
Intermetallic Ni2Si/SiCN as a highly efficient catalyst for the one-pot tandem synthesis of imines and secondary amines†
Xiao
Chen
 ,
Shuhua
Han
,
Dongdong
Yin
and
Changhai
Liang
*
,
Shuhua
Han
,
Dongdong
Yin
and
Changhai
Liang
*
State Key Laboratory of Fine Chemicals & Laboratory of Advanced Materials and Catalytic Engineering, Dalian University of Technology, Dalian 116024, China. E-mail: changhai@dlut.edu.cn
First published on 15th October 2019
Abstract
Based on the isolation of the active sites in the crystal structure as well as the strong alteration of the electronic structure in comparison to elemental metals, metal silicides as typical intermetallic compounds have boosted their applications in catalysis. Heterogeneous Ni2Si/SiCN as a promising and low-cost catalyst alternative to noble-metals has been successfully synthesized via the chemical modification of a polysilazane precursor at a molecular level by the use of metal complexes followed by its temperature-programmed pyrolysis. In the one-pot tandem synthesis of imines and secondary amines via the reductive coupling of nitrobenzene and benzaldehyde, the as-prepared Ni2Si/SiCN-1000 catalyst exhibits excellent catalytic performance (conversion >99% and selectivity to aromatic amine >92%) and excellent recyclability under optimal reaction conditions. Due to the reduction induction support–metal strong interactions and the formation of intermetallic Ni2Si, the electronic structure of the metallic Ni active site has been modified, which may tone the catalytic performance and enhance the stability of the catalyst.
Introduction
Imines and their derivatives as a class of versatile intermediates have multiple applications in dyes, fragrances, organic syntheses, optical materials, agricultural chemicals, pharmaceuticals, and other fine chemicals.1,2 Generally, the synthesis of imines and their derivatives is a basic chemical reaction via the reaction of ketones or aldehydes with amines over Lewis acid catalysts,3 while the preparation of amines is based on the selective reduction of nitroarenes.4–6 Recently, tandem catalysis corresponding to the concept of green chemistry has aroused great interest for increasing the efficiency of chemical synthesis,7 in which multiple reactions have been simultaneously coupled in a single reaction vessel, avoiding intermediate separation and purification steps, and minimizing energy utilization. Therefore, the development of a highly efficient catalyst with the functions of both the selective hydrogenation of nitroarenes and the catalytic coupling of aldehydes and amines is essential for the one-pot tandem synthesis of imines and secondary amines.Over the last few years, few noble metal catalysts have been reported on the one-pot reductive amination synthesis of imines.8 For example, Cao et al. found hydroxyapatite-supported Au as a versatile multifunctional catalyst for the tandem oxidation of alcohols and amines to imines (or oximes) under solvent-free conditions.9 Qin et al. reported that a catalyst with Pt–Fe2O3 interfaces presented high efficiency and >89% selectivity to imines for the reductive coupling of nitrobenzene and furfural.10 Ru complexes and Ir–Pd heterodimetallic complexes have also been studied and shown to be highly active in the one-pot preparation of imines from nitroarenes by a tandem process.8,11 However, rare noble metals are expensive, easily poisoned, and prone to leeching in one-pot tandem reactions. In addition, the application of rare noble metals as catalysts is not in accordance with the conservation of the elemental resources. As a consequence, there is a great interest in developing non-noble metal-based catalysts that show higher activity for the one-pot tandem synthesis of imines. Recently, Beller et al. developed novel non-noble Co and Fe oxide-based nanocatalysts for nitroarene reduction and reductive amination.12–17 Kempe et al. developed novel, reusable, and robust Co and Ni nanocomposite catalysts for the one-pot tandem synthesis of imines from aromatic nitro compounds and aldehydes (or carbonyl compounds), in which functional groups that were easy to hydrogenate were tolerated.18,19 The use of inexpensive, easily handled, and abundantly available non-noble metals is a particularly attractive strategy for the replacement of rare precious metals for the synthesis of imines.20–22
Transition metals incorporated into the specific lattice site function as catalytically active centers that have great potential for modulating the local structure and the electronic properties of active sites and enhancing the catalytic activity and stability.23–28 Due to the active-site isolation by silicon atoms and electron transfer between metals and silicon, intermetallic silicides as promising and low-cost catalyst alternatives to noble-metal catalysts have aroused great interest in selective hydrogenation.29 Hosono et al. reported that a new copper-based intermetallic electride catalyst (LaCu0.67Si1.33) afforded the selective hydrogenation of nitroarenes with above 40-fold higher turnover frequencies than those of well-studied metal-loaded catalysts.30 Beller et al. reported that supported nickel silicide catalysts could be used for the hydrogenation of structurally diverse unsaturated compounds (>100 types), including nitroarenes, carbonyls, nitriles, N-containing heterocycles, and unsaturated carbon–carbon bonds.31 In our previous research, transition metal silicides (Ni2Si, NiSi, Co2Si, and CoSi) have been confirmed to exhibit high selectivity and stability in the hydrogenation of alkynes, unsaturated aldehydes, and aromatics.32–38 The presence of silicon as a secondary component in the metal lattice plays a key role in its properties, and is of crucial importance for improved catalytic activity. However, these earth-abundant materials have never been reported to have catalyst applications in the one-pot tandem synthesis of imines and secondary amines.
In this study, we report a heterogeneous Ni2Si/SiCN-1000 catalyst prepared via the chemical modification of a polysilazane (PSZ) precursor at a molecular level by the use of metal complexes followed by its temperature-programmed pyrolysis for the one-pot tandem synthesis of imines and secondary amines via the reductive coupling of nitrobenzene and benzaldehyde. The growth and structure of nickel silicide and the kinetics of the one-pot reductive coupling of nitrobenzene and benzaldehyde over nickel silicide have been studied extensively. In addition, the stability of the novel intermetallic Ni2Si/SiCN catalyst has been investigated.
Experimental section
Preparation of the catalyst
A nickel-modified polysilazane (PSZ) precursor was prepared by cross-linking under the flow of dry inert gas using standard Schlenk and glove box techniques. PSZ served as a silicon source (Honghai Chemical Technology Co., Ltd Guangzhou, China), nickel acetylacetonate (Ni(acac)2, Aladdin, 99.7%) served as a nickel source, and anhydrous and oxygen-free tetrahydrofuran (THF, Kermel, 99.7%) served as a solvent. Typically, Ni(acac)2 (0.6 g) was dispersed in THF (30 mL) with vigorous stirring in a glove box until a clear and transparent green solution was obtained. PSZ (3.0 g), acting both as a silicon source and as a cross-linking agent, was dripped into the green solution. It was clearly found that the color of the solution changed to black immediately. For promoting the thermal cross-linking, the obtained solution was heated at 60 °C for 2 h with stirring under nitrogen atmosphere. The pyrolyzed precursor was obtained by purging the solvent at 85 °C and then evaporating under vacuum at room temperature. Subsequently, the pyrolyzed precursor was further pyrolyzed at 200 °C for 1 h and then at 1000 °C for 2 h under argon/hydrogen (ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) atmosphere to obtain the intermetallic Ni2Si/SiCN-1000 catalyst. For understanding the pyrolysis process, the effect of the pyrolysis temperature of the nickel-modified PSZ precursor at 400 °C, 600 °C, and 800 °C has also been investigated in-depth.
:1) atmosphere to obtain the intermetallic Ni2Si/SiCN-1000 catalyst. For understanding the pyrolysis process, the effect of the pyrolysis temperature of the nickel-modified PSZ precursor at 400 °C, 600 °C, and 800 °C has also been investigated in-depth.
