. It was found that the optimum oxygen flow rate increased with increasing hydrogen fraction in the carrier gas from 4.5 ml min−1 for 0% of hydrogen to 12, 22, 30 and 40 ml min−1, respectively, for 33, 53, 73 and 100% of hydrogen in the carrier gas. In spite of the dramatic increase of the oxygen demand with increasing hydrogen fraction, sensitivity decreased with the hydrogen fraction only slightly (assuming an optimized oxygen flow rate): the increase of the hydrogen fraction in the carrier gas from zero to 100% resulted in a decline by 20% – from 0.56 to 0.45 s ng−1.
3.2 Preliminary considerations
To reach the target of this work, i.e. to achieve an optimum performance of Se in-atomizer collection, it was required to find conditions compatible with maximum trapping efficiency in the first step and subsequently to optimize the volatilization step by maximization the signal/noise ratio which included a maximization of the volatilization/atomization efficiency.3.3 Determination of the trapping efficiency with the radiotracer
The use of the radiotracer can yield essential information on processes taking place in the trap-and-atomizer device and mainly on the efficiencies of individual steps of the analytical procedure. The focus was put on the trapping efficiency in the Au packed inlet arm with a capillary.After the trapping step of the collection mode procedure, 94% of the analyte delivered from the hydride generator to the trap-and-atomizer device was detected in the Au wire, 2% was found on the quartz capillary and additional 1% was leached from the surface of the bare quartz tube that forms the inlet arm. The remaining 2% was leached from the optical tube. There was no detectable activity in the appended columns. In conclusion, the trapping efficiency was 97%. Fig. 2 illustrates the distribution of the trapped analyte in the device.
 | ||
| Fig. 2 Spatial distribution of 75Se in a modular trap-and-atomizer device (inlet arm setup: Au packed with a capillary) after the trapping step of the collection mode procedure (exposure time: 24 hours). Trapping temperature 375 °C, 75 ml min−1 H2 carrier gas flow rate and 40 ml min−1 O2. Carrier-free 75Se tracer solution employed (2.8 kBq, 2.5 ng ml−1 Se, 4.5 ng Se absolute). | ||
Additional experiments were performed to estimate the influence of analyte concentration on the trapping efficiency. Because of the extreme complexity of experiments with the configuration radio_setup HA, the radio_setup IA was employed that allows much simpler operation. For the “standard” Se concentration of 2.5 ng ml−1, the trapping efficiency of 93%, found in both parallel experiments, was in a reasonable agreement with that found in the configuration radio_setup HA (see above).
The trapping efficiencies for Se concentrations of 21 and 210 ng ml−1, respectively, found in parallel experiments were 91% and 94%, and 25% and 22%. Taking into account that the hydride generation efficiency of non-radioactive selenium is close to 100% in the current hydride generator,2 0.44 μg Se should be converted to hydride and delivered to the trap when working with the standard solution of 210 ng ml−1. Then the observed trapping efficiency corresponds to the trapped analyte mass of 0.10 μg. Assuming a van der Waals Se atom radius of 0.20 nm,58 0.049 μg of Se atoms would form a single atomic layer at the employed Au wire surface area of 0.6 cm2 (a smooth surface of the wire is supposed). It can be concluded that selenium atoms are trapped at the Au surface in a more compact way than corresponding to the van der Waals atom radius: assuming a single atomic layer, 0.10 μg of trapped selenium would correspond to the interatomic distance of 0.28 nm. It should be related to the bond length of 0.23 nm in the Se dimer.58
The radiotracer experiments have revealed the feasibility of quantitative retention of selenium on the gold surface under given trapping conditions. Further optimization of the preconcentration procedure and apparatus arrangement was performed using an AAS as a detector which allows us to monitor the signal development in real time.
3.4 Atomic absorption spectrometry optimization of the collection procedure
It has been previously shown that to reach 70% Se collection efficiency in the compact trap-and-atomizer device with the empty inlet arm, a minimum temperature in the trapping step and an excess hydrogen under elevated temperature in the volatilization step were required. The excess hydrogen made it possible to eliminate slow heating of the trap-volatilization was initiated by switching off oxygen and, after a 5 s delay time, by switching on hydrogen, not before the trap reached the volatilization temperature.8 The delay time served to flush out the atomizer/trap device to prevent spectral interference of molecular oxygen, which could not be compensated by the deuterium background correction.59 Se losses in the trapping step, unavoidable in the presence of hydrogen developed in the hydride generator, could be largely eliminated by burning out of hydrogen in a stoichiometric excess of oxygen.8 The incomplete analyte collection was partially due to persisting losses in the trapping step and partially due to losses in the hydrogen presence during the 5 s delay when the trap already reached the volatilization temperature.8The above treated radiotracer experiments indicated that the trap-and-atomizer device with a Au packed inlet arm with a capillary yielded substantial improvements: lossless Se trapping can be achieved even at a temperature of almost 400 °C and in the presence of hydrogen (Section 3.3). The aim of atomic spectrometry experiments was to optimize all relevant parameters in order to maximize the signal/noise ratio. Consequently, the analyte should be volatilized/atomized with maximum efficiency but also as fast as possible to yield a narrow signal. Additionally, to prevent a serious deterioration of signal repeatability due to the spectral interference of molecular oxygen there should not be a significant change of molecular oxygen concentration in the atomizer atmosphere during the time interval in which the absorbance peak is recorded.
