
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of covalent organic framework derived Fe-COFs composites as a peroxidase-mimicking artificial enzyme†
Liya
Zhou
,
Xucong
Luo
,
Jing
Gao
,
Guanhua
Liu
 ,
Li
Ma
,
Ying
He
,
Zhihong
Huang
and
Yanjun
Jiang
*
,
Li
Ma
,
Ying
He
,
Zhihong
Huang
and
Yanjun
Jiang
*
Hebei University of Technology, China. E-mail: yanjunjiang@hebut.edu.cn
First published on 3rd February 2020
Abstract
An artificial catalyst (Fe-COFs) with peroxidase-like activity was successfully synthesized at room temperature and applied to catalyze the reaction between TMB and H2O2. This catalytic system can be used not only to detect residual hydrogen peroxide (H2O2) in milk efficiently, but also to degrade rhodamine B (RhB) in waste water.
With the deterioration of the environment and the continuous shortage of resources and energy, environment-friendly enzymatic catalysis has become more and more popular. Artificial enzymes are a new kind of synthetic material with enzyme-like properties, which are more stable and economical than natural enzymes, and they can be widely used in biological, agricultural, medical, environmental and other fields as alternatives to enzymes.1,2 To date, a large number of artificial enzymes such as metal oxides,3,4 noble metal nanoparticles,5,6 metal sulfides,7,8 metal–organic frameworks (MOFs),1,9 and their derived nanocomposites10,11 have been explored to exhibit enzyme-like activity.
Covalent organic frameworks (COFs) are constructed with lightweight elements (i.e., H, B, C, N, and O) by covalent bonding, and they have emerged as a new kind of material.12,13 The multiple advantages of COFs make them applicable in diverse fields like sensing,14 gas storage,15 optoelectronics,16 and catalytic fields.17,18 The imine-type ligand was one of the most versatile ligands studied in coordination chemistry. The imine-linked metal-ion-incorporated COF material Pd/COF-LZU1 had been synthesized and used as a catalyst in the Suzuki–Miyaura coupling reaction, which showed excellent catalytic activity, high stability and easy recyclability.18 Co/Co(OH)2 embedded in the pores of COFs was also prepared and exhibited excellent catalytic activity in the hydrogenation of nitro compounds and nitrile to amine reactions.19 According to previous reports,19–22 imine-type (Schiff base) ligands coordinate with a variety of noble metal ions and imine-based metal complexes had also been used as catalysts. Compared with MOF mimics, COF mimics possessed higher chemical stability and outstanding structural stability under different harsh conditions. However, there are fewer reports on the synthesis of non-noble metal-COF materials as an artificial enzyme via a simple method at room temperature (Scheme 1).
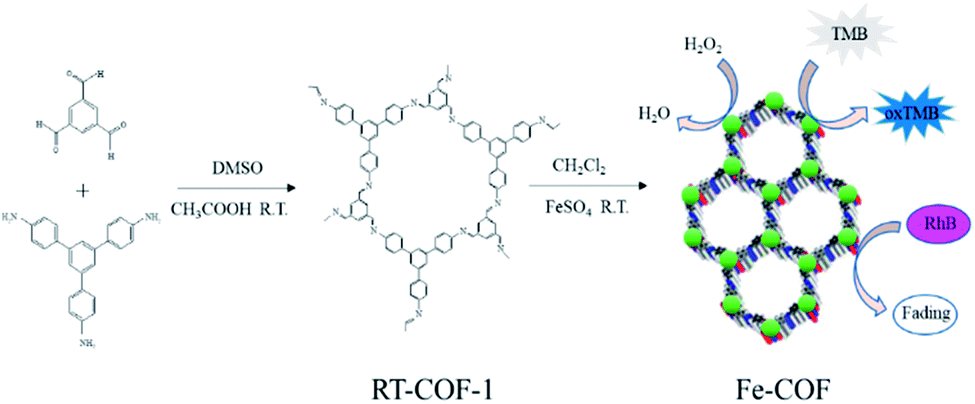 | ||
| Scheme 1 A schematic illustration of the synthesis and catalytic reaction of Fe-COFs. | ||
In this work, Fe based COFs (Fe-COFs) were prepared under mild conditions at room temperature, and the entire operation process was simple and convenient.23 To our surprise, the obtained Fe-COFs possess high peroxidase-like activity and excellent stability under harsh conditions and show great potential in other fields.
According to a previous report, the host COF24 (denoted as RT-COF-1) was synthesized and then the Fe-COFs were prepared. The procedure is described in the ESI.† SEM images showed the flakes of RT-COF-1 (Fig. S1a†), and Fe-COFs showed the formation of uniform aggregates of flakes (Fig. S1b†). Compared to RT-COF-1, the morphology of Fe-COFs remained basically intact, which indicated that the introduction of iron did not break the skeleton of RT-COF-1. The TGA curves of RT-COF-1 showed that its weight loss started from 500 °C, which is the same as that in previous reports24 (Fig. S2†). In addition, the weight loss of Fe-COFs also started at 500 °C, indicating that the introduction of Fe atoms did not have an obvious influence on the framework structure. The pore volume and surface area of RT-COF-1 were measured by nitrogen adsorption–desorption analysis at 77 K (Fig. S11†). The Brunauer–Emmett–Teller (BET) surface areas were found to be 302 m2 g−1 and the pore volume was calculated to be 0.199 cm3 g−1; these results were close to those of previous reports.24 The FT-IR spectra of RT-COF-1 and Fe-COFs are shown in Fig. 1a. Compared to the spectrum of RT-COF-1, the shift in the peak of νC–N for Fe-COFs could be attributed to the coordination bond between Fe and N atoms. The Raman spectra of the RT-COF-1 and Fe-COFs are shown in Fig. S10.† The strongest G+ peak at 1633.1 cm−1 is an indication of a well-ordered sp2 planar structure. The G− peak at 1589.2 cm−1 is related to the distortion. The D peak at 1353.2 cm−1 is related to carbon defects in the layer structure. Comparing the Raman results of RT-COF-1 and Fe-COFs, it can be observed that the G band shifts from 1633.1 to 1631.7 cm−1 and 1589.2 to 1594.7 cm−1, and the D band shifts from 1353.2 to 1349.0 cm−1, which could be attributed to the coordination bond between Fe and N atoms.22,25,26 The XPS results (Fig. S3†) confirmed the existence of the Fe element. The XPS spectra of Fe 2p showed that the peaks of Fe 2p1/2 and Fe 2p3/2, which could be split into two signals, and the two satellite peaks (Fig. 1d). The peaks at 715.9 eV and 727.2 eV were the satellite peaks. The peaks at 724.1 eV and 712.2 eV correspond to Fe(III), the other peaks at 722.6 eV and 709.6 eV are assigned to Fe(II).27–30 The N 1s spectrum with RT-COF-1 indicated that the nitrogen peak was at 398.5 eV (Fig. 1b). The measurements with Fe-COFs showed characteristic peaks at 398.5 eV and 401.3 eV (Fig. 1c). The peak at 398.5 eV could be assigned to the imine bond (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and the peaks at 401.3 eV indicated that iron was coordinatively bonded (Fe–N) to the imine nitrogen atoms of the RT-COF-1.18,31 All these results confirmed that Fe-COFs were successfully prepared.
N) and the peaks at 401.3 eV indicated that iron was coordinatively bonded (Fe–N) to the imine nitrogen atoms of the RT-COF-1.18,31 All these results confirmed that Fe-COFs were successfully prepared.