Characterization of catalyst
PSZ, Ni(acac)2 and nickel-modified PSZ precursors were dissolved in deuterated chloroform (CDCl3), respectively, and chemical shifts were detected using a nuclear magnetic resonance (H NMR) spectrometer (Bruker AVANCE III 500). Thermogravimetric analysis and differential scanning calorimetry coupled with mass spectrometry (TGA/DSC-MS) of the pyrolysis precursors was performed using a TGA/DSC analyser (Mettler-Toledo TGA/SDTA3+) by monitoring temporal mass and heat flow changes with temperature. The samples (ca. 10 mg) were heated from room temperature to 1373 K (heating rate: 10 K min−1) under argon atmosphere. Different functional groups in the samples were detected and analyzed by Fourier-transform infrared spectroscopy (FT-IR, U.S. Thermo Nicolet Corporation, Nexus EURO). X-ray diffraction (XRD) analyses of the samples were performed using a Rigaku D/Max-RB diffractometer equipped with a Cu Kα1 monochromatized radiation source operating at 40 kV and 100 mA. The sample was accurately weighed (0.1 g) and the specific surface area of the sample was analyzed by the Brunauer–Emmett–Teller (BET, U.S. Kangta autosorb iQ) instrument. The loading of nickel or nickel silicide in the sample was analyzed using an inductively coupled plasma emission spectrometer (ICP, PerkinElmer Optima 2000DV). The sample (ca. 10 mg) was dissolved in a mixed acid solution for testing. The morphology of the catalyst was observed using a scanning electron microscope (SEM, equipped with EDX; manufactured by JEOL, JSM-5600LV). The particle size and crystalline nature of the samples were analyzed in detail by transmission electron microscopy (TEM, Philips CM200 FEG). Powder samples were ultrasonicated in ethanol and were dispersed on copper grids covered with a holey carbon film. Surface compositions of the samples were characterized by X-ray photoelectron spectroscopy (XPS, ESCALAB250, Thermo VG). XPS measurements were performed using an X-ray source of Al Kα (1486.6 eV) with a power of 150 W.Catalyst activity evaluation
Catalytic reactions were performed in a 50 mL stainless steel autoclave with stirring at 700 rpm for eliminating the external diffusion effect. Before testing, the autoclave was purged five times with H2 for replacing air. Typically, the reaction was carried out at 170 °C and 5 MPa H2 with nitrobenzene (0.123 g, 1 mmol), benzaldehyde (0.212 g, 2 mmol), catalyst (0.1 g), and cyclohexane (20 mL). For analyzing the reaction path of the one-pot tandem synthesis of imines and secondary amines, nitrobenzene hydrogenation, benzaldehyde hydrogenation and the coupling of benzaldehyde and aniline were also performed on the as-prepared Ni2Si/SCN catalyst under the same conditions. The reaction product composition was analyzed using a gas chromatograph equipment (Echrom A90) equipped with a flame ionization detector and an HP-5 capillary column.Results and discussion
Synthesis of nickel-modified polysilazane
The properties of nickel-modified PSZ and cross-linked thermosets have been studied using 1H NMR. Fig. 1a shows the spectra of PSZ, Ni(acac)2, and nickel-modified PSZ in CDCl3, respectively. The characteristic signals in the spectrum of Ni(acac)2 decrease after the doping of PSZ. Simultaneously, the signals of Si–CH3 and –NH functional groups shift, which evidences that the transfer of nickel to the nitrogen functional groups of PSZ was most probably due to the availability of more coordination sites in the polymer. It is consistent with the report on the dehydrocoupling between Si–CH3 bonds by the reaction of Ru3(CO)12.39–42 Based on the aforementioned observations, the possible cross-linking mechanism of PSZ and Ni(acac)2 in THF has been proposed (Fig. 1b). The cleavage of bonds between nickel and acetylacetone groups occurred initially and then, bonds with –NH and Si–CH3 functional groups were cleaved during the process of cross-linking. As shown in Fig. 1c, PSZ reacted with Ni(acac)2 in THF via an exothermic reaction, resulting in effervescence, and the color of the solution changed from green to black immediately (only 10 s) along with the production of hydrogen in the cross-linking reaction. These phenomena also indicate that nickel modified PSZ successfully, and the process was not a mechanical mixing of the two substances.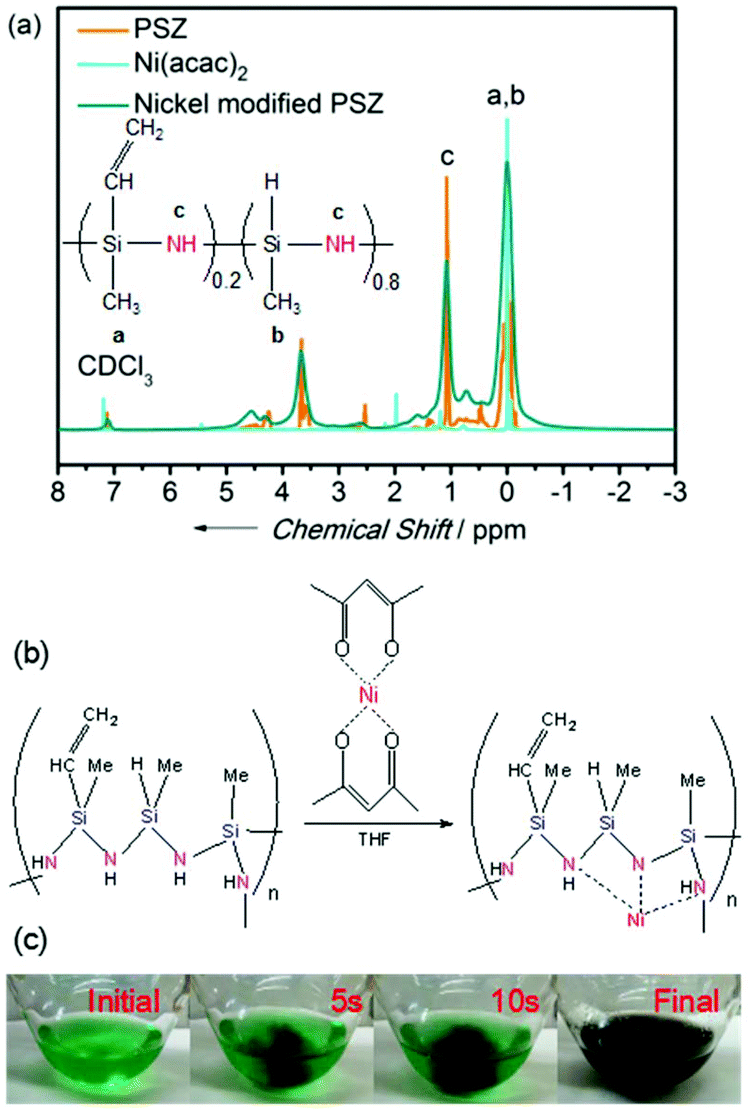 | ||
| Fig. 1 (a) 1H NMR spectra of PSZ, Ni(acac)2, and nickel-modified PSZ precursors. (b) The possible cross-linking mechanism of PSZ and Ni(acac)2 in THF. (c) The images of solution versus the reaction time. | ||
Subsequently, the pyrolysis behavior of nickel-modified PSZ and Ni(acac)2 was investigated using TG/DSC-MS (Fig. 2). The evolution of gaseous CO2 (m/z = 44) and CH4 (m/z = 16), and the fragments of H2O or NH3 (m/z = 17) and H2O (m/z = 18) have been detected by MS. The main mass loss of the nickel-modified PSZ precursor (47%) occurs between 100 and 800 °C (Fig. 2a). In detail, there is ca. 25% mass loss and the evolution of CH4, CO2, and H2O in the temperature range of 100–300 °C, which can be attributed to the pyrolytic volatilization of acetylacetonate (Fig. 2b). At 300–800 °C, there is ca. 22% mass loss with the evolution of CH4 and NH3, which is attributed to the volatilization of oligomer fragments caused by the decomposition of Si–CH3 groups and Si–NH–Si groups and the cleavage of C–C bonds. As a comparison, the main mass loss of the Ni(acac)2 sample (80%) occurs between 100 and 500 °C (Fig. 2c). The smaller weight loss peak in the range of 100–120 °C could be caused by the evaporation of absorbed water in the Ni(acac)2 sample, while the main weight loss (71%) in the range of 200–500 °C could be attributed to the pyrolytic volatilization of a large amount of acetylacetonyl groups (Fig. 2d). Due to the combination of Ni and PSZ, the pyrolysis precursor contains a large number of free acetylacetonate groups. The weight loss peak of the acetylacetonate group in the nickel-modified PSZ precursor shifted toward low temperatures, which further confirmed that the nickel complex chemically modified the PSZ precursor by coordination. For obtaining the SiCN-supported nickel or nickel silicide samples, the nickel-modified cross-linked polymers were heated in a hydrogen atmosphere based on the result of TG/DSC-MS in a furnace at 400 °C, 600 °C, 800 °C, and 1000 °C, respectively.