In all performed collection mode experiments, normalized signal intensities were assessed by relating the peak area of a given signal recorded in the volatilization step of the collection procedure to the peak area of the signal recorded in the on-line atomization mode with the same setup of the trap-and-atomizer device under the same atomizer atmosphere composition and flow rate. For the sake of simplicity, the inlet arm was heated to the temperature of 1020 °C in the on-line atomization mode. Regular checks were performed to prove that signals obtained did not significantly differ from those obtained for corresponding measurements with the trap-and-atomizer device with an empty inlet arm either heated or unheated.
A normalized signal intensity can serve as an approximation of the collection efficiency. Namely in the case of switching between Ar and H2 carriers (see below), it cannot be taken as an exact efficiency value because of the fading down of the Ar fraction in the H2 atmosphere during the peak onset.
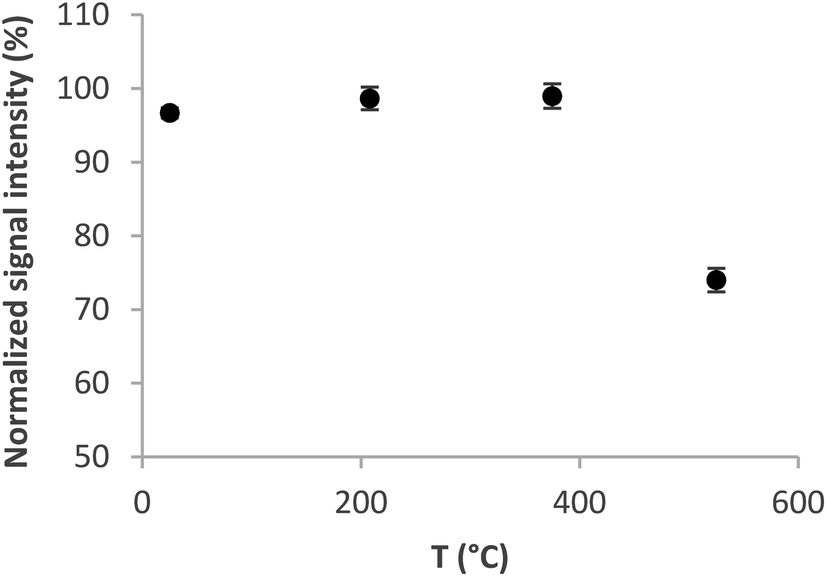 | ||
| Fig. 3 Trapping curve for the Au packed inlet arm without a capillary. Se concentration 5 ng ml−1, volatilization temperature 1020 °C, and H2 flow rate 75 ml min−1. | ||
The optimum volatilization temperature was between 920 °C and 1020 °C (Fig. 4). 1020 °C was employed for further experiments because of the slightly better signal repeatability – RSD 2% versus 3.3%. Lower temperatures were not sufficient to completely volatilize the analyte from the trap.
