
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational insights into selective CO2 hydrogenation to CH3OH catalysed by ZnO based nanocages†
Shyama Charan
Mandal
 and
Biswarup
Pathak
*
and
Biswarup
Pathak
*
Discipline of Chemistry, Indian Institute of Technology Indore, Simrol, Indore 453552, India. E-mail: biswarup@iiti.ac.in
First published on 19th August 2020
Abstract
Cu and ZnO based nanostructures were extensively studied for CO2 hydrogenation reaction. In this study, we have performed density functional theory (DFT) calculations for understanding the CO2 hydrogenation reaction mechanism on ZnO and Cu doped ZnO based nanocages (NCs). Two different ZnO based NCs and three different Cu doped ZnO based NCs have been considered for the investigation. The stabilities of the NCs have been investigated using the formation energy, cohesive energy, phonon dispersion and ab initio molecular dynamics (AIMD) calculations. Our calculated adsorption energy values show that the CO2 hydrogenation reaction intermediates adsorb strongly on the NCs compared to that on the bulk Cu(111), Cu(111) monolayer and Cu nanocluster. Besides, the detailed mechanistic investigation and the calculated ZPE corrected reaction energy values show that the ZnO and Cu doped ZnO based NCs show excellent selectivity for CH3OH. These catalysts also work under very low working potentials (0.55 V for ZnO NC and 0.39 V for Cu doped ZnO NC) compared to the bulk Cu(111), Cu(111) monolayer and Cu nanocluster. Hence, Cu@ZnO based nanocages can be highly efficient and selective catalysts compared to ZnO based nanocages and Cu based catalysts for CO2 hydrogenation to CH3OH. Moreover, the influence of *COOH and *COH coverage for ZnO NC, *COH and *CHOH coverage for Cu@ZnO NC on adsorption energy values show that the catalysts can be used at high surface coverage.
1. Introduction
Carbonaceous fossil fuel combustion increases the amount of CO2 in the environment. This is one of the main reasons for climate change and possible ocean acidification.1,2 Thus, CO2 chemistry has received significant attention among the scientific community to transform CO2 into valuable chemicals and fuels (such as CO, HCOOH, CH2O, CH3OH, CH3OCH3 and so on).3–11 Among all these, CH3OH is highly in demand due to its wide industrial application and fuel-based properties.8,9,12–17 Also, use of CH3OH as a fuel and its formation from CO2 form a carbon-neutral process. Therefore, research efforts are devoted to the hydrogenation of CO2 to CH3OH (CO2 + 3H2 → CH3OH + H2O). Here, the overall standard electrochemical potential for CO2 hydrogenation to CH3OH is −0.38 V, but this process competes with the methanation reaction (CO2 + 4H2 → CH4 + 2H2O).18 Liu et al. have reported the first copper–zinc oxide based catalyst for CO2 hydrogenation to CH3OH where conversion of CO2 and yield of CH3OH are low.19 Thus, efficient and selective catalysts that can activate CO2 and break only one C–O bond of the CO2 molecule are highly useful. An earlier report has shown that the process is dependent on the temperature and pressure of the reaction.20 Keeping all these in mind, several homogeneous and heterogeneous catalysts have been investigated for efficient CO2 hydrogenation to CH3OH.8,9,12,15–31 Besides, most of the industrial catalysts are metals supported by oxide-based nanoparticles.20,32 Industrial CH3OH is also obtained from the syngas mixture (CO, CO2 and H2) over the Cu/ZnO/Al2O3 catalyst at a temperature of 473–573 K and a pressure of 5–10 MPa.33 In this context, Chinchen et al. have reported that the C-atom of CH3OH is obtained from the C of CO2, i.e. CO2 is responsible for the formation of CH3OH.34 Several Cu/ZnO/Al2O3 based catalysts have been reported for the CO2 hydrogenation reaction.35–38 They demonstrated that the synergistic effect plays an important role in improving their catalytic activity, however, the industrial Cu/ZnO/Al2O3 catalyst does not cleave both the C–O bonds of the CO2 molecule and thus shows poor catalytic activity at low temperatures. The CO2 activation can be facilitated by increasing the temperature but undesirable products (CO and H2O) are also formed via the reverse water–gas shift reaction (RWGS). Furthermore, Kattel et al. have concluded that Cu/ZnO facilitates CH3OH formation in comparison to Cu–Zn.38 On this account, it is very important to explore the effect of Cu and ZnO for CH3OH formation from CO2 and H2.Besides, Cu based single-atom catalysts (SACs) have also been identified as promising catalysts for selective CH3OH formation at a low overpotential.39,40 These SACs are highly beneficial due to their low metal use, i.e. maximum atom utilization efficiency. The different coordination numbers and unique electronic structures of SACs increase their catalytic activity in the reaction. In all cases, the nature of the catalyst's active sites and the reaction route are two important factors. Earlier studies have shown that CO2 hydrogenation to CH3OH is achieved via two different pathways: (i) the formate (HCOO) pathway and (ii) the carboxyl (COOH) pathway.41,42 However, several experimentalists have confirmed that carbon monoxide (*CO) and formaldehyde (*CH2O) are important intermediates, where *CO is obtained via the *COOH intermediate only.43,44 So, the CO2 hydrogenation reaction proceeds via the *COOH pathway followed by *CO and *CH2O. However, it is very difficult for experimentalists to identify all the intermediates. Hence, DFT studies can be very influential in understanding the underlying reaction mechanisms of such reactions.
Besides, ZnO based nanostructures are highly abundant and non-toxic. In addition, various ZnO based nanostructures (such as nanocombs, nanosprings, nanorings, nanobows, nanobelts, nanocages and so on) have been synthesised very easily.45,46 Most of these nanostructures are also found to be promising for the hydrogenation reaction and more often than not, they have been identified as an efficient catalyst compared to their bulk catalysts.47,48 Recently, Wu et al. have synthesised (ZnO)n (n = 1–15) based nanostructures.49 Dmytruk et al. have shown that the (ZnO)34, (ZnO)60 and (ZnO)70 nanostructures are highly stable.50 Furthermore, Tian and his co-workers have described that the (ZnO)60 nanocluster is composed of a (ZnO)12 based structure where (ZnO)12 acts as a basic unit for the formation of ZnO based nanocage (NC) structures.51 Thus, we have considered Zn12O12 and Zn24O24 based NCs (Fig. 1) for our study. Between these two NCs, the energetically most stable NC has been considered for the CO2 hydrogenation reaction. The main objective of this study is to understand the catalytic activity of a single Cu-atom based catalyst compared to the previously reported Cu-bulk and Cu-nanostructure based catalysts. Therefore, we have investigated the CO2 hydrogenation reaction over the Cu doped ZnO system and then compared our results with the previously reported Cu-bulk and Cu-nanostructure based catalysts (Cu(111), Cu ML, and Cu NCs). The most favourable pathways for the CO2 hydrogenation reaction have been identified to gain insights into the catalytic activity of the single Cu-atom based catalyst compared to the previously reported catalytic systems.