 | ||
| Fig. 2 TG/DSC-MS of (a and b) the nickel-modified PSZ precursor and (c and d) Ni(acac)2. | ||
Preparation method and structure of Ni2Si/SiCN-1000 catalyst
Powder XRD analyses of Ni or Ni silicide-containing SiCN samples prepared at different pyrolysis temperatures (400 °C, 600 °C, 800 °C, and 1000 °C) were carried out to confirm the amorphous nature of the support as well as the presence of the metallic phase (Fig. 3). It is clearly shown that there is a broad peak for the four samples at ca. 27° due to the presence of the amorphous SiCN matrix (Fig. 3a). Compared with the standard pattern of Ni (JCPD PDF 04-0850), there are two peaks at 44.5° and 51.8° for the samples prepared by pyrolysis at 400 °C and 600 °C, which is assigned to the (111) and (200) crystal faces of metallic Ni. However, it is not difficult to deduce that there is no pure phase of metallic Ni supported on the SiCN matrix for the Ni/SiCN-400 sample due to the incomplete pyrolysis according to the result of TG/DSC. With the increase in the pyrolysis temperature to 800 °C, a mixture peak appeared at ca. 45°, which may be apparent for some new species. When the pyrolysis temperature reached 1000 °C, single-phase Ni2Si supported on the SiCN matrix (Ni2Si/SiCN-1000) was obtained. The pattern of this sample exhibited three diffraction peaks at 43.5°, 45.5°, and 48.8°, which could be assigned to the (021), (121), and (002) crystal faces of Ni2Si (JCPD PDF 48-1339), respectively. Although the main peak of Ni2Si (45.5°) was very close to that of Ni (44.5°), the peak at 52.0° due to Ni (200) disappeared and the peaks attributed to Ni2Si (021) and (002) appeared with the increase in the pyrolysis temperature, which further confirms that metallic Ni reacted with the SiCN matrix to form intermetallic nickel silicide at a high temperature under H2 atmosphere due to the reduction induction support–metal strong interaction (SMSI). In addition, the ICP-OES result indicates that the actual Ni content in the Ni2Si/SiCN-1000 sample is 7.3 wt%, which is consistent with the theoretical value (7.9 wt%). This indicates that there is no loss of metal content during the pyrolysis of the nickel-modified PSZ precursor under H2 atmosphere.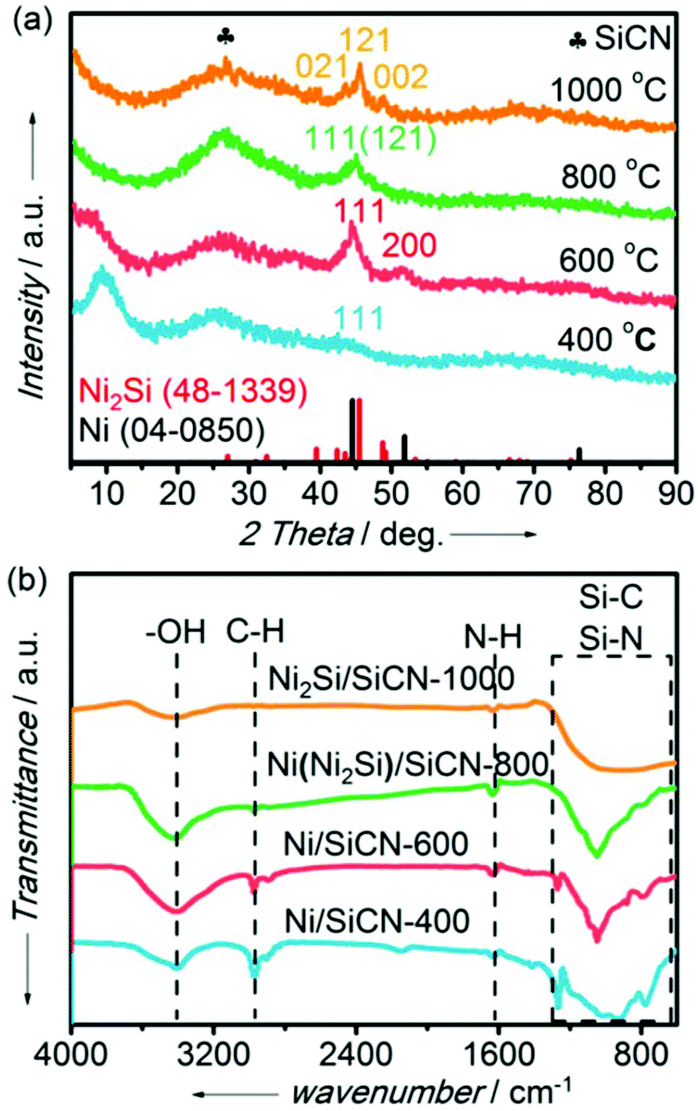 | ||
| Fig. 3 (a) XRD patterns and (b) FT-IR spectra of samples (pyrolysis at 400 °C, 600 °C, 800 °C, and 1000 °C under H2 atmosphere, respectively). | ||
The organic groups in the pyrolysis samples were detected by FT-IR spectra, as shown in Fig. 3b. There were four peaks at 3350 cm−1, 2950 cm−1, 1600 cm−1, and the region of 650–1300 cm−1 for all samples prepared at different pyrolysis temperatures, which could be assigned to the –OH group, C–H group, N–H group, and Si–C or Si–N group, respectively. With the increase in the pyrolysis temperature from 400 °C to 1000 °C, the peak intensity of the –OH group increased initially and then decreased, while the peaks ascribed to C–H group and N–H group generally disappeared, which indicates that the ligands of the nickel-modified PSZ processor decomposed under H2 atmosphere with the increase in temperature, leading to the formation of SiCN matrix-supported metallic nickel or nickel silicide. In addition, the intensity of Si–C and Si–N groups increased at high pyrolysis temperatures, which further indicates the formation of the SiCN matrix. This is consistent with the XRD result.
The specific surface area of the pyrolysed samples was determined by N2 physical adsorption (Fig. S1†), which presents the volcanic type with the increase in the pyrolysis temperature. The specific surface area of Ni/SiCN-600 reached 254 m2 g−1, while that of the single-phase Ni2Si/SiCN-1000 sample was only 19 m2 g−1 (Table S1†). At a lower pyrolysis temperature, the evolution of gaseous NH3, CH4, CO2, etc., created pores in the SiCN matrix, which provided a large surface area. However, the reduction induction SMSI at a higher pyrolysis temperature may lead to the tunnel collapse of the SiCN matrix or the aggregation of metal particles, decreasing the surface area of Ni2Si/SiCN-1000 sample.