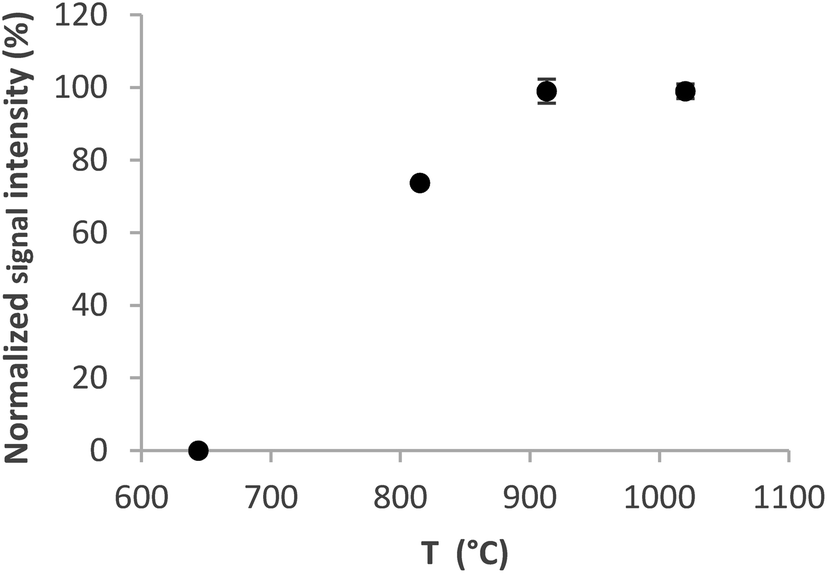 | ||
| Fig. 4 Volatilization curve for the Au packed inlet arm without a capillary. Se concentration 5 ng ml−1, trapping temperature 375 °C, and H2 flow rate 75 ml min−1. | ||
Additional experiments revealed that changing the carrier H2 flow rate from 75 ml min−1 could not bring any positive effect. Even though an increase or decrease of the flow rate by 50% did not change markedly normalized signal intensities, the decrease resulted in unacceptably broad peaks and the increase induced a considerable drop of sensitivity. Consequently, any change of the flow rate of 75 ml min−1 caused significant deterioration of the detection performance.
As illustrated in Fig. 3 and 4, the optimum trapping and volatilization temperatures were compatible with essentially 100% normalized signal intensity. However, the signals were rather broad (Fig. 5) having the full width at half maximum (FWHM) around 9 s so that the minimum integration time required was 30 s. The long integration time is a drawback in terms of the signal/noise ratio. The reason for the relatively broad signals is that the peak evolution is controlled by the rate of heating of the trap: the peak evolution starts when the trap temperature reaches sufficiently high temperature and it is not finished before the temperature reaches 1020 °C.
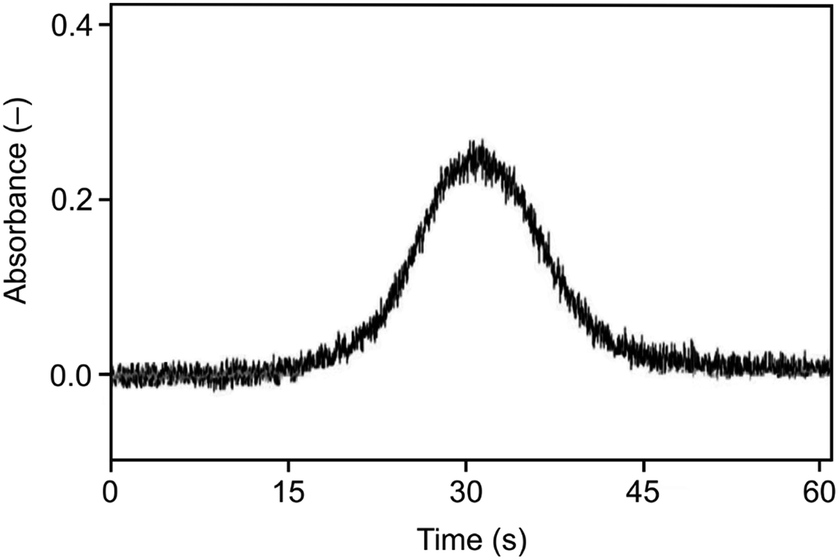 | ||
| Fig. 5 The signal observed for the Au packed inlet arm without a capillary, Se concentration 5 ng ml−1, trapping temperature 375 °C, volatilization temperature 1020 °C, and H2 flow rate 75 ml min−1. | ||
A substantially faster trap heating with the current heating device is not feasible. Consequently, to obtain narrower peaks (and consequently a better signal/noise ratio) it is required to prevent analyte losses from the trap during its heating to the optimum volatilization temperature of 1020 °C and, subsequently, to volatilize all trapped analyte fast in a single shot. Preliminary experiments were performed using Ar as the carrier gas in the trapping step and in the beginning of the following step until reaching the set volatilization temperature so that there was no hydrogen in the trap during increasing its temperature after finishing hydride generation. When the trap temperature reached the optimum value of 1020 °C the carrier Ar flow was replaced by the hydrogen flow. Immediately, the signal of the volatilized and atomized analyte was recorded. In this case, the peak evolution was controlled by the change of the atomizer atmosphere from hydrogen deficient to hydrogen rich. A substantial narrowing of the peak was observed: an integration time of 15 s was sufficient to cover the peak fully – see Fig. 6. The corresponding normalized signal intensity was around 100% indicating that the analyte losses were prevented. However, these experiments could not lead to a better signal/noise ratio because of a marked change of the observed baseline immediately after the carrier Ar flow was replaced by the hydrogen flow. The higher baseline in Ar is due to the oxygen presence in the optical tube of the atomizer. Molecular oxygen under elevated temperatures is the source of spectral interference which cannot be compensated by the deuterium background correction.59 Oxygen is burned down in a hydrogen atmosphere and, consequently, the baseline decreases with decreasing O2 amount in the optical tube.