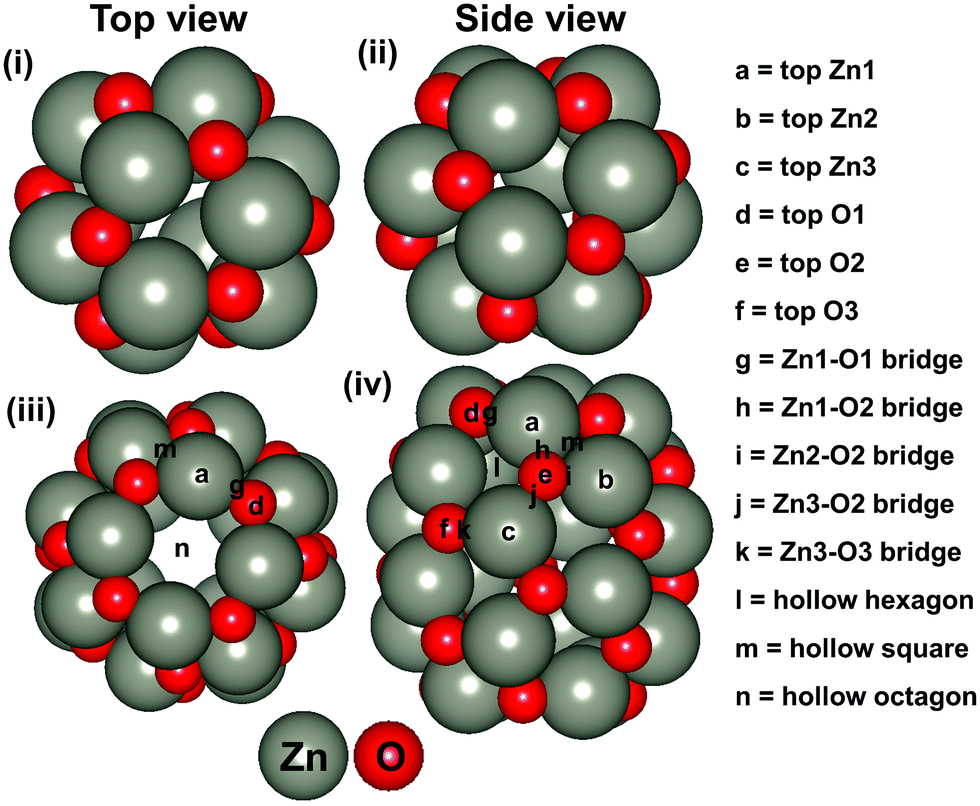 | ||
| Fig. 1 Top and side views of the considered NCs: (i and ii) Zn12O12 and (iii and iv) Zn24O24. | ||
2. Computational details
All the density functional theory (DFT) calculations have been performed using the Vienna ab initio simulation package (VASP) through the projector augmented wave (PAW) method.52–54 The generalized gradient approximation (GGA) for the Perdew–Burke–Ernzerhof (PBE) functional has been used for the description of the exchange–correlation interaction.55,56 The Grimme's D3 semi-empirical dispersion correction has also been included to account for the long-range dispersion forces.57 More than 15 Å of vacuum space has been considered along all three directions to avoid two successive periodic image interactions. We have calculated the energies of the NCs with 3 × 3 × 3 and 1 × 1 × 1 gamma centred k-points and found that the difference in energy is negligible (≈0.0009 eV). Hence, the plane-wave basis cutoff energy has been set to 470 eV with a 1 × 1 × 1 gamma centred k-point. All the structures have been optimised until the electronic energies and forces became less than 1 × 10−4 eV and 0.02 eV Å−1, respectively. The adsorption energy (Ead) of the intermediates has been calculated using the following equation:| Ead = ENC + adsorbate − (ENC + Eadsorbate) |
| ΔG = ΔE + ΔZPE − TΔS |
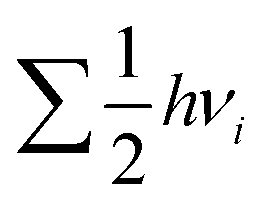 term, where h and νi denote the Planck's constant and the vibrational frequencies of the intermediates, respectively. All the calculations have been performed at 0 K temperature (T = 0 K). So, the TΔS term is zero. Therefore, the reaction energy (ΔE0) reported in the manuscript is actually the ZPE corrected reaction energy (ΔE + ΔZPE). On the other hand, if the considered temperature is non-zero, entropy will have contributions from translation, rotational and vibrational degrees of freedom of each atom. In general, translation and rotational entropies are negligible for intermediates adsorbed on solid material-based catalysts whereas the vibrational entropy contribution can be calculated using the following statistical thermodynamics equation:59
term, where h and νi denote the Planck's constant and the vibrational frequencies of the intermediates, respectively. All the calculations have been performed at 0 K temperature (T = 0 K). So, the TΔS term is zero. Therefore, the reaction energy (ΔE0) reported in the manuscript is actually the ZPE corrected reaction energy (ΔE + ΔZPE). On the other hand, if the considered temperature is non-zero, entropy will have contributions from translation, rotational and vibrational degrees of freedom of each atom. In general, translation and rotational entropies are negligible for intermediates adsorbed on solid material-based catalysts whereas the vibrational entropy contribution can be calculated using the following statistical thermodynamics equation:59In the above equation, Svib is the vibrational entropy, kB is the Boltzmann constant, T is the temperature and ħ = h/2π. From the above equation, it is clear that the vibrational entropy will be less for solid-state materials, i.e. the change in entropy can be neglected during reaction energy calculations. Moreover, in the presence of an external potential (U), the chemical potential of the reaction shifts by −eU where e is the elementary potential charge of the considered step.60 Besides, vibrational frequencies have been used for the characterisation of the reaction intermediates. Furthermore, we have calculated Bader atomic charges of some important intermediates using Henkelman code with the near-grid algorithm refine-edge method for the comparison of the adsorption energies of intermediates.61,62 Hereafter, all the adsorbed intermediates in the manuscript have been represented by an asterisk sign (*).
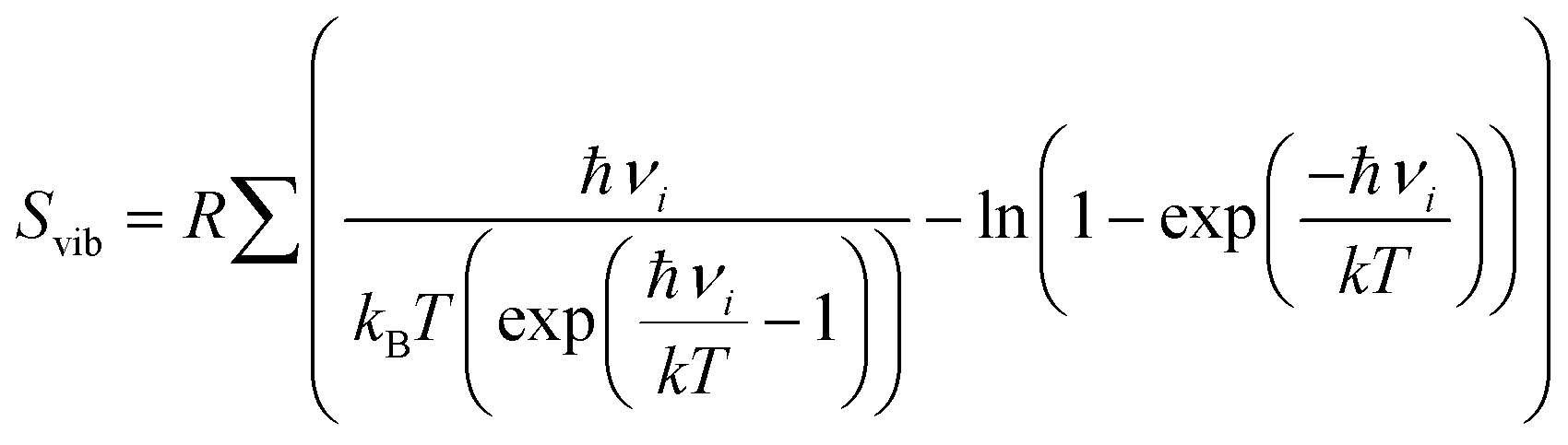
3. Results and discussion
In the beginning of this section, we have checked the stability of the considered NCs through energetic, dynamic and thermal stability calculations. Based on the stability calculations, we have considered the most stable NC for the CO2 hydrogenation reaction. We have compared our results with earlier reports to understand the catalytic activity of the NC.3.1. Stability of the NCs
The stability of the considered NC based structures has been explored in various ways. In this context, we have considered the energetic stability of Zn12O12 and Zn24O24 NCs through the calculation of formation and cohesive energies, which are listed in Table 1. Our calculated formation energy values indicate that the formation of the Zn24O24 NC is 0.2 eV more favourable compared to the formation of the Zn12O12 NC. Similarly, the cohesive energy values also show that the Zn24O24 NC is more stable compared to the Zn12O12 NC. For this reason, we have considered the Zn24O24 NC for further studies. Moreover, it is worth mentioning that the formation energy of the bulk ZnO has been calculated and found to be −2.96 eV per formula unit, which is comparable to the previously reported −3.04 eV per formula unit.63 Therefore, the level of theory used in this study is good enough for further study.| ZnO systems | Formation energy (eV per formula unit) | Cohesive energy (eV per formula unit) |
|---|---|---|
| Zn12O12 | −1.70 | −6.39 |
| Zn24O24 | −1.90 | −6.59 |
| bulk ZnO | −2.96 (−3.04) | −7.64 (—) |
Next, the energetically stable Zn24O24 NC has been considered for further study. Hereafter, all the possible Zn sites of the Zn24O24 NC have been substituted with Cu atoms to understand the effect of Cu doping on ZnO for the CO2 hydrogenation reaction. The Zn24O24 NC has three different Zn sites that have been replaced by Cu atoms (Fig. 2a–f) and the energetically stable structure has been considered. Fig. 2a, b, c, d and e, f show the first-, second- and third-layer Cu doped structures, respectively. We have considered all the three possible Zn sites for Cu doping in the modelled ZnO NC. The calculated total energies show that the Cu doping at the top layer is 0.02 and 0.19 eV more stable compared to the doping in the second and third layer, respectively. Moreover, we have noticed that the doping with the Cu atom in the ZnO NC does not change the skeleton of the nanocage, which could be due to the similar atomic radius of Cu and Zn atoms (Cu: 1.28 and Zn: 1.34 Å). This could also be one of the reasons why Cu/ZnO/Al2O3 based catalysts have been found to be some of the best catalysts for such reactions. So, the first layer Cu doped ZnO NC has been considered for the CO2 hydrogenation reaction. From here on, the Zn24O24 NC and the most stable Cu doped Zn23CuO24 NC have been represented as ZnO NC and Cu@ZnO NC, respectively. Furthermore, dynamic stability calculations were performed for ZnO and Cu@ZnO using phonon calculation as implemented in VASP.64 Our phonon results show a small imaginary frequency up to 9i cm−1 for ZnO NC and up to 7i cm−1 for Cu@ZnO NC. Previous studies have reported that clusters showing very small imaginary frequencies can be considered as a dynamically stable structure.65
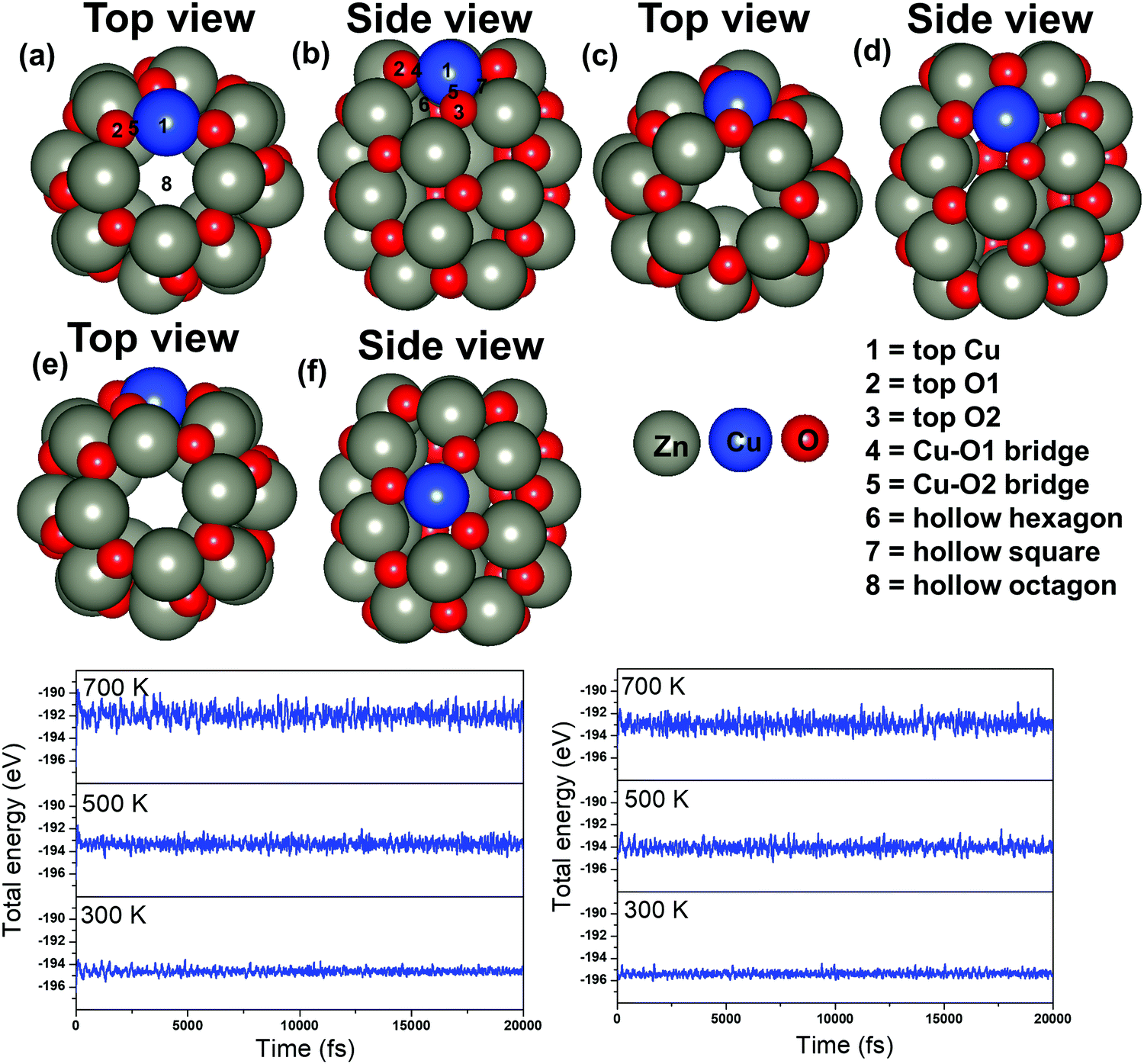 | ||
| Fig. 2 Top and side views of considered Cu doped ZnO NCs: (a and b) first layer Cu doped Zn24O24, (c and d) second layer Cu doped Zn24O24, and (e and f) third layer Cu doped Zn24O24. (g and h) Ab initio molecular dynamics simulations of (g) ZnO and (h) Cu@ZnO NCs at 300, 500 and 700 K. | ||
In general, CO2 hydrogenation reactions are carried out in the temperature range of 473–573 K.33 Hence, the catalyst must be stable in this temperature range. So, we have performed thermal stability calculations of ZnO and Cu@ZnO to find out the structural stability at the operational temperature. Ab initio molecular dynamics (AIMD) simulations with a Nosé thermostat model have been used to check the thermal stability of the considered NCs.66 The simulations have been performed with the NVT ensemble at temperatures of 300 K, 500 K and 700 K, with a time step of 1 femtosecond (fs) for 20 picoseconds (ps). Our simulations show that there are no significant changes in the energy throughout the simulation at 300 K for both the NCs (Fig. 2g and h). Also, at a temperature of 500 K and 700 K, the overall energy fluctuation is less for the considered NCs. So, the possibility of interconversion of ZnO and Cu@ZnO NCs into other local minimum energy structures is not possible within the 300–700 K temperature range. Thus, we can say that ZnO and Cu@ZnO NCs are thermally stable up to 700 K and can be used for the CO2 hydrogenation reaction.
3.2. Adsorption of different intermediates
The applicability of ZnO and Cu@ZnO NCs has been investigated in detail through the adsorption of different CO2 hydrogenation reaction intermediates on the NCs. We have considered all the possible adsorption sites on the ZnO NC (Fig. 1) and also adsorption sites close to the Cu atom on the Cu@ZnO NC (Fig. 2a and b). We have chosen adsorption sites close to the Cu atom to determine the role of the single atom catalyst in the catalytic activity. Therefore, all the possible top, bridge and hollow sites of the ZnO NC and Cu@ZnO have been taken into consideration. We could not compare our calculated results with the Cu/ZnO/Al2O3 catalyst as the previously reported values were not calculated using the same level of theory. For example, Liu and his co-workers theoretically investigated the CO2 hydrogenation reaction on a Cu/ZnO/Al2O3 based catalyst to support their experimental findings. However, they used the GGA/PW91 level of theory for their theoretical calculations.38 Therefore, we have compared our results only with the Cu-based catalytic systems that were calculated at the GGA/PBE level of theory. Therefore, the adsorption energies of the intermediates on ZnO and Cu@ZnO NCs have been compared with the adsorption energies of the intermediates on bulk Cu(111), hexagonal Cu(111) monolayer (Cu(111) ML), and a Cu nanocluster and compared our results with the synergistic effects of the Cu/ZnO based catalyst.38,67,68 The most stable adsorption patterns of the intermediates on ZnO and Cu@ZnO NCs are given in Fig. S1 and S2 (ESI†) and their respective adsorption energies are given in Table 2. We find that the most stable adsorption sites of the considered intermediates are different on both the NCs. Here, the first intermediate, i.e. the CO2 molecule, adsorbs strongly on both the NCs whereas CO2 interacts weakly with the extensively studied Cu based materials.67–69 Therefore, the considered NCs can be promising for the activation of CO2 molecules such that the following reaction steps are facilitated.| Adsorbates | ZnO NC | Cu@ZnO NC | Bulk Cu(111)67 | Cu(111) ML68 | Cu nanocluster67 |
|---|---|---|---|---|---|
| *CO2 | −3.27 (e) | −3.83 (2) | Not adsorbed | −0.21 | −1.14 |
| *COOH | −5.28 (i) | −2.45 (1) | −1.72 | −1.84 | −2.29 |
| *CO | −2.06 (g) | −2.05 (5) | −0.91 | −0.98 | −1.10 |
| *CHO | −4.63 (h) | −3.79 (2) | −1.44 | −1.57 | −2.21 |
| *COH | −6.86 (i) | −7.80 (4) | −2.89 | −2.97 | −3.10 |
| *CHOH | −4.99 (h) | −6.56 (7) | −2.00 | −2.41 | −2.35 |
| *CH2O | −3.79 (i) | −3.27 (5) | −0.04 | −0.35 | −1.66 |
| *CH2OH | −3.48 (h) | −3.28 (2) | −1.24 | −1.54 | −1.85 |
| *CH3O | −2.77 (l) | −1.92 (1) | −2.41 | −2.62 | −2.88 |
| *CH3OH | −0.87 (b) | −0.78 (1) | −0.12 | −0.39 | −0.49 |
| *CH2 | −5.02 (h) | −4.82 (5) | −3.37 | −3.58 | −3.87 |
| *H2O | −0.71 (a) | −0.35 (1) | −0.16 | −0.33 | −0.38 |
| *H | −3.13 (e) | −3.48 (2) | −2.50 | −3.52 | −2.71 |
| *O | −3.28 (n) | −4.07 (8) | −4.79 | −5.26 | −5.21 |
| *OH | −2.28 (n) | −3.06 (8) | −3.10 | −3.83 | −3.84 |
Besides, we find that the calculated adsorption energies of the considered intermediates are higher on the ZnO and Cu@ZnO NCs compared to those on bulk Cu(111), Cu(111) ML, Cu nanocluster and Cu–ZnO based catalysts.38,67,68 Furthermore, our calculated adsorption energy values show that the *COOH intermediate adsorbs strongly on the ZnO NC compared to that on the Cu@ZnO NC, whereas the adsorption energies of the other considered intermediates are almost the same on ZnO and Cu@ZnO NCs. To understand the reason behind the strong *COOH adsorption on ZnO NC compared to that on Cu@ZnO NC, we have calculated the Bader atomic charges, which showed that 0.03 |e| charge is transferred from the Zn atom of ZnO NC to the O of *COOH, whereas 0.06 |e| charge is gained by the O atom of ZnO NC from the C atom of *COOH. Moreover, there is no charge transfer from Cu of Cu@ZnO NC to the C atom of *COOH, whereas 0.05 |e| charge is gained by the O atom of Cu@ZnO NC from the H atom of *COOH. The amount of transferred charge is more in the case of ZnO NC compared to Cu@ZnO NC. So, the adsorption energy of the *COOH intermediate is higher on ZnO NC compared to Cu@ZnO NC. However, intermediates *COOH, *CO, *CHO, *CH2O, *CH2OH, *CH3O, *CH3OH, *CH2 and *H2O adsorb more strongly on ZnO NC compared to Cu@ZnO NC whereas intermediates *CO2, *COH, *CHOH, *H, *O and *OH adsorb more strongly on Cu@ZnO NC compared to ZnO NC. On the other hand, the adsorption energies of the products should be lower so that they can be removed easily from the catalytic surface for further steps. In our study, *CH3OH and *CH2 are two expected products and the calculated adsorption energy values of the *CH3OH and *CH2 intermediates are −0.87 eV and −5.02 eV on ZnO NC whereas −0.78 eV and −4.82 eV are the adsorption energies on Cu@ZnO NC. As a result, the removal of *CH3OH is easier compared to *CH2.
3.3. CO2 hydrogenation reaction mechanism
In this section, we have considered different CO2 hydrogenation reaction pathways that are discussed in detail with their calculated ZPE corrected reaction energies. Here, Scheme 1 represents the possible reaction pathways with their ZPE corrected reaction energies whereas Tables S1 and S2 (ESI†) give the details of the energy.At first, the CO2 molecule has been examined for the adsorption on ZnO and Cu@ZnO NCs. Our calculated ZPE corrected reaction energies for the CO2 adsorption steps are found to be exergonic. The calculated ZPE corrected reaction energies are −0.36 and −0.42 eV for ZnO and Cu@ZnO NCs, respectively. However, the ZPE corrected reaction energy values show that the CO2 adsorption is almost the same on both NCs. This may be due to the similar adsorption energy of CO2 on the NCs. In the next step, adsorbed CO2 can be reduced to *CO via direct and/or indirect reduction pathways. However, earlier reports have suggested that the direct *CO2 reduction to *CO is unfavourable, whereas indirect *CO2 reduction to *CO is favourable via the *COOH intermediate. Therefore, we have studied the indirect CO2 reduction procedure.
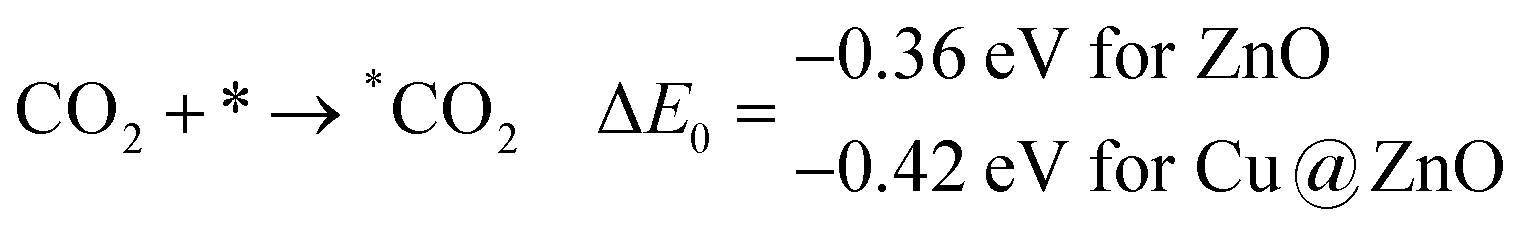
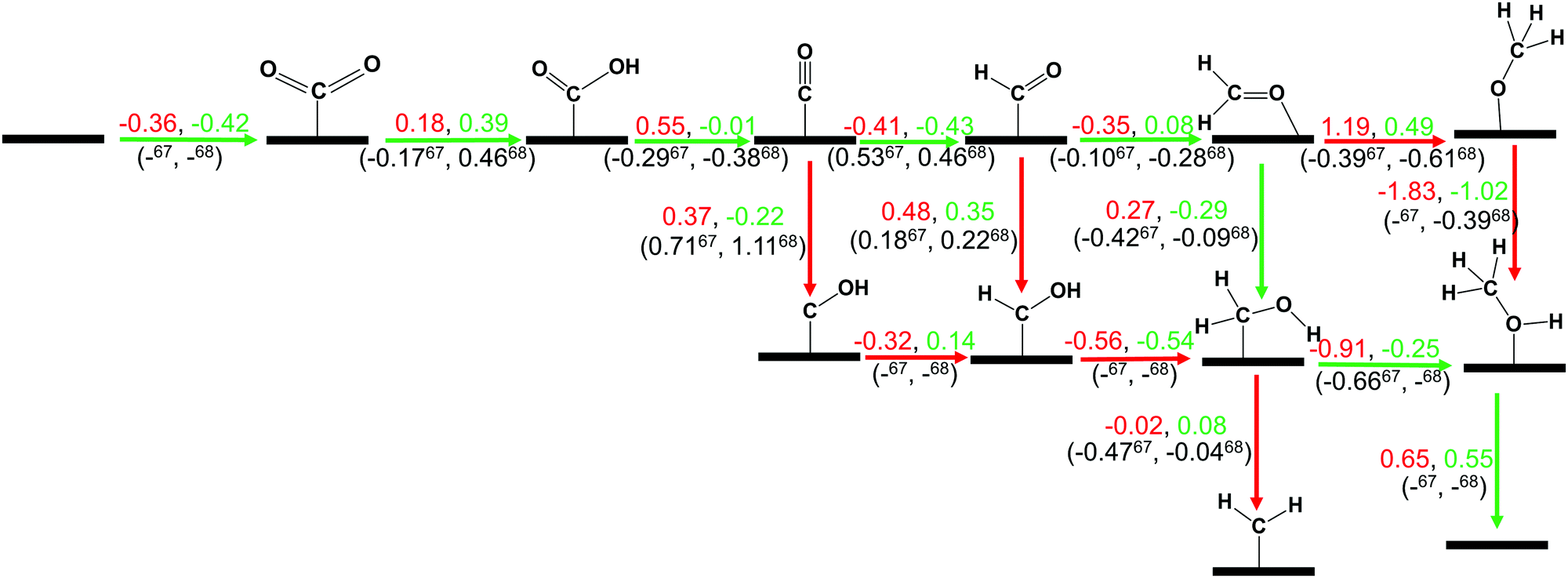 | ||
| Scheme 1 The calculated ZPE corrected reaction energies (eV) for electrochemical CO2 hydrogenation reaction on ZnO (red) and Cu@ZnO (green) NCs. Our calculated ZPE corrected reaction energies are compared with those of the previous reports on the Cu nanocluster and Cu(111) ML.67,68 Here, (–) means that the energy is not available for this step. The green arrow shows the most favourable pathway for the CO2 hydrogenation reaction to CH3OH. | ||
In the indirect mechanism, *CO2 reacts with the adsorbed/free H atom for the hydrogenation, and forms *COOH via weakening of one of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds. This hydrogenation step is highly dependent on the proton source and transfer mechanism of the proton. In most of the cases, hydrogen can react via the Langmuir–Hinshelwood (LH) or Eley–Rideal (ER) type of mechanism. In general, CO2 hydrogenation reaction proceeds via the Langmuir–Hinshelwood (LH) mechanism.67–69 Therefore, we have considered the Langmuir–Hinshelwood mechanism for the *CO2 hydrogenation to *COOH.
O bonds. This hydrogenation step is highly dependent on the proton source and transfer mechanism of the proton. In most of the cases, hydrogen can react via the Langmuir–Hinshelwood (LH) or Eley–Rideal (ER) type of mechanism. In general, CO2 hydrogenation reaction proceeds via the Langmuir–Hinshelwood (LH) mechanism.67–69 Therefore, we have considered the Langmuir–Hinshelwood mechanism for the *CO2 hydrogenation to *COOH.
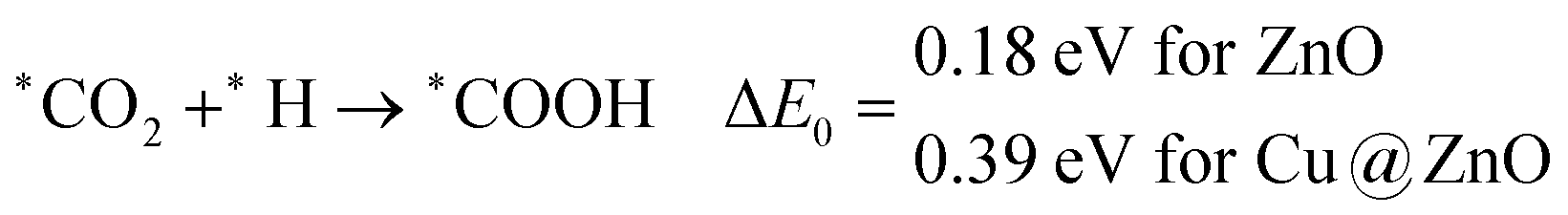
In most of the cases, the formation of *CO from *COOH is a downhill process. In this case, the calculated ZPE corrected reaction energies are endergonic compared to those of the Cu NC and Cu(111) ML-based catalysts for the formation of *CO from *COOH.67,68 This may be due to the strong adsorption energy of *COOH on ZnO NC compared to Cu@ZnO NC, which makes the step endergonic for ZnO NC and reversible for Cu@ZnO. Furthermore, the intermediate *CO can be hydrogenated to *CHO and *COH via hydrogenation at the C and O centres of *CO. Earlier reports have concluded that *CHO forms CH3OH whereas *COH forms CH4.67–70 Hence, this step is highly important for selective product formation (CH3OH vs. CH4).
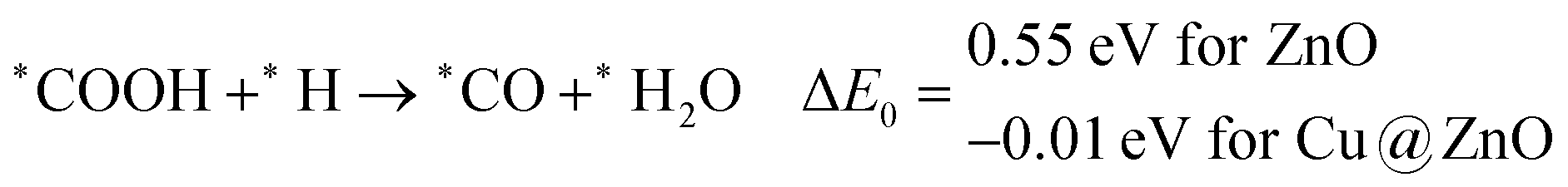
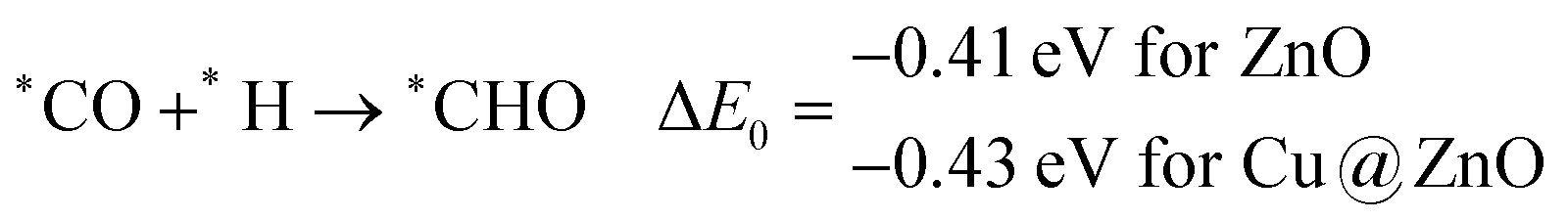
The calculated ZPE corrected reaction energies are −0.41 and −0.43 eV on ZnO and Cu@ZnO NCs, respectively, for the formation of the *CHO intermediate. Besides, the calculated ZPE corrected reaction energies for *COH formation are 0.37 and −0.22 eV on ZnO and Cu@ZnO NCs, respectively. Therefore, the formation of the *CHO intermediate is highly favourable compared to the formation of the *COH intermediate on both the NCs. Thus, we have considered *CHO as an important intermediate for our further study. Furthermore, *CHO can be converted to *CH2O and *CHOH through hydrogenation at the C and O centres of *CHO, respectively.
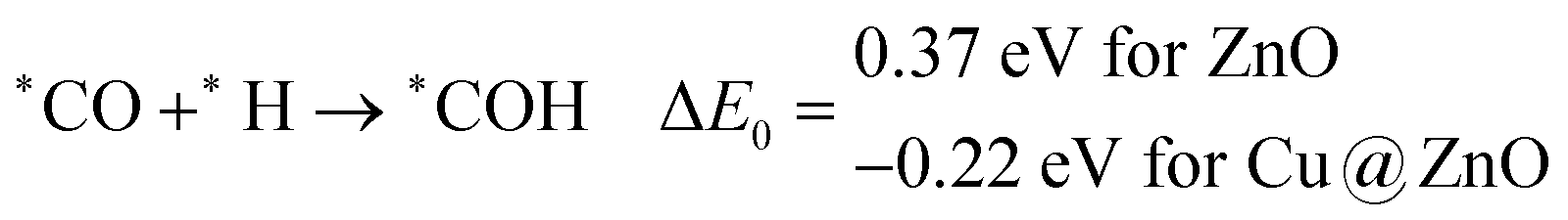
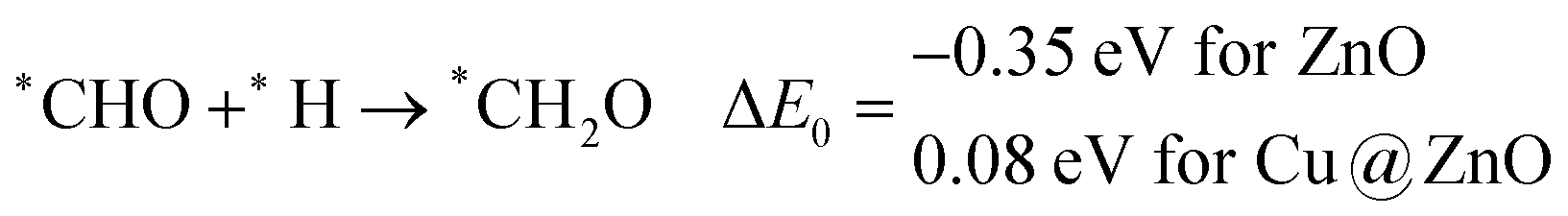
Our calculated ZPE corrected reaction energies suggest that the formation of *CH2O is 0.83 and 0.27 eV more favourable compared to the formation of *CHOH on ZnO and Cu@ZnO NCs, respectively. The calculated ZPE corrected reaction energies reveal that *CH2O formation is exergonic on ZnO NC and endergonic on Cu@ZnO NC. Therefore, the reaction will proceed via the *CH2O intermediate, which is in agreement with the earlier reports.67–70 Afterwards, *CH2O can form *CH2OH and *CH3O via hydrogenation at the O and C centres of *CH2O for the formation of O–H and C–H bonds, respectively.
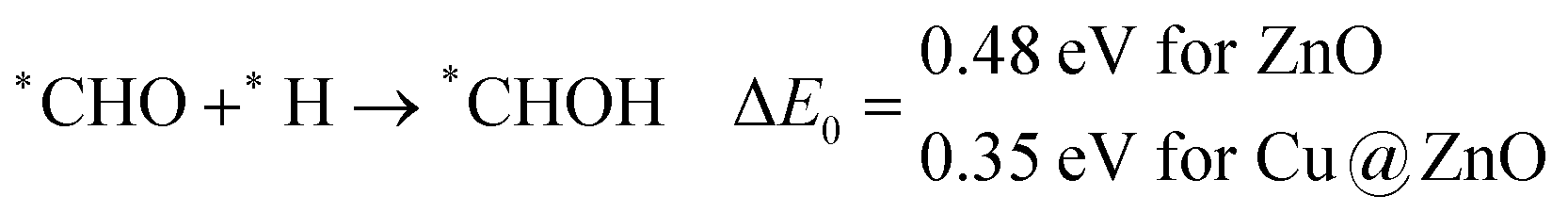
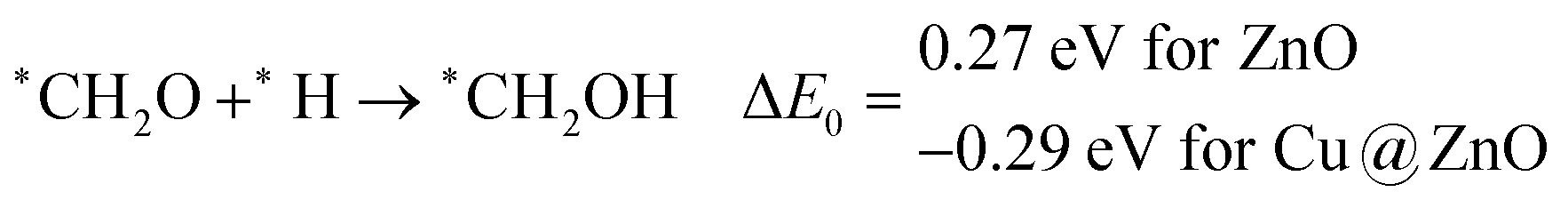
In this case, the formation of *CH2OH is 0.92 and 0.78 eV more favourable on ZnO and Cu@ZnO NCs compared to *CH3O. Besides, it is also clear that the formation of the O–H bond is 0.56 eV more favourable on ZnO NC compared to Cu@ZnO NC. Earlier reports have suggested that the formation of *CH3O is more favourable on the Cu(111) ML, whereas the formation of *CH2OH is more favourable on the Cu nanocluster.67,68 However, our calculated ZPE corrected reaction energies show that Cu@ZnO NC can be considered as an efficient catalyst for *CH2OH formation.
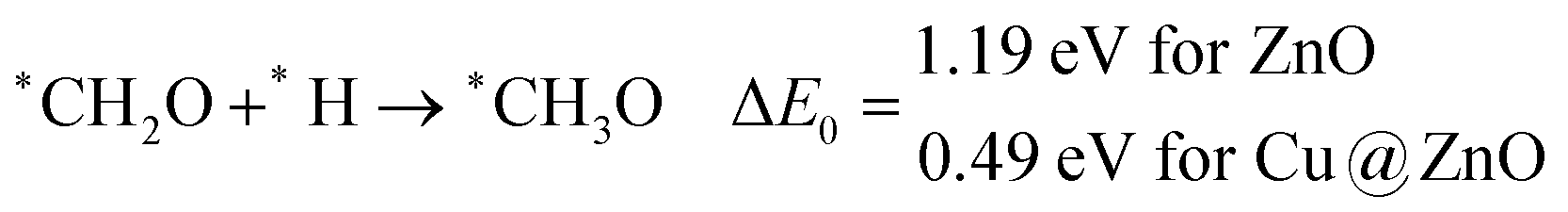
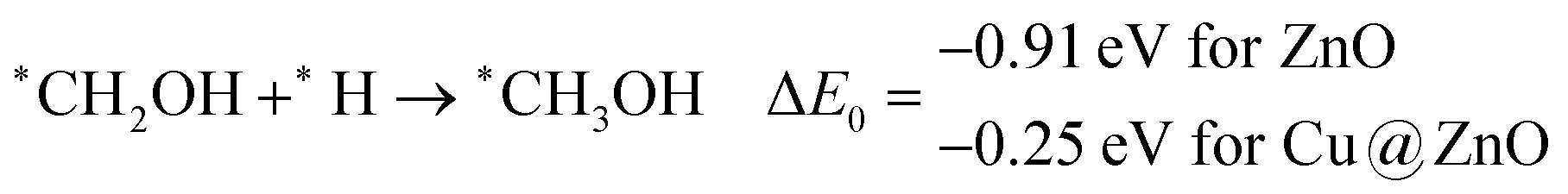
These steps are highly important for the formation of selective CO2 hydrogenated products such as CH3OH and CH4. Here, CH2OH may react with *H at the C and O centre of *CH2OH for the formation of *CH3OH via hydrogenation at the C centre of *CH2OH or *CH2 and *H2O via cleavage of the C–O bond, followed by O–H bond formation. If the formation of *CH2 is favourable, the final product will be CH4. The calculated ZPE corrected reaction energies show that the formation of the *CH3OH intermediate is highly exergonic on both the NCs and formation of the *CH2 intermediate is reversible on ZnO NC and endergonic on Cu@ZnO NC. Moreover, *CH3OH formation on ZnO NC is 0.66 eV more favourable compared to Cu@ZnO NC. This can also be explained by the adsorption energies of *CH3OH and *CH2. Therefore, the formation of CH4 is not favourable on both the considered NCs.
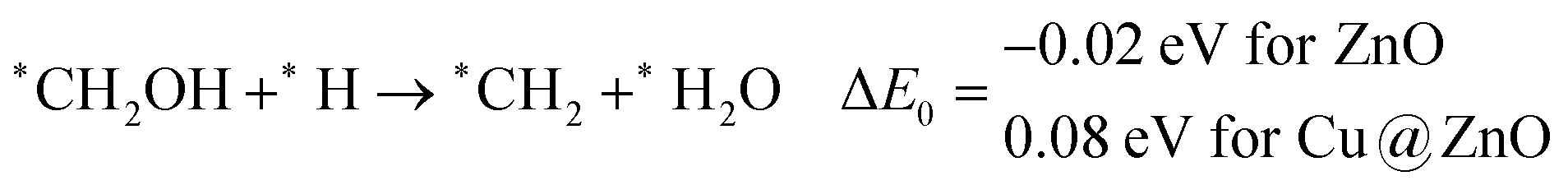
In the last step, *CH3OH desorbs from the catalytic surface and NCs are regenerated for the next catalytic cycle. The calculated ZPE corrected reaction energies for this desorption step are 0.65 and 0.55 eV on ZnO and Cu@ZnO NCs, respectively. Hence, the desorption of the *CH3OH intermediate is 0.10 eV more favourable on Cu@ZnO NC compared to ZnO NC.
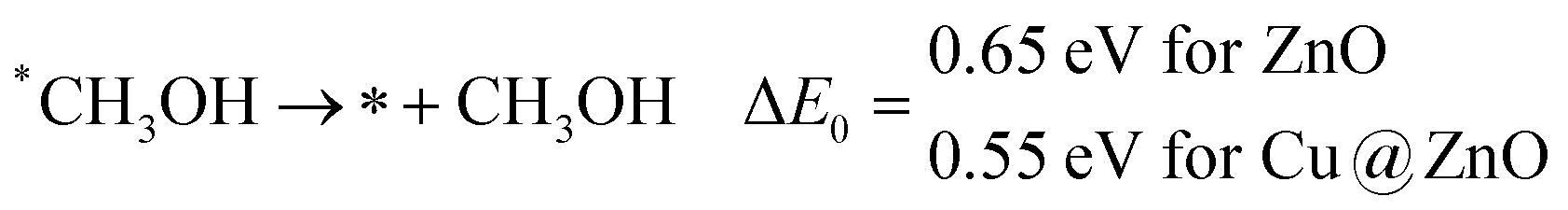
Thus, our calculated ZPE corrected reaction energy values suggest that ZnO and Cu@ZnO NCs can be considered as highly active and selective catalysts for CH3OH formation. Moreover, the CO2 hydrogenation reaction to CH3OH on ZnO and Cu@ZnO NCs proceeds via * → *CO2 → *COOH → *CO → *CHO → *CH2O → *CH2OH → *CH3OH → CH3OH. Here, the *CO2 + *H → *COOH, *CHO + *H → *CH2O and *CH2OH + *H → *CH3OH steps are favourable on ZnO NC whereas the * + CO2 → *CO2, *COOH + *H → *CO + *H2O, *CO + *H → *CHO, *CH2O + *H → *CH2OH and *CH3OH → * + CH3OH steps are favourable on Cu@ZnO NC. In this study, we have calculated the Bader atomic charges of Zn and Cu atoms in ZnO and Cu@ZnO NCs to understand the role of the single atom catalyst. Our calculated Bader atomic charges show that the charge on the Zn atom of ZnO NC is +1.16 |e| whereas the charge on the Cu atom of Cu@ZnO NC is +0.90 |e|. Therefore, the Zn atom in ZnO NC possesses a high positive charge compared to the Cu atom in Cu@ZnO. So, this indicates that the single Cu atom is available for the CO2 reduction reaction compared to the Zn site as Cu can be oxidised easily compared to the Zn atom. Therefore, Cu doping of ZnO NC increases the catalytic activity, which could be due to the synergistic effects between the metal atoms. Similarly, Wang and his co-workers have reported that synergetic effects between Cu and zinc oxides are responsible for efficient CO2 hydrogenation to methanol.71 Similarly, many other previous studies have shown that a synergic effect plays an important role in high methanol production.37,38
3.4. Comparison between ZnO and Cu@ZnO NCs
Our considered ZnO and Cu@ZnO NCs can selectively hydrogenate CO2 for the formation of CH3OH. In this context, the most endergonic elementary step of the reaction is the potential limiting step.60 These potential limiting steps are highly important to find out the applied electrode potential of the reaction. On the basis of the energies obtained above, Fig. 3 presents the calculated ZPE corrected reaction energies of the intermediates and their dependence on the applied electrode potential. Considering the electrochemical steps of CO2 hydrogenation reaction to CH3OH, the formation of *CO from *COOH is the potential limiting step (Fig. 3) for ZnO NC and *COOH formation from *CO2 is the potential limiting step for Cu@ZnO NC.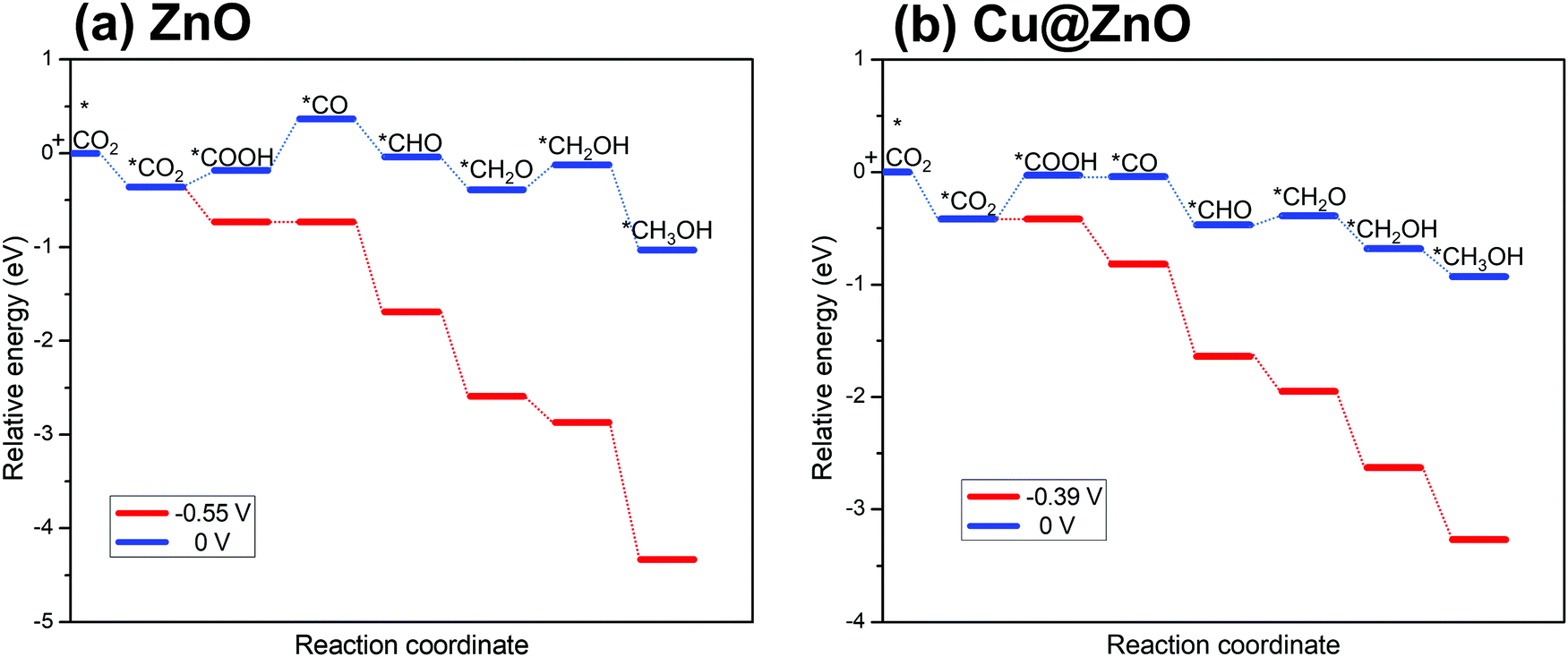 | ||
| Fig. 3 Relative CO2 hydrogenation ZPE corrected reaction energies and their dependence on the applied electrode potential for (a) ZnO NC, and (b) Cu@ZnO NC. | ||
The calculated ZPE corrected reaction energies of the considered potential limiting steps are 0.55 and 0.39 eV on ZnO and Cu@ZnO NCs, respectively. Therefore, at applied potentials of 0.55 V for ZnO NC and 0.39 V for Cu@ZnO NC, all the CO2 hydrogenation reaction steps become exergonic/reversible. Hence, Cu@ZnO NC requires 0.16 V less potential compared to ZnO NC for CH3OH formation. Our earlier reports have concluded that *CO to *CHO is the potential limiting step for a Cu nanocluster with an applied electrode potential of 0.53 V whereas *CO2 to *COOH and *CO to *CHO are the potential limiting steps for Cu(111) ML with an applied electrode potential of 0.46 V.67,68 Besides, the bulk Cu(111) surface has been reported to have an applied electrode potential of 0.71 V.69 Therefore, the calculated results show that Cu@ZnO NC can be a very active catalyst for CH3OH formation compared to the previously reported bulk Cu(111), Cu nanocluster, Cu(111) ML, ZnO NC and Cu@ZnO NC. Besides, the catalytic performance of ZnO NC is comparable with that of the Cu nanocluster.
In this study, we have considered intermediate coverages of ZnO and Cu@ZnO based NCs. We have focused on the coverage study of *COOH and *COH intermediates on ZnO NC, and *COH and *CHOH intermediates on Cu@ZnO NC based on their respective adsorption energy values. Here, the average adsorption energies of the important intermediates were calculated at different coverages and the calculated adsorption energies are summarised in Table 3. Our calculated results show that the average adsorption energy value mostly reduces at high coverage. However, the adsorption energy values are still significant at high surface coverage, which indicates that the catalytic activity of the NCs may not change significantly due to surface coverage. The skeleton of the NCs also remains the same even at high surface coverage, i.e. there is no surface reconstruction. This indicates that the attraction between Zn and O atoms is quite strong in the NC for the CO2 hydrogenation reaction even at high surface coverage. Therefore, the NCs can be used as an active catalyst for CO2 hydrogenation reaction even at high surface coverage.
| Coverages (ML) on ZnO NC | Average adsorption energy (eV) | Coverages (ML) on Cu@ZnO NC | Average adsorption energy (eV) | ||
|---|---|---|---|---|---|
| *COOH | 0.042 | −5.28 | *COH | 0.042 | −7.80 |
| 0.083 | −4.72 | 0.083 | −6.83 | ||
| 0.125 | −5.40 | 0.125 | −6.95 | ||
| 0.167 | −5.41 | 0.167 | −4.89 | ||
| *COH | 0.042 | −6.86 | *CHOH | 0.042 | −6.56 |
| 0.083 | −4.04 | 0.083 | −5.44 | ||
| 0.125 | −5.38 | 0.125 | −4.95 | ||
| 0.167 | −4.02 | 0.167 | −4.84 | ||
4. Conclusion
In conclusion, DFT calculations have been performed on the CO2 hydrogenation reaction to CH3OH on ZnO and Cu@ZnO NCs. Different adsorption possibilities of all the considered intermediates and various possible reaction pathways have been considered to understand the catalytic activity of ZnO and Cu@ZnO NCs. Our calculated adsorption energy values show that the considered intermediates bind strongly on both the NCs. However, intermediate *COOH binds strongly on ZnO NC compared to Cu@ZnO NC. Moreover, *CHO formation is favourable over *COH on both the considered NCs for selective CO2 hydrogenation to CH3OH via a *CHO intermediate. Our calculated ZPE corrected reaction energy values show that *CHO formation is 0.78 and 0.21 eV more favourable over *COH on ZnO and Cu@ZnO NCs, respectively. Hence, the catalysts are selective for CH3OH formation. Furthermore, the considered reaction mechanisms show that *CO2 hydrogenation to *COOH is the potential limiting step for ZnO NC, whereas formation of *CO from *COOH is the potential limiting step for Cu@ZnO NC. Here, the calculated applied electrode potential to make all the steps exergonic and/or reversible is 0.55 and 0.39 V for ZnO and Cu@ZnO NCs, respectively, which is lower compared to earlier Cu based catalysts. Moreover, the adsorption energy values are still significant even at high surface coverage. Therefore, the NCs can be used as an efficient catalyst at high coverage. Therefore, our detailed mechanistic study shows that ZnO and Cu@ZnO NCs can be efficient and selective catalysts for the CO2 hydrogenation reaction to CH3OH. Moreover, Cu@ZnO NC was found to be a more promising catalyst compared to ZnO NC.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank IIT Indore for the lab and computing facilities. This work is supported by DST-SERB [Project Number: CRG/2018/001131] and SPARC [Project Number: SPARC/2018-2019/P116/SL]. S. C. M. thanks MHRD for the research fellowship.References
- T. R. Knutson and R. E. Tuleya, J. Clim., 2004, 17, 3477–3495 CrossRef.
- J. Hansen, M. Sato, R. Ruedy, K. Lo, D. W. Lea and M. Medina-Elizade, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14288–14293 CrossRef CAS.
- X. Zhang, X. Zhu, L. Lin, S. Yao, M. Zhang, X. Liu, X. Wang, Y.-W. Li, C. Shi and D. Ma, ACS Catal., 2017, 7, 912–918 CrossRef CAS.
- K. Tsuchiya, J.-D. Huang and K.-I. Tominaga, ACS Catal., 2013, 3, 2865–2868 CrossRef CAS.
- S. C. Mandal, K. S. Rawat and B. Pathak, Phys. Chem. Chem. Phys., 2019, 21, 3932–3941 RSC.
- T. Maihom, S. Wannakao, B. Boekfa and J. Limtrakul, J. Phys. Chem. C, 2013, 117, 17650–17658 CrossRef CAS.
- A. M. Bahmanpour, A. Hoadley and A. Tanksale, Green Chem., 2015, 17, 3500–3507 RSC.
- S. C. Mandal, K. S. Rawat, S. Nandi and B. Pathak, Catal. Sci. Technol., 2019, 9, 1867–1878 RSC.
- T. Toyao, S. Kayamori, Z. Maeno, S. M. A. H. Siddiki and K.-I. Shimizu, ACS Catal., 2019, 9, 8187–8196 CrossRef CAS.
- H. Bahruji, R. D. Armstrong, J. R. Esquius, W. Jones, M. Bowker and G. J. Hutchings, Ind. Eng. Chem. Res., 2018, 57, 6821–6829 CrossRef CAS.
- W. Li, H. Wang, X. Jiang, J. Zhu, Z. Liu, X. Guo and C. Song, RSC Adv., 2018, 8, 7651–7669 RSC.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- C. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS.
- X. Xiaoding and J. A. Moulijn, Energy Fuels, 1996, 10, 305–325 CrossRef CAS.
- M. D. Porosoff, B. Yan and J. G. Chen, Energy Environ. Sci., 2016, 9, 62–73 RSC.
- G. A. Olah, T. Mathew and G. K. S. Prakash, J. Am. Chem. Soc., 2017, 139, 566–570 CrossRef CAS.
- J. Ye, C. Liu, D. Mei and Q. Ge, ACS Catal., 2013, 3, 1296–1306 CrossRef CAS.
- C. V. Miguel, M. A. Soria, A. Mendes and L. M. J. Madeira, J. Nat. Gas Sci. Eng., 2015, 22, 1–8 CrossRef CAS.
- G. Liu, D. Willcox, M. Garland and H. H. Kung, J. Catal., 1984, 90, 139–146 CrossRef CAS.
- L. Torrente-Murciano, D. Mattia, M. D. Jones and P. K. Plucinski, J. CO2 Util., 2014, 6, 34–39 CrossRef CAS.
- J. Zhong, X. Yang, Z. Wu, B. Liang, Y. Huang and T. Zhang, Chem. Soc. Rev., 2020, 49, 1385–1413 RSC.
- K. W. Ting, T. Toyao, S. M. A. H. Siddiki and K.-I. Shimizu, ACS Catal., 2019, 9, 3685–3693 CrossRef CAS.
- R.-P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell, Q. Li, M. Fan and Y.-G. Yao, Nat. Commun., 2019, 10, 5698 CrossRef CAS.
- S. Kar, R. Sen, A. Goeppert and G. K. S. Prakash, J. Am. Chem. Soc., 2018, 140, 1580–1583 CrossRef CAS.
- J. Kothandaraman, A. Goeppert, M. Czaun, G. A. Olah and G. K. S. Prakash, J. Am. Chem. Soc., 2016, 138, 778–781 CrossRef CAS.
- S. Kar, A. Goeppert, J. Kothandaraman and G. K. S. Prakash, ACS Catal., 2017, 7, 6347–6351 CrossRef CAS.
- E. Lam, K. Larmier, P. Wolf, S. Tada, O. V. Safonova and C. Coperet, J. Am. Chem. Soc., 2018, 140, 10530–10535 CrossRef CAS.
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, J. Am. Chem. Soc., 2016, 138, 12440–12450 CrossRef CAS.
- C. Liu, B. Yang, E. Tyo, S. Seifert, J. DeBartolo, B. von Issendorff, P. Zapol, S. Vajda and L. A. Curtiss, J. Am. Chem. Soc., 2015, 137, 8676–8679 CrossRef CAS.
- S. Tada, S. Kayamori, T. Honma, H. Kamei, A. Nariyuki, K. Kon, T. Toyao, K. Shimizu and S. Satokawa, ACS Catal., 2018, 8, 7809–7819 CrossRef CAS.
- X. Nie, X. Jiang, H. Wang, W. Luo, M. J. Janik, Y. Chen, X. Guo and C. Song, ACS Catal., 2018, 8, 4873–4892 CrossRef CAS.
- S. G. Jadhav, P. D. Vaidya, B. M. Bhanage and J. B. Joshi, Chem. Eng. Res. Des., 2014, 92, 2557–2567 CrossRef CAS.
- K. C. Waugh, Catal. Today, 1992, 15, 51–75 CrossRef CAS.
- G. C. Chinchen, P. J. Denny, D. G. Parker, M. S. Spencer and D. A. Whan, Appl. Catal., 1987, 30, 333–338 CrossRef CAS.
- T. Lunkenbein, J. Schumann, M. Behrens, R. Schlçgl and M. G. Willinger, Angew. Chem., Int. Ed., 2015, 54, 4544–4548 CrossRef CAS.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893–897 CrossRef CAS.
- S. Kuld, M. Thorhauge, H. Falsig, C. F. Elkjær, S. Helveg, I. Chorkendorff and J. Sehested, Science, 2016, 352, 969–974 CrossRef CAS.
- S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 355, 1296–1299 CrossRef CAS.
- S. Back, J. Lim, N.-Y. Kim, Y.-H. Kim and Y. Jung, Chem. Sci., 2017, 8, 1090–1096 RSC.
- Z. Zhao and G. Lu, J. Phys. Chem. C, 2019, 123, 4380–4387 CrossRef CAS.
- H. Nakano, I. Nakamura, T. Fujitani and J. Nakamura, J. Phys. Chem. B, 2001, 105, 1355–1365 CrossRef CAS.
- I. Nakamura, H. Nakano, T. Fujitani, T. Uchijima and J. Nakamura, J. Vac. Sci. Technol., A, 1999, 17, 1592–1595 CrossRef CAS.
- W. Wang, Z. Qu, L. Song and Q. Fu, J. Catal., 2020, 382, 129–140 CrossRef CAS.
- G. C. Chinchen, K. C. Waugh and D. A. Whan, Appl. Catal., 1986, 25, 101–107 CrossRef CAS.
- Z. L. Wang, Mater. Today, 2004, 7, 26–33 CrossRef CAS.
- Ü. Özgür, Y. I. Alivov, C. Liu, A. Teke, M. A. Reshchikov, S. Doğan, V. Avrutin, S.-J. Cho and H. Morkoç, J. Appl. Phys., 2005, 98, 041301 CrossRef.
- A. L. Dent and R. J. Kokes, J. Phys. Chem., 1969, 73, 3772–3780 CrossRef CAS.
- X. Liu, L. Ye, S. Liu, Y. Li and X. Ji, Sci. Rep., 2016, 6, 38474 CrossRef CAS.
- S. Wu, N. Yuan, H. Xu, X. Wang and Z. A. Schelly, Nanotechnology, 2006, 17, 4713–4718 CrossRef CAS.
- A. Dmytruk, I. Dmitruk, Y. Shynkarenko, R. Belosludovc and A. Kasuyad, RSC Adv., 2017, 7, 21933–21942 RSC.
- S. Zhang, Y. Zhang, S. Huang, H. Liu, P. Wang and H. Tian, J. Mater. Chem., 2011, 21, 16905–16910 RSC.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- D. A. McQuarrie, Statistical Mechanics, Harper & Row, 1973 Search PubMed.
- M. Karamad, H. A. Hansen, J. Rossmeisl and J. K. Nørskov, ACS Catal., 2015, 5, 4075–4081 CrossRef CAS.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, J. Comput. Chem., 2007, 28, 899–908 CrossRef CAS.
- D. Q. Fang and R. O. Zhang, J. Appl. Phys., 2011, 109, 044306 CrossRef.
- S. Baroni, P. Giannozzi and A. Testa, Phys. Rev. Lett., 1987, 58, 1861–1864 CrossRef CAS.
- A. Mahata, P. Garg, K. S. Rawat, P. Bhauriyal and B. Pathak, J. Mater. Chem. A, 2017, 5, 5303–5313 RSC.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- K. S. Rawat, A. Mahata and B. Pathak, J. Catal., 2017, 349, 118–127 CrossRef CAS.
- S. C. Mandal, K. S. Rawat, P. Garg and B. Pathak, ACS Appl. Nano Mater., 2019, 2, 7686–7695 CrossRef CAS.
- X. Nie, W. Luo, M. J. Janik and A. Asthagiri, J. Catal., 2014, 312, 108–122 CrossRef CAS.
- X. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Angew. Chem., Int. Ed., 2013, 52, 2459–2462 CrossRef CAS.
- Y. Wang, S. Kattel, W. Gao, K. Li, P. Liu, J. G. Chen and H. Wang, Nat. Commun., 2019, 10, 1166 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00208a |
| This journal is © The Royal Society of Chemistry 2020 |