The SEM images and maps of the Ni2Si/SiCN-1000 sample and the corresponding energy-dispersive X-ray spectrum are presented in Fig. 4, demonstrating the typical morphology of platelet-like shapes with unordered crystallite aggregation. The EDX spectrum confirms the existence of Ni, Si, C, and N elements in the sample. The Ni content is ca. 7.03%, which is in accordance with the ICP-OES result. As shown in the corresponding elemental maps, homogeneous Ni, Si, C, and N distributions are found in the Ni2Si/SiCN-1000 sample. In order to investigate the structural properties and the composition distribution, representative TEM images were obtained (Fig. 5). The (inter)metallic nanoparticles are homogenously distributed on the SiCN matrix. The corresponding size distribution shows the presence of dispersed nanoparticles with a diameter of ca. 7.05 nm (Fig. 5a). There is no aggregation of nanoparticles at high pyrolysis temperatures (1000 °C), indicating the strong interaction between (inter)metallic nanoparticles and the SiCN matrix. The high-resolution TEM image shown in Fig. 5b demonstrates that the measured lattice spacing of the nanoparticle is 0.341 nm and 0.214 nm, which is in accordance with the (101) and (310) planes of orthorhombic Ni2Si, respectively.43 To further identify the structure of (inter)metallic nanoparticles, the selected area electron diffraction (SAED) is shown in Fig. 5c. It clearly shows the polycrystalline diffraction ring and the single crystallinity structure of Ni2Si.43 High-resolution EDX elemental mappings show that the elemental map of Ni matched fairly well with that of Si in the nanoparticle distribution, which further confirms that Ni–Si intermetallic nanoparticles are highly dispersed throughout the SiCN support.
 | ||
| Fig. 4 SEM image, maps, and the corresponding energy-dispersive X-ray spectrum of the Ni2Si/SiCN-1000 sample. | ||
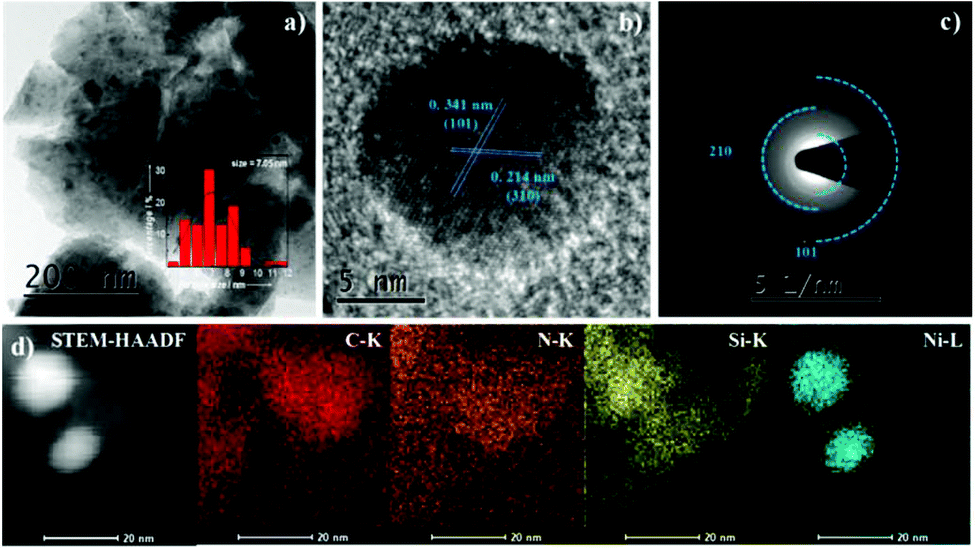 | ||
| Fig. 5 (a) Representative TEM image (the inset shows the corresponding size distribution), (b) HRTEM image, (c) selected area electron diffraction (SAED) pattern, and (d) high-resolution EDX elemental mappings of C, N, Si, and Ni of the Ni2Si/SiCN-1000 sample. | ||
The surface information on the Ni, Si, and C states was obtained by using XPS (Fig. 6). With the increase in reduction temperatures, the nickel-modified PSZ precursor gradually pyrolyzed. As shown in the Ni 2p region of samples, metallic Ni emerges until the reduction temperature reached 600 °C (Fig. 6a). On further increasing the temperature, due to the strong interactions between metallic Ni and the Si-containing support, the Ni–Si bonds at 853.0 eV were formed at 1000 °C.28,44–46 Unfortunately, the Ni–O bonds appeared at 855.9 eV for all samples because of the formation of oxide layers when exposed to air.31,47 In the Si 2p region, the peak due to the bond in the Si-containing polymer at 100.8 eV is missing after the reduction temperature reached 600 °C (Fig. 6b). Moreover, it clearly shows that the peaks at 101.6 eV, 102.4 eV, and 103.2 eV, respectively, could be attributed to the Si–C bonds, Si–N bonds, and Si–O bonds. This evidences the formation of the SiCN support during pyrolysis at high temperatures. This result is further confirmed by the peaks ascribed to C–N, C–C, and C–Si bonds in C 1s spectra (Fig. 6c). Therefore, SiCN-supported intermetallic Ni2Si can be synthesized by pyrolyzing the nickel-modified PSZ precursor at 1000 °C. There is an electronic structure modification of metallic Ni active sites due to the SMSI and the formation of intermetallic Ni2Si, which may tone the catalytic performance and enhance the stability of the catalyst.
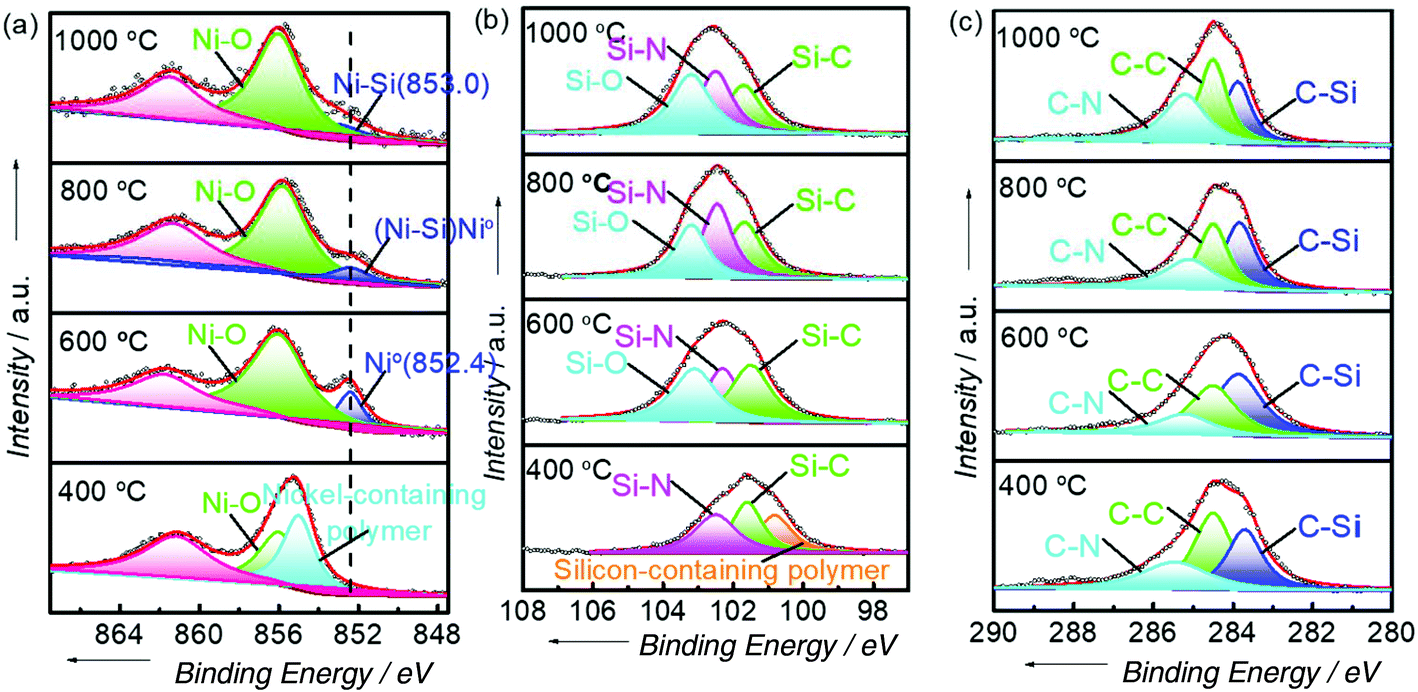 | ||
| Fig. 6 XPS of (a) Ni 2p region, (b) Si 2p region, and (c) C 1s region of Ni/SiCN-400, Ni/SiCN-600, Ni(Ni2Si)/SiCN-800, and Ni2Si/SiCN-1000 samples. | ||