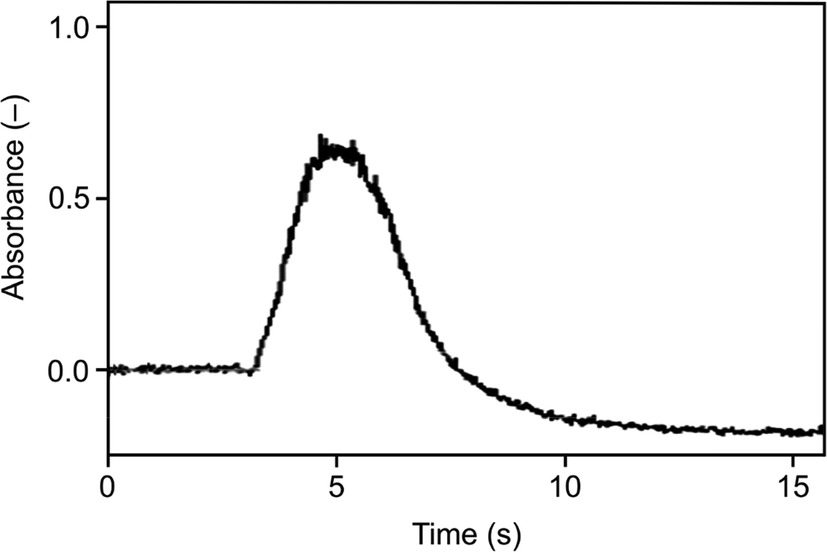 | ||
| Fig. 6 The signal observed for the Au packed inlet arm without a capillary using Ar as the carrier gas in the trapping step, Se concentration 5 ng ml−1, trapping temperature 375 °C, volatilization temperature 1020 °C, and H2 flow rate in the volatilization step 75 ml min−1. | ||
As illustrated in Fig. 7, narrow peaks were obtained, comparable to those observed with the arrangement without a capillary using Ar as the carrier gas in the trapping step (Fig. 6), but in contrast to those, there was no baseline shift. As expected, the influence of trapping temperature on the signal corresponded to the situation for the arrangement without a capillary shown in Fig. 3. The optimization of volatilization temperature made for the trapping temperature of 375 °C showed a plateau of the normalized signal intensity of 0.99 ± 0.06 for temperatures between 850 °C and the maximum temperature tested of 1020 °C. Volatilization temperatures below 850 °C were insufficient to completely release all the trapped analyte as reflected by decreased normalized signal intensities. Virtually the same behavior was observed for the trapping temperature of 200 °C.
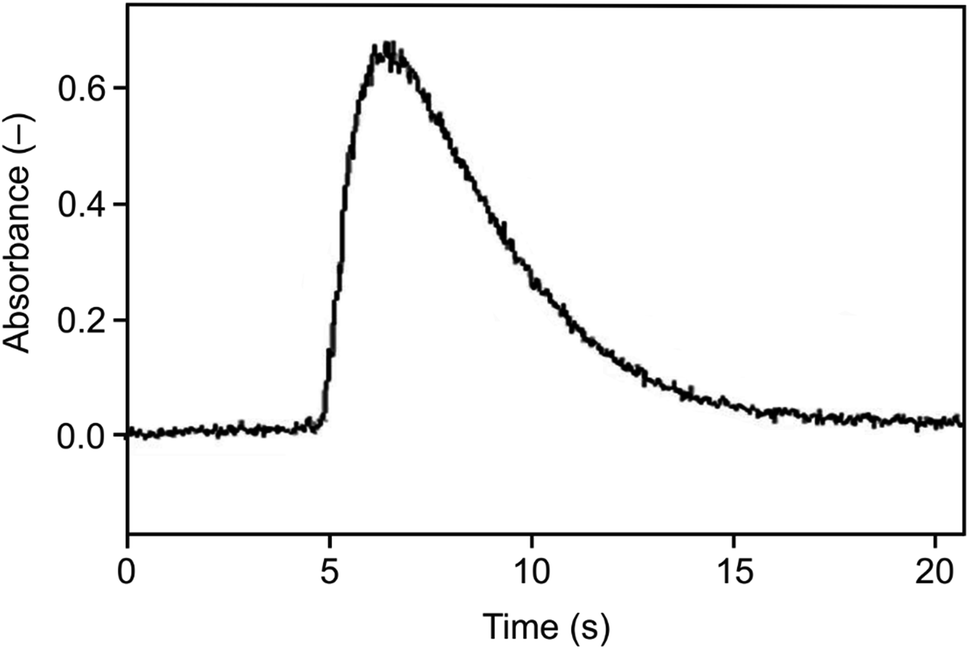 | ||
| Fig. 7 The signal observed for the Au packed inlet arm with a capillary, Se concentration 5 ng ml−1, trapping temperature 375 °C, volatilization temperature 1020 °C, and H2 flow rate 75 ml min−1. | ||
In summary, the optimum trapping and volatilization temperatures, respectively, were 200 °C to 375 °C and 850 °C to 1020 °C. It should be highlighted that the length of the time interval between start of the heating and redirecting H2 flow to the generator was critical. Increasing the interval from the minimum of 20 s to 60 s caused losses of 5%, 9% and 27%, respectively, at volatilization temperatures of 675 °C, 750 °C and 850 °C.
