
Utilizing ballistic nanoparticle impact to reconfigure the metal support interaction in Pt–TiN electrocatalysts†
Abheek
Datta‡
*,
Zakaria
Ziadi‡
 ,
Pawan
Kumar
,
Toshio
Sasaki
,
Eric
Danielson
and
Alexander J.
Porkovich
*
,
Pawan
Kumar
,
Toshio
Sasaki
,
Eric
Danielson
and
Alexander J.
Porkovich
*
Okinawa Institute of Science and Technology (OIST) Graduate University, 1919-1 Tancha, Onna-Son, Okinawa 904-0495, Japan. E-mail: datta.abheek@oist.jp; alexander.porkovich@oist.jp
First published on 30th July 2020
Abstract
Tuning the metal support interaction (MSI) in heterogeneous catalysts is of utmost importance for various applications in different catalysis reactions. Pt–TiN systems are strong contenders for commercial catalysts, although the charge screening of Pt and non-involvement of N reduces their effective MSI and limits it to the Pt–Ti interface. Here, the bias driven landing of gas phase synthesized Pt nanoparticles (NPs) is used to change the nature of the MSI and enhance the charge transfer phenomenon. Bias driven landing of the Pt NPs translates their impact energies to the TiN surface, resulting in a weakening of the Ti–N bonds. This facilitates a new interaction between the Pt and N atoms, resulting in an electronic equilibration in the N–Pt–Ti triumvirate, nullifying the charge screening of Pt. This change in the nature of the MSI enables long range charge transfer throughout the catalyst surface and an increase in the electrocatalytic hydrogen evolution reaction (HER) activity of the Pt–TiN system.
New conceptsMetal–support interactions (MSIs) are well known in heterogeneous catalysis. This typically describes the transfer of charge between a metallic nanoparticle (NP) and its non-metal support. Utilising the support to modulate the electronic structure of the NPs through MSIs is an evolving area of interest in surface science. However, less attention has been paid to the ways that MSIs between the NPs and support may be modulated from the NP side. This work demonstrates the effect on the chemical and surface charge of embedding NPs into a non-metal (nitride) support through ballistic impact. Importantly, it was found that the charge transfer from NP to nitride could be increased with greater impact energy during deposition. Given that electron rich supports (e.g. nitrides, sulphides, and carbides) have difficulties in transferring charge from the support to the NPs (due to a lack of polarisation and charge screening), this strategy of synthesis presents a possible way to unlock the great potential of these supports in catalysis and possibly beyond. |
Introduction
Metal nanoparticles (NPs) are instrumental in various nanotechnological applications, due to their unique properties, which are dependent on their size, morphology and composition.1,2 Thus, different techniques have been employed to design and control these features of NPs to improve their applicability.3,4 Immobilizing nanoparticles on a support was found to be one of the most useful techniques to enhance their activity and stability towards various catalysis reactions.2,5,6 However, while employing this technique, researchers also found that the supports can have a special type of interaction with the NPs leading to unique interface phenomena, i.e. electronic metal–support interactions (EMSIs).2,7,8 Such EMSIs have significant effects on the catalytic performances of supported metal NPs towards various heterogeneous catalysis reactions.2,5–8 Supported metal NP catalysts also exhibit a great influence of EMSIs on their electrocatalytic activities in fuel cell reactions.9,10According to previously published research, EMSI phenomena in supported NP catalysts are typically associated with charge transfer, the interfacial perimeter, and the NP morphology and chemical composition. Since the EMSIs have a huge impact on the catalytic activity of supported NP catalysts, various methods have been employed to tune or exploit the EMSI for better catalytic output.2 These methods include varying the composition, morphology, and synthesis method of the metal and support material to instigate chemical or structural changes in order to tune the EMSIs, as described in recent studies.2,9–11
Since the interfacial sites are of utmost importance for EMSIs in supported nanoparticle electrocatalysts,12,13 the exposure of the interfacial perimeter to different chemical environments can also influence the catalytic activity of supported NPs.2 Recent reports have revealed that different degrees of NP embedding into the support can offer different environments to the interfacial perimeters, leading to modulation of the chemical and physical properties.14,15 Palmer and his co-workers have reported the ‘pinning down’ of various NPs into well-defined support surfaces by changing the bias during gas phase synthesis of NPs.16–18 It has also been shown that variation in bias during deposition led to different impact energies of the particles, which in turn interacted differently with the substrate surface.19,20 It was also reported that the deposition of size selected nanoclusters with higher impact energy created a higher degree of damage to the support and its chemical bonds.21,22 However, no attempt has been made to study the electronic interactions between NPs deposited with different impact energies and the substrates nor their effect on potential catalytic activities.
The EMSI phenomenon has been extensively studied for various electrocatalytic reactions important in fuel cell applications, e.g. the oxygen evolution reaction (OER), oxygen reduction reaction (ORR), hydrogen evolution reaction (HER), etc.23–25 It was also found that EMSI phenomena are frequently observed for nitride support materials, which themselves are active catalysts, due to the interplay between the support molecules and the metal atoms.26–28 Among various metal and non-metal nitride support materials, titanium nitride (TiN) has caught researchers’ attention due to its durability, activity and unique interaction with supported metal NPs.29,30 It has also been found that the interaction between Pt and TiN under electrocatalytic fuel cell conditions can help TiN supported Pt (Pt–TiN) to dramatically enhance its activity.31 However, the majority of the EMSIs in Pt–TiN composites take place between Pt and Ti atoms, as Pt–Ti interactions are stronger than Pt–N interactions which effectively pushes the Pt atoms away from the N atoms and draws them closer to the Ti atoms.32,33 Some studies have also found that multi-atom nanoparticle or bulk Pt has less interaction with Ti regarding charge transfer.34 Thus, the interactions between Pt NPs and TiN are deprived of Pt–N interaction and full extent Pt–Ti interactions. Presently, no attempts have been made to change the EMSIs present in Pt–TiN electrocatalysts to strengthen both the Pt–N and Pt–Ti interactions, obtaining enhanced charge transfer at the interface in order to reach the catalysts’ full potential in electrocatalysis reactions.
We report the effect of Pt NP deposition on atomically layered TiN surfaces with different impact energies using three different biases (0 kV, 2 kV and 6 kV). These samples were characterized by transmission electron microscopy to understand the distribution and size variation of the NPs. Atomic force microscopy (AFM) and X-ray photo electron spectroscopy (XPS) were employed to determine the changes of the TiN surface and Ti–Pt–N interactions after deposition of NPs with different impact energies. Peak force Kelvin probe force microscopy (PF-KPFM) investigations show an enhancement in surface potential for NPs implanted at different biases, indicating increased charge transfer though better EMSI. Lastly, an electrochemical HER performance study revealed that the Pt–TiN sample with Pt NPs deposited with 6 kV bias acted as a superior catalyst due to enhanced EMSIs with high impact energy.