Catalytic property of the Ni2Si/SiCN-1000 catalyst
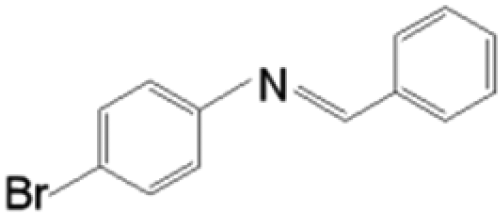
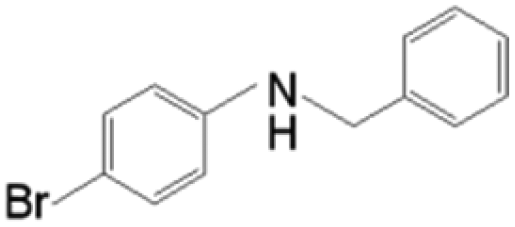
The as-prepared Ni/SiCN and Ni2Si/SiCN catalysts were tested for the one-pot tandem synthesis of imines and secondary amines via reductive coupling of nitrobenzene and benzaldehyde. The reaction network for the reductive coupling of nitrobenzene and benzaldehyde is complex and includes the hydrogenation of nitrobenzene to aniline and the coupling of aniline and benzaldehyde (Scheme 1).48,49 The key challenge for this reaction is to inhibit the hydrogenation of benzaldehyde to benzylalcohol, which means that the hydrogenation active site of the catalyst should be controlled. The reaction network shown in Scheme 1 is consistent with that of the aromatic amine product identified via GC-MS of the stainless steel autoclave effluent. As shown in Fig. 7, the catalytic activities of Ni/SiCN-400, Ni/SiCN-600, Ni(Ni2Si)/SiCN-800, and Ni2Si/SiCN-1000 samples have been compared. With the increase in the reduction temperature, the conversion of nitrobenzene over different catalysts presented the following sequence: Ni/SiCN-600 ≈ Ni(Ni2Si)/SiCN-800 > Ni/SiCN-400 > Ni2Si/SiCN-1000. With the increased interactions between Ni nanoparticles and the SiCN support through the reduction, the conversion of nitrobenzene decreased to some extent, which could be attributed to the aggregation of metals and the formation of intermetallic Ni2Si. Due to the high hydrogenation activity of metallic Ni, the feedstock of benzaldehyde is also converted into benzylalcohol, which leads to the low selectivity to arylamines (benzylidene and benzylaniline). As shown in Fig. 7b, the Ni2Si/SiCN-1000 catalyst presents much higher selectivity to arylamines (ca. 92%) than the other catalysts. With the extension of the reaction time, the selectivity to arylamines over Ni2Si/SiCN-1000 catalysts remained almost unchanged. However, it decreased remarkably over SiCN-supported Ni-based catalysts, which could be attributed to the decomposition of benzylidene and benzylaniline to aniline (detected by GC). Therefore, compared with Ni/SiCN catalysts, the Ni2Si/SiCN catalyst presents high selectivity to imines and secondary amines in the one-pot reductive coupling of nitrobenzene and benzaldehyde.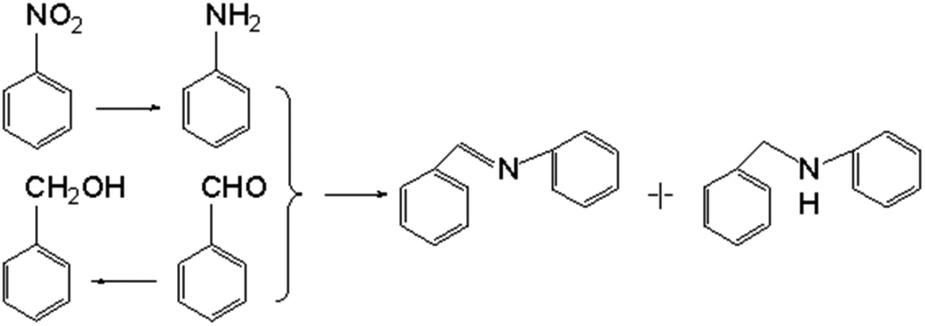 | ||
| Scheme 1 Reaction routes of the reductive coupling of nitrobenzene and benzaldehyde to imines and secondary amines. | ||
 | ||
| Fig. 7 (a) Conversion of nitrobenzene and (b) selectivity to arylamine over Ni/SiCN-400, Ni/SiCN-600, Ni(Ni2Si)/SiCN-800, and Ni2Si/SiCN-1000 catalysts in the reductive coupling of nitrobenzene and benzaldehyde. (The reaction was performed at 170 °C and 5.0 MPa H2 with 1 mmol nitrobenzene, 2 mmol benzaldehyde, 0.1 g catalyst, and 20 mL cyclohexane.) | ||
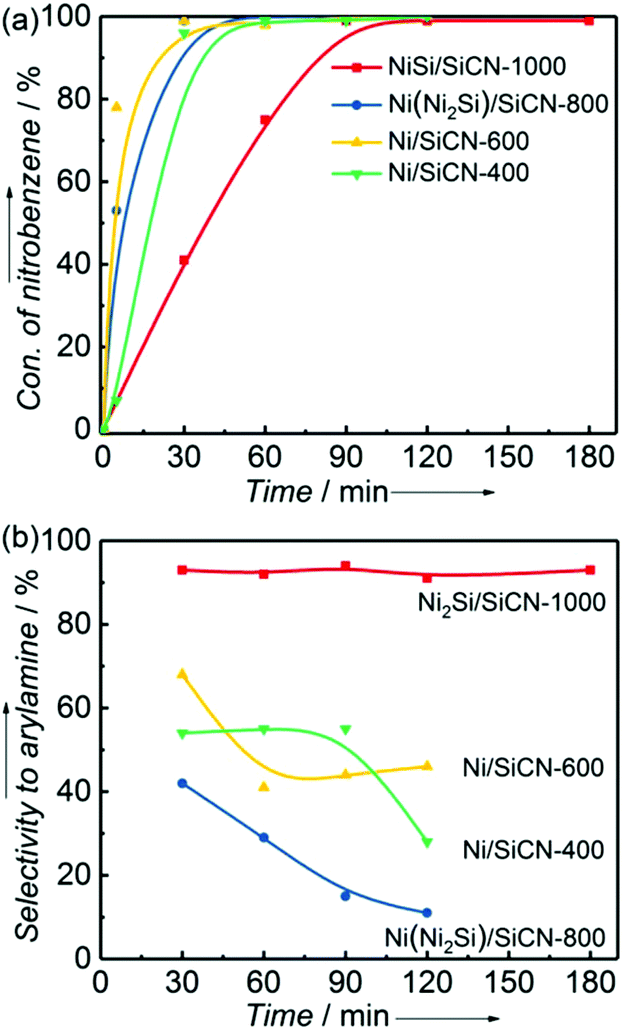
Fig. 8 shows the product distribution and selectivity to arylamine versus the reaction time in the reductive coupling of nitrobenzene and benzaldehyde to imine over the Ni2Si/SiCN-1000 sample. With the increase in the reaction time, both nitrobenzene and benzaldehyde were consumed, while benzylidene and aniline were produced (Fig. 8a). At 90 min, the conversion of nitrobenzene reached 99% and stoichiometric benzaldehyde was consumed equally. The selectivity to benzylidene and benzylaniline can reach 82% and 10%, respectively (Fig. 8b). In addition, a small amount of aniline with selectivity of 8% did not couple with arylamines, which could be attributed to the hydrogenolysis of benzylaniline. Interestingly, excess benzaldehyde was not hydrogenated over the Ni2Si/SiCN-1000 catalyst when nitrobenzene was added, and only trace benzylalcohol was detected, which indicates that the hydrogenation of benzaldehyde to benzylalcohol was inhibited over the Ni2Si/SiCN-1000 catalyst.
 | ||
| Fig. 8 (a) Product distribution and (b) selectivity to arylamine versus the reaction time in the reductive coupling of nitrobenzene and benzaldehyde to imine over the Ni2Si/SiCN-1000 sample. (The reaction was performed at 170 °C and 5.0 MPa H2 with 1 mmol nitrobenzene, 2 mmol benzaldehyde, 0.1 g catalyst, and 20 mL cyclohexane.) | ||
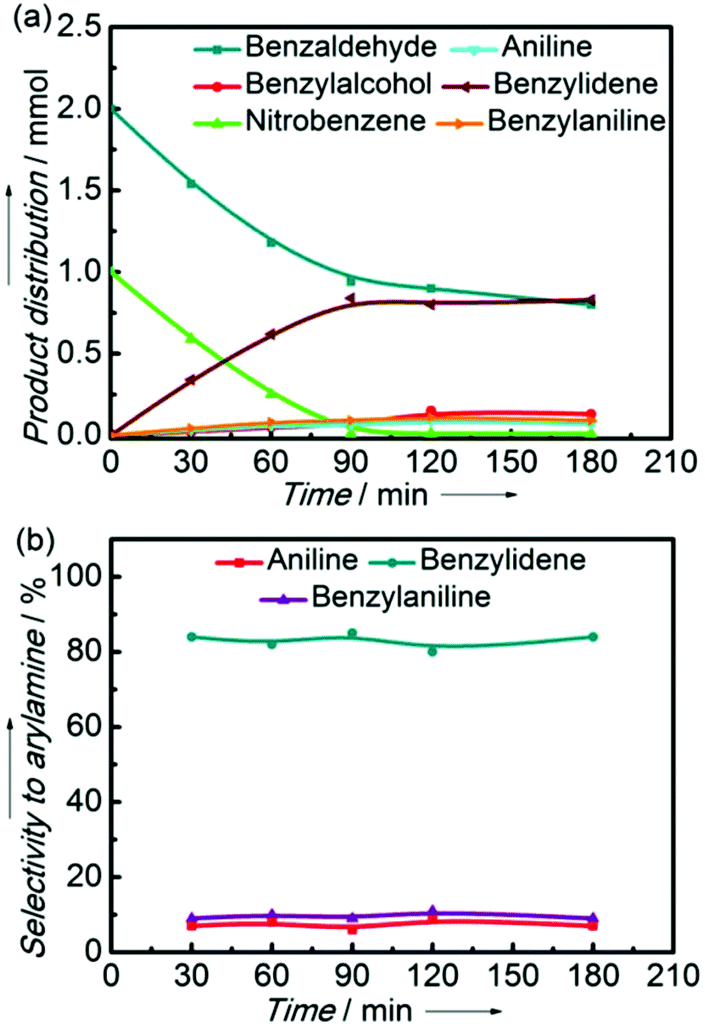
For understanding the process of the one-pot reductive coupling of nitrobenzene and benzaldehyde over the Ni2Si/SiCN-1000 catalyst, benzaldehyde hydrogenation, nitrobenzene hydrogenation, and coupling of benzaldehyde and aniline over the Ni2Si/SiCN-1000 catalyst have been tested. The key factor for the one-pot tandem synthesis of imines and secondary amines via the reductive coupling of nitrobenzene and benzaldehyde is that the reaction rate of the coupling of benzaldehyde and aniline is much higher than that of benzaldehyde hydrogenation, inhibiting the product benzyl alcohol. As shown in Fig. 9, the reaction rate of nitrobenzene hydrogenation and coupling of benzaldehyde and aniline over the Ni2Si/SiCN-1000 catalyst is 1.68 and 3.17, respectively, which is much faster than that of the hydrogenation of benzaldehyde (0.49), due to the repulsive force between the electronegative silicon atoms in the intermetallic Ni2Si and oxygen atoms in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of aldehyde, which lead to the low activity in the hydrogenation of aldehyde to alcohol.32 In addition, electron transfer occurs between the Ni and Si atoms due to the difference in the electronegativity. The active site of Ni isolated by the interposition of Si in nickel silicide has been confirmed previously.23,38,50 Therefore, the SMSI and the formation of intermetallic Ni2Si modify the metallic Ni active sites, which may change the reaction route significantly, enhancing the one-pot tandem synthesis of imines and secondary amines via the reductive coupling of nitrobenzene and benzaldehyde.
O bond of aldehyde, which lead to the low activity in the hydrogenation of aldehyde to alcohol.32 In addition, electron transfer occurs between the Ni and Si atoms due to the difference in the electronegativity. The active site of Ni isolated by the interposition of Si in nickel silicide has been confirmed previously.23,38,50 Therefore, the SMSI and the formation of intermetallic Ni2Si modify the metallic Ni active sites, which may change the reaction route significantly, enhancing the one-pot tandem synthesis of imines and secondary amines via the reductive coupling of nitrobenzene and benzaldehyde.
 | ||
| Fig. 9 Conversion versus reaction time in the benzaldehyde hydrogenation, the nitrobenzene hydrogenation, and the coupling of benzaldehyde and aniline over the Ni2Si/SiCN-1000 sample, respectively. (The reaction was performed at 170 °C and 5.0 MPa H2 with 0.1 g catalyst, 20 mL cyclohexane as solvent, and 1 mmol nitrobenzene, 2 mmol benzaldehyde, and the mixtures of 1 mmol benzaldehyde and 1 mmol aniline as substrates, respectively.) | ||
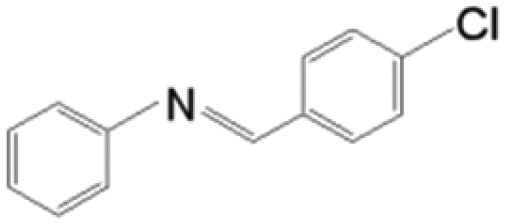
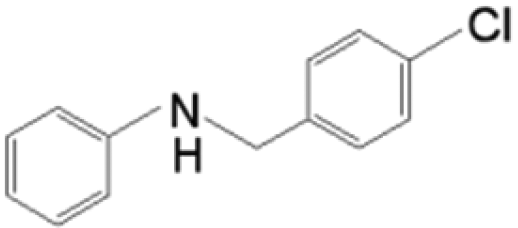
The reductive coupling of various aromatic aldehydes and nitroarenes under optimal conditions was investigated to demonstrate the general applicability of the intermetallic Ni2Si/SiCN-1000 catalyst. As shown in Table 1, despite the different electronic nature of the substituent groups, the nitro compounds were reduced smoothly with the conversion >90% in the one-pot reductive coupling of substituted nitrobenzenes and benzaldehyde, and preferable yields of imines and secondary amines were obtained. The electronic effects of substituents in aromatic carbonyl compounds have no significant effect on the conversion and selectivity. Halogenated substrates afford the corresponding imines in 71%–83% selectivity of the isolated product. The yield of halogenated imines has the following sequence: F– > Br– > Cl–. This can be attributed to the electron donor effect. In addition, alkyl substitutes afford the corresponding imines in 62–67% selectivity of the isolated product. The steric hindrance effect was also not observed in the one-pot reductive coupling of alkyl-substituted nitrobenzene and benzaldehyde over the intermetallic Ni2Si/SiCN-1000 catalyst. Chloric substrate benzaldehyde has also been tested to couple with nitrobenzene (entry 7 in Table 1), which presents 76% selectivity to the corresponding imines and has no negative effect on the catalyst result. In the reductive coupling of nitrobenzene and furfural, the intermetallic Ni2Si/SiCN-1000 catalyst still presents high activity (100% conversion) and 76% selectivity to the corresponding imine, which closely matches the activity of the Fe2O3–Pt core–shell structure catalyst.10 Therefore, intermetallic Ni2Si/SiCN-1000 as a novel catalyst is promising for the reductive coupling of various aromatic aldehydes and nitroarenes.
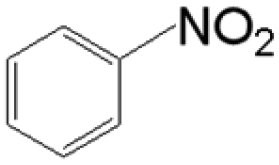
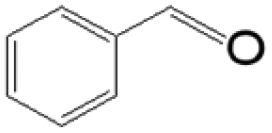
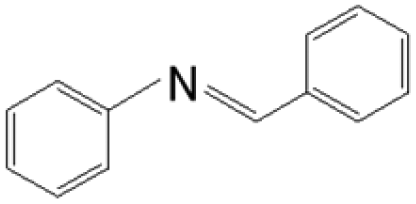
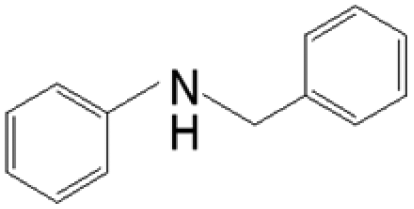
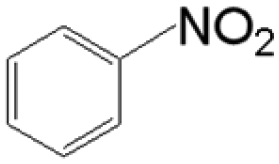
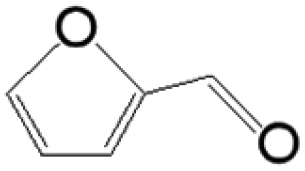
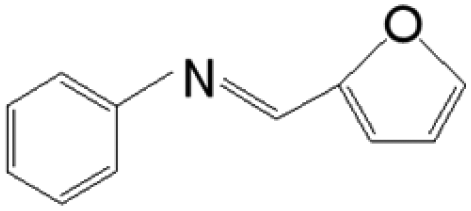
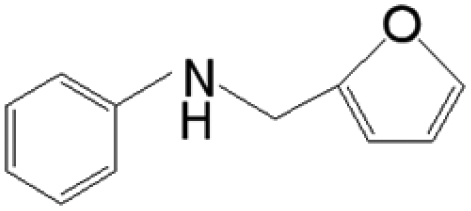
The one-pot tandem synthesis of imines and secondary amines via the reductive coupling of nitrobenzene and benzaldehyde was also chosen as a test reaction to demonstrate its reusability. The Ni2Si/SiCN-1000 catalyst exhibits excellent stability with ca. 90% conversion of nitrobenzene and ca. 92% selectivity to aromatic amine, mainly benzylidene, after cycling five times under the optimize conditions: cyclohexane as solvent under 5.0 MPa H2 pressure at 170 °C and 90 min (Fig. 10). Through detecting the content of metals in the reaction solution by ICP-OES, no Ni leaching was observed (lower than the limit of detection). The spent catalyst has been characterized by XRD and TEM. As shown in Fig. S3,† the XRD patterns show that the structure of Ni2Si is almost invariant after the stability test, whereas the intensity of peaks slightly decrease. In addition, the TEM image of the spent Ni2Si/SiCN-1000 catalyst (Fig. S4†) shows that the Ni2Si particles with a particle size of 7.29 nm are dispersed on the SiCN support, which is slightly larger than that of the fresh catalyst (7.05 nm), indicating that Ni2Si particles are partially aggregated after recycling. These results indicate that the SiCN-supported nickel silicide catalyst derived from the metal-containing polymer sustains the robust stability in the one-pot tandem synthesis of imines and secondary amines owing to the special geometric and electronic structure of intermetallic nickel silicide and the strong interaction between nickel silicide and the SiCN matrix.39,51,52
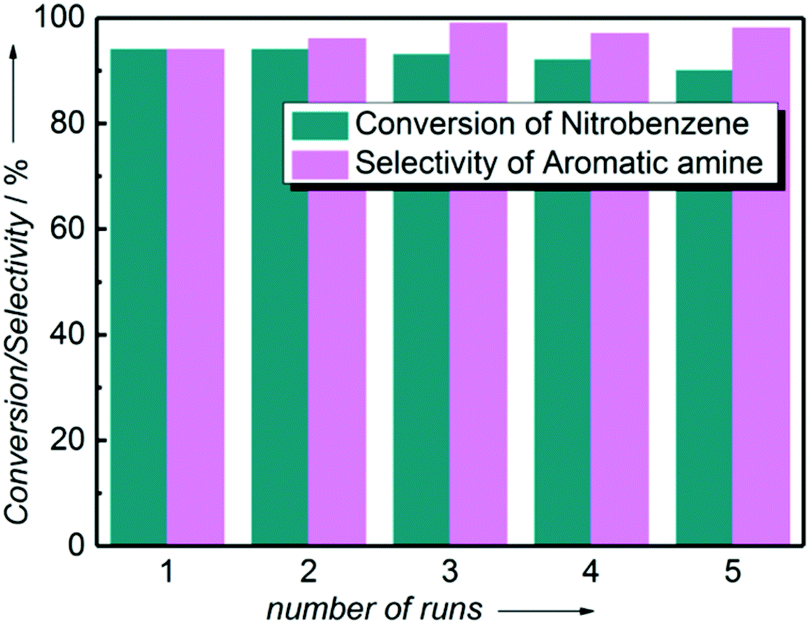 | ||
| Fig. 10 Catalytic conversion and the corresponding selectivity versus recycling times over Ni2Si/SiCN-1000 without the addition of new catalysts. (The reaction was performed at 170 °C, 90 min, and 5.0 MPa H2 with 1 mmol nitrobenzene, 2 mmol benzaldehyde, 0.1 g catalyst, and 20 mL cyclohexane.) | ||
Conclusions
For enhancing the catalytic activity and stability by modulating the local structure and the electronic properties of active sites, the intermetallic Ni2Si/SiCN-1000 catalyst has been synthesized through the temperature-programmed pyrolysis of a nickel-modified polysilazane precursor according to the concept of support–metal strong interaction. The reaction rate of the coupling of benzaldehyde and aniline over the Ni2Si/SiCN-1000 catalyst is much higher than that of benzaldehyde hydrogenation, inhibiting the product, benzyl alcohol. In the one-pot reductive coupling of nitrobenzene and benzaldehyde, the intermetallic Ni2Si/SiCN-1000 catalyst presents high efficiency with >99% conversion of nitrobenzene and >92% selectivity to aromatic amine, and sustains the robust stability. In addition, intermetallic Ni2Si/SiCN-1000 as a novel catalyst is promising for the reductive coupling of various aromatic aldehydes and nitroarenes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support provided by the National Natural Science Foundation of China (21373038 and 21403026) and the Fundamental Research Funds for the Central Universities (DUT18GJ206).References
- A. W. Bosman, H. M. Janssen and E. W. Meijer, Chem. Rev., 1999, 99, 1665–1688 CrossRef CAS.
- V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283–316 CrossRef CAS.
- S. Ichikawa, M. Tada, Y. Iwasawab and T. Ikariya, Chem. Commun., 2005, 924–926 RSC.
- H. Blaser, H. Steiner and M. Studer, ChemCatChem, 2009, 1, 210–221 CrossRef CAS.
- R. S. Downing, P. J. Kunkeler and H. van Bekkum, Catal. Today, 1997, 37, 121–136 CrossRef CAS.
- H. K. Kadam and S. G. Tilve, RSC Adv., 2015, 5, 83391–88347 RSC.
- T. Higuchi, R. Tagawa, A. Iimuro, S. Akiyama, H. Nagae and K. Mashima, Chem. – Eur. J., 2017, 23, 12795–12804 CrossRef CAS.
- M. Sankar, Q. He, S. Dawson, E. Nowicka, L. Lu, P. Bruijnincx, A. M. Beale, C. J. Kiely and B. M. Weckhuysen, Catal. Sci. Technol., 2016, 6, 5473–5482 RSC.
- H. Sun, F. Su, J. Ni, Y. Cao, H. He and K. Fan, Angew. Chem., Int. Ed., 2009, 48, 4390–4393 CrossRef CAS.
- B. Zhang, X. Guo, H. Liang, H. Ge, X. Gu, S. Chen, H. Yang and Y. Qin, ACS Catal., 2016, 6, 6560–6566 CrossRef CAS.
- A. Zanardi, J. A. Mata and E. Peris, Chem. – Eur. J., 2010, 16, 10502–10506 CrossRef CAS.
- F. A. Westerhaus, et al. , Nat. Chem., 2013, 5, 537–543 CrossRef CAS.
- R. V. Jagadeesh, T. Stemmler, A. Surkus, M. Bauer, M. Pohl, J. Radnik, K. Junge, H. Junge, A. Brückner and M. Beller, Nat. Protoc., 2015, 10, 916–926 CrossRef.
- L. Liu, P. Concepción and A. Corma, J. Catal., 2016, 340, 1–9 CrossRef CAS.
- R. V. Jagadeesh, A. Surkus, H. Junge, M. Pohl, J. Radnik, J. Rabeah, H. Huan, V. Schünemann, A. Brückner and M. Beller, Science, 2013, 342, 1073–1075 CrossRef CAS.
- R. V. Jagadeesh, T. Stemmler, A. Surkus, H. Junge, K. Junge and M. Beller, Nat. Protoc., 2015, 10, 548–557 CrossRef CAS.
- Y. Li, Y. Zhou, X. Ma and H. Jiang, Chem. Commun., 2016, 52, 4199–4202 RSC.
- T. Schwob and R. Kempe, Angew. Chem., Int. Ed., 2016, 55, 15175–15179 CrossRef CAS.
- G. Hahn, J. Ewert, C. Denner, D. Tilgner and R. Kempe, ChemCatChem, 2016, 8, 2461–2465 CrossRef CAS.
- P. Zhou, Z. Zhang, L. Jiang, C. Yu, K. Lv, J. Sun and S. Wang, Appl. Catal., B, 2017, 210, 522–532 CrossRef CAS.
- T. Song, P. Ren, Y. Duan, Z. Wang, X. Chen and Y. Yang, Green Chem., 2018, 20, 4629–4637 RSC.
- C. Bäumler and R. Kempe, Chem. – Eur. J., 2018, 24, 8989–8993 CrossRef.
- X. Chen, A. Zhao, Z. Shao, C. Li, C. T. Williams and C. Liang, J. Phys. Chem. C, 2010, 114, 16525–16533 CrossRef CAS.
- A. L. Schmitt, J. M. Higgins, J. R. Szczech and S. Jin, J. Mater. Chem., 2010, 20, 223–235 RSC.
- Y. Lin, Y. Chen and Y. Huang, Nanoscale, 2012, 4, 1412–1421 RSC.
- L. Zhang, X. Chen, S. Jin, X. Di, C. T. Williams, Z. Peng and C. Liang, Mater. Chem. Phys., 2016, 180, 89–96 CrossRef CAS.
- C. Liang, A. Zhao, X. Zhang, Z. Ma and R. Prins, Chem. Commun., 2009, 2047–2049 RSC.
- Z. Dong, T. Wang, J. Zhao, T. Fu, X. Guo, L. Peng, B. Zhao, N. Xue, W. Ding and Z. Xie, Appl. Catal., A, 2016, 520, 151–156 CrossRef CAS.
- X. Chen and C. Liang, Catal. Sci. Technol., 2019, 9, 4785–4820 RSC.
- T. Ye, Y. Lu, J. Li, T. Nakao, H. Yang, T. Tada, M. Kitano and H. Hosono, J. Am. Chem. Soc., 2017, 139, 17089–17097 CrossRef CAS.
- P. Ryabchuk, G. Agostini, M. Pohl, H. Lund, A. Agapova, H. Junge, K. Junge and M. Beller, Sci. Adv., 2018, 4, 2375–2548 Search PubMed.
- X. Chen, M. Li, J. Guan, X. Wang, C. T. Williams and C. Liang, Ind. Eng. Chem. Res., 2012, 51, 3604–3611 CrossRef CAS.
- X. Chen, M. Zhang, K. Yang, C. T. Williams and C. Liang, Catal. Lett., 2014, 144, 1118–1126 CrossRef CAS.
- K. Yang, X. Chen, J. Guan and C. Liang, Catal. Today, 2015, 246, 176–183 CrossRef CAS.
- L. Zhang, X. Chen, C. Li, M. Armbrüster, Z. Peng and C. Liang, ChemistrySelect, 2018, 3, 1658–1666 CrossRef CAS.
- L. Zhang, X. Chen, Z. Peng and C. Liang, Mol. Catal., 2018, 449, 14–24 CrossRef CAS.
- L. Zhang, X. Chen, Y. Chen, Z. Peng and C. Liang, Catal. Sci. Technol., 2019, 9, 1108–1116 RSC.
- K. Yang, X. Chen, L. Wang, L. Zhang, S. Jin and C. Liang, ChemCatChem, 2017, 9, 1337–1342 CrossRef CAS.
- D. Seyferth, H. Lang, C. A. Sobon, J. Borm, H. J. Tracy and N. Bryson, J. Inorg. Organomet. Polym., 1992, 2, 59–77 CrossRef CAS.
- X. Zhang, L. Chen, C. Luo and J. Kong, J. Am. Ceram. Soc., 2016, 99, 1485–1493 CrossRef CAS.
- T. Schmalz, T. Kraus, M. Günthner, C. Liebscher, U. Glatzel, R. Kempe and G. Motz, Carbon, 2011, 49, 3065–3072 CrossRef CAS.
- G. Glatz, T. Schmalz, T. Kraus, F. Haarmann, G. Motz and R. Kempe, Chem. – Eur. J., 2010, 16, 4231–4238 CrossRef CAS.
- Y. Song, A. L. Schmitt and S. Jin, Nano Lett., 2007, 7, 965–969 CrossRef CAS.
- M. García-Méndez, F. Castillón, G. Hirata, M. Farías and G. Beamson, Appl. Surf. Sci., 2000, 161, 61–73 CrossRef.
- P. Tam and L. Nyborg, Surf. Coat. Technol., 2009, 203, 2886–2890 CrossRef CAS.
- S. Pérez-García and L. Nyborg, Surf. Interface Anal., 2006, 38, 859–862 CrossRef.
- S. Valeri, U. Del Pennino and P. Sassaroli, Surf. Sci., 1983, 134, L537–L542 CrossRef CAS.
- A. Corma and P. Serna, Science, 2006, 313, 332–334 CrossRef CAS.
- O. Verho, K. P. J. Gustafson, A. Nagendiran, C. Tai and J. Bäckvall, ChemCatChem, 2014, 6, 3153–3159 CrossRef CAS.
- H. Itahara, W. F. Simanullang, N. Takahashi, S. Kosaka and S. Furukawa, Inorg. Chem., 2019, 58, 5406–5409 CrossRef CAS.
- D. Forberg, J. Obenauf, M. Friedrich, S. Hühne, W. Mader, G. Motz and R. Kempe, Catal. Sci. Technol., 2014, 4, 4188–4192 RSC.
- S. M. Sachau, M. Zaheer, A. Lale, M. Friedrich, C. E. Denner, U. B. Demirci, S. Bernard, G. Motz and R. Kempe, Chem. – Eur. J., 2016, 22, 15508–15512 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional nitrogen physical adsorption of samples and the stability test result over Ni2Si/SiCN-1000 catalyst at low conversion. See DOI: 10.1039/c9qi01077g |
| This journal is © the Partner Organisations 2020 |