3.5 Analytical figures of merit
As shown above, the Au packed inlet arm with a capillary provided the best potential for analytical application. Therefore, the figures of merit were evaluated for this modular trap-and-atomizer setup and compared with the on-line atomization mode.See Table 1 for a comparison of interference extent in the collection mode with the on-line atomization mode. Interference of As, Sb, Bi and Hg on Se determination was investigated since volatile species of these elements are effectively co-generated2,5 with selenium hydride. Other hydride forming elements, such as Sn or Pb, were not investigated since the efficiency of generation of the corresponding hydrides under actual experimental conditions is very low.2,28 The extent of interference of As, Sb and Bi observed in the on-line atomization mode compares very well with the values previously reported (for on-line atomization in the multiatomizer).30,60,61 The on-line interference extent of Hg is not substantial. Only atomization interference can be responsible for the observed effects at least up to 300 ng ml−1 since no interference to selenium hydride generation should occur in the presence of As, Sb and Bi at concentrations of 500 ng ml−1 (ref. 62) and in the case of Hg, no interference was observed at concentrations at least up to 10 μg ml−1.63,64 However, collection mode interferents are much more serious (Table 1). Their extent, even more pronounced than observed for on-line atomization in a conventional externally heated QTA,3 could lead to a competition between the analyte and interferents for the trapping surface resulting in deteriorated trapping efficiency of the analyte. This should be compared with the trapping capacity of around 100 ng of Se of the employed Au wire (see Section 3.3). Additionally, the investigated interferents could be more effective in blocking (or poisoning) the Au surface than the analyte.
| Interferent | Mode | Recoveries (%) in the presence of an interferent concentrationa (ng ml−1) | |||
|---|---|---|---|---|---|
| 3 | 30 | 300 | 3000 | ||
| a Analyte concentration of 3 ng ml−1. | |||||
| AsIII | On-line | 96 ± 6 | 91 ± 8 | 90 ± 8 | 55 ± 9 |
| Collection | 87 ± 5 | 58 ± 7 | 40 ± 5 | 0 ± 5 | |
| SbIII | On-line | 100 ± 6 | 97 ± 5 | 72 ± 3 | 62 ± 4 |
| Collection | 100 ± 5 | 50 ± 3 | 25 ± 3 | 1 ± 3 | |
| BiIII | On-line | 100 ± 2 | 98 ± 3 | 51 ± 15 | 30 ± 3 |
| Collection | 80 ± 7 | 79 ± 6 | 49 ± 5 | 0 ± 4 | |
| HgII | On-line | 96 ± 4 | 91 ± 5 | 91 ± 5 | 85 ± 5 |
| Collection | 80 ± 8 | 71 ± 7 | 66 ± 8 | 39 ± 8 | |
Calibration curves in the on-line atomization mode and in the collection mode with a sample introduction time of 30 s were measured up to 5 ng ml−1. With a sample introduction time of 300 s, calibration curves were measured up to 0.5 ng ml−1. In these concentration ranges, linearity was statistically confirmed in all cases. There was a marked calibration curvature at higher concentrations.
See Table 2 for a limit of detection (LOD) and accuracy. Regarding LOD values for on-line atomization, the slightly better value for the Ar carrier than for H2 corresponds to the higher sensitivity in the Ar atmosphere (Section 3.1). The integrated signal in the volatilization step of the collection mode with a sample introduction time of 30 s corresponds to the analyte mass from the same sample volume as in the on-line atomization mode. However, the signal integration time in the collection mode is more than four times shorter than in the on-line atomization mode (Section 2.5). Consequently, a better LOD improvement than the only 1.3 times better LOD of the collection mode compared to the on-line atomization mode (Table 2) was expected. The substantial LOD improvement with a longer sample introduction time in the collection mode is rather clear from Table 2. Further extension of the sample introduction time was not tested. However, the extent of the LOD improvement (more than 10 times for a 10 times longer sample introduction time) suggests that the LOD observed is not controlled by the contamination of reagents so that the longer sample introduction time could improve the LOD even more. LODs observed can be compared to those for other in-atomizer collection methods with AAS detection: 12 pg ml−1 found for the DBD atomizer34 and to values down to pg ml−1 reported in the literature for the mature and well-established in situ collection in GF.3 LODs down to pg ml−1 were reported also for HG coupled to the most sensitive detectors – AFS3 and ICPMS.65,66
| Mode | Inlet arm setup | Carrier gas | Sample introduction time (s) | LODa (ng ml−1) | TMRAIN-04b,c (ng ml−1) | SRM 1643eb,d (ng ml−1) |
|---|---|---|---|---|---|---|
| a 3 × SD, n = 20. b n = 6. c Certified value: 0.84 ± 0.07 ng ml−1. d Certified value: 11.97 ± 0.14 ng ml−1. | ||||||
| On-line | Empty | Ar | 30 | 0.09 | 0.82 ± 0.07 | 12.1 ± 0.3 |
| On-line | Empty | H2 | 30 | 0.13 | 0.81 ± 0.05 | 12.3 ± 0.3 |
| Collection | Au packed without a capillary | H2 | 30 | 0.10 | — | — |
| Collection | Au packed with a capillary | H2 | 300 | 0.007 | 0.78 ± 0.02 | 12.7 ± 0.4 |
Results of analysis of certified reference materials TMRAIN-04 and SRM 1643e (Table 2) consisting of rain water and trace elements in water, respectively, illustrate the good accuracy and precision yielded by both operation modes.
4 Conclusions
The performance of a novel design of a device serving to trap hydrides and to volatilize the analyte to be detected by AAS, a modular trap-and-atomizer device, was found to compare well with the performance of the traditional compact design of the trap-and-atomizer device described previously.8 The substantial advantage of the modular design is that it allows an easy and fast replacement of the inlet arm which serves as the trap. This makes it feasible to work with various modifications of the trap surface (also by liquid reagents) or with various absorbers (metal wires, foils, quartz wool, etc.) placed inside the trap for testing effective preconcentration of hydride forming elements.A modular trap-and-atomizer device with a gold wire absorber was employed for selenium collection. 75Se radiotracer experiments proved that selenium hydride was trapped quantitatively in the inlet arm of the device which served as the trap at a temperature of almost 400 °C and in the presence of hydrogen. The applicability of this approach is limited for samples containing high concentrations of elements whose volatile forms are effectively co-generated with selenium hydride. Atomic absorption spectrometry experiments resulted in finding optimum trapping and volatilization temperatures as well as peak shape profiles. Two arrangements of the trap packed with Au wire were tested in order to maximize the signal/noise ratio. The arrangement with a capillary provided the best potential for analytical application. The observed LOD is not controlled by the contamination of reagents so that a longer sample introduction time resulted in better LOD – 7 pg ml−1 for an introduction time of 300 s. An even lower LOD can be expected for longer times.
The modular design of the trap-and-atomizer device appears to be promising for collection of other hydride forming elements, namely As. In addition, the idea to bypass the trap after the trapping step before signal recording can be useful generally.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has been supported by the Czech Science Foundation under contract 18-01116S and by the Institute of Analytical Chemistry of the Czech Academy of Sciences (Institutional Research Plan no. RVO: 68081715). This work used instruments provided by C4Sys infrastructure. I. Machado was supported by Agencia Nacional de Investigación e Innovación (ANII), PEDECIBA-Química and Comisión Sectorial de Investigación Científica (CSIC-UdelaR). The authors are obliged to M. Svoboda for preparation of Fig. 2.References
- J. S. Becker, Inorganic Mass Spectrometry, John Wiley & Sons Ltd., Chichester, 2007 Search PubMed.
- J. Dědina and D. L. Tsalev, Hydride Generation Atomic Absorption Spectrometry, John Wiley & Sons, Inc., Chichester, 1995 Search PubMed.
- J. Dědina, Spectrochim. Acta, Part B, 2007, 62, 846–872 CrossRef.
- O. Y. Ataman, Spectrochim. Acta, Part B, 2008, 63, 825–834 CrossRef.
- J. Kratzer, M. Vobecký and J. Dědina, J. Anal. At. Spectrom., 2009, 24, 1222–1228 RSC.
- J. Kratzer, S. Musil, M. Vobecký and J. Dědina, J. Anal. At. Spectrom., 2013, 28, 344–353 RSC.
