
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDefect induced “super mop” like behaviour of Eu3+-doped hierarchical Bi2SiO5 nanoparticles for improved catalytic and adsorptive behaviour†
Debashrita
Sarkar
,
Sagar
Ganguli
 ,
Athma E.
Praveen
and
Venkataramanan
Mahalingam
*
,
Athma E.
Praveen
and
Venkataramanan
Mahalingam
*
Nanomaterials Research Lab, Department of Chemical Sciences, IISER Kolkata, Mohanpur Campus, Nadia, West Bengal 741246, India. E-mail: mvenkataramanan@yahoo.com
First published on 28th July 2020
Abstract
Development of “super-mops”, i.e., materials that are able to rapidly adsorb and degrade pollutants from water bodies, is a necessity for sustaining the marine ecosystem. But, most materials can either adsorb or degrade pollutants leading to a sluggish pollutant removal process. With the aim of developing a “super-mop”, we have synthesized Eu3+ incorporated Bi2SiO5 with high surface area. Eu3+ incorporation led to the rapid formation and stabilization of orthorhombic/monoclinic phase Bi2SiO5 by inducing asymmetry in the structure at significantly lower temperatures compared to earlier reports. Furthermore, partial reduction of Eu3+ to Eu2+ due to reaction conditions resulted in defects in the system by creation of oxygen deficient regions on the surface. These promoted the self-assembly of the initially formed nanoflakes into hierarchical microflowers. The as-synthesized materials, particularly after 1.5 percent Eu3+ incorporation, showed high efficiency in the removal of a wide range of pollutants from aqueous media through the adsorption process with an initial rate as high as 3.25 mg g−1 min−1. To illustrate, the material was able to adsorb >60 percent of pollutants within the first five minutes of contact with the aqueous solution. Eu3+ incorporation also significantly improved (∼5 times enhancement in rate constant) the photocatalytic activity of Bi2SiO5 by suppressing the degree of recombination as Eu3+ present in the material can effectively trap the photo-excited electrons to produce Eu2+, which thereafter rapidly de-traps to efficiently produce reactive radicals, and the oxygen defects, which are present in large number, acted as excellent sites for temporary trapping of electrons to generate the reactive radical species responsible for photocatalytic degradation.
Introduction
Direct discharge of effluents containing hazardous organic materials, such as dyes, pesticides, etc. by industries and farming communities is a major concern for the sustainability of marine ecosystems. In recent years, a lot of research is being pursued around the world for removing these pollutants.1,2 One of the research directions for this purpose is the development of porous materials that can adsorb the pollutants from water.3 For example, Mao et al. reported a one-step synthesis of a cationic poly(epichlorohydrin)-ethylenediamine hydrogel that has shown remarkable activity towards dye removal from aqueous solutions.4 Ayad et al. evaluated the dye adsorption capability of polyaniline nanotubes towards methylene blue adsorption and studied the corresponding physical processes in detail.5 Unfortunately, the majority of the research studies on the development of adsorbents focus only on improving the maximum adsorption capacity of materials. The other aspect that is often ignored but is of pivotal importance is the rate of removal, which must be very high for practical application purposes.6 Another major limitation of adsorbent materials is their possibility of subsequent leaching of the dye molecules.7 This demands the development of a “super-mop” material that would be able to rapidly adsorb as well as degrade the pollutants.In this context, inorganic semiconductor nanomaterials may be potential candidates due to their controllable morphology, high surface area, tuneable bandgap, dimensionality, etc. Through a prudent choice of reaction conditions, it is possible to prepare inorganic materials with superior microstructural properties that are capable of removing pollutants from aqueous solutions through an adsorption process.8 Furthermore, due to the semiconducting nature of these materials, they can also participate in photocatalysis reactions. In the presence of sunlight, excitons get produced in these materials and they subsequently produce reactive radical species with the ability to degrade the organic pollutants.9 The conversion of toxic dyes or pollutant molecules into relatively non-toxic intermediates and by-products via photocatalysis has been studied to validate the effectiveness of the photocatalytic degradation pathway.10,11 Doping of metal centres into inorganic nanomaterials, particularly in oxide-based nanostructures, has recently gained significant attention as the resulting materials often show improved photocatalytic performance due to the modulation of the band structure, band position, stabilization of metastable phase, etc.12–14 The structure of the photocatalyst also plays a key role in facilitating the course of the photocatalytic reaction.15 Among the diverse forms of structural variations reported so far, hierarchical nanostructures have shown efficacious activity over conventional structures. The improved photocatalytic behaviour is attributed to the acceleration of the reaction kinetics due to enhanced light absorption, high surface area and improved diffusion of the generated reactive ions.16,17
Among inorganic nanomaterials, bismuth silicate based semiconductor materials are particularly promising for dye removal through both adsorption and photocatalysis processes due to their high surface area. Bi2SiO5 (BSO) is one of the emerging lead free substitutes of ferroelectric as well as luminescent probes in thermometry.18–24 In particular, BSO is a potential material because of its layered structure made up by intergrowth of the [SiO3]2− pyroxene layer and [Bi2O2]2+ layer. The [Bi2O2]2+ layer is structurally built up of an oxygen bound square pyramidal layer with Bi3+ ions sitting at the top of the pyramid. The opposite directionality of the Bi3+ lone pairs and the O dipole generates an internal electric field that is able to separate the photo-induced exciton with enhanced efficiency.25 Furthermore, BSO is known to exist in different polymorphs, tetragonal, cubic, monoclinic, and orthorhombic.26 Among them, the monoclinic and orthorhombic phases would be able to efficiently separate the photo-generated electrons and holes leading to suppressed recombination because of their lower symmetry.27 As this results in an enhanced photocatalytic performance, the synthesis of monoclinic/orthorhombic BSO is actively pursued by researchers. However, due to its metastable character, the synthesis of pure monoclinic or orthorhombic phase BSO is a challenging step. The available reports show that high temperatures, long reaction times (∼48 hours), and tedious post-synthesis protocols have been employed.28–33 This has motivated us to develop a low-cost strategy to produce monoclinic or orthorhombic BSO with superior structural properties at lower reaction temperatures and time. Moreover, the as-synthesized BSO must be a “super-mop”, i.e. it should be able to remove pollutants rapidly from water via adsorption and subsequently degrade them effectively through a photocatalysis process.
In this study, we have developed pure phase monoclinic BSO, which is otherwise difficult to stabilise, through asymmetry generation by Eu3+-doping. Furthermore, the Eu3+-doped BSO (Eu-BSO) nanoparticles are able to demonstrate “super-mop” behaviour, i.e., they exhibit excellent adsorption as well as photocatalytic behaviour. Briefly, the slight doping of Eu3+ into the BSO matrix led to the emergence of a range of interesting changes. This encompasses asymmetric phase stabilization, formation of hierarchical self-assembled microstructures, higher surface area, pore size and pore volume, oxygen deficiency sites, redox couple, etc. Time dependent crystallisation of Eu-BSO was studied to understand the growth mechanism. Partial reduction of Eu3+ to Eu2+ took place due to the reaction conditions resulting in the generation of defect sites. Oxygen deficient regions, as defect sites, at the surface lead to the self-assembly of the initially formed nanoflakes into hierarchical microflowers with improved surface area as compared to BSO. The as-synthesized materials, particularly after 1.5 percent Eu3+ incorporation, showed high efficiency in the removal of a wide range of pollutants via adsorption and photocatalysis. The effectiveness of Eu3+-incorporation to enhance the rapid removal of a wide range of organic pollutants from aqueous solutions through adsorption and photocatalytic degradation processes compared to the available reports on BSO material validates it to be a promising strategy to develop “super-mop” materials for practical application purposes.
Results and discussion
Phase analysis and growth mechanism
The as-obtained BSO materials synthesized via solvothermal methods at different time intervals were subjected to PXRD analysis to obtain information regarding their phases as well as the growth mechanism. The material was found to first crystallize into a tetragonal phase in 2 hours (as shown in Fig. S1c, ESI†). With an increase in the reaction time (6 h), a peak at 2θ ∼ 12° was found to appear. The exact origin of this peak is debatable as this peak is reported to appear for tetragonal BSO with I4/mmm symmetry only upon heteroatom doping.20,25 Moreover, this is also present in the standard pattern of both monoclinic and orthorhombic phases of BSO. As in our case, this peak appeared for undoped BSO, and we believe that this indicates the formation of monoclinic/orthorhombic phases. Recently, the exact phase identification between monoclinic and orthorhombic BSO, particularly at room temperature, has received great attention. Taniguchi et al. have reported that the orthorhombic BSO is stable only at temperatures higher than 500 °C and with lower temperatures, the SiO4 units twist making 172.2° angle leading to the stabilization of the monoclinic crystal structure.18 Girard and co-workers have however suggested that it is difficult to ascertain the exact crystal phase as the monoclinic distortion is very small and is less than the uncertainties of both diffraction measurements and theoretical calculations.23 Kim and co-workers even calculated the energy difference between the two phases to be 23 meV f.u.−1, which is very small for interconversion.24 Therefore, though the new peak suggests the partial conversion of the tetragonal phase into a phase of relatively lower symmetry, its exact identification is very difficult. For consistency, we have referred to this new phase as monoclinic in the ensuing discussion as all characterization methods and experiments were carried out at room temperature. With the aim to synthesize pure monoclinic BSO due to its advantages in practical applications (discussed in the Introduction section), the hydrothermal reactions were carried out for longer times (8 h, 10 h and 24 h). However, pure phase monoclinic BSO remained unattainable at any of the reaction duration and resulted in mixed tetragonal and monoclinic phases. In fact, the material obtained after 24 h of reaction was found to consist of cubic Bi4Si3O12 along with its tetragonal and monoclinic counterparts thereby suggesting that the monoclinic phase was getting transformed into the cubic phase with increasing reaction duration (Fig. S1b, ESI†). Furthermore, such transformation into the cubic phase with increasing reaction time indicates the preference of the system to adopt a crystal structure of higher symmetry and underlines the requirement of introducing asymmetry-inducing factors for stabilizing the monoclinic phase.In this context, Eu3+ at various concentrations (0.5, 1, 1.5, and 2 percent with respect to Bi3+) was introduced into the reaction medium and the solvothermal reactions were carried out for different time intervals. Eu3+ was chosen because of its identical charge (III) but slightly smaller ionic radius (108.7 pm) than that of Bi3+ (117 pm), making it compatible for occupying the lattice positions of BSO and thereby inducing the desired asymmetry in the crystal structure. As shown in Fig. S1a (ESI†), all four characteristic peaks belonging to (200), (020), (002), and (022) of monoclinic BSO became prominent after 8 h and pure monoclinic BSO was obtained after 10 h of reaction for the material synthesized using 1.5% of Eu3+ w.r.t. Bi3+ (Eu1.5-BSO). A closer inspection of the PXRD patterns recorded for materials synthesized at lower reaction times (2 h, 6 h, etc.) indicates that the formation of the final monoclinic phase may be proceeding through the generation of some Bi2O3 polymorphs (probably cubic delta or tetragonal beta phases) and a small amount of silicate. Such an observation was earlier reported by Back et al.20 The peaks in PXRD for the other three materials (Eu0.5-BSO, Eu1-BSO, and Eu2-BSO) obtained after 10 h also correspond to pure monoclinic BSO without the presence of any impurity phase (Fig. 1).
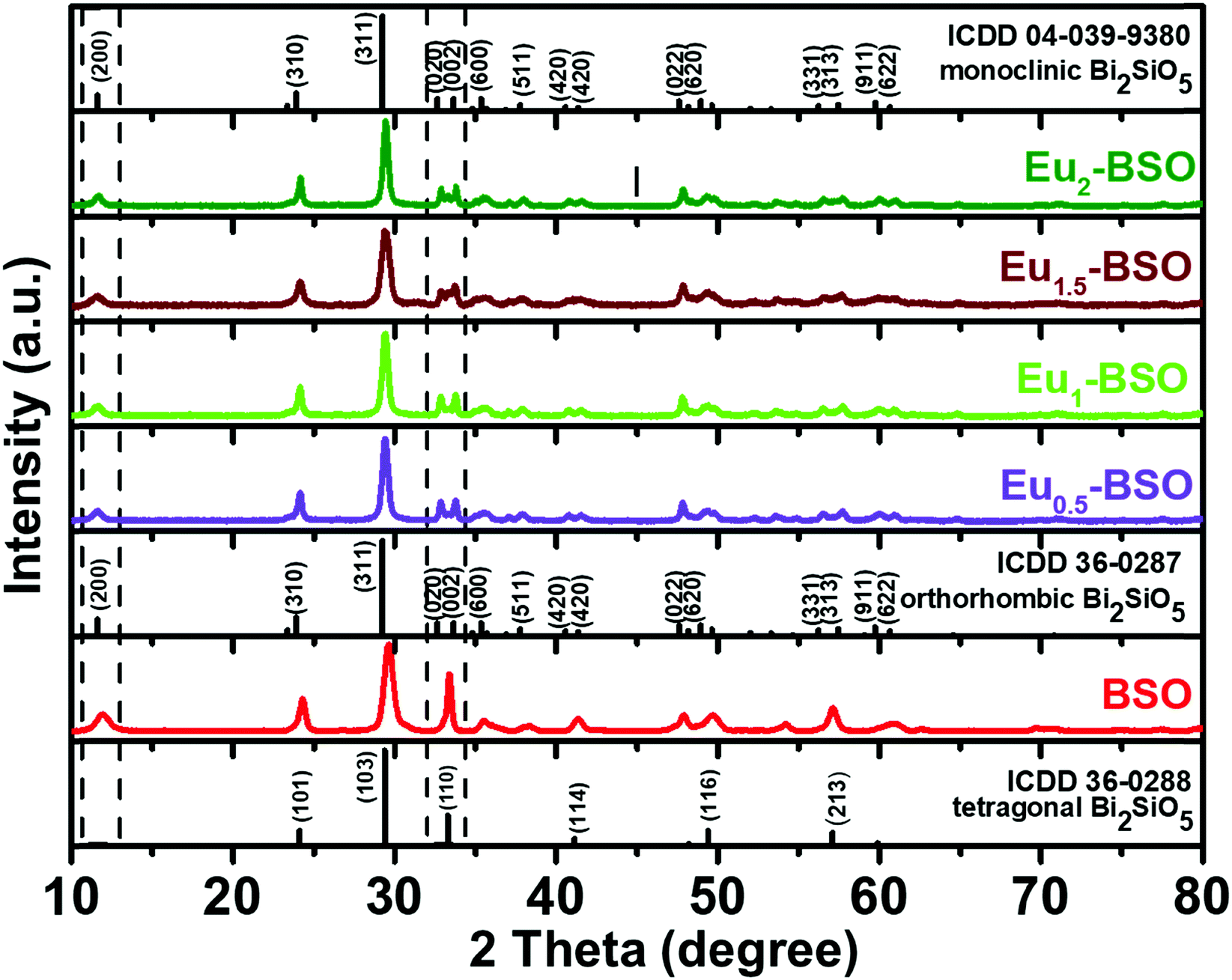 | ||
| Fig. 1 The PXRD pattern of Bi2SiO5 (BSO) and Eu3+-doped Bi2SiO5 (Eu-BSO) nanoparticles. | ||
Thereafter to further confirm the phase, the Rietveld refinement process was performed using GSASII software to determine the crystallographic information of Eu1.5-BSO nanoparticles. The monoclinic phase BSO (ICDD 04-039-9380) was used as the model for refinement. As shown in Fig. S2 (ESI†), the experimental, calculated, difference and Bragg's positions indicate that Eu1.5-BSO belongs to the monoclinic phase of BSO class of materials. The crystal structure parameters and fitting reliability of GOF 2.4% and Rwp 10.2% further confirm that doping Eu3+ into BSO stabilises the BSO structure in the pure phase monoclinic system as summarised in Table S1 (ESI†). We believe that such Eu3+ doping-induced faster phase transformation to stabilize the asymmetric crystal structure clearly indicates that Eu3+ doping has lowered the energy barrier for the transition from the tetragonal crystal system to a pure phase monoclinic system. To our knowledge, this is the first report where pure monoclinic BSO has been prepared within such a short duration and at such a low temperature.
FTIR analysis
To further understand the role of Eu3+ in stabilizing the monoclinic phase, FT-IR analysis of all materials synthesized after 10 h of solvothermal reactions was carried out (Fig. 2a). The obtained bands with corresponding bond vibrations are tabulated in the ESI† (Table S2). The most distinct feature observed after Eu3+ incorporation is the intensification of the peak around 945 cm−1. Zhu et al. have assigned this band to the stretching vibration mode of isolated (SiO5)6− groups forming a distorted tetrahedron.31,34 Thus, the intensification of this band in Eu-BSO compared to BSO strongly validates our claim that the incorporation of Eu3+ has induced asymmetry in the crystal structure leading to the faster generation and stabilization of the monoclinic BSO phase. | ||
| Fig. 2 (a) FT-IR spectra of BSO and Eu-BSO, XPS peaks of (b) O 1s for BSO (c) Eu 3d for Eu1.5-BSO and (d) O 1s for BSO. | ||
Elemental composition analysis
To quantify the exact amount of incorporated Eu3+ and its relative ratio to the amount of Bi3+ in the as-synthesized materials, ICP-AES and EDS analyses were performed and the obtained results are presented in Tables S3, S4 and Fig. S4 (ESI†). Both data suggest that the relative percentage of incorporated Eu3+ w.r.t. Bi3+ is in accordance with that of the corresponding reactant concentration. For example, the Eu3+ present in Eu1.5-BSO was found to be 1.4 percent w.r.t. Bi3+ from ICP-AES, respectively.Chemical state analysis
To understand the chemical nature of different elements in the as-synthesized materials, XPS analysis was performed for BSO and Eu1.5-BSO and the results are shown in Fig. S3 (ESI†) (calibrated to the C1s peak at 284.6 eV).35 For BSO, the peaks at 159.1 eV and 164.3 eV corresponding to the binding energies of Bi 4f7/2 and Bi 4f5/2 core levels, respectively, are attributed to Bi3+ in the short Bi–O bonds of Bi2SiO5 (Fig. S3a, ESI†).36 Moreover, the two smaller peaks at 163.0 and 157.9 eV can be ascribed to the complex formed between the free Bi3+ present on the surface with ethylene glycol used as the solvent during synthesis. The absence of any significant change in the observed binding energy of Bi3+ in Eu1.5-BSO suggests that the chemical nature around the Bi3+ ions remains the same even after Eu3+ doping (Fig. S3b, ESI†).The basis of peak separation of 5.2 eV is in agreement with the studies by Yu and co-workers where they observed a similar difference in binding energy for Bi 4f7/2 and Bi 4f5/2 levels.37 The peak at 101.2 eV in both materials corresponds to the presence of Si4+ and this stems from the Si–O bond of the silicate tetrahedra (Fig. S3c and d, ESI†). Interestingly, though Eu was found to primarily exist in its (+III) oxidation state as confirmed from the strong peaks at 1163.6 eV and 1133.96 eV, the presence of small amounts of Eu2+ was also evident from the appearance of its characteristic peaks at 1155.2 eV and 1125.8 eV (Fig. 2c).38 In one of our previous articles, we have observed that ethylene glycol is capable of reducing Eu3+ into Eu2+ at high temperatures.39 Therefore, we believe that the usage of ethylene glycol during synthesis is responsible for the partial reduction of Eu3+ into Eu2+ in the as-prepared Eu-BSO materials. The effect of the formation of Eu2+ is also reflected in the O 1s spectrum as shown in Fig. 2b and d. For BSO, a peak at a binding energy of 529.2 eV arises. Interestingly, for Eu1.5-BSO, while the lattice O peak appeared at 529.8 eV, a significant contribution in the overall spectrum was observed at 531.77 eV. In recent past, several research groups have attributed this to the O atoms present in the oxygen deficient regions in a material.40–42 The relative percentage of the oxygen vacancies was determined following the protocol developed by Wang et al. where the ratio of the area under the curves was found to be ∼10.3% w.r.t. lattice oxygen present in Eu1.5-BSO.43 Therefore, the presence of Eu2+ in the Eu1.5-BSO results in the formation of a higher number of oxygen deficiencies, thus causing defect sites in semiconducting BSO. We believe that this may be due to the necessity of overall charge neutralization in the final material.
Morphology analysis
The as-synthesized materials (BSO and Eu-BSO obtained after 10 h of reaction) were subjected to electron microscopy (FE-SEM and TEM) analyses to understand their morphologies. Both SEM and TEM images for BSO suggest the formation of nanoflakes like structures with an average length of 200 nm and an average width of 20–30 nm (Fig. 3 and Fig. S5, ESI†). The appearance of 0.263 nm lattice spacing in the HR-TEM image reveals the presence of the (002) plane associated with the monoclinic phase of BSO and this is further supported by the corresponding FFT pattern. Interestingly, for all Eu-BSO nanoparticles, a self-assembly of such nanoflakes via interweaving into hierarchical microspheres was observed.44 | ||
| Fig. 3 FE-SEM images of (a1 and a2) BSO nanoparticles, (b1 and b2) Eu0.5-BSO, (c1 and c2) Eu1-BSO, (d1 and d2) Eu1.5-BSO and (e) Eu2-BSO. | ||
Nanoplate to microflower formation
To better understand the evolution of respective morphologies, a time dependent microscopy analysis was carried out. For the precursor of BSO (0 min) as well as BSO (30 min), spherical particles with a diameter of ∼7–8 nm were found to assemble into irregular structures with a roughened surface (Fig. 4b). Interestingly, after 2 h, these rough and irregular structures were transformed into nanoflakes and were found to grow radially into relatively larger flakes. A similar observation of irregular assembly of small spherical particles was observed for the precursor of Eu1.5-BSO and Eu1.5-BSO (30 min). But, for the Eu1.5-BSO obtained from reactions carried out for longer times, self-assembly of nanoplates into hierarchical microspheres resembling that of the corresponding final product was observed (Fig. 4i–l). | ||
| Fig. 4 Time dependent evolution of (a–f) BSO nanoparticles depicting plate like structures; (g–l) Eu1.5-BSO depicting assembled structures. | ||
We believe that the tendency of the nanoplates to self-assemble only in Eu-doped materials arises from the relatively higher population of oxygen deficiencies due to partial reduction of Eu3+ into Eu2+ at the surface. To confirm this, Sm3+-doped BSO was prepared through a solvothermal method and was characterized to be phase pure monoclinic by PXRD analysis (Fig. S6a, ESI†). Sm3+ was chosen because it is known to strongly resist reduction into the Sm2+ state and therefore the generation of oxygen deficiencies at the surface would be minimum.45 FE-SEM analysis confirmed that while the material is plate-like in morphology, no self-assembly has taken place despite the presence of a lanthanide dopant (Fig. S6b, ESI†). Therefore, we propose strongly that oxygen-deficiency was the primary driving force for the self-assembly process. The oxygen-deficiency resulted in localized charges at the surface resulting in high surface energy and in order to lower the overall surface energy of the system, the nanoflakes self-assembled into hierarchical structures. Thereafter, all materials (BSO and Eu-BSO) were subjected to EDS mapping analysis and as shown in Fig. S7 (ESI†), Eu was found to be evenly distributed in the materials.
Surface area analysis
To quantitatively understand the porosity features of the as-synthesized materials, nitrogen adsorption–desorption analysis was performed. For BSO, the isotherm was type IV in nature with a H3 hysteresis loop characterizing the formation of slit-like pores from plate-like particles (Fig. S8, ESI†).46 Interestingly, the hysteresis in the isotherm for Eu0.5-BSO and Eu1.5-BSO corresponded to a combination of H3 and H4, which is typically symbolic for narrow slit-like pores having an irregular shape and broad size distribution. The surface area, average pore size and pore volume for all materials are presented in Table S5 (ESI†). While the BET surface area increased from 27 m2 g−1 for BSO to 70 m2 g−1 for Eu1.5-BSO, a sudden decrease to 18 m2 g−1 was observed for Eu2-BSO. We believe that the initial improvement is a result of the self-assembly of the nanoplates into microflowers as the latter consisted of higher number of pores due to their hierarchical structure. Despite similar hierarchy, the steep decrease of surface area in Eu2-BSO probably arose due to the substantial lowering of pore volume for this material when compared to others. We believe that such a decreased pore volume in the presence of a higher dopant concentration was a result of the “doping induced pore-collapse” phenomenon.47,48Optical property analysis
Upon 312 nm excitation, a broad emission peak centred at 470 nm was observed for all materials. This broad emission may be attributed to the bandgap emission from bismuth silicate.49 According to reports, the valence band of Bi2SiO5 was comprised of Bi 6s and O 2p orbitals, whereas the conduction band was comprised of Bi 6p orbitals. From Fig. S9a (ESI†), it is observed that the emission intensity of the Eu1.5-BSO nanoparticles is the least among the materials signifying a lower degree of radiative recombination. We believe that such lowering of recombination is primarily due to the trapping of the excitons in the defects evolved after Eu-doping. This also suggests the applicability of this material in applications that require charge extraction, such as photocatalysis.The presence of Eu3+ ions in Bi2SiO5 is advantageous as it is a suitable optical probe in understanding the crystallinity of the local environment. To confirm the presence of Eu3+-doping, photoluminescence spectra were recorded in solid-state by 396 nm excitation. The emission spectra of Eu-doped BSO nanoparticles display five bands around 580 nm, 595 nm, 616 nm, 653 nm and 703 nm wavelength regions corresponding to 5D0 → 7FJ=0–4, transitions in Eu3+, respectively (Fig. S9b, ESI†). The higher intensity of the hypersensitive 5D0 → 7F2 (616 nm) electric-dipole transition as compared to that of the 5D0 → 7F1 (595 nm) magnetic dipole transition indicates that the metal sites are of low symmetry and possess no inversion centre.50 It is interesting to note that while the emission bands corresponding to all the transitions from 5D0 → 7F1,2,3,4 increase in intensity up to Eu1.5-BSO, a decrease was observed for the Eu2-BSO. The observed decrease in intensity with an increase in Eu3+ is attributed to increased cross-relaxation and concentration quenching.51 To probe the presence of Eu2+ in BSO nanoparticles, the solid state materials were excited at a wavelength of 465 nm,52 and the corresponding photoluminescence spectra are shown in Fig. S10 (ESI†). A broad peak centred at 496 nm and 514 nm corresponding to the transition 4f65d1 → 4f7 has been observed corresponding to Eu2+.53–55 As the amount of Eu doping was very miniscule, the doping amount was increased to 2.5% to obtain a relatively higher intensity of emission. Furthermore, relatively weaker peaks at 595 nm, 616 nm, 653 nm and 703 nm, characteristic of Eu3+, were also observed in the photoluminescence spectra of the materials. The high intensity of peaks arising from Eu2+ compared to those arising from Eu2+ may be ascribed to the allowed inter-configuration nature of the transitions. In fact, the intensity ratio of the 496 nm peak belonging to Eu2+ and 714 nm belonging to Eu3+ was found to be highest in case of Eu1.5-BSO implying that the proportion of conversion of Eu3+ into Eu2+ is maximum in this material.
Band gap analysis
The band gap of all materials was determined using diffuse reflectance spectroscopy. Fig. S11a (ESI†) shows the absorption spectra of the different Eu-BSO nanomaterials. The Eu-BSO materials showed a blue-shift with an increase in the dopant amount. This may be either due to an increase in the Fermi energy of the conduction edge leading to band gap enhancement or due to reduction of particle size with an increase in Eu3+ doping.56 In general, band gaps of semiconductors are determined using Tauc plots according to the following formula| αhν = A(hν − Eg)2/n | (1) |
| F(R) = (1 − R)2/2R | (2) |
Band position analysis
The band positions of the undoped and doped BSO nanoparticles were determined using the Mott Schottky plot obtained at different frequencies as shown in Fig. S12 (ESI†). The analysis was performed thrice and the average potential and the standard deviation were calculated (Table S6, ESI†). The positive slope obtained for BSO and Eu-BSO suggests that the synthesised nanoparticles retain their n-type character even after doping.57 As shown in Fig. S12 (ESI†), the negative flat band potentials of −0.97 V, −1.02 V, −1.01 V, −0.61 V, and −0.61 V (vs. NHE) corresponding to BSO, Eu0.5-BSO, Eu1-BSO, Eu1.5-BSO, and Eu2-BSO were directly used as the conduction band (CB) potential and the band gap value calculated using the Kubelka–Munk plot was subsequently used to determine the valence band (VB) position of the nanoparticles. As shown in Fig. S13 (ESI†), the valence band potentials of BSO, Eu0.5-BSO, Eu1-BSO, Eu1.5-BSO, and Eu2-BSO were calculated to be 2.65 V, 2.61 V, 2.63 V, 3.22 V, and 3.19 V, respectively, using the formula:| Eg = CB − VB | (3) |
Water decontamination
The improved BET surface area, stabilized monoclinic phase, and lower degree of radiative recombination in Eu-BSO compared to BSO motivated us to employ the as-synthesized materials for water decontamination. First, the adsorption ability of the materials was explored.| qe = (C0 − Ce)V/m | (4) |
log(qe − qt) = log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) qe − (k1t/2.303) qe − (k1t/2.303) | (5) |
| t/qt = (1/k2qe2) + (t/qe) | (6) |
| qt = kit0.5 + C | (7) |
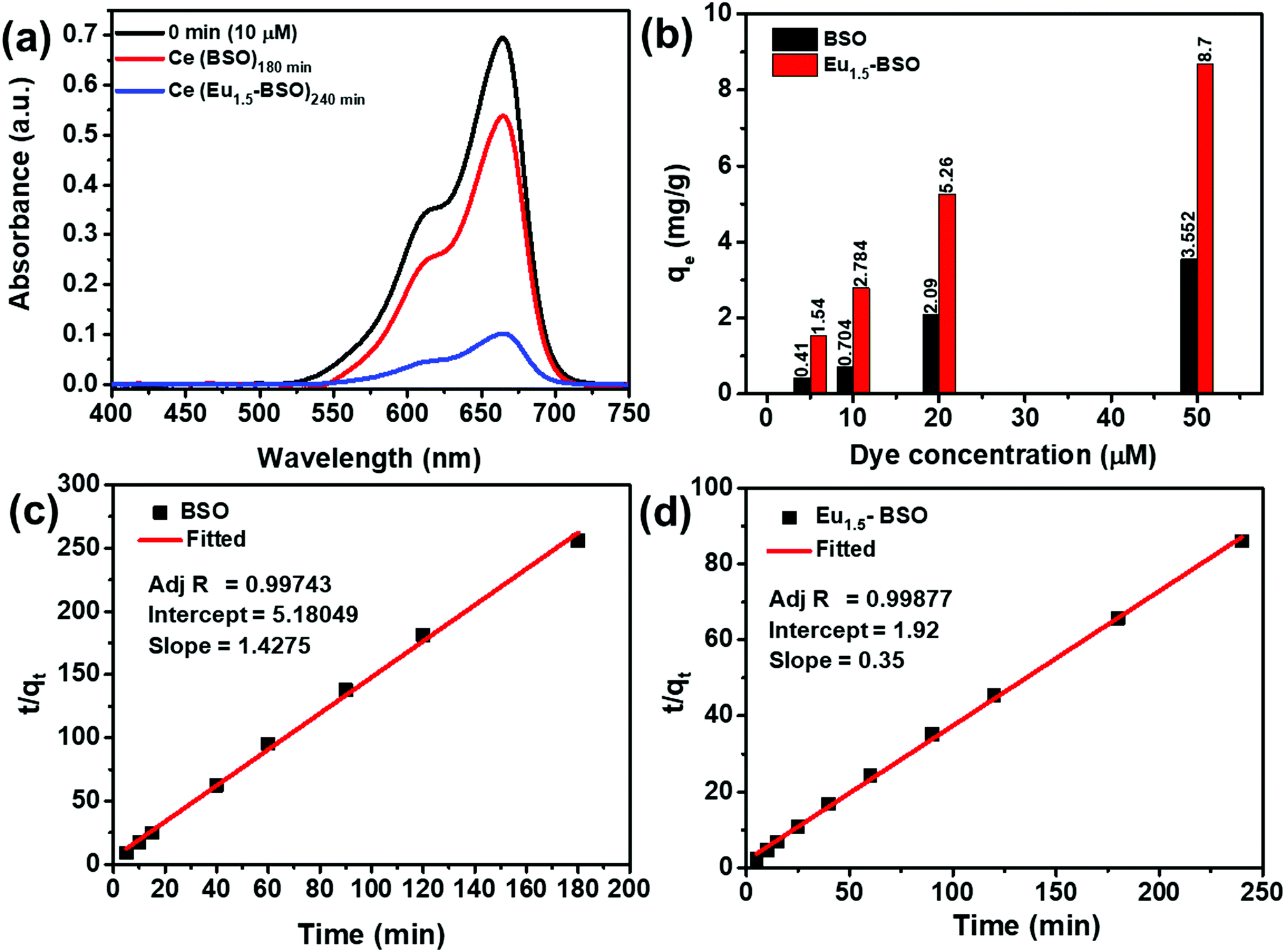 | ||
| Fig. 5 (a) Absorption spectra of methylene blue after attaining equilibrium adsorption by BSO and Eu1.5-BSO, respectively (b) relative adsorption rate for methylene blue. The pseudo-second order fitting for determining the kinetic rate of methylene blue adsorption by (c) BSO and (d) Eu1.5-BSO. | ||
It is clearly evident from Fig. 5c, d and Fig. S14 and S15 (ESI†) that the R2 of the pseudo-second order kinetic model is much higher than that of the other two models and therefore, we may conclude that the dye adsorption processes for both BSO and Eu1.5-BSO at different dye concentrations are primarily proceeding through the chemisorption process.15,58–60
As mentioned in the introduction section, the adsorption process must be fast for practical application purposes. In this context, the as-synthesized materials were found to be excellent candidates as majority of the pollutant removal took place within the first five minutes of contact period for both BSO and Eu1.5-BSO (Fig. S14, ESI†). This also gets reflected in the k2 values (Table S7, ESI†), which represent the time required to attain an equilibrium state. A higher k2 value indicates faster attainment of the equilibrium state.61 While the majority of recent articles report this value to be in the order of 10−3 or 10−4, the values shown by BSO and Eu1.5-BSO were found to be almost 2–3 orders higher in magnitude (∼10−2), thereby suggesting the ability of the as-synthesized materials to adsorb the pollutants quickly.
Moreover, the decrease in k2 values with an increase in the concentration of dyes indicates the dependence of attaining the equilibrium on the initial dye concentration and this is in accordance with the theoretical interpretations of this parameter.62,63 To our knowledge, the k2 values alone cannot present the true capability of the materials in terms of rate of dye removal. This is because, while in the first five minutes of contact in an aqueous solution of 10 μM methylene blue, BSO showing a k2 value of 39.3 × 10−2 g mg−1 min−1 removed ∼18 percent of the dye, and Eu1.5-BSO showing a k2 value of 6.5 × 10−2 g mg−1 min−1 removed ∼66 percent.
Thus, despite lower k2, Eu1.5-BSO was able to remove much higher amount of pollutant from aqueous solutions in the initial five minutes. In this context, the initial sorption rate (h = k2qe2) is a more appropriate parameter to represent the true potential of materials in faster removal of pollutants and was calculated for BSO and Eu1.5-BSO.64 As shown in Table S7 (ESI†), the h of Eu1.5-BSO was ∼2.46 and ∼2.41 times higher than that of BSO for 10 and 50 μM dye solutions, respectively. Upon comparing the values with literature reports (Table S8, ESI†), it is quite evident that Eu1.5-BSO with an h value of 3.25 mg g−1 min−1 is a promising material for faster dye removal from aqueous solutions. Thereafter, to understand the maximum adsorption capacity (qm) of BSO and Eu1.5-BSO as well as to garner further information regarding the interaction between the sorbate and the adsorbent, the obtained data were fitted into linear expressions of the Langmuir and Freundlich isotherms. The validity of the models was determined from the corresponding coefficient of determination (R2) values. As shown in Fig. S16 (ESI†), BSO and Eu1.5-BSO were found to follow the Freundlich and Langmuir isotherms, respectively. Moreover, the maximum adsorption capacity (qm) of Eu1.5-BSO was found to be ∼9.53 mg g−1 from the corresponding Langmuir isotherm analysis (Table S9, ESI†).
 | ||
| Fig. 6 (a) The photodegradation of methylene blue dye (50 μM) in the presence of BSO under a solar simulator. (b) Rate of degradation of methylene blue assuming first order kinetics (c) Recyclability study of the photocatalytic process with Eu1.5-BSO, and (d) photocatalytic experiments performed in the presence of active species scavengers. | ||
Upon calculating the rate of dye degradation of methylene blue using first order kinetics, the rate constant of Eu-BSO materials improved by ∼5 times compared to BSO (as shown in Fig. 6b). The above data clearly suggest that upon Eu3+ incorporation, not only the dye adsorption but also the dye degradation properties of the materials got significantly enhanced. Different amounts of methylene blue dye solution were studied to evaluate the effectiveness of Eu1.5-BSO in the degradation of contaminants. As depicted in Fig. S17 (ESI†), the relative adsorption is nearly 40% for 75 μM dye solution and upon light irradiation, the catalyst was able to remove ∼99% of the dye. The removal of pollutants and subsequent degradation efficiency of Eu1.5-BSO were further evaluated using aqueous solutions containing other common organic pollutants such as, rhodamine B (RhB), methyl orange (MO), chicago sky blue (CSB), rose Bengal (RBX) dye (Fig. S18 and S21a, ESI†).
These dyes were chosen as they are considered as the model cationic dye, anionic dye, neutral azo dye and neutral dye with attached halogen atoms. It was observed that the amount of dye degradation was 60, 55 and 80 percent for MO, CSB and RBX, respectively. The practical use of Eu1.5-BSO was further evaluated by studying its ability to degrade industrial and pharmaceutical wastes such as phenol and antibiotics. It was found to be able to remove ∼80% of phenol, ∼65% of tetracycline hydrochloride and ∼60% of chloramphenicol (as shown in Fig. S21b, ESI†), which are considered as leading causes of superbug problems65 associated with dumping of pharmaceutical waste in river bodies.
The stability of the Eu1.5-BSO as a photocatalyst was determined by carrying out recyclability studies and it was found to be able to retain ∼95 percent of its initial dye removal activity after five consecutive cycles (Fig. 6c). The slight reduction in the photocatalytic activity of Eu1.5-BSO can be attributed to the pore collapse phenomenon which is known to occur for oxide based semiconductors after annealing them at higher temperatures that was employed in this work between consecutive cycles for complete removal of dye molecules from the materials.66
To understand the reactive species that are primarily responsible for dye degradation, the photocatalytic experiments were performed in the presence of Na2EDTA, AgNO3, isopropanol, and Ar as the hole scavenger, electron scavenger, hydroxide ion scavenger, and superoxide ion scavenger, respectively. The impeded degradation in the presence of EDTA and Ar suggests that holes and superoxide ions are the active radicals involved (Fig. 6d). To further confirm this, photocatalytic reactions were performed in the presence of a different set of scavenging ions. Ammonium oxalate, p-benzoquinone, potassium persulphate, and tertiary butyl alcohol were employed as hole, superoxide, electron, and hydroxide radical scavengers, respectively (as shown in Fig. S22, ESI†). This also complements the earlier inference that holes are the primary radical species responsible for the degradation of dyes during photocatalysis closely followed by superoxide ions. This is because scavenger species such as Na2EDTA and (NH4)2C2O4 have a tendency to quickly form intermediates with holes to generate glyoxylic acid, iminodiacetic acid, etc., which does not allow the holes to take part in the degradation mechanism any further.67 Superoxide radicals are not able to carry out oxidation in the presence of benzoquinones, as the radical involves itself in a cyclic process of redox reactions. Similarly, the constant purging of Ar hinders the formation of superoxide ions, which hampers the degradation rate.68
Degradation mechanism
To understand the reaction mechanism, the rhodamine B solution at different time intervals (60 min, 120 min and 180 min) was collected during photocatalysis and subjected to ESI-MS analysis to identify the various intermediates. As shown in Fig. S19a (ESI†), the initial m/z denoted as −60 is referred to as the rhodamine solution without any catalyst. The peak at 443 is referred to as the rhodamine B molecule. After 60 min of light irradiation, the peaks observed at m/z values of 413, 387, and 359 are identified with de-ethylated fragments of rhodamine B. The peaks at m/z values of 274, 244, and 230 are various decarboxylated intermediates as observed at 120 min and 180 min, respectively (Fig. S19c and d, ESI†). It is interesting to note that with an increase in light irradiation, the intensity of the N-de-ethylated products further decreased. Based on the above observation, a probable fragmentation mechanism of rhodamine B is shown in Fig. S20 (ESI†) The intermediates identified at different time intervals are similar to the previously reported literature.69–72The effectiveness of the Eu1.5-BSO material towards the rapid removal and photocatalytic degradation of such a wide range of organic pollutants from aqueous solutions compared to the already available reports (Table S10, ESI†) clearly validates it to be a potent material for practical application purposes.
Conclusions
In conclusion, the incorporation of Eu3+ aided in the rapid formation and stabilization of Bi2SiO5 in its monoclinic phase by inducing asymmetry in the structure during material growth. Partial reduction of Eu3+ to Eu2+ due to reaction conditions resulted in the generation of defect sites with oxygen deficient regions on the surface. To lower the surface energy, the initially formed nanoflakes self-assembled into hierarchical microflowers with substantial improvement in surface area as compared to Bi2SiO5. The as-synthesized materials, particularly after Eu3+ incorporation showed high efficiency in adsorbing a wide range of pollutants from aqueous medium through the chemisorption process with an initial adsorption rate as high as 3.25 mg g−1 min−1. Further, Eu3+ incorporation was found to significantly improve the photocatalytic activity of Bi2SiO5 (∼5 times enhancement in rate constant). This was primarily due to the lower degree of recombination as (i) Eu3+ present in the material can effectively trap the photo-excited electrons to produce Eu2+, which thereafter rapidly de-traps and transfers the electrons to O2 adsorbed on the catalyst surface leading to the efficient production of reactive O2−˙ radicals, and (ii) the presence of higher oxygen deficient defects acted as excellent sites for temporary trapping of electrons to generate the reactive radical species responsible for photocatalytic degradation. The effectiveness of Eu3+-incorporation to enhance the rapid removal of such a wide range of organic pollutants from aqueous solutions through adsorption and photocatalytic degradation processes compared to the available reports obviously validate it to be a promising strategy to develop “super-mop” materials for practical application purposes.Experimental
Materials
Bismuth nitrate pentahydrate, ethylene glycol, concentrated monomethyl ether, concentrated nitric acid, and sodium hydroxide pellets were purchased from Merck. Sodium metasilicate, methylene blue, rhodamine B, Chicago sky blue, and rose Bengal dye were purchased from Loba chemicals. Europium oxide was purchased from Sigma Aldrich. Crystalline phenol was purchased from SRL chemicals. All chemicals were used without further purification.Synthesis of Bi2SiO5 (BSO) nanoparticles
Bismuth silicate nanoparticles (NPs) were synthesised via a solvothermal method. Initially, for the undoped samples, Bi(NO3)3·5H2O (2 mmol = 970.14 mg) was taken in a 100 ml glass beaker with 20 ml of ethylene glycol to dissolve the nitrate salts. After stirring the beaker on a magnetic stirrer for 20 min at room temperature, white crystals of (Bi(NO3)3) were dissolved. Following this, the sodium silicate solution (1 mmol = 284.2 mg dissolved in 20 ml) was added dropwise and stirred for another 30 min. This was succeeded by dropwise addition of 2 M solution of NaOH till the pH reaches 11. The white opaque basic solution was stirred for 30 min before transferring it to a Teflon based autoclave and heated at 200 °C for 10 h. After cooling, the resulting product was centrifuged 3–4 times with deionised water and ethanol, and dried overnight at 60 °C in an oven.Synthesis of Eu(x%) doped-Bi2SiO5 (Eux-BSO) nanoparticles
Eu3+ doped samples were prepared in the same manner. Typically for 0.5% of Eu3+ doping, 0.005 mmol of Eu2O3 was converted to nitrates by stirring and heating in HNO3. After that 1.99 mmol of Bi(NO)3·5H2O was dissolved in ethylene glycol. The europium nitrate solution dissolved in 20 ml of water was added to the bismuth nitrate solution. After stirring for 30 min, 1 mmol of sodium metasilicate dissolved in 20 ml of water was added dropwise to the solution containing nitrate solution of Bi3+ and Eu3+. After 30 min of stirring 2 M NaOH solution was added till the pH reaches 11. The whole dispersion was transferred to an autoclave vessel and heated at 200 °C for 10 h. After cooling, the solution was centrifuged 3–4 times with deionised water and ethanol and dried overnight at 60 °C in an oven. Similarly, 1.0%, 1.5%, and 2.0% Eu3+-doped Bi2SiO5 were prepared. The samples were abbreviated as BSO for Bi2SiO5 nanoparticles and Eux-BSO for x (=0.5, 1, 1.5, and 2)% Eu3+ doped Bi2SiO5 nanoparticles.Characterization
The crystallinity and phase analysis of nanoparticles was carried out using powder X-ray diffraction (PXRD) measurements using a Rigaku-Smartlab diffractometer with Cu-Kα 1.54059 radiation operating at 70 kV and 35 mA at a scanning rate of 1° min−1 in the range of 10–80°. The samples were completely well powdered and spread evenly on a quartz slide. The morphology of the nanoparticles was characterized by field emission scanning electron microscopy (FESEM). FESEM images were taken using a JEOL (JSM 6390 LV) instrument and the samples were coated with a thin film of gold in order to avoid charging effects prior to loading of the samples into the chamber. EDS was performed on a SUPRA 55-VP instrument with patented GEMINI column technology. The surface area measurement was carried out with a Micromeritics Gemini VII surface area analyser. The nitrogen adsorption–desorption isotherms were reported by BJH (Barret-Joyner-Halenda) surface/volume mesopore analysis. Each sample was degassed at 150 °C for 12 h. Fourier transform infrared studies were performed with a PerkinElmer Spectrum RX1 with KBr pellets. Diffuse reflectance UV-vis spectra (DRS) of the samples were recorded with a Varian Cary 100 spectrophotometer equipped with a diffuse reflectance accessory in the region of 250–500 nm, with barium sulphate as the reference. The reflectance spectra were converted into Kubelka–Munk function [F(R)] which is proportional to the absorption coefficient for low values of F(R). The PL spectra were recorded on a Horiba Fluoromax 4 spectrometer equipped with a tungsten xenon lamp. The excitation and emission light was dispersed using a Czerny Turner monochromator with an optical resolution of 1 nm. The emitted photons were detected using a Himamatsu R928 detector. The output signal was recorded using a computer. Room temperature optical UV- visible absorption spectra of all samples were recorded on a Lab India spectrophotometer. TEM images were collected on a JEM 2100F field emission transmission electron microscope operating at 200 kV. ICP-AES analysis was carried out in an ACROS, simultaneous ICP spectrometer manufactured by SPECTRO Analytical Instruments GmbH, Germany. XPS analysis was performed using a PHI 5000 Versa Probe II, FEI Inc. XPS system. Mott Schottky plots were obtained at different frequencies using a Biologic 300 electrochemical station with a three electrode system.Adsorption
Adsorption was performed by mixing 1 mg/1 ml solution of catalyst (BSO and Eu1.5-BSO) in methylene blue dye taken at different concentrations (5 μM, 10 μM, 20 μM, and 50 μM). Initially, 3 mg of adsorbent was taken in a cuvette with 3 ml of solution. The solution was magnetically stirred for 1 min to allow mixing of the adsorbate and adsorbent using a rice pellet. Then the cuvette was kept inside the spectrophotometer and the absorbance was measured at different time intervals to determine the kinetic rate of removal of the dye. The measurement was carried out until no change in the absorption spectrum of the dye was recorded to establish the equilibrium adsorption.Photocatalysis
Photocatalysis was performed by taking the catalyst to the dye solution ratio as 1 mg:1 ml. Typically, 20 mg of catalyst was dissolved in 20 ml of solution (50 μM unless mentioned separately) taken in a 100 ml beaker. Before adding the catalyst, 1 ml of the dye solution was separated to be used for determining the initial concentration of the dye. The catalyst dispersed in the solution was stirred for 1 h under dark conditions by covering the beaker in aluminium foil. This was done to establish the adsorption–desorption equilibrium between the adsorbent and solution. After this, a 1 ml aliquot was taken to determine the concentration of the solution after the equilibrium has been established to see if any solution is adsorbed by the catalyst. Then the solution was kept under a simulated solar Xe lamp (Newport, Stanford) with 140 W power (70 mA). 1 ml aliquots were taken after a time interval of 30 min. The aliquots taken at different time intervals were diluted by adding 3 ml of deionised water and the catalyst present in the dispersion was removed by centrifugation. The supernatant of the solution was taken for analysis by UV-vis absorption spectroscopy. The rate of degradation was calculated by the following equation assuming pseudo first order kinetics.| ln(C/C0) = kt | (8) |
Electrochemical measurement
The flat band potential was determined using the Mott Schottky plot using a three electrode system with material coated glassy carbon as the working electrode, platinum as the counter electrode, and Ag/AgCl (satd. KCl) as the reference electrode. 5 mg of catalyst was mixed with 50 μL of Nafion as a binder and 500 μL of ethanol to prepare the ink, which was subsequently drop cast on the working electrode. 1 M Na2SO4 solution was used as an electrolyte. The potential measured was calibrated to the normal hydrogen electrode (NHE) using the equation:| ENHE = EAg/AgCl + 0.0591 × pH + 0.22 V | (9) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge DST, India, for funding the project (DST/TMD/MES/2K17/70) and IISER Kolkata for instrumental facilities. The authors thank “TEM, DST-FIST, IISER, Kolkata” for providing TEM facility and “DST and SAIF, IIT Bombay” for ICP-AES analysis. We are thankful to NCL Pune for XPS analysis. For fellowship, D. S. and A. E. P. are thankful to IISER Kolkata and DST-INSPIRE, respectively.Notes and references
- S. Karak, K. Dey, A. Torris, A. Halder, S. Bera, F. Kanheerampockil and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 7572–7581 CrossRef CAS PubMed.
- H. Liu, J. B. Joo, M. Dahl, L. Fu, Z. Zeng and Y. Yin, Energy Environ. Sci., 2015, 8, 286–296 RSC.
- P. Samanta, A. V. Desai, S. Let and S. K. Ghosh, ACS Sustainable Chem. Eng., 2019, 7, 7456–7478 CrossRef CAS.
- D. Li, Q. Li, N. Bai, H. Dong and D. Mao, ACS Sustainable Chem. Eng., 2017, 5, 5598–5607 CrossRef CAS.
- M. M. Ayad and A. A. El-Nasr, J. Phys. Chem. C, 2010, 114, 14377–14383 CrossRef CAS.
- Q. Sui, J. Huang, Y. Liu, X. Chang, G. Ji, S. Deng, T. Xie and G. Yu, J. Environ. Sci., 2011, 23, 177–182 CrossRef CAS.
- X. Xing, H. Qu, R. Shao, Q. Wang and H. Xie, Water Sci. Technol., 2017, 76, 1243–1250 CrossRef CAS PubMed.
- C. X. Gui, Q.-Q. Wang, S.-M. Hao, J. Qu, P. P. Huang, C. Y. Cao, W. G. Song and Z.-Z. Yu, ACS Appl. Mater. Interfaces, 2014, 6, 14653–14659 CrossRef CAS PubMed.
- S. Ganguli, C. Hazra, T. Samanta and V. Mahalingam, RSC Adv., 2015, 5, 45611–45617 RSC.
- J. Li, X. Dong, G. Zhang, W. Cui, W. Cen, Z. Wu, S. C. Lee and F. Dong, J. Mater. Chem. A, 2019, 7, 3366–3374 RSC.
- J. Li, W. Cui, P. Chen, X. Dong, Y. Chu, J. Sheng, Y. Zhang, Z. Wang and F. Dong, Appl. Catal., B, 2020, 260, 118130 CrossRef CAS.
- C. K. Rastogi, S. K. Sharma, A. Patel, G. Parthasarathy, R. G. S. Pala, J. Kumar and S. Sivakumar, J. Phys. Chem. C, 2017, 121, 16501–16512 CrossRef CAS.
- Y. Gao, Y. Huang, Y. Li, Q. Zhang, J. Cao, W. Ho and S. C. Lee, ACS Sustainable Chem. Eng., 2016, 4, 6912–6920 CrossRef CAS.
- S. Usai, S. Obregón, A. I. Becerro and G. Colón, J. Phys. Chem. C, 2013, 117, 24479–24484 CrossRef CAS.
- C. O. Panão, E. L. S. Campos, H. H. C. Lima, A. W. Rinaldi, M. K. Lima-Tenório, E. T. Tenório-Neto, M. R. Guilherme, T. Asefa and A. F. Rubira, J. Mol. Liq., 2019, 276, 204–213 CrossRef.
- Y. Li, G. Chen, Q. Wang, X. Wang, A. Zhou and Z. Shen, Adv. Funct. Mater., 2010, 20, 3390–3398 CrossRef CAS.
- X. Li, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2016, 45, 2603–2636 RSC.
- H. Taniguchi, A. Kuwabara, J. Kim, Y. Kim, H. Moriwake, S. Kim, T. Hoshiyama, T. Koyama, S. Mori, M. Takata, H. Hosono, Y. Inaguma and M. Itoh, Angew. Chem., 2013, 125, 8246–8250 CrossRef.
- M. Back, E. Trave, G. Zaccariello, D. Cristofori, P. Canton, A. Benedetti and P. Riello, Nanoscale, 2019, 11, 675–687 RSC.
- M. Back, E. Casagrande, C. A. Brondin, E. Ambrosi, D. Cristofori, J. Ueda, S. Tanabe, E. Trave and P. Riello, ACS Appl. Nano Mater., 2020, 3, 2594–2604 CrossRef CAS.
- H. Taniguchi, S. Tatewaki, S. Yasui, Y. Fujii, J.-I. Yamaura and I. Terasaki, Phys. Rev. Mater., 2018, 2, 45603 CrossRef CAS.
- A. Girard, M. Stekiel, W. Morgenroth, H. Taniguchi, V. Milman, A. Bosak and B. Winkler, Phys. Rev. B, 2019, 99, 064116 CrossRef CAS.
- A. Girard, H. Taniguchi, S. M. Souliou, M. Stekiel, W. Morgenroth, A. Minelli, A. Kuwabara, A. Bosak and B. Winkler, Phys. Rev. B, 2018, 98, 134102 CrossRef CAS.
- J. Park, B. G. Kim, S. Mori and T. Oguchi, J. Solid State Chem., 2016, 235, 68–75 CrossRef CAS.
- S. Georges, F. Goutenoire and P. Lacorre, J. Solid State Chem., 2006, 179, 4020–4028 CrossRef CAS.
- C. C. Chen, C. T. Yang, W. H. Chung, J. L. Chang and W. Y. Lin, J. Taiwan Inst. Chem. Eng., 2017, 78, 157–167 CrossRef CAS.
- J. Ke, M. Adnan Younis, Y. Kong, H. Zhou, J. Liu, L. Lei and Y. Hou, Nano-Micro Lett., 2018, 10, 1–27 CrossRef PubMed.
- J. Wang, G. Zhang, J. Li and K. Wang, ACS Sustainable Chem. Eng., 2018, 6, 14221–14229 CrossRef CAS.
- W. Gu, F. Teng, Z. Liu, Z. Liu, J. Yang and Y. Teng, J. Photochem. Photobiol., A, 2018, 353, 395–400 CrossRef CAS.
- H. Lu, Q. Hao, T. Chen, L. Zhang, D. Chen, C. Ma, W. Yao and Y. Zhu, Appl. Catal., B, 2018, 237, 59–67 CrossRef CAS.
- W. Q. Li, Z. H. Wen, S. H. Tian, L. J. Shan and Y. Xiong, Catal. Sci. Technol., 2018, 8, 1051–1061 RSC.
- Z. Wan and G. Zhang, J. Mater. Chem. A, 2015, 3, 16737–16745 RSC.
- L. Zhang, W. Wang, S. Sun, D. Jiang and E. Gao, CrystEngComm, 2013, 15, 10043–10048 RSC.
- D. Liu, J. Wang, M. Zhang, Y. Liu and Y. Zhu, Nanoscale, 2014, 6, 15222–15227 RSC.
- W. Z. Wang, G. H. Wang, X. S. Wang, Y. J. Zhan, Y. K. Liu and C. L. Zheng, Adv. Mater., 2002, 14, 67–69 CrossRef CAS.
- Y. Guo, G. Zhang, J. Liu and Y. Zhang, RSC Adv., 2013, 3, 2963–2970 RSC.
- K. Le Jia, J. Qu, S. M. Hao, F. An, Y. Q. Jing and Z. Z. Yu, J. Colloid Interface Sci., 2017, 506, 255–262 CrossRef PubMed.
- D. Kim, Y. H. Jin, K. W. Jeon, S. Kim, S. J. Kim, O. H. Han, D. K. Seo and J. C. Park, RSC Adv., 2015, 5, 74790–74801 RSC.
- C. Hazra, T. Samanta, S. Ganguli and V. Mahalingam, ChemistrySelect, 2017, 2, 5970–5977 CrossRef CAS.
- P. T. Hsieh, Y. C. Chen, K. S. Kow and C. M. Wang, Appl. Phys. A: Mater. Sci. Process., 2008, 90, 317–321 CrossRef CAS.
- S. Jaiswar and K. D. Mandal, J. Phys. Chem. C, 2017, 121, 19586–19601 CrossRef CAS.
- M. J. Jackman, A. G. Thomas and C. Muryn, J. Phys. Chem. C, 2015, 119, 13682–13690 CrossRef CAS.
- A. Thøgersen, M. Rein, E. Monakhov, J. Mayandi and S. Diplas, J. Appl. Phys., 2011, 109, 113532 CrossRef.
- M. X. Song, M. Du, Q. Liu, F. Xing, C. Huang and X. Qiu, Catal. Today, 2019, 335, 193–199 CrossRef CAS.
- J. Kim, B. O. Jeong, E. H. Lee, D. H. Ahn, Y. Jung and S. W. Paek, Int. J. Electrochem. Sci., 2012, 7, 11257–11263 Search PubMed.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CAS.
- S. Liu, E. Guo and L. Yin, J. Mater. Chem., 2012, 22, 5031–5041 RSC.
- M. Zhou, J. Yu and B. Cheng, J. Hazard. Mater., 2006, 137, 1838–1847 CrossRef CAS PubMed.
- L. Dou, J. Zhong, J. Li, J. Luo and Y. Zeng, Mater. Res. Bull., 2019, 116, 50–58 CrossRef CAS.
- D. Sarkar, B. Meesaragandla, T. Samanta and V. Mahalingam, ChemistrySelect, 2016, 1, 4785–4793 CrossRef CAS.
- N. J. J. Johnson, S. He, S. Diao, E. M. Chan, H. Dai and A. Almutairi, J. Am. Chem. Soc., 2017, 139, 3275–3282 CrossRef CAS PubMed.
- S. Kasturi and V. Sivakumar, RSC Adv., 2016, 6, 98652–98662 RSC.
- J. S. Kim, Y. H. Park, S. M. Kim, J. C. Choi and H. L. Park, Solid State Commun., 2005, 133, 445–448 CrossRef CAS.
- Z. Wang, X. Hou, Y. Liu, Z. Hui, Z. Huang, M. Fang and X. Wu, RSC Adv., 2017, 7, 52995–53001 RSC.
- S. Qi, Y. Huang, T. Tsuboi, W. Huang and H. J. Seo, Opt. Mater. Express, 2014, 4, 396–402 CrossRef CAS.
- P. P. Pal and J. Manam, J. Rare Earths, 2013, 31, 37–43 CrossRef CAS.
- A. E. Praveen, T. Samanta, S. Ganguli and V. Mahalingam, ChemPhotoChem, 2019, 3, 204–210 CrossRef CAS.
- H. N. Tran, S. J. You, A. Hosseini-Bandegharaei and H. P. Chao, Water Res., 2017, 120, 88–116 CrossRef CAS PubMed.
- G. Blanchard, M. Maunaye and G. Martin, Water Res., 1984, 18, 1501–1507 CrossRef CAS.
- S. Bhattacharya and R. Shunmugam, ACS Omega, 2020, 5, 2800–2810 CrossRef CAS PubMed.
- W. Plazinski, W. Rudzinski and A. Plazinska, Adv. Colloid Interface Sci., 2009, 152, 2–13 CrossRef CAS PubMed.
- J. Febrianto, A. N. Kosasih, J. Sunarso, Y. H. Ju, N. Indraswati and S. Ismadji, J. Hazard. Mater., 2009, 162, 616–645 CrossRef CAS PubMed.
- B. K. Nandi, A. Goswami and M. K. Purkait, J. Hazard. Mater., 2009, 161, 387–395 CrossRef CAS PubMed.
- M. Streat, L. A. Sweetland and D. J. Horner, Process Saf. Environ. Prot., 1998, 76, 142–150 CrossRef CAS.
- B. Aslam, W. Wang, M. I. Arshad, M. Khurshid, S. Muzammil, M. H. Rasool, M. A. Nisar, R. F. Alvi, M. A. Aslam, M. U. Qamar, M. K. F. Salamat and Z. Baloch, Infect. Drug Resist., 2018, 11, 1645–1658 CrossRef CAS PubMed.
- T. López, F. Rojas, R. Alexander-Katz, F. Galindo, A. Balankin and A. Buljan, J. Solid State Chem., 2004, 177, 1873–1885 CrossRef.
- P. A. Babay, C. A. Emilio, R. E. Ferreyra, E. A. Gautier, R. T. Gettar and M. I. Litter, Int. J. Photoenergy, 2001, 3, 193–199 CrossRef CAS.
- M. Rajendran, Photodiagn. Photodyn. Ther., 2016, 13, 175–187 CrossRef CAS PubMed.
- B. R. V. Ferreira, D. N. Correa, M. N. Eberlin and P. H. Vendramini, J. Braz. Chem. Soc., 2017, 28, 136–142 CAS.
- H. Yang and J. Yang, RSC Adv., 2018, 8, 11921–11929 RSC.
- T. S. Natarajan, M. Thomas, K. Natarajan, H. C. Bajaj and R. J. Tayade, Chem. Eng. J., 2011, 169, 126–134 CrossRef CAS.
- Z. He, C. Sun, S. Yang, Y. Ding, H. He and Z. Wang, J. Hazard. Mater., 2009, 162, 1477–1486 CrossRef CAS PubMed.
- I. F. Teixeira, E. C. M. Barbosa, S. Chi, E. Tsang and P. H. C. Camargo, Chem. Soc. Rev., 2018, 47, 7783 RSC.
- Z. Xu, M. Quintanilla, F. Vetrone, A. O. Govorov, M. Chaker and D. Ma, Adv. Funct. Mater., 2015, 25, 2950–2960 CrossRef CAS.
- A. Sarkar, A. B. Ghosh, N. Saha, A. K. Dutta, D. N. Srivastava, P. Paul and B. Adhikary, Catal. Sci. Technol., 2015, 5, 4055–4063 RSC.
- M. Förster, R. J. Potter, Y. Ling, Y. Yang, D. R. Klug, Y. Li and A. J. Cowan, Chem. Sci., 2015, 6, 4009–4016 RSC.
- J. Gan, X. Lu, J. Wu, S. Xie, T. Zhai, M. Yu, Z. Zhang, Y. Mao, S. C. I. Wang, Y. Shen and Y. Tong, Sci. Rep., 2013, 3, 1–7 Search PubMed.
- B. Wang, M. Zhang, X. Cui, Z. Wang, M. Rager, Y. Yang, Z. Zou, Z. L. Wang and Z. Lin, Angew. Chem., Int. Ed., 2020, 59, 1611–1618 CrossRef CAS PubMed.
- Y. Liu, B. Wei, L. Xu, H. Gao and M. Zhang, ChemCatChem, 2015, 7, 4076–4084 CrossRef CAS.
- X. Dong, W. Cui, H. Wang, J. Li, Y. Sun, H. Wang, Y. Zhang, H. Huang and F. Dong, Sci. Bull., 2019, 64, 669–678 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00363h |
| This journal is © The Royal Society of Chemistry 2020 |