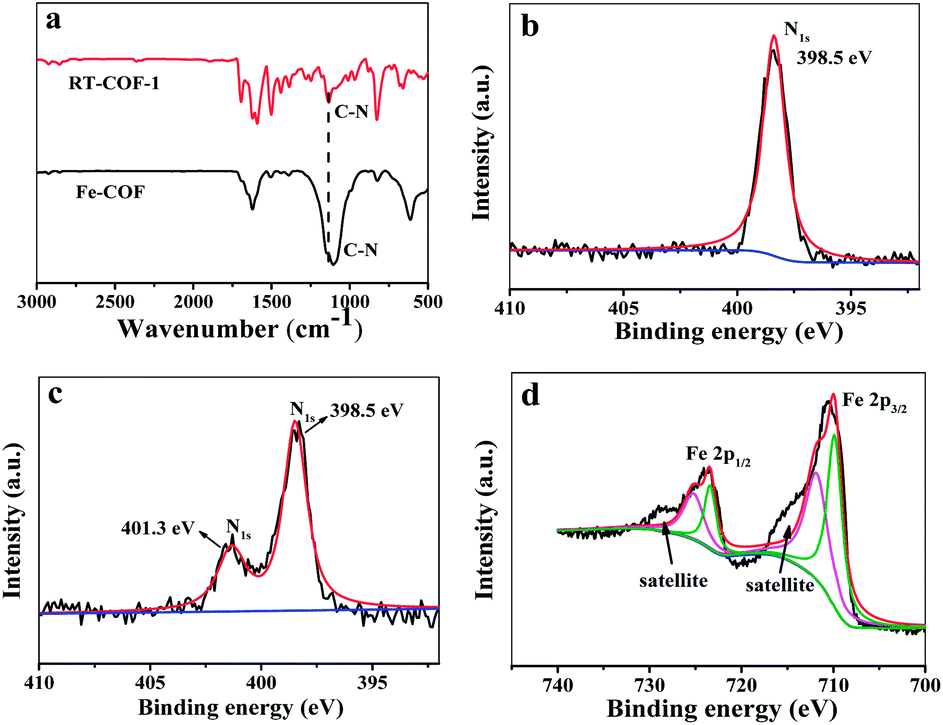 | ||
| Fig. 1 (a) FT-IR spectra of RT-COF-1 and Fe-COFs. (b) N 1s XPS spectra of RT-COF-1. (c) N 1s XPS spectra of Fe-COFs. (d) Fe 2p XPS spectra of Fe-COFs. | ||
The peroxidase-like activity of Fe-COFs was demonstrated by using TMB as the substrate. The catalytic activities of Fe-COFs used in five different reaction systems are shown in Fig. S4.† It could be seen that the color change occurred only in the TMB + H2O2 + Fe-COFs reaction system and the absorbance was intense at 652 nm. However, the others were colorless and exhibited no absorption peak, demonstrating that Fe-COFs possessed peroxidase-like activity and could oxidize TMB to produce blue oxTMB,32 while RT-COF-1 has no catalytic function. According to previous reports,33 the peroxidase mimics could catalyze H2O2 to generate hydroxyl radicals (˙OH), which were able to oxidize TMB to produce oxTMB. To prove the catalytic mechanism of Fe-COFs, terephthalic acid (TA) was selected as a fluorescent probe that could connect with ˙OH to form a fluorescent 2-hydroxyterephthalic acid.34 As shown in Fig. 2a, the fluorescence intensity increased with the increase in the concentration of the Fe-COFs, which demonstrated that Fe-COFs could catalyze H2O2 to produce a large amount of ˙OH. However, when Fe-COFs weren't added to the reaction system, the fluorescence peak could not be observed at 425 nm. For further verifying the role of ˙OH in the catalytic system, sulfite was selected as the radical inhibitor, which could combine with reactive oxygen species to prevent the oxidation of TMB.35 As shown in Fig. 2b, the reaction system turned into blue in the absence of sulfite and a distinct absorption peak at 652 nm appeared. However, after sulfite was added, the reaction solution became colorless and no absorption peak was observed at 652 nm. These results showed that ˙OH plays an important role in this catalytic system and the catalytic mechanism of Fe-COFs could be attributed to the ˙OH, decomposed by H2O2.
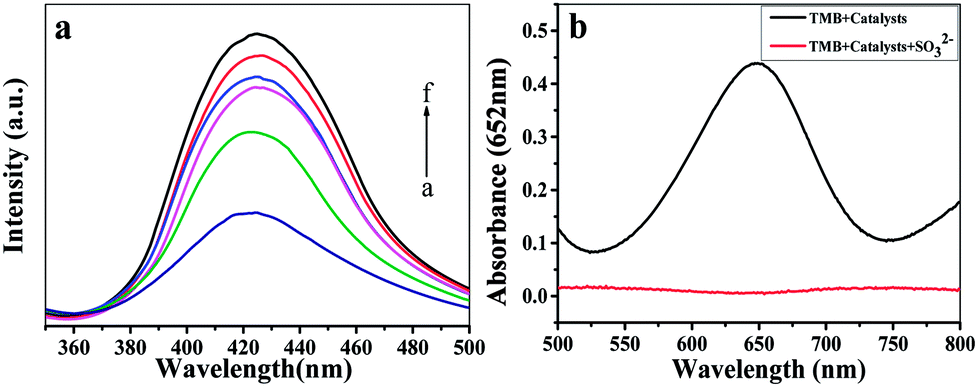 | ||
| Fig. 2 (a) The fluorescence emission spectra of terephthalic acid in the presence of H2O2 and different concentrations of Fe-COFs ((a–f) 400, 500, 600, 700, 800, and 900 μg mL−1). (b) UV-vis absorption spectra of the peroxidase-like catalytic system with Fe-COFs in the absence and the presence of 300 μM sodium sulfite. | ||
Like the natural peroxidase, the reaction condition was crucial for the chemical reactivity of Fe-COFs. As shown in Fig. S5a,† the optimal pH for the catalytic activity of Fe-COFs was 3.5. However, iron ions could be released from the Fe-COFs in the buffer solutions of pH < 5.0. In view of the stability and catalytic activity of the catalyst, 5.0 was selected for the following experiments.23 The absorbance reached the maximum when the incubation temperature was 35 °C (Fig. S5b†). Moreover, it could be seen (Fig. S5c†) that the absorbance of oxTMB increased when more H2O2 was added to the reaction system, but it decreased when an excessive amount of substrate was involved. Thus, the optimal concentration of H2O2 was 100 mM. As seen in Fig. S5d,† the catalytic activity of Fe-COFs was improved with the increase of the reaction concentration of Fe-COFs and reached the maximum at 800 μg mL−1. When the concentration was higher than 800 μg mL−1, the catalytic activity reached a stable value. Accordingly, the optimal conditions such as a pH of 5, incubation temperature of 35 °C, catalyst concentration of 800 μg mL−1 and H2O2 concentration of 100 mM were selected and used in subsequent experiments.
The peroxidase-catalyzed steady-state kinetics of Fe-COFs were investigated by varying the concentration of H2O2 and TMB. As shown in Fig. 3a–d, the data were calculated with the Lineweaver–Burk plot, and the results indicated that the reaction followed the typical Michaelis–Menten equation. The steady-state kinetics fitting parameters of Km and Vmax values for H2O2 and TMB were calculated and are listed in Table S1.† It is known that a smaller Km value means that there is a greater affinity between the enzyme mimics and substrate.36 From Table S1,† it can be seen that the Km values of Fe-COFs were much lower than the native HRP and the other artificial enzymes, indicating that Fe-COFs had a higher affinity to TMB and H2O2 than the natural enzymes and the other enzyme mimics. Moreover, the larger Vmax values further confirmed the enhanced affinity of the composite.
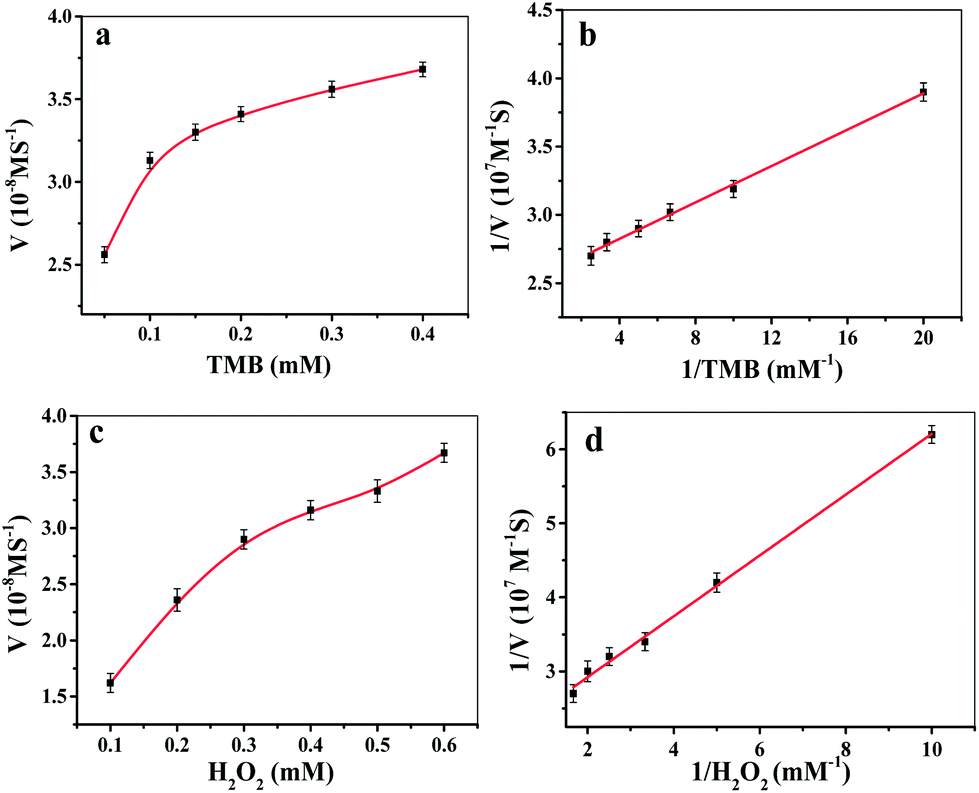 | ||
| Fig. 3 Steady-state kinetic assay of the Fe-COFs composite by (a) changing the concentrations of TMB, and (b) changing the concentrations of H2O2, and the double-reciprocal model of the concentration of H2O2 (c) and TMB (d). | ||
Under the optimal experimental conditions, a simple colorimetric method was developed for the detection of H2O2 by using Fe-COFs as the catalyst. As shown in Fig. S6,† the absorbance of oxTMB at 652 nm increased gradually with increasing the concentration of H2O2 from 10 μM to 2000 μM (R2 = 0.9998). The limit of detection (LOD) was 5.6 μM (S/N = 3), lower than that of the other peroxidase mimics (Table S2†). The stability of Fe-COFs under different harsh conditions was also evaluated.37 The results indicated that more than 60% of the catalytic activity of Fe-COFs was retained after 2 h of incubation under different pHs (3–9), suggesting the good pH stability (Fig. S7a†). The effects of temperature (25–55 °C) were also examined. After 2 h of incubation, more than 59% of the catalytic activity of Fe-COFs was retained (Fig. S7b†). Furthermore, more than 80% of activity can be retained for Fe-COFs even when soaked in aqueous solution for 30 days at room temperature (Fig. S7c†).
The catalytic system of Fe-COFs could be used to detect residual H2O2 in milk samples.9,38,39 As shown in Table S3,† the recoveries of H2O2 in milk products were in the range from 96.27 to 100.70% and the relative standard deviation (RSD) was in the range from 0.71 to 4.55%, which indicated that no residual H2O2 could be detected in milk samples. These results indicated that this catalytic system exhibited a high credibility and could be applied to detection of H2O2 in milk. In order to investigate whether the potential interfering substances including glucose, urea, alanine, glycine, L-cysteine, glutathione, ascorbic acid, K+, Ca2+, Na+, Zn2+, Cl−, SO42− and NO3− might affect the determination of H2O2 in milk samples, the selectivity of Fe-COFs for the determination of H2O2 was tested. The results are shown in Fig. S8,† and the system with added H2O2 had higher absorbance than that with these interfering substances together with the blank sample at 652 nm. These interferents had negligible effects on the determination of H2O2 in milk samples, so this proved that the colorimetric sensor of Fe-COFs had an excellent selectivity.
The Fe-COFs catalytic system could be used not only to detect H2O2 but also to degrade RhB.40,41 Fenton oxidation was usually reported to have great potential in degrading dyes. The mechanism was used to accelerate the formation of ˙OH, which could oxidize and degrade organic matter. As shown in Fig. S9,† the RhB solution turned almost colorless after adding Fe-COFs to the system, and no absorption peak appeared at 554 nm. Fig. 4a shows that the RhB degradation efficiency increased with the increase of reaction time. After adding Fe-COFs, the degradation efficiency of RhB reached 89.45% within 30 min, which indicated that Fe-COFs could effectively degrade dyes. The natural logarithm of the real-time concentration and the initial concentration ratio was fitted, which showed that the kinetics of RhB degradation followed the pseudo-first-order kinetic model:
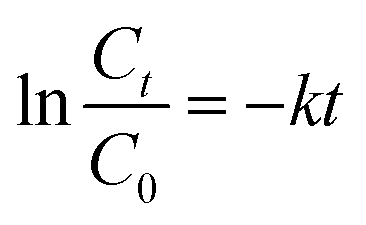
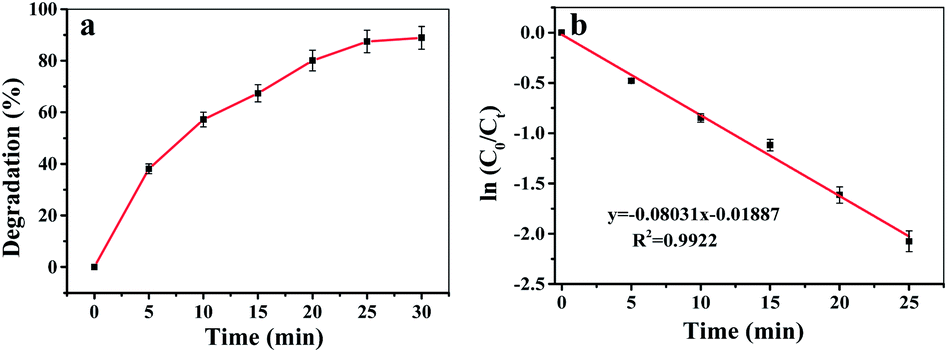 | ||
| Fig. 4 (a) Degradation efficiency of RhB at the times of 0, 5, 10, 15, 20, 25 and 60 min by the Fe-COFs catalytic system. (b) RhB degradation kinetics fitting curve. | ||
In summary, Fe-COFs with superior peroxidase-like activity was first synthesized at room temperature and were effective, fast, and simple and could save energy and costs. Fe-COFs exhibited a better stability and a stronger affinity to substrates. Based on this peroxidase mimic, a simple, highly sensitive and low-cost colorimetric assay method was established for H2O2 detection with a wide linear range (10 μM to 2000 μM) and a low LOD of 5.6 μM. This peroxidase mimic was further applied to detect H2O2 in milk with superior recovery and RSD values. In addition, the Fe-COFs catalytic system was also used to degrade RhB, and the degradation efficiency reached 88.9% within 30 min. We believed that the Fe-COFs catalytic system has great potential in environmental chemistry and food chemistry and might replace natural enzymes in some fields in the future.
Conflicts of interest
There are no conflicts to declare.Notes and references
- L. Ai, L. Li, C. Zhang, J. Fu and J. Jiang, Chem.–Eur. J., 2013, 19, 15105–15108 CrossRef CAS PubMed.
- H. Chen, Q. Qiu, S. Sharif, S. Ying, Y. Wang and Y. Ying, ACS Appl. Mater. Interfaces, 2018, 10, 24108–24115 CrossRef CAS PubMed.
- L. Han, P. Liu, H. Zhang, F. Li and A. Liu, Chem. Commun., 2017, 53, 5216–5219 RSC.
- L. Gao, J. Zhuang, L. Nie, J. Zhang, Y. Zhang, N. Gu, T. Wang, J. Feng, D. Yang, S. Perrett and X. Yan, Nat. Nanotechnol., 2007, 2, 577–583 CrossRef CAS PubMed.
- Y. Jv, B. Li and R. Cao, Chem. Commun., 2010, 46, 8017–8019 RSC.
- M. Ma, Y. Zhang and N. Gu, Colloids Surf., A, 2011, 373, 6–10 CrossRef CAS.
- Z. Dai, S. Liu, J. Bao and H. Ju, Chem.–Eur. J., 2009, 15, 4321–4326 CrossRef CAS PubMed.
- W. He, H. Jia, X. Li, Y. Lei, J. Li, H. Zhao, L. Mi, L. Zhang and Z. Zheng, Nanoscale, 2012, 4, 3501–3506 RSC.
- Z. Qi, L. Wang, Q. You and Y. Chen, Biosens. Bioelectron., 2017, 96, 227–232 CrossRef CAS PubMed.
- M. Chi, S. Chen, M. Zhong, C. Wang and X. Lu, Chem. Commun., 2018, 54, 5827–5830 RSC.
- Y. Jiang, N. Song, C. Wang, N. Pinna and X. Lu, J. Mater. Chem. B, 2017, 5, 5499–5505 RSC.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- S. Zhang, Q. Yang, C. Wang, X. Luo, J. Kim, Z. Wang and Y. Yamauchi, Adv. Sci., 2018, 5, 1801116 CrossRef PubMed.
- S. Y. Ding, M. Dong, Y. W. Wang, Y. T. Chen, H. Z. Wang, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2016, 138, 3031–3037 CrossRef CAS PubMed.
- Y. Zeng, R. Zou, Z. Luo, H. Zhang, X. Yao, X. Ma, R. Zou and Y. Zhao, J. Am. Chem. Soc., 2015, 137, 1020–1023 CrossRef CAS PubMed.
- M. Dogru and T. Bein, Chem. Commun., 2014, 50, 5531–5546 RSC.
- F. Li, L.-G. Ding, B.-J. Yao, N. Huang, J.-T. Li, Q.-J. Fu and Y.-B. Dong, J. Mater. Chem. A, 2018, 6, 11140–11146 RSC.
- S. Y. Ding, J. Gao, Q. Wang, Y. Zhang, W. G. Song, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef CAS PubMed.
- D. Mullangi, D. Chakraborty, A. Pradeep, V. Koshti, C. P. Vinod, S. Panja, S. Nair and R. Vaidhyanathan, Small, 2018, 14, e1801233 CrossRef PubMed.
- F. Pan, M. Wang, H. Ding, Y. Song, W. Li, H. Wu, Z. Jiang, B. Wang and X. Cao, J. Membr. Sci., 2018, 552, 1–12 CrossRef CAS.
- X. Gao, X. Zou, H. Ma, S. Meng and G. Zhu, Adv. Mater., 2014, 26, 3644–3648 CrossRef CAS PubMed.
- L. Su, Z. Zhang and Y. Xiong, Nanoscale, 2018, 10, 20120–20125 RSC.
- J. Wang, X. Yang, T. Wei, J. Bao, Q. Zhu and Z. Dai, ACS Appl. Bio Mater., 2018, 1, 382–388 CrossRef CAS.
- A. de la Pena Ruigomez, D. Rodriguez-San-Miguel, K. C. Stylianou, M. Cavallini, D. Gentili, F. Liscio, S. Milita, O. M. Roscioni, M. L. Ruiz-Gonzalez, C. Carbonell, D. Maspoch, R. Mas-Balleste, J. L. Segura and F. Zamora, Chem.–Eur. J., 2015, 21, 10666–10670 CrossRef CAS PubMed.
- J. Xie, S. A. Shevlin, Q. Ruan, S. J. A. Moniz, Y. Liu, X. Liu, Y. Li, C. C. Lau, Z. X. Guo and J. Tang, Energy Environ. Sci., 2018, 11, 1617–1624 RSC.
- Y. Zhu, M. Qiao, W. Peng, Y. Li, G. Zhang, F. Zhang, Y. Li and X. Fan, J. Mater. Chem. A, 2017, 5, 9272–9278 RSC.
- W. Liu, L. Zhang, X. Liu, X. Liu, X. Yang, S. Miao, W. Wang, A. Wang and T. Zhang, J. Am. Chem. Soc., 2017, 139, 10790–10798 CrossRef CAS PubMed.
- D. Zhou, X. Cao, Z. Wang, S. Hao, X. Hou, F. Qu, G. Du, A. M. Asiri, C. Zheng and X. Sun, Chem.–Eur. J., 2017, 23, 5214–5218 CrossRef CAS PubMed.
- E. de Smit, M. M. van Schooneveld, F. Cinquini, H. Bluhm, P. Sautet, F. M. de Groot and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2011, 50, 1584–1588 CrossRef CAS PubMed.
- G. Chen, Y. Zhao, G. Fu, P. N. Duchesne, L. Gu, Y. Zheng, X. Weng, M. Chen, P. Zhang, C. W. Pao, J. F. Lee and N. Zheng, Science, 2014, 344, 495–499 CrossRef CAS PubMed.
- R. S. B. Gonçalves, A. B. V. de Oliveira, H. C. Sindra, B. S. Archanjo, M. E. Mendoza, L. S. A. Carneiro, C. D. Buarque and P. M. Esteves, ChemCatChem, 2016, 8, 743–750 CrossRef.
- B. Li, Y. Du, T. Li and S. Dong, Anal. Chim. Acta, 2009, 651, 234–240 CrossRef CAS PubMed.
- Y. Chen, H. Cao, W. Shi, H. Liu and Y. Huang, Chem. Commun., 2013, 49, 5013–5015 RSC.
- K.-i. Ishibashi, A. Fujishima, T. Watanabe and K. Hashimoto, J. Photochem. Photobiol., A, 2000, 134, 139–142 CrossRef CAS.
- W. Lu, J. Shu, Z. Wang, N. Huang and W. Song, Mater. Lett., 2015, 154, 33–36 CrossRef CAS.
- L. Y. Chau, Q. He, A. Qin, S. P. Yip and T. M. H. Lee, J. Mater. Chem. B, 2016, 4, 4076–4083 RSC.
- J. Sun, J. Ge, W. Liu, M. Lan, H. Zhang, P. Wang, Y. Wang and Z. Niu, Nanoscale, 2014, 6, 255–262 RSC.
- Y. Lu, W. Ye, Q. Yang, J. Yu, Q. Wang, P. Zhou, C. Wang, D. Xue and S. Zhao, Sens. Actuators, B, 2016, 230, 721–730 CrossRef CAS.
- H. Liu, Y.-N. Ding, B. Yang, Z. Liu, X. Zhang and Q. Liu, ACS Sustainable Chem. Eng., 2018, 6, 14383–14393 CrossRef CAS.
- S. Wang, Y. Jia, L. Song and H. Zhang, ACS Omega, 2018, 3, 18456–18465 CrossRef CAS PubMed.
- Z. Ai, L. Lu, J. Li, L. Zhang, J. Qiu and M. Wu, J. Phys. Chem. C, 2007, 111, 4087–4093 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00025f |
| This journal is © The Royal Society of Chemistry 2020 |