- M. Rybínová, S. Musil, V. Červený, M. Vobecký and P. Rychlovský, Spectrochim. Acta, Part B, 2016, 123, 134–142 CrossRef.
- J. Kratzer and J. Dědina, Anal. Bioanal. Chem., 2007, 388, 793–800 CrossRef CAS PubMed.
- H. Matusiewicz and R. E. Sturgeon, Spectrochim. Acta, Part B, 1996, 51, 377–397 CrossRef.
- X. M. Guo and X. W. Guo, J. Anal. At. Spectrom., 2001, 16, 1414–1418 RSC.
- Z. Furdíková and B. Dočekal, Spectrochim. Acta, Part B, 2009, 64, 323–328 CrossRef.
- A. A. Shaltout, I. N. B. Castilho, B. Welz, E. Carasek, I. B. G. Martens, A. Martens and S. M. F. Cozzolino, Talanta, 2011, 85, 1350–1356 CrossRef CAS PubMed.
- M. Rybínová, V. Červený and P. Rychlovský, J. Anal. At. Spectrom., 2015, 30, 1752–1763 RSC.
- A. M. Martinez, S. Vazquez, R. Lara, L. D. Martinez and P. Pacheco, Spectrochim. Acta, Part B, 2018, 140, 22–28 CrossRef.
- M. da Luz Potes, F. Venâncio Nakadi, C. F. Grasel Frois, M. G. Rodrigues Vale and M. Messias da Silva, Microchem. J., 2019, 147, 324–332 CrossRef CAS.
- D. K. Korkmaz, N. Ertas and O. Y. Ataman, Spectrochim. Acta, Part B, 2002, 57, 571–580 CrossRef.
- D. K. Korkmaz, J. Dědina and O. Y. Ataman, J. Anal. At. Spectrom., 2004, 19, 255–259 RSC.
- D. Korkmaz, C. Demir, F. Aydin and O. Y. Ataman, J. Anal. At. Spectrom., 2005, 20, 46–52 RSC.
- J. Kratzer and J. Dědina, Spectrochim. Acta, Part B, 2005, 60, 859–864 CrossRef.
- J. Kratzer and J. Dědina, J. Anal. At. Spectrom., 2006, 21, 208–210 RSC.
- I. Menemenlioglu, D. K. Korkmaz and O. Y. Ataman, Spectrochim. Acta, Part B, 2007, 62, 40–47 CrossRef.
- J. Kratzer and J. Dědina, Spectrochim. Acta, Part B, 2008, 63, 843–849 CrossRef.
- M. Dessuy, J. Kratzer, M. G. R. Vale, B. Welz and J. Dědina, Talanta, 2011, 87, 255–261 CrossRef CAS PubMed.
- J. Kratzer, Spectrochim. Acta, Part B, 2012, 71–72, 40–47 CrossRef CAS.
- S. Musil, J. Kratzer, M. Vobecký and T. Matoušek, J. Anal. At. Spectrom., 2012, 27, 1382–1390 RSC.
- S. Musil and J. Dědina, J. Anal. At. Spectrom., 2013, 28, 593–600 RSC.
- P. Novotný and J. Kratzer, Spectrochim. Acta, Part B, 2013, 79–80, 77–81 CrossRef.
- L. Průša, J. Dědina and J. Kratzer, Anal. Chim. Acta, 2013, 804, 50–58 CrossRef PubMed.
- J. Kratzer, J. Boušek, R. E. Sturgeon, Z. Mester and J. Dědina, Anal. Chem., 2014, 86, 9620–9625 CrossRef CAS PubMed.
- O. Duben, J. Boušek, J. Dědina and J. Kratzer, Spectrochim. Acta, Part B, 2015, 111, 57–63 CrossRef CAS.
- J. Kratzer, O. Zelina, M. Svoboda, R. E. Sturgeon, Z. Mester and J. Dědina, Anal. Chem., 2016, 88, 1804–1811 CrossRef CAS PubMed.
- X. Mao, Y. Qi, J. Huang, J. Liu, G. Chen, X. Na, M. Wang and Y. Qian, Anal. Chem., 2016, 88, 4147–4152 CrossRef CAS PubMed.
- P. Novák, J. Dědina and J. Kratzer, Anal. Chem., 2016, 88, 6064–6070 CrossRef PubMed.
- J. Kratzer, S. Musil and J. Dědina, J. Anal. At. Spectrom., 2018, 34, 193–202 RSC.
- O. Cankur and O. Y. Ataman, J. Anal. At. Spectrom., 2007, 22, 791–799 RSC.