Results and discussion
Pinning down the nanoparticles with different bias driven sputtering
Pt–TiN composites were obtained by sputtering Pt NPs via a gas phase synthesis method on the surface of atomic layer deposited (ALD) 10 nm TiN thin film (studied using atomic force microscopy (Fig. S1, ESI†)). The Pt NPs were sputtered with a DC magnetron using substrate biases of 0, 2 and 6 kV to create three separate samples, hereafter referred to as Pt–TiN(A), Pt–TiN(B), and Pt–TiN(C), respectively. Details of the synthesis method and characterization techniques are discussed in the ESI†of this article.Scanning electron microscopy (SEM) and atomic force microscopy (AFM) were used to examine the effect of Pt atom bombardment on the TiN. Fig. 1(a) shows SEM images of 45° tilted sample surfaces. The Pt–TiN(A) surface has an even distribution of Pt NPs (white arrows), while Pt–TiN(B) and Pt–TiN(C) have many crater-like defects (dotted yellow arrows). Pt NPs are localized at the rim of these craters. Pt NPs are more prevalent on Pt–TiN(C) and the surface craters are also greater in number and deeper than those on Pt–TiN(B).
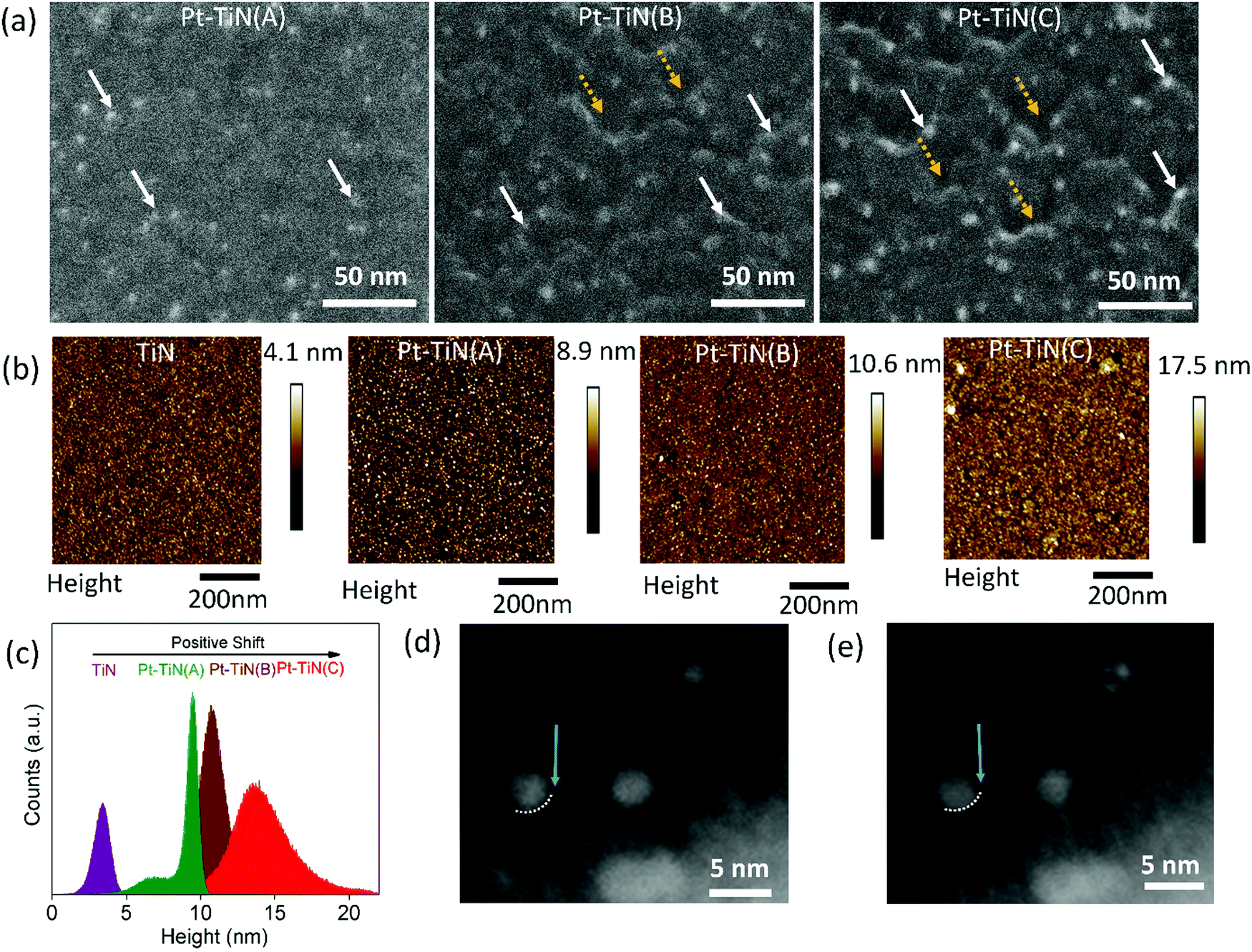 | ||
| Fig. 1 (a) 45° tilted SEM images of the Pt–TiN samples. The solid white arrows mark the presence of Pt NPs and the dotted yellow arrows mark the presence of craters created by the impact of bias landed particles. With the increase in bias, deeper craters are formed and push more particles to the crater floors in the TiN surface. (b) AFM topography for bare TiN film and all the Pt–TiN samples shows an increase in roughness with the deposition of NPs at higher biases. (c) Quantitative analyses of height profiles of the samples in (b). (d and e) Two cross-sectional STEM images of a TiN coated CuO nanowire with Pt NPs deposited at 6 kV. The dotted white line and blue arrow in both images mark the interface between the Pt NP and amorphous TiN at the viewing angle in (d). Extension of the Pt NP beyond the white line and a larger separation at the blue arrow in (e) indicate NP implantation into the TiN. | ||
The sputtering of metal NPs at high biases exerts a high impact energy on the surface during collision, as Pt atoms in Pt–TiN(C) could diffuse through the surface with high momentum and create deep craters in the TiN substrate before collecting at the crater floor.16–18,21,22 The same occurred in Pt–TiN(B), but with less bias and lower impact energy the craters are shallow and fewer Pt atoms reach the floor. By tilting the sample during SEM imaging the Pt NPs appeared to be at the crater rim. As expected, more Pt NPs were visible at the apparent crater rims in Pt–TiN(C) than Pt–TiN(B). Without any applied bias during sputtering, the Pt NPs in Pt–TiN(A) did not form craters and remain on the surface.
Fig. 1(b) shows topography images of all Pt–TiN samples along with the bare TiN film before NP deposition. The TiN film is smooth with an rms roughness of 0.8 nm. Deposition of Pt NPs on the TiN surface without bias did not alter the surface structure much as the rms roughness for Pt–TiN(A) was found to be 0.82 nm. Applying bias during NP sputtering did change the rms roughness of the surface to 1.2 and 2.1 nm for Pt–TiN(B) and Pt–TiN(C), respectively. Fig. 1(c) shows that the peak of the height distribution also shift from 3.5 nm to 8.5 nm after Pt NP deposition on the bare TiN surface without any bias. Applying bias voltage during NP sputtering caused further shifts to 11 and 14 nm in Pt–TiN(B) and Pt–TiN(C). Since the surface morphology was altered by the impact energy of incident particles,35 the positive shift of the height distribution peak can directly be associated with the bias applied during Pt NP sputtering and the corresponding roughening of the surface with the creation of nanoscale craters in the TiN substrate.
NP implantation was also observed after depositing Pt NPs at a landing bias of 6 kV (Pt–TiN(C)) on TiN coated CuO nanowires, which were mechanically transferred to a SiN TEM grid. This allowed the sample to be tilted to focus on multiple cross sections of the film. Two such cross sections can be seen in Fig. 1(d) and (e). In Fig. 1(d) a clear interface between a Pt NP and the TiN surface can be seen, marked with a dotted white line and blue arrow. An image of the same area at a new angle is shown in Fig. 1(e), with the dotted white line and arrow at the same position as Fig. 1(d). Now the nanoparticle extends under the white dotted line and there is a larger separation at the interface near the blue arrow. This is indicative of nanoparticle implantation into the amorphous TiN.
These observed changes are due to the different impact energies of the nanoparticles, as increasing the deposition bias from 2 kV to 6 kV did not affect the nanoparticle distribution, size, or structure of the Pt NPs (Fig. S2(a and b), ESI†). Scanning transmission electron microscopy (STEM) was used to characterize the Pt NPs bombarded on TiN supports. Both populations of Pt NPs show a log-normal size distribution with similar mean particle diameters: 3.3 ± 1.5 nm for Pt–TiN(B) and 3 ± 1 nm for Pt–TiN(C) (Fig. S2(c and d), ESI†). Plane d-spacings consistent with the literature values of bulk face-centre cubic Pt (0.227 nm, 0.196 nm, 0.139 nm) with a lattice constant of a = 0.392 nm were consistently observed for a variety of particles across both samples (Fig. S2(e and f), ESI†). Fig. S3 (ESI†) shows the elemental mapping of Pt–TiN(C) where an extensive presence of Ti and N was observed along with a significant amount of oxygen on the surface. The elemental mapping of Pt also suggests that the Pt atoms infiltrated the surface during synthesis under high bias.
Change in the MSI; KPFM study
To understand the effect of particle impact energies on the electronic environment at the Pt–TiN interface, peak force Kelvin probe force microscopy (PF-KPFM) studies were performed for all samples. Fig. 2(a) and (b) show the resulting surface potential images and corresponding surface potential distribution profiles. A large negative shift from 50 to −50 mV was observed in the surface potential after the 0 kV deposition of Pt NPs on TiN. This was caused by the interfacial charge transfer phenomenon at the Pt–TiN interface due to the difference in work functions of TiN and Pt.36,37 When Pt NPs were deposited on TiN a junction is formed and electrons from TiN moved to the higher work function Pt until the Fermi levels lined up at equilibrium. An energy band diagram of the process is shown in Fig. S4 (ESI†). Further negative shift in the surface potential occurred with the application of bias during Pt NP deposition with Pt–TiN(B) and Pt–TiN(C) having −140 and −180 mV surface potentials, respectively. This indicates a greater amount of MSI via higher electron transfer from TiN to Pt.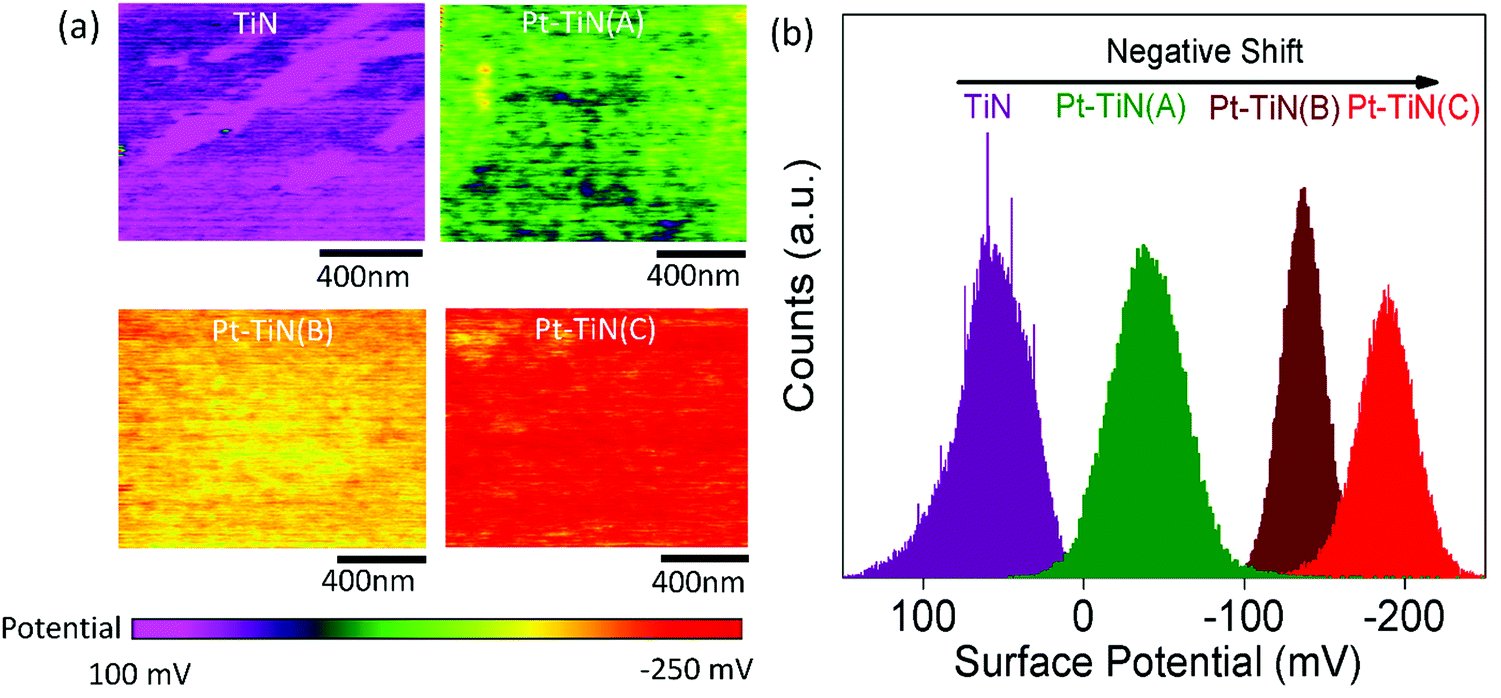 | ||
| Fig. 2 (a) Surface potential images of bare TiN film and all Pt–TiN samples from PF-KPFM measurements. The surface potential undergoes a negative shift upon deposition of Pt NPs, which increases with deposition bias, indicating an enhancement in charge transfer. (b) Surface potential profile of the same samples presented in (a) showing the quantitative analysis of the change in potential. | ||
Change in the nature of MSI: XPS study
Binninger et al. reported the contribution of atomic orbitals to effective long range MSI phenomena,37 which made the study of the chemical environment necessary to understand the MSI in supported metal catalysts. Extensive XPS studies were carried out for all the catalyst surfaces and the results are summarized in Fig. 3(a)–(d). The core spectra were deconvoluted using Fityk 0.9.8 software. Fig. 3(a) shows the Pt 4f core spectrum for all samples. Conventional Pt(0) (71.7 ± 0.1 eV (4f7/2) and 75.0 ± 0.1 eV (4f5/2)) peaks were observed in all cases.38 In addition, two other peaks were found in each sample. Along with Pt(0) in Pt–TiN(A) and Pt–TiN(B), was a peak of Pt(4+) (73.5 ± 0.1 eV (4f7/2) and 76.8 ± 0.1 eV (4f5/2))38 and a third species for Pt appearing at 72.2 ± 0.1 eV (4f7/2) and 75.5 ± 0.1 eV (4f5/2), negatively shifted from that of a classical Pt(2+)/PtO,38,39 indicating a reduction of Pt species. However, it is higher than the reported Pt–Ti bond,40 therefore although there is no alloy formation between Pt and Ti, a strong electronic flux to Pt from the surrounding Ti/TiOx species is responsible for this negative shift. This peak is referred to as Pt*Ti in Fig. 3(a).41 Pt–TinN(C), however, shows an overall negative shift in the 4f spectrum. While Pt(0) and Pt*Ti still exist, a more negative peak has evolved (71.1 ± 0.1 eV (4f7/2) and 74.5 ± 0.1 eV (4f7/2)). This feature (δ−) indicates an increase of electrons on the Pt.42 Since the Pt–Ti interaction is significantly stronger than Pt–N,32,33 the Pt–Ti interaction was 44% of the total Pt species population in Pt–TiN(A), whereas Pt(0) and Pt(4+) were 32 and 24%, respectively (Fig. 3(d) and Table S1, ESI†). With the increase in sputtering bias, Pt–Ti interaction decreased to 41 and 26% and metallic Pt increased to 38 and 53% for Pt–TiN(B) and Pt–TiN(C). At the same time, due to the increasing negative charge, δ− went from no significant concentration in Pt–TiN(A) and Pt–TiN(B), to 21% in Pt–TiN(C). Fig. 3(b) shows the Ti 2p core spectra for the catalysts, which were deconvoluted considering the contribution from nitride (455.6 ± 2.1 eV and 462.6 ± 1.6 eV), oxonitride (457.4 ± 1.8 eV and 462.1 ± 1.7 eV) and oxide/hydroxide (458.6 ± 1 eV and 464.1 ± 1 eV) species.43 A significant decrease in the concentration of nitride from 28 to 15 to 11% was observed with increasing NP sputtering bias. The oxonitride concentration values followed the same trend, whereas Ti-oxide/hydroxide species concentrations increased with sputtering bias (Fig. 3(d) and Table S1, ESI†). This implies significant weakening of the Ti–N bond along with the corresponding oxidation of Ti with increasing sputtering bias. N 1s core spectral analyses (Fig. 3(c)) also support this conclusion. With increasing sputtering bias the concentration of Ti–N species decreased and the amount of N–O species increased linearly (Fig. 3(d) and Table S1, ESI†). O 1s core spectra analyses agreed with these observations as the amount of both Ti–O and oxonitride species increase in the following order Pt–TiN(A) < Pt–TiN(B) < Pt–TiN(C) (Fig. S5 and Table S1, ESI†). These trends clearly suggest the weakening of the Ti–N bond with increasing degree of oxidation from higher sputtering bias of Pt NPs.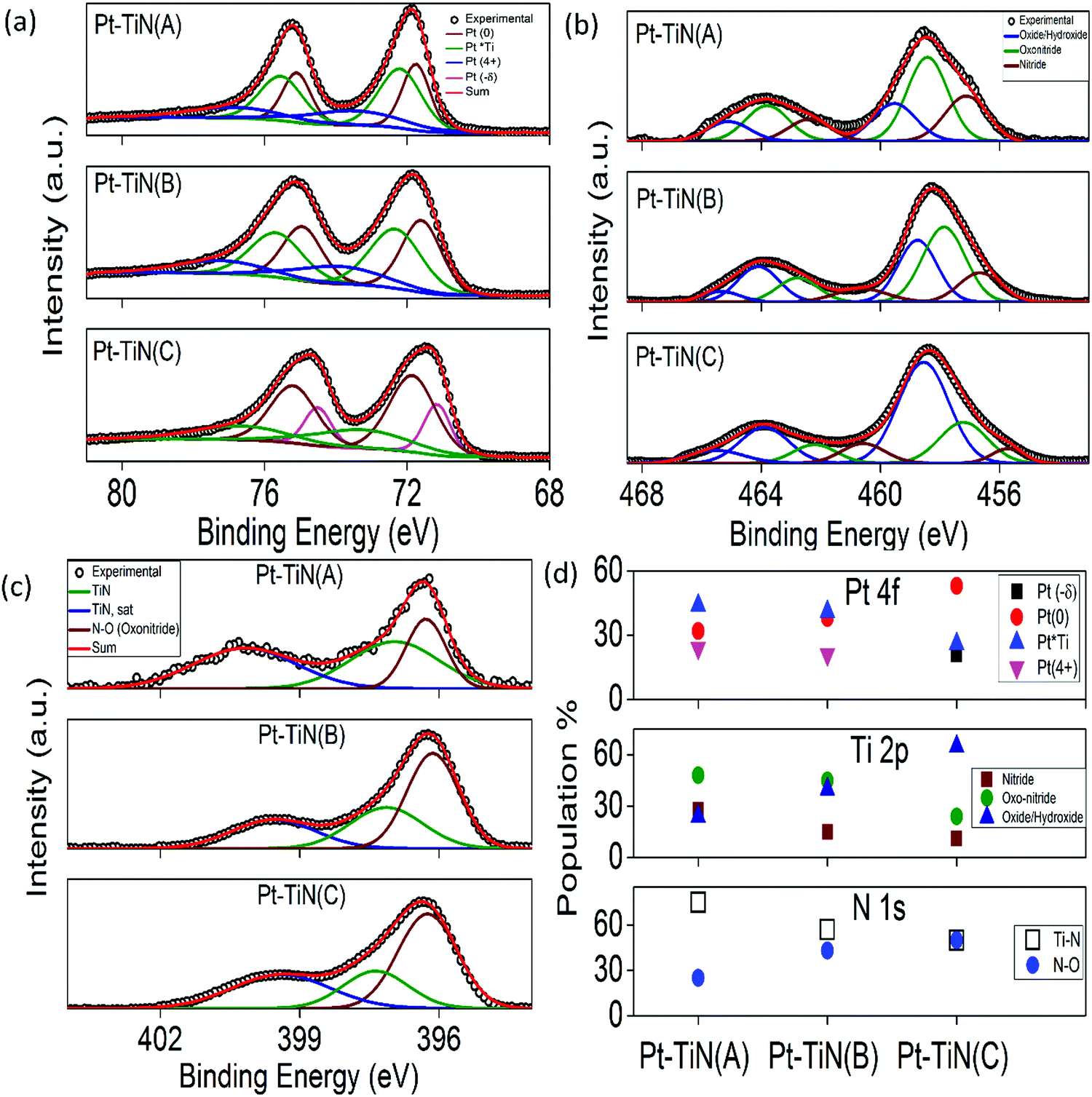 | ||
| Fig. 3 XPS core spectra of Pt 4f (a), Ti 2p (b), and N 1s (c) for all Pt–TiN samples. (d) Study of the relative contributions of different species of Pt, Ti and N calculated from (a), (b) and (c). Shirley background correction was used for fitting the curves. For calibration, the carbon 1s peak at 284.8 eV was considered standard. | ||
When the Pt NPs are deposited on the TiN surface at 0 kV, the Pt atoms interacted mainly with the surrounding Ti atoms while N atoms were pushed away from the Pt atoms, generating a localized MSI at the Pt–Ti interface where electrons flow from Ti to Pt atoms.31–33 Due to charge screening from Pt atoms inhibiting the charge transfer, the MSI is limited.37,40 With applied bias, the particles collided with the surface at higher impact energies and ruptured the TiN bond. Hence surface N atom mobility increased and they could build a stronger interaction with Pt. This new set of interactions consists of a tricenter system of N–Pt–Ti where electrons flowed from Ti to N via Pt, as Pt has an electronegativity value of 2.28, between those of Ti (1.54) and N (3.04). The charge transferred to N via Pt nullified the effect of charge screening in MSI and generated an electronic equilibration. With higher bias leading to more mobile N atoms, the strong MSI between Pt and Ti became electronic MSI with the active involvement of N. Long range charge transfer reached the outer surface of atoms rendering more metallic sites of Pt (as observed in XPS study) and creating a globalized MSI phenomenon throughout the surface.37 This modified MSI scaled up the work function between Pt and TiN further and charge transfer became easier, as shown by PF-KPFM.
XPS depth profile studies were also performed to understand the effect of the different applied biases on the change in the MSI (Fig. 4(a) and (b)). For both Pt–TiN(B) and Pt–TiN(C), Ti, N, and O atomic% values were constant after an initial increase due to etching of surface contamination. Magnified plots of Pt concentration vs. etching time are shown in the inset of the figures. Atomic percentage of Pt in Pt–TiN(B) decreased to 0.48% after 2 hours of etching, while in Pt–TiN(C) it only decreased to 0.63%. Pt atoms diffuse deeper into the TiN substrate when Pt NPs impact the surface at higher biases.
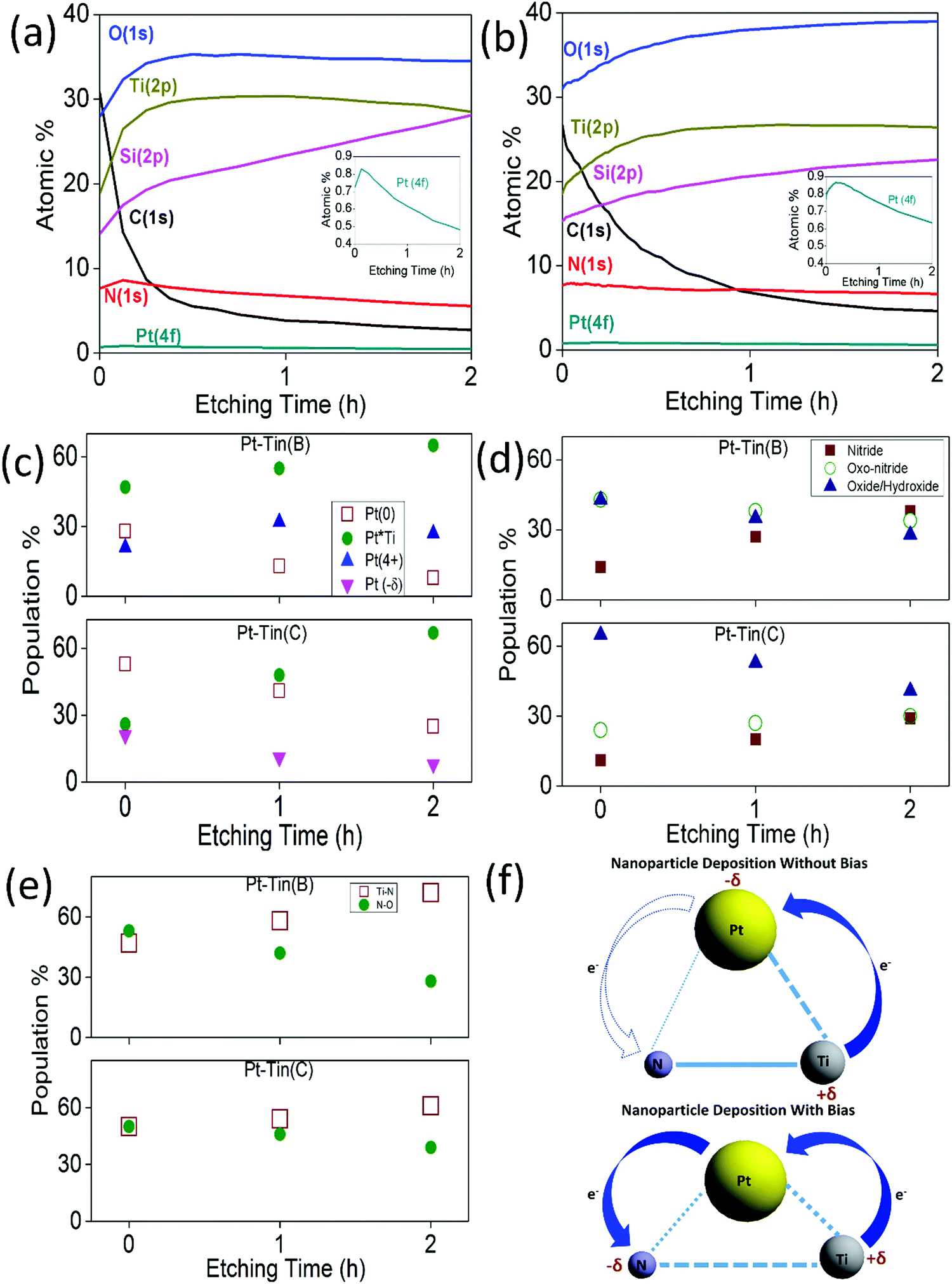 | ||
| Fig. 4 XPS depth profile studies for (a) Pt–TiN(B) and (b) Pt–TiN(C) over 2 h of etching. The insets show the magnified depth profile studies for Pt 4f in the samples. The relative contributions of different species of (c) Pt 4f, (d) Ti 2p and (e) N 1s were calculated from observed core XPS spectra after 0, 1, and 2 h of etching. (f) Schematic of the MSI in Pt–TiN with and without bias driven landing of Pt NPs. With higher bias, Pt atoms interact with N more effectively, creating an enhanced electron flow from Pt to N which leads to long range charge transfer. | ||
Pt 4f, Ti 2p, and N 1s core spectra for Pt–TiN(B) and Pt–TiN(C) were measured at 1 hour intervals during etching (Fig. 4(c–e)). Both samples show a linear increase in the Pt–Ti interactions and a linear decrease in metallic Pt at greater depths (Table S2, ESI†). As Pt atoms lose momentum traveling deeper in the TiN, they cannot rupture the Ti–N bonds at impact. This leads to the retention of strong MSI and less charge transfer between TiN and Pt. The concentration of Pt(4+) remains stable throughout the etching process (Table S2, ESI†) indicating that the MSI change was not caused by any chemical change but by the electronic equilibration mentioned earlier. Pt NPs deposited at 6 kV travelled deeper into the TiN layers, expanding the enhanced MSI phenomenon throughout the support. This is supported by the observation that at the core of the TiN support, the concentration of Ti–N species was found to be less for Pt–TiN(C) than Pt–TiN(B) (Fig. 4(d), (e) and (Table S2, ESI†)). Fig. 4(f) shows a schematic of the metal support interaction between Pt and TiN with and without bias sputtering.
Effect of nanoparticle impact on electrocatalytic activity
To examine the effect of this MSI enhancement on the catalytic activity of Pt–TiN systems, the electrochemical HER performance of all the catalysts was evaluated (Fig. 5). All samples were found to be active in the HER as shown by the linear sweep voltammograms (LSV) in Fig. 5(a). Pt–TiN(C) was found to be the best with an overpotential of 31 mV@10 mA cm−2 whereas Pt–TiN(A) and Pt–TiN(B) showed 46 and 71 mV overpotential at the same current density. Tafel plots for all catalysts are shown in Fig. 5(b) and the corresponding slopes were 68, 47 and 31 mV dec−1 for Pt–TiN(A), Pt–TiN(B) and Pt–TiN(C), respectively, confirming superior activity of the 6 kV biased sample. The Tafel slope is lower than that of a commercial Pt/C catalyst, which agrees well with previous studies.44 As the electrochemically active surface area (ECSA) for an electrocatalyst is directly proportional to the double layer capacitance (Cdl) values,45Cdl values were calculated from the slopes of the straight-line fits of the differences between anodic and cathodic current densities (δJ) against varying scan rates from 10 to 100 mV s−1 at 0.10 V vs. RHE (Fig. 5(c)). δJ values were obtained from CVs recorded in the non-faradaic region i.e. 0–0.2 V vs. RHE (Fig. S6, ESI†). The Cdl values were 22, 43 and 59 mF for Pt–TiN(A), Pt–TiN(B) and Pt–TiN(C). As discussed earlier, Pt NPs sputtered with bias weakens the Ti–N bond to form an N–Pt–Ti interaction where the Pt centers become more negative due to enhanced electron flow from the support. This phenomenon prompts H+ ions to be adsorbed readily on (111) sites of Pt. Enhanced electron flow from the support to the metal also facilitates the recombination of H+ ions with the electrons of the substrate surface.46 Pt–TiN(C) had the best performance of all Pt–TiN samples in the electrochemical HER, as expected from its greater MSI. Fig. 5(d) shows chronoamperometric studies for all samples. Pt–TiN(B) and Pt–TiN(C) retained their activity for a period of 4 h proving the change in nature of MSI via bias driven landing of NPs is not a metastable phenomenon but a permanent one. The charge transfer resistance (RCT) values calculated from the fitted electrochemical impedance spectroscopy (EIS) studies for the catalysts were 5, 4 and 2.8 Ohm for Pt–TiN(A), Pt–TiN(B) and Pt–TiN(C) (Fig. S7, ESI†). Since the charge transfer at the liquid–electrode interface varies inversely with the RCT, the degrees of interfacial charge transfer kinetics should be in the order of Pt–TiN(A) < Pt–TiN(B) < Pt–TiN(C), respectively. This specific order suggests the highest interfacial charge transfer in the case of Pt–TiN(C), indicating interaction between the catalyst surface and HER intermediates of highest order favoring the reaction most. This activity of the Pt–TiN(C) catalyst towards the reaction intermediates suggests the presence of a great deal of charge polarization throughout the composite's surface. As discussed earlier, this extensive charge polarization is a result of the enhanced MSI in the Pt–TiN(C).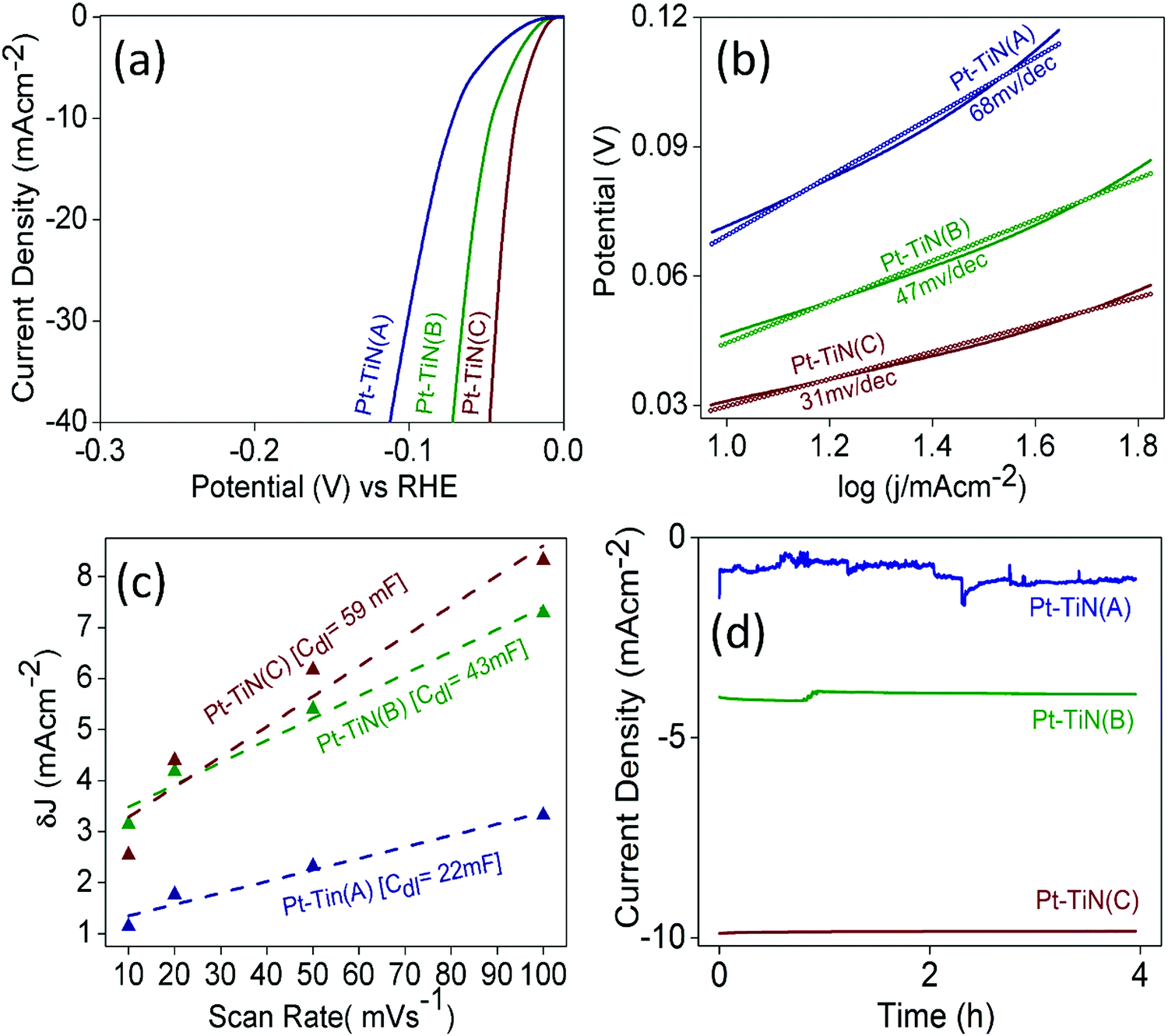 | ||
| Fig. 5 Electrochemical HER activity of Pt–TiN catalysts. (a) LSV curves, (b) Tafel plots, (c) double layer capacitance study and (d) chrono-amperometric curves. | ||
The comparison of theoretically calculated and experimentally obtained values of the amount of evolved hydrogen gas for the best catalyst, i.e. Pt–TiN(C), is given in Fig. S8 (ESI†). The faradaic efficiency was calculated to be 97.8%. Such a high value indicates an almost complete transformation of electrical energy into chemical energy by Pt–TiN(C).
Conclusions
In summary, we report an effective strategy to change the nature of MSI in Pt–TiN interfaces for an enhanced charge transfer from TiN to Pt. Pt NPs were deposited on TiN film via a gas phase synthesis method and the substrate bias was varied from 0 to 6 kV. The MSI in conventional TiN supported Pt is limited by the charge screening of Pt and non-involvement of N atoms. By imposing a bias on the substrate, the impact energies of Pt NPs to the TiN surface result in the disruption of the Ti–N bond, increasing the atomic mobility of N to form a new interaction between Pt and N atoms. This interaction is a triumvirate of N, Pt and Ti atoms at the metal support interface in which the strong electron pull from N nullifies the charge screening by Pt. This establishes an electronic equilibrium and charge transfer from the support to the metal is dramatically enhanced. Charge transfer increased with respect to substrate bias, with the strongest MSI corresponding to Pt NPs deposited at 6 kV. Modifying the MSI using bias landed NPs also had a significant effect on the HER activity of Pt–TiN samples. The involvement of N into MSI phenomenon renders Pt more negative and facilitates H+ ion absorption at Pt catalytic sites, which prompts the HER reaction. We believe that this general method of NP bias landing can be used to alter the MSI phenomenon in supported metal catalysts to provide remarkable performance of heterogeneous catalysis reactions.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by funding from the Okinawa Institute of Science and Technology Graduate University (OIST). The authors acknowledge Ms Noriko Ishizu from OIST Mech. Eng. & Microfabrication Support Section for the SEM and XPS experiments.Notes and references
- W. Gao, D. Wen, J. C. Ho and Y. Qu, Mater. Today Chem., 2019, 12, 266–281 CrossRef.
- T. W. van Deelen, C. H. Mejía and K. P. de Jong, Nat. Catal., 2019, 2, 955–970 CrossRef.
- M. M. Modena, B. Rühle, T. P. Burgand and S. Wuttke, Adv. Mater., 2019, 31, 1901556 CrossRef PubMed.
- T. K. Sau, A. L. Rogach, F. Jäckel, T. A. Klar and J. Feldmann, Adv. Mater., 2010, 22, 1805–1825 CrossRef PubMed.
- J. Zhang, L. Wang, B. Zhang, H. Zhao, U. Kolb, Y. Zhu, L. Liu, Y. Han, G. Wang, C. Wang, D. S. Su, B. C. Gates and F.-S. Xiao, Nat. Catal., 2018, 1, 540–546 CrossRef.
- F.-Z. Song, Q.-L. Zhu, X. Yang, W.-W. Zhan, P. Pachfule, N. Tsumori and Q. Xu, Adv. Energy Mater., 2018, 8, 1701416 CrossRef.
- Q. Zhang, X.-X. Qin, F.-P. Duan-Mu, H.-M. Ji, Z.-R. Shen, X.-P. Han and W.-B. Hu, Angew. Chem., Int. Ed., 2018, 57, 9351–9356 CrossRef CAS PubMed.
- J. Zhang, Q. Zhang and X. Feng, Adv. Mater., 2019, 31, 1808167 CrossRef PubMed.
- H. Li, C. Chen, D. Yan, Y. Wang, R. Chen, Y. Zou and S. Wang, J. Mater. Chem. A, 2019, 7, 23432–23450 RSC.
- J. Zhang, C. Liu and B. Zhang, Small Methods, 2019, 3, 1800481 CrossRef.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- I. S. Metcalfe, Catal. Today, 1991, 9, 145–152 CrossRef CAS.
- B. Qiu, C. Wang, N. Zhang, L. Cai, Y. Xiong and Y. Chai, ACS Catal., 2019, 9, 64846490 Search PubMed.
- J. S. Du, T. Bian, J. Yu, Y. Jiang, X. Wang, Y. Yan, Y. Jiang, C. Jin, H. Zhang and D. Yang, Adv. Sci., 2017, 4, 1700056 CrossRef PubMed.
- S. Liu, A. Pandey, J. Duvigneau, J. Vancso and J. H. Snoeijer, Macromolecules, 2018, 51, 24112–24417 Search PubMed.
- R. E. Palmer, S. Pratnotep and H.-G. Boyen, Nat. Mater., 2003, 2, 443–448 CrossRef PubMed.
- M. Di Vece, S. Palomba and R. E. Palmer, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 073407 CrossRef.
- S. J. Carroll, S. Pratontep, M. Streun, R. E. Palmer, S. Hobday and R. Smith, J. Chem. Phys., 2000, 113, 7723–7727 CrossRef.
- C. Xirouchaki and R. E. Palmer, Vacuum, 2002, 66, 167–173 CrossRef.
- S. J. Carroll, P. D. Nellist and R. E. Palmer, Phys. Rev. Lett., 2000, 84, 2654–2657 CrossRef.
- S. Vučković, J. Samela, K. Nordlund and V. N. Popok, Eur. Phys. J. D, 2009, 52, 107–110 CrossRef.
- V. N. Popok, S. Vučković, J. Samela, T. T. Järvi, K. Nordlund and E. E. B. Campbell, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 205419 CrossRef.
- H. He, J. Chen, D. Zhang, F. Li, X. Chen, Y. Chen, L. Bian, Q. Wang, P. Duan, Z. Wen and X. Lv, ACS Catal., 2018, 8, 6617–6626 CrossRef.
- I. Jiménez-Morales, S. Cavaliere, D. Jones and J. Rozière, Phys. Chem. Chem. Phys., 2018, 20, 8765–8772 RSC.
- X. Wu, B. Feng, W. Li, Y. Niu, Y. Yu, S. Lu, C. Zhong, P. Liu, Z. Tian, L. Chen, W. Hu and C. M. Li, Nano Energy, 2019, 62, 117–126 CrossRef.
- J. Peng, X. Chen, W.-J. Ong, X. Zhao and N. Li, Chem, 2019, 5, 18–50 Search PubMed.
- Q. Liu, L. Du, G. Fu, Z. Cui, Y. Li, D. Dang, X. Gao, Q. Zheng and J. B. Goodenough, Adv. Energy Mater., 2019, 9, 1803040 CrossRef.
- I. M. Patil, A. Swami, R. Chavan, M. Lokanathan and B. Kakade, ACS Sustainable Chem. Eng., 2018, 6, 16886–16895 CrossRef.
- Y.-M. Chi, M. Mishra, T.-K. Chin, W.-S. Liu and T.-P. Perng, ACS Appl. Energy Mater., 2019, 2, 398–405 CrossRef.
- F. Liu, X. Yang, D. Dang and X. Tian, ChemElectroChem, 2019, 6, 2208–2214 CrossRef.
- R.-Q. Zhang, T.-H. Lee, B.-D. Yu, C. Stampfl and A. Soon, Phys. Chem. Chem. Phys., 2012, 14, 16552–16557 RSC.
- J. A. Kwon, M.-S. Kim, D. Y. Shin, J. Y. Kim and D.-H. Lim, J. Ing. Eng. Chem., 2017, 49, 69–75 CrossRef.
- R. Yue, M. Xia, M. Wang, P. Chen, W. Gong, S. Liao, Z. Li, F. Gao, L. Zhang and J. Wang, Appl. Surf. Sci., 2019, 495, 143620 CrossRef.
- W. Chen, L. Severin, M. Göthelid, M. Hammar, S. Cameron and J. Paul, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 5620–5627 CrossRef PubMed.
- N. Jiang, H. J. Zhang, S. N. Bao, Y. G. Shen and Z. F. Zhou, Physica B, 2004, 352, 118–126 CrossRef.
- M. M. Ottakam Thotiyl, T. Ravikumar and S. Sampath, J. Mater. Chem., 2010, 20, 10643–10651 RSC.
- T. Binninger, T. J. Schmidt and D. Kramer, Phys. Rev. B, 2017, 96, 165405 CrossRef.
- J. H. Kim, J. Cheon, T. J. Shin, J. Y. Park and S. H. Joo, Carbon, 2016, 101, 449–457 CrossRef CAS.
- M. A. Matin, E. Lee, H. Kim, W.-S. Yoon and Y.-U. Kwon, J. Mater. Chem. A, 2015, 3, 17154–17164 RSC.
- B. C. Beard and P. N. Ross, J. Phys. Chem., 1986, 90, 6811–6817 CrossRef CAS.
- A. Dauscher, L. Hilaire, J. C. Spirlet, W. Muller and G. Maire, Surf. Sci., 1988, 204, 161–173 CrossRef CAS.
- Y.-T. Kim, M. A. Matin and Y.-U. Kwon, Carbon, 2014, 66, 691–698 CrossRef CAS.
- B. Avasarala and P. Haldar, Electrochim. Acta, 2010, 55, 9024–9034 CrossRef CAS.
- C. Wang, H. Shi, H. Liu, J. Fu, D. Wei, W. Zeng, Q. Wan, G. Zhang and H. Duan, Electrochim. Acta, 2018, 292, 727–735 CrossRef CAS.
- A. Sahasrabudhe, H. Dixit, R. Majee and S. Bhattacharyya, Nat. Commun., 2018, 9, 2014 CrossRef PubMed.
- J. N. Tiwari, S. Sultan, C. W. Myung, T. Yoon, N. Li, M. Ha, A. M. Harzandi, H. J. Park, D. Y. Kim, S. S. Chandrasekaran, W. G. Lee, V. Vij, H. Kang, T. J. Shin, H. S. Shin, G. Lee, Z. Lee and K. S. Kim, Nat. Energy, 2018, 3, 773–782 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nh00344a |
| ‡ A. Datta and Z. Ziadi contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |