
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Antibacterial efficacy from NO-releasing MOF–polymer films†
Morven J.
Duncan
 a,
Paul S.
Wheatley
a,
Emma M.
Coghill
b,
Simon M.
Vornholt
a,
Stewart J.
Warrender
*a,
Ian L.
Megson
b and
Russell E.
Morris
a
a,
Paul S.
Wheatley
a,
Emma M.
Coghill
b,
Simon M.
Vornholt
a,
Stewart J.
Warrender
*a,
Ian L.
Megson
b and
Russell E.
Morris
a
aSchool of Chemistry, University of St Andrews, North Haugh, St Andrews, KY16 9ST, UK. E-mail: sjw9@st-andrews.ac.uk
bFree Radical Research Facility, Division of Biomedical Sciences, University of the Highlands & Islands, Inverness, IV2 3JH, UK
First published on 7th October 2020
Abstract
The formulation and antibacterial efficacy of nitric oxide (NO)-releasing MOF–polyurethane films are reported for the first time. Uniform standalone films were successfully prepared containing 1, 5, 10, and 15 wt% CPO-27 (Ni). The MOF within each film was successfully activated and loaded with NO. Adsorption and release profiles are reported for the films and show that while the polymer influences the quantity of NO adsorbed and stored by the MOF and the time scale of release; the proportion of stored NO that is released is dependent on wt% loading of MOF in the film. In a cytotoxicity assay, the formulations exhibited very low toxicity (<20% cell death), and this toxicity was attributed to the NO rather than the MOF. Antibacterial data against E. coli and S. aureus indicated that bactericidal efficacy can be achieved in 5 hours from 1 wt% films, and within 1 hour for films containing at least 5 wt% MOF.
Introduction
Healthcare Associated Infections (HAIs) are a major problem for modern society bringing a significant financial burden and death toll.1 In Europe, 4.1 million patients are affected each year resulting in 16 million extra days of hospital stay, 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths and an economic impact of €7 billion. Similarly, in the US, where 1.7 million patients are affected annually, HAIs account for 99000 deaths and $6.5 billion in additional costs. A major source of infection is the prolonged use of indwelling and invasive devices. For example, approximately one in four infections are a result of catheterization.2 Catheters are long, thin, tube-like devices that are employed to access arteries, veins and organs to permit the removal or delivery of fluids, the administration of medication, and the conducting of surgical procedures, dialysis and sample collection. They are typically manufactured from polymeric materials such as silicones, polyurethanes and poly vinyl chloride. Bacteria can be carried into the body during the insertion of the device and microbes can adhere and grow on their surface. Resulting conditions include catheter-related blood stream infections (CRBSI) and catheter-associated urinary tract infections (CAUTI). It is estimated that an additional $3700–36441 per patient is spent in the US treating each CRBSI,3 while total annual additional costs of CAUTIs in the UK have been estimated to be £1–2.5 billion.4 The problem is also augmented by the inappropriate use of antibiotics leading to antibiotic resistance. Indeed, a recent study reports that bacterial resistance is consistently higher for device-associated HAIs than for the same bacteria isolated from surgical site infections.5 Current approaches to addressing catheter-related HAIs include impregnating the devices with antibiotics, silver nanoparticles and/or antiseptics.6 However, the aforementioned prevalence of antibiotic resistance, concerns over the effectiveness and possible resistance towards silver,7 and growing sensitization towards common antiseptics8 drives the development of new approaches.
000 deaths and an economic impact of €7 billion. Similarly, in the US, where 1.7 million patients are affected annually, HAIs account for 99000 deaths and $6.5 billion in additional costs. A major source of infection is the prolonged use of indwelling and invasive devices. For example, approximately one in four infections are a result of catheterization.2 Catheters are long, thin, tube-like devices that are employed to access arteries, veins and organs to permit the removal or delivery of fluids, the administration of medication, and the conducting of surgical procedures, dialysis and sample collection. They are typically manufactured from polymeric materials such as silicones, polyurethanes and poly vinyl chloride. Bacteria can be carried into the body during the insertion of the device and microbes can adhere and grow on their surface. Resulting conditions include catheter-related blood stream infections (CRBSI) and catheter-associated urinary tract infections (CAUTI). It is estimated that an additional $3700–36441 per patient is spent in the US treating each CRBSI,3 while total annual additional costs of CAUTIs in the UK have been estimated to be £1–2.5 billion.4 The problem is also augmented by the inappropriate use of antibiotics leading to antibiotic resistance. Indeed, a recent study reports that bacterial resistance is consistently higher for device-associated HAIs than for the same bacteria isolated from surgical site infections.5 Current approaches to addressing catheter-related HAIs include impregnating the devices with antibiotics, silver nanoparticles and/or antiseptics.6 However, the aforementioned prevalence of antibiotic resistance, concerns over the effectiveness and possible resistance towards silver,7 and growing sensitization towards common antiseptics8 drives the development of new approaches.
Nitric oxide (NO) is a biological signalling molecule involved in many physiological and pathological processes.9 Amongst its many properties, NO has potent antimicrobial activity and offers a potential solution to catheter related HAIs if it can be released in efficacious concentrations from the surface of the devices.10–12 The gaseous and radical nature of NO, however, presents challenges in terms of its handling, its storage within the device surface and its release in a controlled manner exactly when and where required. Moreover, high concentrations of NO are toxic to mammalian cells, therefore precisely controlled release is essential. Several approaches are being studied to achieve NO release from catheters including direct impregnation of NO13 and attachment and/or incorporation of NO-binding compounds (such as NONOates and S-nitrosothiols) to the polymer employed to manufacture the catheter.14–16
We are interested in developing metal organic frameworks (MOFs) as NO releasing agents. MOFs are crystalline microporous materials comprised of metal ion or cluster nodes connected by organic ligands.17,18 Their three-dimensionally connected scaffold-like structures possess vast internal surface areas and porosity making them ideal for many applications, not least gas storage19 and drug delivery.20,21 Indeed, the potential of MOFs to act as reservoirs and delivery systems for antimicrobial agents to help address the problems of infection and bacterial resistance is now receiving much attention. For example, MOFs can be employed as delivery agents for antimicrobial metal ions such as Zn, Cu and Ag with improved efficiency;22,23 they offer controlled and potentially more targeted delivery of antibiotics and antiseptics (for example, ciprofloxacin,24 ceftazidime,25 vancomycin,26 metronidazole,27 rifampicin28 and tinidazole29); and they can offer combination treatments by delivering multiple agents simultaneously or over different time scales.30,31 Some reports suggest synergistic effects can be achieved through careful selection of metal ion and antibiotic to heighten drug potency.32
In addition to their porosity and storage/delivery capacities, MOFs offer unrivalled flexibility in terms of surface functionality modification and tailoring of chemical properties. We and our collaborators have shown how MOFs with coordinatively unsaturated metal sites (or “CUSs”, achieved in some MOFs following activation) are highly efficient at storing NO and releasing it in antimicrobial concentrations when in contact with a moist environment.33–37 Alternatively NO can also be attached to and released from functional moieties incorporated into the organic ligands of MOFs.38,39 With regards to employing CUSs as the binding mechanism, we have shown that the CPO-27 family of MOFs is particularly effective in this regard. NO binds to the CUSs via a metal–nitrogen coordinate bond and is released from the MOF following substitution by water (Fig. 1). Remarkably, when Ni and Co are employed in the framework of CPO-27 the NO binding is almost completely reversible when exposed to moisture.33 We have also shown that NO release capacity can be tuned by carefully altering the ratio of metallic species in mixed-metal MOFs.40 One of the major advantages of using MOFs to deliver NO compared to, for example S-nitrosothiols and NONOates, is their ability to release large concentrations of NO. Indeed, insufficient delivery is often a factor that results in low, ineffective and/or slow antimicrobial efficacy from many systems and warrants the combination of NO with other agents such as silver nanoparticles.15,16,41,42 Our previous studies have confirmed that concentrations of NO released from MOF powders can be effective against a wide range of bacteria, including antibiotic resistant strains, such as MRSA.43 However, to date, our reported studies have focused on the function of and efficacy from MOF powders. In order to achieve industrial adoption, it is essential to understand and develop the performance of the NO-releasing MOF powders when formulated into polymers. There are many studies in the literature regarding MOF–polymer composites (referred to as mixed matrix membranes or MMMs) for applications such as gas separation44 and water purification.45 This growing body of work confirms the excellent compatibility of MOFs with a wide variety of polymers, and highlights the ease with which MOFs can be formulated. Of course, the incorporation of inorganic powder additives into polymers is a well understood field, and this knowledge helps to progress MOF development and application. This is a further advantage of using MOFs as potential NO-delivery agents. Many other approaches are often incompatible with polymeric articles (e.g. liquid phase agents) or would require significant alterations to currently employed polymer formulations.46 Furthermore, MOFs can be incorporated via far less protracted procedures and require much easier NO-loading conditions (i.e. exposure to only 1 atm NO for a matter of seconds) compared to other systems.47 Once the specific processing and formulation protocols are refined, MOFs could offer a more facile “plug and play” solution. There are, however, very few studies reporting the formulation of MOFs into polymeric articles for antimicrobial purposes; and those which exist mostly employ metal ion release as the antimicrobial mechanism.48–52 In this contribution we report the successful formulation of NO-releasing CPO-27 into a model medical polyurethane using solvent casting to generate standalone films. We show that NO gas can be successfully loaded into the composite and released in antibacterial concentrations when in a moist atmosphere. We also show how the polymer matrix attenuates NO release and why this must be carefully considered when developing formulations for medical device application.
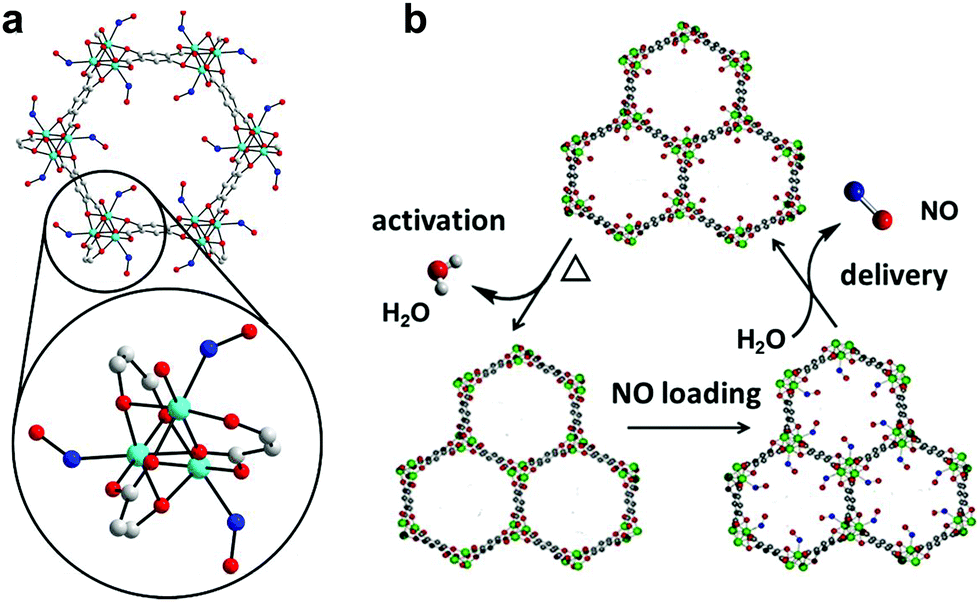 | ||
| Fig. 1 NO adsorption and release by CPO-27 MOFs. Detail of the structural environment surrounding CUSs in CPO-27 MOFs with bound NO (refined from powder X-ray diffraction data): cyan – divalent metal ion (Co2+ in this particular case), blue – nitrogen, red – oxygen, grey – carbon, hydrogen atoms have been removed for clarity (a); and schematic illustration of the NO-adsorption/release cycle (b).33 | ||
Experimental
All reagents were commercially available and used without further purification. 2,5-Dihydroxyterephthalic acid (DHTP) was purchased from Zhejian Dragon, Ni(II) acetate tetrahydrate was purchased from Acros, while THF was purchased from Fisher. Polyurethane (poly[4,4′-methylenebis(phenyl isocyanate)-alt-1,4-butanediol/di(propylene glycol)/polycaprolactone]) was purchased from Aldrich. NO (99.5% purity, 35 bar) and NO calibration gas (∼95 ppm NO in nitrogen) were purchased from BOC and Air Products, respectively.Synthesis of CPO-27 (Ni)53
DHTP (7.43 g, 37.5 mmol) was dissolved in THF (125 mL) under stirring at room temperature within the Teflon liner of a 600 mL stainless steel Parr autoclave. Separately, Ni acetate (18.66 g, 75 mmol) was dissolved in deionised water (125 mL) under stirring at room temperature. Once each had completely dissolved, the Ni acetate solution was added to the DHTP solution under stirring. The autoclave was sealed and heated to 110 °C for 3 days. The product was recovered by filtration after having gradually cooled to room temperature. Yield 16.8 g (91%).Preparation of MOF–polymer films
In a typical preparation, 6 g of polyurethane pellets were dissolved in 40 mL THF overnight under stirring at room temperature. Separately, the required quantity of MOF powder was pre-dispersed in THF (∼10 mL) using a hand held manual homogeniser to break down agglomerates. The dispersion was assessed visually before being added to the polymer/THF solution. Once the polymer had completely dissolved, the MOF–THF suspension was added to the polymer solution and mixed until homogenous. MOF–polymer solution mixtures were cast into films approximately 8 cm × 15 cm using a doctor blade and allowed to cure at room temperature. Cured films were subsequently cut into 4 cm × 2.5 cm (∼100 mg) sections ready for activation and NO loading.Characterisation
Powder X-ray diffraction data collection was conducted using a Stoe STADI P diffractometer operated in transmission mode and employing monochromated CuKα1 radiation. Thermogravimetric analysis (TGA) was performed using a Netzsch TGA 209 instrument, in which ∼5 mg of sample was heated to 500 °C at a rate of 5 °C min−1 in air. BET measurements were carried out at 77 K using a Micromeritics ASAP 2020 surface area and porosity analyser. Samples were prepared by removal of solvent by heating to 150 °C at a rate of 10 °C min−1 under vacuum for 16 hours. Nitrogen gas was then incrementally dosed onto the sample and the volume of gas adsorbed was measured as a function of partial pressure. Scanning electron microscopy (SEM) was performed using an FEI Scios Dualbeam SEM. The microscope was operated at low voltages (2–5 kV) and beam currents (50 pA) to prevent degradation of the polymer films. Multiple detectors allowed for a relatively short working distance of 7 mm; images recorded by the backscattered electron detector were used throughout this study. Samples were supported on an aluminium stub using an adhesive carbon tab. To prevent charging and to improve conductivity, samples were brushed with an epoxy silver paste and further gold sputter coated using a Quorum Q150R ES Au-sputter coater operated at 10 mA for 30 s. In order to visualise the distribution of the MOF particles throughout the depth of the film, a small strip of a 15 wt% MOF–polymer film was prepared using common scissors and mounted edge-on to the beam.Thermodynamic NO adsorption/release isotherms were measured gravimetrically using bespoke apparatus described previously.35 Prior to exposure to NO, samples of MOF powder and MOF–polymer films (4 cm × 2.5 cm) were activated at 423 K under 1 × 10−4 mbar until no further mass loss was observed. Isotherms were collected at 298 K.
To load MOF powders and MOF–polymer composites with NO for kinetic release measurements, cytotoxicity and antimicrobial testing, samples were placed into glass ampules within a Schlenk flask, the flask was connected to a Schlenk line and the samples were heated to 403 K overnight, under vacuum (1 × 10−4 mbar). The samples were exposed to dry NO (2 bar) for 1 hour and sealed under dry argon.
Kinetic NO release measurements were recorded using a Sievers NOA 280i chemiluminescence NO analyser under flowing humid nitrogen (11% relative humidity), as described previously.35 Data collection was stopped at 20 ppb (nearing the sensitivity limit of the analyser), and the data were collated to generate plots of the concentration of NO (in ppm) released over time, and of the cumulative total number of moles of NO released per gram of film and per gram of MOF.
Cell viability assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay was used to evaluate the cytotoxicity of the MOF–polymer films. Human umbilical vein endothelial cells (HUVECs) were cultured in an endothelial growth medium (Medium 199), composed of Earle's salts (Gibco, Fisher Scientific) and supplemented with 20% foetal bovine serum (FBS; Gibco, Fisher Scientific), 1% penicillin–streptomycin (Gibco, Fisher Scientific), 0.67% endothelial cell growth supplement from bovine neural tissue (Merck), 2% 1 M HEPES solution (Merck), 2% 100 mM sodium pyruvate solution (Merck), and 1% heparin sodium (Tesco Pharmacy). The cells were seeded in 12 well culture plates at 1 × 105 cells per well in 1 mL of growth medium. The plates were incubated for 24 hours at 37 °C in a 5% CO2/95% air humidified atmosphere.After the incubation period, the medium was refreshed (including a phosphate buffered saline (PBS) wash) before the cells were exposed to sections (1.5 cm2) of the MOF–polymer films, containing 0, 5, 10 and 15 wt% MOF. The NO loading process served to sterilise the NO-containing test samples while films containing only MOF (without NO) and films of blank polymer were sterilised by lightly spraying with 70% ethanol (VWR) and allowing to dry.
The control wells on the plate included cells in medium only, cells in medium covered with a glass coverslip and medium only. The films were placed in each well using forceps and then covered with a glass coverslip to facilitate contact with the cells. The cells were exposed to the films for 30 minutes (37 °C, 5% CO2). After careful removal of the films, the medium was refreshed and the cells incubated for a further 24 hours prior to assessment of cell viability.
The cultured medium was removed from the well plate, each well was washed with PBS and replaced with a 10% MTT solution in 1 mL of phenol red free medium (Gibco, Fisher Scientific) supplemented with 10% foetal bovine serum (FBS; Gibco, Fisher Scientific), 1% penicillin–streptomycin (Gibco, Fisher Scientific), 0.67% endothelial cell growth supplement from bovine neural tissue (Merck), 2% 1 M HEPES solution (Merck), 2% 100 mM sodium pyruvate solution (Merck), and 1% heparin sodium (Tesco Pharmacy). The plate was incubated in the dark at 37 °C for 3 hours before addition of the MTT solubilisation solution. Cell viability was assessed by detection of formazan using a platereader (Varioscan, Thermo) set to λ = 570 nm. Absorbance readings were normalised to those for untreated (control cells) and viability expressed as % control. The experiment was conducted on 6 separate occasions (N = 6) and data were assessed using 2-factor ANOVA with Bonferroni post-test; P < 0.05 was considered to be statistically significant.
Antibacterial assay
Determination of the antibacterial activity of the MOF–polymer films against Escherichia coli (strain ATCC 8739) and Staphylococcus aureus (strain ATCC 6538P) was carried out in accordance with ISO 22196:2007 Plastics – Measurement of Antimicrobial Activity on Plastics Surfaces, with slight modifications. In summary, 0.1 mL of test inoculum (targeted concentration 6 × 105 cells per mL, estimated spectrophotometrically at 620 nm), was pipetted onto the surface of test films measuring 2 cm × 2 cm. Polythene cover slips measuring 4 cm × 4 cm were employed as per the standard procedure. Test specimens were incubated at 36 ± 1 °C for 0, 1 and 5 hours at a relative humidity of >90%. Bacteria were recovered from the test specimens by washing with 5 × 1.0 mL SCDLP broth, and viable cells were enumerated following serial dilution, plating and incubation (36 ± 1 °C for 40–48 hours) as per the standard protocol. Results were expressed as number of bacteria colony forming units (cfu) per cm2, and as a log reduction. The latter was calculated in relation to the zero hour time point for each specimen as per eqn (I):| R = log10N0 − log10Nx | (I) |
Six specimens (N = 6) of each film type (i.e. each wt% MOF) were tested for each time point, including 6 blank film controls. The conditions used for film activation and NO loading with the subsequent storage under argon were sufficient to sterilise the test articles prior to testing. The control films were sterilised by swabbing with 70% alcohol.
Results and discussion
MOF and film preparation
To ensure consistency within the study a single batch of CPO-27 (Ni) was prepared, as above, and employed in the preparation of all films. Powder XRD data, TGA profiles, nitrogen adsorption isotherms and SEM images collected from the as-prepared MOF powder are given in Fig. S1–S4 (ESI†). The XRD pattern shows close agreement with the simulated pattern generated from literature data,54 indicating that the correct MOF structure was prepared. The TGA profile exhibits an initial mass loss of ∼30% due to the evolution of water from the structure, and a further mass loss of 40% at ∼270 °C corresponding to structural collapse. A BET surface area of 913 m2 g−1 was recorded for the material. The TGA profile, the inferred thermal stability and activation requirements, and the surface area measurements are in line with previous observations for this material.35 SEM images of the MOF powder indicate that the primary particle size is approximately 100–200 nm, and that these form agglomerates measuring approximately 3 μm × 5 μm in size.Polyurethanes are commonly employed in the manufacture of medical devices such as catheters. The polyurethane selected for this study was chosen based on its wide applicability (including medical devices) reported by the manufacturer. MOF–polymer films were prepared at four MOF concentrations: 1, 5, 10 and 15 wt% (based on polymer weight). Cast films were free of bubbles, occlusions and visible signs of agglomeration. X-ray diffraction patterns collected from the films (Fig. S5, ESI†) consist of a superposition of sharp reflections, corresponding to the crystalline MOF, on an amorphous background resulting from the polymer. The intensity of the crystalline CPO-27 (Ni) reflections increases with wt% MOF in the films. These data indicate that the MOF structure is stable towards the solvent casting methodology employed in the creation of the films. SEM micrographs of representative sections of film surfaces are shown in Fig. 2. The blank polymer film (0 wt%, Fig. 2a) exhibits smooth surface texture and is translucent (evidenced in the SEM image by the irregularities of the carbon adhesive shining through from beneath). The surface images of MOF-containing samples (Fig. 2b–e) illustrate an even distribution of MOF particles in each film, with the MOF fully embedded in the polymer. Those particles nearest the film surface are observed to protrude slightly. The dimensions of the MOF particles within the films can be estimated from the surface images and appear to be in line with the proportions of the agglomerates mentioned above. This indicates that the MOF is present predominantly as small agglomerates rather than nano-scale primary particles. As expected, the density of MOF particles within the films (and visible at the surface) increases with wt% loading. The thickness of the films can be visualised from the cross-sectional images and measured to be ∼110 μm (Fig. 2f and g). At higher magnification (Fig. 2h) MOF particles become visible throughout the whole cross-section. Taken together, the visual appearance and SEM analysis of the films suggest that the MOF mixes well with the polymer and that homogenous distributions of MOF particles have been achieved throughout the length, breadth and depth of the polyurethane matrix. Computational simulations of the interface between the CPO-27 MOF surface and the polyurethane employed in this study indicate a remarkably strong compatibility between both components.55 The analysis suggests that the polymer changes its conformation to fit the profile of the MOF surface, preventing the formation of voids. Further, it is found to participate in hydrogen bonding with the MOF and penetrate the MOF pore to a previously unobserved depth of 14 Å. Such strong interaction undoubtedly contributes to the driving force leading to excellent MOF dispersion.
 | ||
| Fig. 2 SEM micrographs of the surfaces of polyurethane films containing 0 wt% (a), 1 wt% (b), 5 wt% (c), 10 wt% (d) and 15 wt% (e) MOF and the cross section of polyurethane film containing 15 wt% MOF (f–h). | ||
The films were self-supporting when peeled away from their glass supports (Fig. S6, ESI†), and they exhibit good pliability (as demonstrated in Fig. 2f) and comparable physico-tactile properties to the blank polyurethane film. Indeed, analysis of the mechanical properties of films prepared from the same components (conducted in a separate study reported elsewhere55) indicates that ductility and tensile strength are not adversely affected at MOF loadings up to 20 wt%. Beyond this level ductility and tensile strength reduce, indicating the existence of a maximum loading threshold that should not be exceeded if the mechanical properties of the base polymer are to be maintained.
NO adsorption and storage
NO adsorption isotherms were recorded for sections of each film (∼100 mg) as outlined above. Fig. 3a illustrates the resulting data per gram of film sample. Please also refer to Table S1 (ESI†) for key metrics. The data shown in Fig. 3a indicate that each film adsorbs NO and the quantity that is adsorbed increases with increasing wt% of MOF, as would be expected. This confirms that the MOF has been activated within the film and that NO is able to penetrate the polymer matrix, access the MOF pores and bind to the coordinatively unsaturated metal sites. Importantly, upon reduction of the pressure, very little of the adsorbed NO is desorbed, suggesting almost all adsorbed NO is chemisorbed. Previous studies of NO storage in CPO-27 powders suggest that this chemisorbed NO can be stably stored within the MOF under dry conditions, until exposure to moisture triggers its release.33Fig. 3b illustrates the same data but this time normalised per gram of MOF present in each film to account for the different MOF contents in each sample. Analysing the data in this way reveals the quantity of NO adsorbed per gram of MOF is very similar in each film and is ∼3.10–3.61 mmol g−1. The quantity that is chemisorbed (stored) is also in this region. This suggests that the adsorption/storage efficiency of the MOF powder within the film, as well as the number of accessible adsorption sites within each gram of MOF present, is equal in each case and is not affected by the wt% of MOF incorporated into the film. This appears logical as there is no reason for the adsorption/storage performance of the MOF within a given matrix to vary with wt% loading. However, comparison of the data to the adsorption isotherm of CPO-27 (Ni) powder (Fig. 3b) indicates that the quantity of NO adsorbed and stored by the MOF inside the films (per gram of MOF present) is approximately half that adsorbed and stored by the as-made powder alone. This suggests that only ∼50% of adsorption sites in the MOF are available for NO coordination when formulated into the films. It also indicates that the reduction in the number of accessible sites is the same no matter how much MOF is present in the film. This reduction could be a result of the polymer blocking access to some of the MOF pores (favoured by the excellent compatibility of the MOF and polymer), some MOF particles being buried inaccessibly deep within the film, slow diffusion of NO through the polymer, and/or incomplete activation of the MOF when within the polymer matrix. Nevertheless, the data confirm for the first time that CPO-27 (Ni) can be activated and loaded with NO whilst formulated into polyurethane.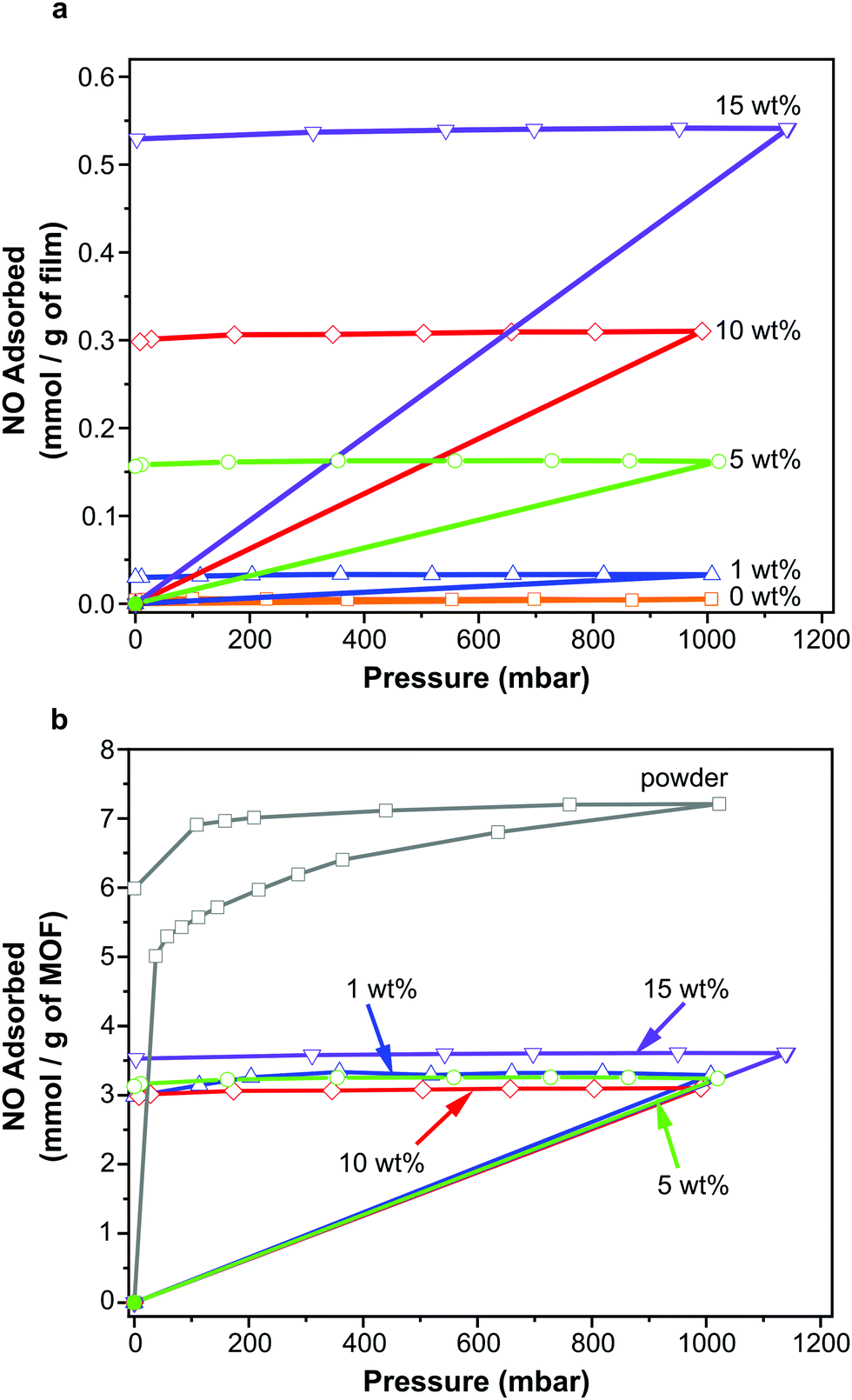 | ||
| Fig. 3 NO adsorption/desorption isotherms measured gravimetrically for polyurethane films containing 1, 5, 10 and 15 wt% CPO-27 (Ni) along with blank film (0 wt%) and CPO-27 (Ni) powder expressed per gram of film (a) and per gram of MOF (b). | ||
A further, qualitative, indication of the successful adsorption of NO by the films is the change in colour observed after exposure of the activated films to NO. The as-prepared films exhibit colours ranging from very light golden yellow at 1 wt% loading, through deeper shades of yellow at 5 and 10 wt%, to mustard colour at 15 wt%; the intensity of colour being indicative of MOF loading (Fig. 4). After activation and exposure to NO the colour of the films darkened markedly to shades ranging from light caramel to dark brown-black. These colour changes are consistent with those observed previously for CPO-27 (Ni) powder35 and indicate that MOF activation and NO loading has occurred.
 | ||
| Fig. 4 Photographs of CPO-27 (Ni)-containing polyurethane film sections as prepared (a) and after activation and NO loading (b). | ||
NO release
Representative 4 cm × 2.5 cm sections of each film were prepared and loaded with NO as outlined above. The slightly lower activation temperature of 403 K was employed to prevent the films adhering to the glass ampules. These sections were exposed to 11% relative humidity upon which measurable quantities of NO were released from each film.Fig. 5 illustrates the concentration of NO (in ppm) measured from each film over the first five hours of release. An initial spike in measured concentration at time zero resulting from loss of physisorbed NO has been removed from the data. The data shown represents release of chemisorbed NO, which depends on the ingression of water through the polymer and into the MOF pores as well as the egression of NO through the polymer. The profiles of the concentration-time charts are influenced by these processes and show an obvious increase in concentration over the first hour as water penetrates the matrix and triggers release. Thereafter the concentration reduces as the stored NO is depleted.
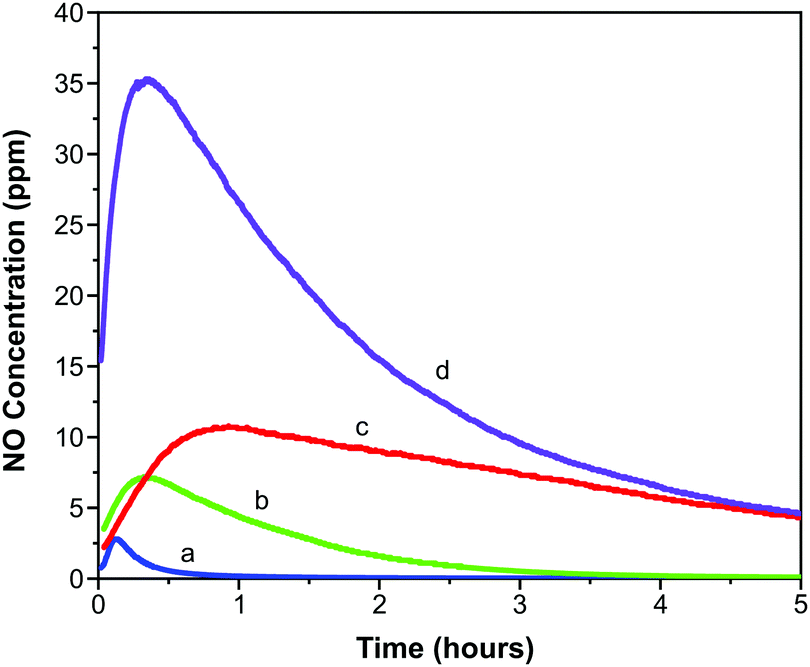 | ||
| Fig. 5 Concentration of NO released from polyurethane films containing 1 wt% (a), 5 wt% (b), 10 wt% (c) and 15 wt% (d) CPO-27 (Ni). | ||
The fast release kinetics in the early stages following exposure to 11% relative humidity may be predicted to increase when the formulations are deployed in a patient setting (where the relative humidity will be much higher than 11%). This, however, is not necessarily counterproductive because it is the early stages of catheter deployment that carry the highest risk of bacterial transmission to the patient. A high initial burst of NO should act to kill the bacteria on the surface of the catheter (one of the main root causes of infection). A sustained release of lower concentrations will allow maintenance of a low bioburden.
It is observed that the maximum NO release concentration increases with MOF wt% loading (as might be expected). The time to reach the maximum concentration also increases with MOF loading up to 10 wt%. However, the 15 wt% film breaks this trend in that the time to reach maximum NO release concentration is shorter than the trend would predict. This may be suggestive of macroscopic alterations in the polymer film structure as MOF loading increases. These factors are analysed in more detail in a further study.55
While each sample exhibits the same general release profile, the maximum concentration released from the films and the longevity of release is dependent on MOF wt% (Fig. 6). As a result of the diffusion processes mentioned above, the release of NO is sustained at levels greater than 20 ppb for much longer (up to 50–60 hours) than in the case of MOF powder (typical values of 15 hours for 30 mg MOF, much more than is present in the films studied here, are reported33). This can be very useful in terms of end application where release at low levels may be required for several days to prevent re-colonisation of the device. A further manifestation of the influence of the polymeric matrix is the separation of physisorbed and chemisorbed NO release; typically these occur in a single event beginning at time zero for powder samples.56 As indicated above, the release of chemisorbed NO from the films occurs later than that of physisorbed NO.
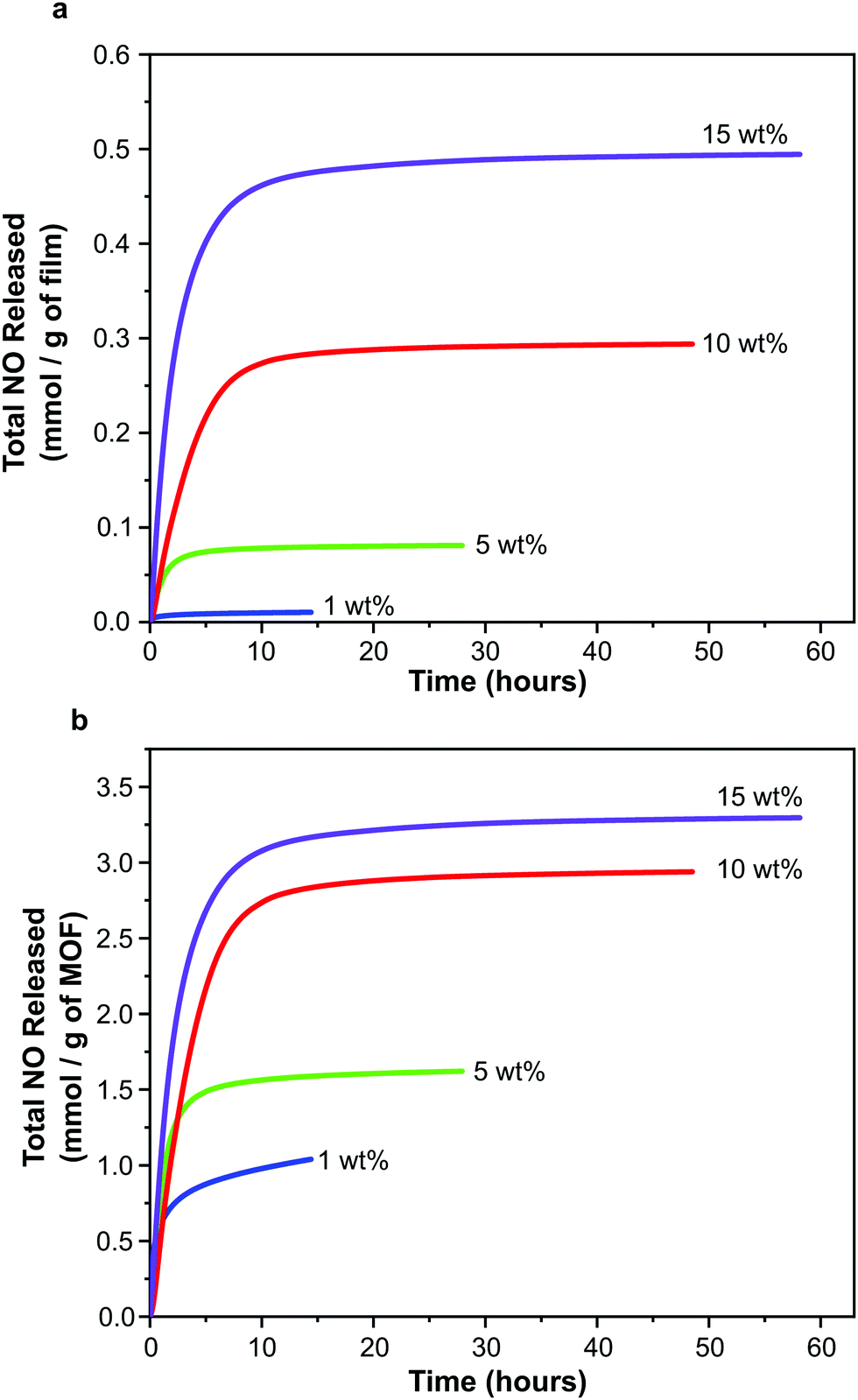 | ||
| Fig. 6 NO release profiles for polyurethane films containing 1, 5, 10 and 15 wt% CPO-27 (Ni) expressed per gram of film (a) and per gram of MOF (b). | ||
In line with the observed increase in NO adsorption per gram of film on increasing MOF wt%, the total quantity of NO released per gram of film also increases with MOF wt% (Fig. 6a). Total release values range from ∼0.01 to 0.49 mmol NO per gram of film. Interestingly, the trend of increasing NO release with MOF wt% is not linear.
Further insight can be gained by examining the data plotted per gram of MOF present in each film (Fig. 6b). In contrast to the observations regarding adsorption of NO discussed above (where the quantity of adsorbed NO per gram of MOF is equal for all films), the quantity of NO released per gram of MOF changes with wt%. Films containing 15 wt% MOF release a similar quantity of NO per gram of MOF to those containing 10 wt%, however films containing 1 and 5 wt% release much lower levels. It should also be noted that the quantity of NO released per gram of MOF from films containing 10 and 15 wt% MOF is approximately equal to the quantity stored per gram of MOF (i.e. ∼3 mmol), suggesting almost complete reversibility of adsorption and near 100% release (consistent with the performance of powder-form MOF). However the quantity released per gram of MOF from films containing 1 and 5 wt% MOF is much lower than the quantity stored (for 1 wt% MOF-containing films 2.98 mmol of NO are stored per gram of MOF but only 1.04 mmol are released; and for 5 wt% MOF-containing films 3.13 mmol per gram of MOF are stored and 1.62 mmol are released), perhaps indicating that adsorption is less reversible at these MOF loadings under the conditions studied. This would suggest that, while the wt% of MOF does not influence the efficiency of NO adsorption and storage by the films, it does influence the efficiency of NO release from the films, with 10 and 15 wt% being more efficient. Variations in the efficiency of releasing stored NO may be a result of changes in guest transport through the composite. This may be caused by alterations in MOF particle connectivity within the films as wt% is varied. These unexpected findings are the subject of further in-depth analysis, the details of which are presented elsewhere.55
Cytotoxicity evaluation
A cell viability (MTT) assay was conducted using HUVECs to assess the level of toxicity imparted by MOF–polymer films containing 5, 10 and 15 wt% CPO-27 (Ni), with and without NO loading. This colourimetric assay determines cell viability as a function of mitochondrial activity. Respiring cells cleave the tetrazolium ring, yielding purple formazan crystals, which are insoluble in aqueous solutions. The colour intensity of the solubilised crystals, determined by photometric measurements, gives a measure of respiratory function in living cells. Endothelial cells were used for this assay because, in our hands, they have proved to be the most sensitive to noxious stimuli and therefore provide a worst-case scenario. An advantage of HUVECs is that they are primary cells as opposed to immortalised cells that constitute cell lines: many such carcinoma-derived cells show substantial resistance to potential toxins. A glass coverslip was required to weight the films to facilitate good contact with the cells. To confirm that this addition was not deleterious to the growth of the cells, a control well containing only cells covered with a coverslip was included on the plate. An additional control on the plate was blank polymer film (containing no MOF), also weighed down with a coverslip.Fig. 7 shows the results from the cell assay and illustrates that all sample viability is greater than 80%. This cell reduction of less than 20% is well within the limit accepted by the sector for cytotoxicity (30% cell death). The utilisation of the coverslip did not significantly influence the viability of the cells (86.8% and 97.7% viability). As to be expected from a medical grade polyurethane, the viability of the cells was also not affected by the blank film. Moreover, the MOF polymer films without NO did not have a significant impact on cell viability in this assay (5 wt%: 97.6%, 10 wt%: 103.2% and 15 wt%: 87.9% viability). It is apparent, therefore, that neither the polymer nor MOF components up to a 15 wt% composition were toxic under the conditions of this experiment. This is not unexpected given that previous studies have shown that neither CPO-27 (Ni) powder nor its components (free Ni2+ metal ions and organic linker, DHTP) showed cytotoxicity against human hepatocytes (HepG2) nor breast cancer cell line (MCF7).57
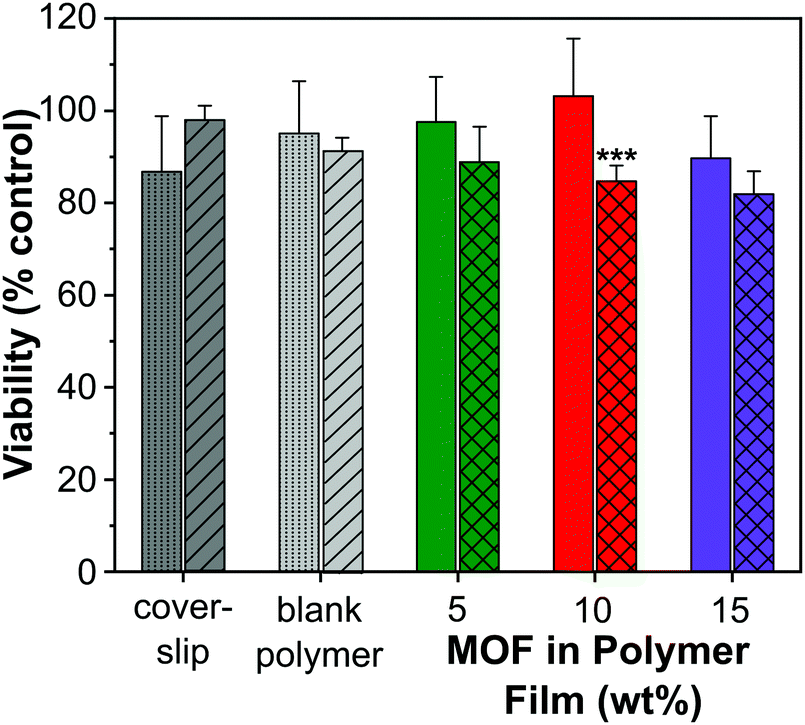 | ||
| Fig. 7 In vitro cytotoxicity test results for polyurethane films containing 5, 10 and 15 wt% CPO-27 (Ni) with (crosses) and without (solid) NO loading in HUVEC cells (N = 6). Controls (coverslip and blank polymer) for experiments employing NO-free (dots) and NO-loaded (hashes) films. There was a significant difference (***) between NO-free and NO-loaded datasets (2-factor ANOVA; P < 0.01), with significance (P < 0.001; Bonferroni post-test) for the 10 wt % MOF polymer film. | ||
However, the data show that there is a gradual downward drift of cell viability with increasing wt% of NO-loaded MOF (88.8%, 84.6% and 81.9% for 5, 10 and 15 wt% MOF loading, respectively). This apparent cytotoxic effect of NO-loaded MOF is statistically significantly different from the equivalent unloaded control specifically at the 10 wt% MOF composition. It is worth noting, however, that the difference in viability between cells treated with 10 wt% NO-loaded MOF (84.6% viability) and those exposed to coverslip alone (86.8% viability) is very small. Ultimately, our inference from the data as a whole is that any toxicity associated with the films was due to NO itself, but that the effect is not significant at 5 wt% MOF composition and very low, even at 15% composition (81.9% viability).
Taken together, the toxicology data presented indicate that the concept of NO-loaded MOFs constitutes a relatively non-toxic intervention at the compositions required for antimicrobial effects. It is important to register nevertheless, that delivery of too much NO has the potential to induce cellular toxicity (as is known58), highlighting the importance of optimal tuning of NO loading and release kinetics for antimicrobial effects without concomitant cellular toxicity.
Antibacterial performance
Antibacterial efficacy was assessed against both Gram negative and Gram positive bacteria (E. coli and S. aureus, respectively) as described in the Experimental section. Both bacterial strains are highly relevant as they are readily transmitted during procedures using medical devices such as catheters. For example, urinary tract infections are the most common type of HAI, usually caused by E. coli (a 90% prevalence rate). One third of the population carries S. aureus, it is often transferred from skin into the bloodstream during the catheterization procedure. Furthermore, S. aureus is the most prevalent organism in hospitals that cause most of the cases of HAI.Results, taken as the average of six replicates and expressed as the number of bacterial colony forming units per area (cfu cm−2), are illustrated in Fig. 8. Numerical data, including log reduction and standard deviation values, are tabulated in Tables S2 and S3 (ESI†). A log reduction of 3 or greater in bacterial cell counts compared to a control is required for the agent to be considered active.
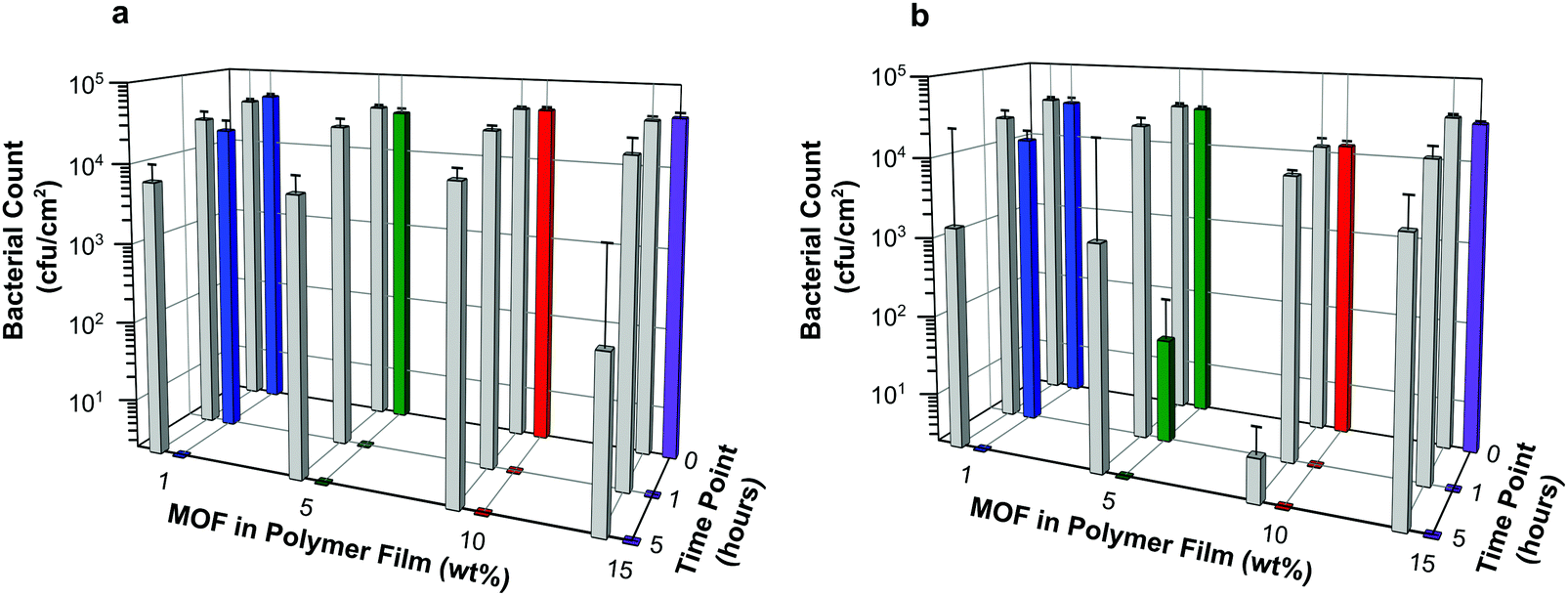 | ||
| Fig. 8 Results of antibacterial assay conducted against E. coli (a) and S. aureus (b) represented as average bacterial counts (cfucm−2) versus time (0, 1 and 5 hours) for each film composition. Each data point is the average of six replicates (i.e. N = 6). Grey bars illustrate data for blank polymer films (containing 0 wt% MOF). | ||
The data suggest that the concentrations of NO being released from the films are indeed able to elicit antibacterial activity. Regardless of bacterial species tested in these experiments, the number of colony forming units per cm2 at time 0 for each sample is similar or equal to that observed for the control containing no MOF. This suggests that antibacterial action is not instantaneous in this test system. However, within the first hour of exposure, a dramatic reduction in bacterial counts is observed for all samples with MOF loading levels >1 wt%. Furthermore after 5 hours, no bacterial colony forming units were counted on any of the test plates indicating that all the bacteria had been killed. This demonstrates that the test films have a potent bactericidal effect.
The control for this assay were blank polymer films containing no MOF. The data appear to show that these films impart some antibacterial activity at the longer contact times. However, this is a result of there being complete cell death on a few of the control films caused by the inoculum inadvertently drying out in those particular wells. This results in a marked reduction in averaged cell counts, as well as large standard deviations for these controls. Nevertheless, on comparing the bacterial counts of control versus sample at each time point, the difference in log reduction between controls and test articles is significant (>log 3) and indicates a clear added benefit of incorporating NO releasing MOFs into the polymer.
Against both E. coli and S. aureus, films containing MOF at 10 wt% or higher show no bacterial counts on the plates at the 1 hour time point. This equates to a >log 4 reduction in cell counts compared with time 0 and can be regarded as bactericidal activity (or “complete kill”). There are subtle differences in the efficacy of these NO releasing films towards Gram negative and Gram positive bacteria. Against E. coli, films containing MOF at 5 wt% or higher show bactericidal activity within 1 hour, see Fig. 8a. A time dependant response is observed for films containing 1 wt% MOF; in this case log reductions of 0.33 and >4 are recorded at 1 hour and 5 hours, respectively against time 0. Against S. aureus, a similar time dependent response is observed for films containing 1 wt% MOF (log reductions of 0.36 and >4 at 1 hour and 5 hours, respectively), and perhaps also from films containing 5 wt% (log reductions of 2.78 and >4 at 1 hour and 5 hours, respectively). Complete kill is observed for all other concentrations at time points 1 and 5 hours (log reductions of >3.7 and >4 for films containing 10 wt% and 15 wt% MOF, respectively, at both time points).
The antibacterial testing data has shown that for both Gram negative and Gram positive bacteria, bacteriostatic or bactericidal efficacy can be achieved by relatively non-toxic formulations containing low loading levels of the active agent. As indicated above, achieving antibacterial efficacy from less than 20 wt% MOF loading level is important to avoid any detriment to mechanical properties. These data indicate that this is indeed possible.
Conclusion
We have shown that CPO-27 (Ni) can be readily formulated into standalone polyurethane films using facile solvent casting methods, and that these films can be activated and loaded with NO gas. Our results have shown that antibacterial concentrations of NO can be released from the films when in contact with a moist environment, and that antibacterial performance can be achieved within 1 hour of contact time employing films containing 5 wt% MOF and higher. NO release profiles akin to that of 5 wt% MOF films (i.e. a maximum total release of 0.04 mmol g−1 of film and single digit ppm concentrations of NO delivered over one hour under 11% RH) may therefore be regarded as an indirect predictor of 1 hour antibacterial activity from test samples under 100% RH. It is notable that this strong performance (>log 3 reduction at 1 hour) is achieved from formulations that show relatively low toxicity which is not always the case for NO releasing formulations.The data indicate that care should be taken when formulating NO-releasing MOFs for antibacterial applications as the polymer matrix will influence the total concentration of NO that can be adsorbed/released, as well as the duration of release. Our findings also suggest that the efficiency of NO release is affected by the wt% of MOF within the film. While elongation of release may be welcomed, it is important that such factors governed by the formulation are carefully considered to ensure the desired end product performance is achieved and achieved efficiently. Furthermore, there is a balance to be found between antibacterial efficacy, mechanical integrity and cytotoxicity for these NO loaded MOF films. It is evident that the NO-adsorption/release performance of MOF–polymer films is not trivial and warrants much further investigation. Nevertheless, the present study is an important step forward in the development and understanding of NO-releasing MOFs for medical applications as it confirms they can perform effectively when included in a product-relevant formulation and highlights key aspects that must be considered during their application.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Miss Katie Ridley for assisting in the preparation and analysis of the films, and BluTest Laboratories Ltd for conducting the antibacterial testing. We also gratefully thank the following funding sources for financial support: Scottish Enterprise (POC13), EPSRC (EP/K005499/1 and Capital for Great Technologies grant EP/L017008/1), European Social Fund Studentship and the University of St Andrews School of Chemistry.References
- World Health Organisation, Report on the Burden of Endemic Health Care-Associated Infection Worldwide, World Health Organisation, Geneva, 2011, ISBN 978-92-4-150150-7.
- S. S. Magill, J. R. Edwards, W. Bamberg, Z. G. Beldavs, G. Dumyati, M. A. Kainer, R. Lynfield, M. Maloney, L. McAllister-Hollod, J. Nadle, S. M. Ray, D. L. Thompson, L. E. Wilson and S. K. Fridkin, N. Engl. J. Med., 2014, 370(13), 1198 CrossRef CAS.
- The Joint Commission, Joint Commission Resources and Joint Commission International, Preventing Central Line–Associated Bloodstream Infections: A Global Challenge, a Global Perspective, Joint Commission Resources, Oak Brook, IL, 2012.
- R. C. L. Feneley, I. B. Hopley and P. N. T. Wells, J. Med. Eng. Technol., 2015, 39(8), 459 CrossRef.
- L. M. Weiner-Lastinger, S. Abner, J. R. Edwards, A. J. Kallen, M. Karlsson, S. S. Magill, D. Pollock, I. See, M. M. Soe, M. S. Walters and M. A. Dudeck, Infect. Control Hosp. Epidemiol., 2019, 1 Search PubMed.
- P. Singha, J. Locklin and H. Handa, Acta Biomater., 2017, 50, 20 CrossRef CAS.
- A. Panáček, L. Kvítek, M. Smékalová, R. Večeřová, M. Kolář, M. Röderová, F. Dyčka, M. Šebela, R. Pruce1, O. Tomanec and R. Zbořil, Nat. Nanotechnol., 2018, 13, 65 CrossRef.
- G. F. Calogiuri, E. Di Leo, A. Trautmann, E. Nettis, A. Ferrannini and A. Vacca, J. Allergy Ther., 2013, 4(4), 1000141 Search PubMed.
- (a) T. M. Dawson and V. L. Dawson, Neuroscientist, 1995, 1(1), 7 CrossRef CAS; (b) A. Butler and R. Nicholson, Life, Death and Nitric Oxide, Royal Society of Chemistry, Cambridge, 2003, ISBN 978-0-85404-686-7 Search PubMed.
- M. L. Jones, J. G. Ganopolsky, A. Labbé, C. Wahl and S. Prakash, Appl. Microbiol. Biotechnol., 2010, 88, 401 CrossRef CAS.
- D. O. Schairer, J. S. Chouake, J. D. Nosanchuk and A. J. Friedman, Virulence, 2012, 3(3), 271 CrossRef.
- D. Margel, M. Mizrahi, G. Regev-Shoshani, M. Ko, M. Moshe, R. Ozalvo, L. Shavit-Grievink, J. Baniel, D. Kedar, O. Yossepowitch, D. Lifshitz, A. Nadu, D. Greenberg and Y. Av-Gay, PLoS One, 2017, 12(4), e0174443 CrossRef.
- G. Regev-Shoshani, M. Ko, C. Miller and Y. Av-Gay, Antimicrob. Agents Chemother., 2010, 54(1), 273 CrossRef CAS.
- M. M. Reynolds and V. B. Damodaran, US Pat., 20150004257A1, 2015 Search PubMed.
- J. Pant, M. J. Goudie, S. M. Chaji, B. W. Johnson and H. Handa, J. Biomed. Mater. Res., Part B, 2018, 106B(8), 2849 CrossRef.
- K. H. Homeyer, M. J. Goudie, P. Singha and H. Handa, ACS Biomater. Sci. Eng., 2019, 5, 2021 CrossRef CAS.
- C. Janiak and J. K. Vieth, New J. Chem., 2010, 34(11), 2366 RSC.
- P. Silva, S. M. F. Vilela, J. P. C. Tome and F. A. A. Paz, Chem. Soc. Rev., 2015, 44(19), 6774 RSC.
- (a) D. DeSantis, J. A. Mason, B. D. James, C. Houchins, J. R. Long and M. Veenstra, Energy Fuels, 2017, 31, 2024 CrossRef CAS; (b) R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47(27), 4966 CrossRef CAS.
- R. C. Huxford, J. D. Rocca and W. Lin, Curr. Opin. Chem. Biol., 2010, 14, 262 CrossRef CAS.
- A. A. Simagina, M. V. Polynski, A. V. Vinogradov and E. A. Pidko, Russ. Chem. Rev., 2018, 87, 831 CrossRef CAS.
- R. K. Alavijeh, S. Beheshti, K. Akhbari and A. Morsali, Polyhedron, 2018, 156, 257 CrossRef.
- G. Wyszogrodzka, B. Marszałek, B. Gil and P. Dorozynski, Drug Discovery Today, 2016, 21(6), 1009 CrossRef CAS.
- (a) M. Esfahanian, M. Ali Ghasemzadeh and S. M. H. Razavian, Artif. Cells Blood Substit. Biotechnol., 2019, 47(1), 2024 CAS; (b) H. Nabipour, M. H. Sadra and G. R. Bardajee, New J. Chem., 2017, 41, 7364 RSC.
- D. F. Sava Gallis, K. S. Butler, J. O. Agola, C. J. Pearce and A. A. McBride, ACS Appl. Mater. Interfaces, 2019, 11, 7782 CrossRef CAS.
- A. Karakeçili, B. Topuz, S. Korpayev and M. Erdek, Mater. Sci. Eng., C, 2019, 105, 110098 CrossRef.
- G. Kumar, A. Kant, M. Kumar and D. T. Masram, Inorg. Chim. Acta, 2019, 496, 119036 CrossRef CAS.
- Z. Song, Y. Wu, Q. Cao, H. Wang, X. Wang and H. Han, Adv. Funct. Mater., 2018, 28, 1800011 CrossRef.
- H. Zhao, S. Hou, X. Zhao and D. Liu, J. Chem. Eng. Data, 2019, 64, 1851 CrossRef CAS.
- A. C. McKinlay, P. K. Allan, C. L. Renouf, M. J. Duncan, P. S. Wheatley, S. J. Warrender, D. Dawson, S. E. Ashbrook, B. Gil, B. Marszalek, T. Düren, J. J. Williams, C. Charrier, D. K. Mercer, S. J. Teat and R. E. Morris, APL Mater., 2014, 2, 124108 CrossRef.
- X. Zhang, L. Liu, L. Huang, W. Zhang, R. Wang, T. Yue, J. Sun, G. Li and J. Wang, Nanoscale, 2019, 11, 9468 RSC.
- N. Bhardwaj, S. K. Pandey, J. Mehta, S. K. Bhardwaj, Ki-H Kim and A. Deep, Toxicol. Res., 2018, 7, 931 CrossRef CAS.
- A. C. McKinlay, B. Xiao, D. S. Wragg, P. S. Wheatley, I. L. Megson and R. E. Morris, J. Am. Chem. Soc., 2008, 130, 10440 CrossRef CAS.
- E. D. Bloch, W. L. Queen, S. Chavan, P. S. Wheatley, J. M. Zadrozny, R. Morris, C. M. Brown, C. Lamberti, S. Bordiga and J. R. Long, J. Am. Chem. Soc., 2015, 137, 3466 CrossRef CAS.
- D. Cattaneo, S. J. Warrender, M. J. Duncan, R. Castledine, N. Parkinson, I. Haley and R. E. Morris, Dalton Trans., 2016, 45, 618 RSC.
- A. C. McKinlay, J. F. Eubank, S. Wuttke, B. Xiao, P. S. Wheatley, P. Bazin, J.-C. Lavalley, M. Daturi, A. Vimont, G. De Weireld, P. Horcajada, C. Serre and R. E. Morris, Chem. Mater., 2013, 25, 1592 CrossRef CAS.
- S. T. Gregg, Q. Yuan, R. E. Morris and B. Xiao, Mater. Today Commun., 2017, 12, 95 CrossRef CAS.
- M. J. Ingleson, R. Heck, J. A. Gould and M. J. Rosseinsky, Inorg. Chem., 2009, 48, 9986 CrossRef CAS.
- A. H. Khan, K. Peikert, F. Hoffmann, M. Fröba and M. Bertmer, J. Phys. Chem. C, 2019, 123, 4299 CrossRef CAS.
- D. Cattaneo, S. J. Warrender, M. J. Duncan, C. J. Kelsall, M. K. Doherty, P. D. Whitfield, I. L. Megson and R. E. Morris, RSC Adv., 2016, 6, 14059 RSC.
- M. Choi, S. Park, K. Park, H. Jeong and J. Hong, ACS Biomater. Sci. Eng., 2019, 5, 1378 CrossRef CAS.
- A. L. Urzedo, M. C. Goncalves, M. H. M. Nascimento, C. B. Lombello, G. Nakazato and A. B. Seabra, ACS Biomater. Sci. Eng., 2020, 6, 2117 CrossRef CAS.
- R. E. Morris, US Pat., 20130171228A1, 2013 Search PubMed.
- L. Ma, F. Svec, Y. Lv and T. Tan, Chem. – Asian J., 2019, 14, 3502 CrossRef CAS.
- A. Elrasheedy, N. Nady, M. Bassyouni and A. El-Shazly, Membranes, 2019, 9, 88 CrossRef CAS.
- M. C. Jen, M. C. Serrano, R. van Lith and G. A. Ameer, Adv. Funct. Mater., 2012, 22, 239 CrossRef CAS.
- J. Park, J. Kim, K. Singha, D.-K. Han, H. Park and W. J. Kim, Biomaterials, 2013, 34, 8766 CrossRef CAS.
- J. Redfern, L. Geerts, J. Won Seo, J. Verran, L. Tosheva and L. H. Wee, ACS Appl. Nano Mater., 2018, 1, 1657 CrossRef CAS.
- (a) T. A. Rickhoff, E. Sullivan, L. K. Werth, D. S. Kissel and J. J. Keleher, J. Appl. Polym. Sci., 2019, 46978 CrossRef; (b) H. S. Rodriguez, J. P. Hinestroza, C. Ochoa-Puentes, C. A. Sierra and C. Y. Soto, J. Appl. Polym. Sci., 2014, 40815 Search PubMed.
- H. E. Emam, O. M. Darwesh and R. M. Abdelhameed, Colloids Surf., B, 2018, 165, 219 CrossRef CAS.
- J. Quirós, K. Boltes, S. Aguado, R. G. de Villoria, J. J. Vilatela and R. Rosal, Chem. Eng. J., 2015, 262, 189 CrossRef.
- M. Liu, Lei Wang, X. Zheng and Z. Xie, ACS Appl. Mater. Interfaces, 2017, 9, 41512 CrossRef CAS.
- P. D. C. Dietzel, V. Besikiotis and R. Blom, J. Mater. Chem., 2009, 19, 7362 RSC.
- P. D. C. Dietzel, B. Panella, M. Hirscher, R. Blom and H. Fjellvag, Chem. Commun., 2006, 959 RSC.
- S. M. Vornholt, M. J. Duncan, S. J. Warrender, R. Semino, N. A. Ramsahye, G. Maurin, M. W. Smith, J. C. Tan, D. N. Miller and R. E. Morris, manuscript submitted, 2020.
- R. E. Morris and P. S. Wheatley, US Pat., 8486451B2, 2013 Search PubMed.
- À. Ruyra, A. Yazdi, J. Espín, A. Carné-Sánchez, N. Roher, J. Loernzo, I. Imaz and D. Maspoch, Chem. – Eur. J., 2015, 21, 2508 CrossRef.
- J. B. Hibbs Jr., R. R. Taintor, Z. Vavrin and E. M. Rachlin, Biochem. Biophys. Res. Commun., 1988, 157(1), 87 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00650e |
| This journal is © The Royal Society of Chemistry 2020 |