- H. Matusiewicz and M. Krawczyk, Chem. Anal., 2007, 52, 565–578 CAS.
- H. Matusiewicz and M. Krawczyk, J. Braz. Chem. Soc., 2007, 18, 304–311 CrossRef CAS.
- S. S. Souza, D. Santos, F. J. Krug and F. Barbosa, Talanta, 2007, 73, 451–457 CrossRef.
- O. Alp and N. Ertas, J. Anal. At. Spectrom., 2008, 23, 976–980 RSC.
- N. Ertas, Z. Arslan and J. F. Tyson, J. Anal. At. Spectrom., 2008, 23, 223–228 RSC.
- I. Kula, Y. Arslan, S. Bakirdere and O. Y. Ataman, Spectrochim. Acta, Part B, 2008, 63, 856–860 CrossRef.
- H. Matusiewicz and M. Krawczyk, J. Anal. At. Spectrom., 2008, 23, 43–53 RSC.
- H. Matusiewicz and M. Krawczyk, Chem. Anal., 2008, 53, 905–925 CAS.
- S. Titretir, E. Kenduezler, Y. Arslan, I. Kula, S. Bakirdere and O. Y. Ataman, Spectrochim. Acta, Part B, 2008, 63, 875–879 CrossRef.
- I. Kula, Y. Arslan, S. Bakirdere, S. Titretir, E. Kenduezler and O. Y. Ataman, Talanta, 2009, 80, 127–132 CrossRef CAS PubMed.
- X. Wu, P. Wu, S. P. He, W. S. Yang and X. D. Hou, Spectrosc. Lett., 2009, 42, 240–245 CrossRef CAS.
- O. Alp and N. Ertas, Talanta, 2010, 81, 516–520 CrossRef CAS PubMed.
- H. Matusiewicz and M. Krawczyk, Anal. Lett., 2010, 43, 2543–2562 CrossRef CAS.
- M. Y. Xi, R. Liu, P. Wu, K. L. Xu, X. D. Hou and Y. Lv, Microchem. J., 2010, 95, 320–325 CrossRef CAS.
- Y. Arslan, E. Kenduzler and O. Y. Ataman, Talanta, 2011, 85, 1786–1791 CrossRef CAS PubMed.
- H. Matusiewicz and M. Krawczyk, Cent. Eur. J. Chem., 2011, 9, 648–659 CAS.
- E. Kilinc, S. Bakirdere, F. Aydin and O. Y. Ataman, Spectrochim. Acta, Part B, 2012, 73, 84–88 CrossRef CAS.
- E. Kilinc, S. Bakirdere, F. Aydin and O. Y. Ataman, Spectrochim. Acta, Part B, 2013, 89, 14–19 CrossRef CAS.
- P. P. Chen, Y. J. Deng, K. C. Guo, X. M. Jiang, C. B. Zheng and X. D. Hou, Microchem. J., 2014, 112, 7–12 CrossRef CAS.
- T. Matoušek, M. Johansson, J. Dědina and W. Frech, Spectrochim. Acta, Part B, 1999, 54, 631–643 CrossRef.
- T. Matoušek, J. Dědina and A. Selecká, Spectrochim. Acta, Part B, 2002, 57, 451–462 CrossRef.
- J. Kratzer, S. Musil, K. Marschner, M. Svoboda, T. Matoušek, Z. Mester, R. E. Sturgeon and J. Dědina, Anal. Chim. Acta, 2018, 1028, 11–21 CrossRef CAS PubMed.
- R. C. Weast, Handbook of Chemistry and Physics, CRC Press, Cleveland, 1982 Search PubMed.
- J. Kratzer, B. Dočekal, U. Heitmann and J. Dědina, J. Anal. At. Spectrom., 2011, 26, 2230–2237 RSC.
- J. Dědina and T. Matoušek, J. Anal. At. Spectrom., 2000, 15, 301–304 RSC.
- É. M. M. Flores, A. M. Nunes, V. L. Dressler and J. Dědina, Spectrochim. Acta, Part B, 2009, 64, 173–178 CrossRef.
- M. Walcerz, E. Bulska and A. Hulanicki, Fresenius. J. Anal. Chem., 1993, 346, 622–626 CrossRef CAS.
- K. Petrick and V. Krivan, Fresenius' Z. für Anal. Chem., 1987, 327, 338–342 CrossRef CAS.
- J. Dědina, Anal. Chem., 1982, 54, 2097–2102 CrossRef.
- C. Moor, R. E. Sturgeon and J. W. Lam, J. Anal. At. Spectrom., 2000, 15, 143–149 RSC.
- K. X. Yang and L. Husain, Spectrosc. Lett., 2006, 39, 187–201 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |