
Pb3Ba3Zn6(BO3)8 and α-BaZn2(BO3)2: new members of the zincoborates containing two different dimensional Zn–O units†
Xiaobo
Pan
a,
Hongping
Wu
 *b,
Shichao
Cheng
c and
Zhengping
Wang
*a
*b,
Shichao
Cheng
c and
Zhengping
Wang
*a
aState Key Laboratory of Crystal Materials, Shandong University, Jinan 250100, China
bTianjin University of Technology, Tianjin 300384, China. E-mail: wuhp@ms.xjb.ac.cn
cSchool of Physical Science and Technology, Xinjiang University, Urumqi 830046, China
First published on 10th October 2019
Abstract
Two new zincoborates, namely, Pb3Ba3Zn6(BO3)8 and α-BaZn2(BO3)2, were synthesized by the high-temperature solution method and their structures were determined by single-crystal X-ray diffraction for the first time. The structure of Pb3Ba3Zn6(BO3)8 can be described as a three-dimensional framework consisting of [Zn4B2O15]∞ chains and isolated ZnO4 tetrahedra and BO3 triangles, which contains three different types of tunnels with Pb2+ and Ba2+ cations residing among them. α-BaZn2(BO3)2 exhibits a two-dimensional layered structure with a [Zn2B2O6] layer, which is formed by [ZnBO4] chains and [Zn2B2O8] chains. Interestingly, both Pb3Ba3Zn6(BO3)8 and α-BaZn2(BO3)2 both contain two different dimensional Zn–O configurations in their structures, which has not been found in other zincoborates. In addition, thermal property analysis, and UV-vis-NIR diffuse reflectance and infrared spectroscopy were also performed. First-principles theoretical studies were also conducted to aid the understanding of their band structures and densities of states.
Introduction
Numerous research interests have been directed towards borates due to their abundant structural features and important technical applications as second-order nonlinear optical materials,1,2 birefringent materials,3–5 catalysts,6,7 ion conductors8,9 and phosphors.10,11 Generally, a boron atom can be in either triangular or tetrahedral coordination with oxygen atoms to form BO3 or BO4 groups, and these two groups can connect with each other through sharing O atoms to construct different degrees of polymerization of the B–O units, such as B2O5,12 B3O6,13 B5O10,14 B6O1215 and B18O36.16 It is remarkable that B6O12 was the first B–O unit consisting of edge-sharing BO4 that was synthesized at ambient pressure. Also, a boron atom can connect with fluorine atoms forming [BOxF4−x](x+1)− units, which can connect with BO3 or BO4 anion groups to construct various B–O–F clusters, such as B2O5F2, B3O5F3, B4O8F2, B5O9F3, B6O11F2 and B6O11F3.17–28 Such rich B–O or B–O–F groups lead to the abundant structural chemistry of borates. For example, Q18Mg6(B5O10)3(B7O14)2F (Q = Rb and Cs)29 and Ban+2Znn(BO3)n(B2O5)Fn (n = 1, 2)12 contain two kinds of B–O groups. However, these plentiful borates cannot completely satisfy the requirements of practical application with the development of science or theoretical research. It is necessary to synthesize a vast number of borates with different topological arrangements of structural configuration to enrich the structure of borates and meet the needs of scientific research.Recently, many investigations have revealed that introduction of comparatively strong covalent ZnO4 tetrahedra is a feasible strategy for obtaining new compounds.30–35 For example, Yu et al. synthesized Cs3Zn6B9O21 and Ba3ZnB5O10PO4, which are new members of the KBe2BO3F2 (KBBF) family and they exhibit excellent NLO properties. Other Zn-containing KBBF-like compounds, such as AZn2BO3X2 (A = K, Rb, NH4; X = Cl, Br), have also been synthesized by Ye's group and Li's group.13,36,37 These preserve the structural merits of KBBF and possess large second harmonic generation response. Similarly, the polymorph zincoborates α-, β- and γ-Pb2Ba4Zn4B14O31 have been obtained,38 which all crystallize in the polar space group. Thus, we continued to explore new compounds in the PbO–BaO–ZnO–B2O3 system and obtained two new compounds: Pb3Ba3Zn6(BO3)8 and α-BaZn2(BO3)2. It is worth noting that Pb3Ba3Zn6(BO3)8 and α-BaZn2(BO3)2 both contain two different types of Zn–O units: 0D (zero-dimensional) ZnO4 tetrahedra and 1D (one-dimensional) Zn4O12 chains for Pb3Ba3Zn6(BO3)8 and 0D Zn2O6 dipolymers and 1D Zn2O7 chains for α-BaZn2(BO3)2 (named according to the calculated density and β-BaZn2(BO3)2 reported in 1992).39 In this paper, the syntheses, crystal structures, optical properties and theoretical calculations of the two title compounds are reported. In addition, we compared the structure of Pb3Ba3Zn6(BO3)8 and α-BaZn2(BO3)2 with other zincoborates containing different types of Zn–O units.
Experimental section
Single-crystal preparation and compound syntheses
A single crystal of Pb3Ba3Zn6(BO3)8 was obtained by high-temperature solution reaction with a spontaneous crystallization method using H3BO3 and PbF2 as the flux. The raw materials of BaCO3, ZnO, H3BO3, PbO and PbF2 in the original ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:15:5:5 were mixed homogeneously before being transferred to a platinum crucible. Then, the mixtures were heated to 850 °C and kept at this temperature for 10 h to make the mixture homogeneous. The temperature was then reduced to 400 °C at a rate of 5 °C h−1 before the furnace was switched off. With this procedure, a few transparent, colourless Pb3Ba3Zn6(BO3)8 crystals were obtained by mechanical separation from the crucible. A single crystal of α-BaZn2(BO3)2 was also obtained by a spontaneous crystallization method using H3BO3 and PbO as the flux.
:4:15:5:5 were mixed homogeneously before being transferred to a platinum crucible. Then, the mixtures were heated to 850 °C and kept at this temperature for 10 h to make the mixture homogeneous. The temperature was then reduced to 400 °C at a rate of 5 °C h−1 before the furnace was switched off. With this procedure, a few transparent, colourless Pb3Ba3Zn6(BO3)8 crystals were obtained by mechanical separation from the crucible. A single crystal of α-BaZn2(BO3)2 was also obtained by a spontaneous crystallization method using H3BO3 and PbO as the flux.
Polycrystalline powders of Pb3Ba3Zn6(BO3)8 were synthesized by solid-state reactions. The stoichiometric ratio of PbO, BaCO3, ZnO and H3BO3 was heated at a temperature of 300 °C for 5 h to release gas and then sintered at 650 °C for 72 h, with several intermediate grindings. In this way, the powder sample of Pb3Ba3Zn6(BO3)8 was obtained. As shown in Fig. 1, the experimental powder X-ray diffraction (XRD) pattern agreed well with the calculated one. Unfortunately, we could not obtain the polycrystalline powders of α-BaZn2(BO3)2 though we made many efforts.
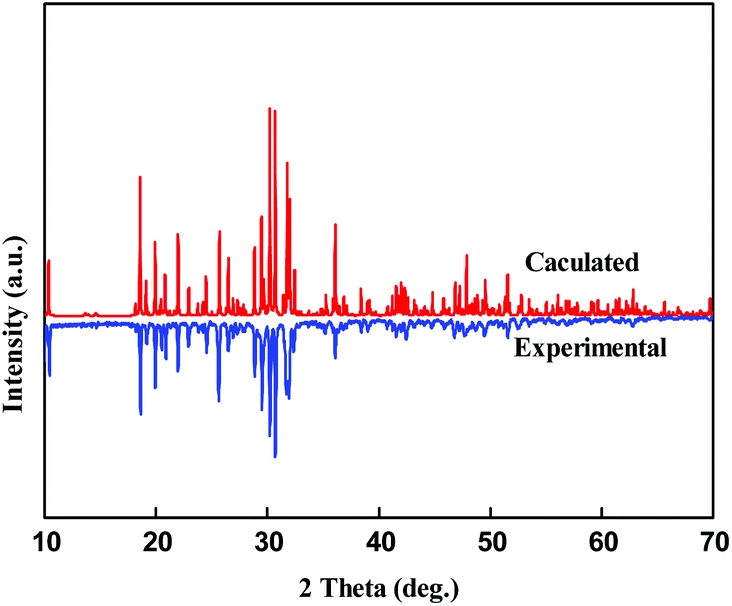 | ||
| Fig. 1 Experimental and calculated XRD patterns of Pb3Ba3Zn6(BO3)8. | ||
Structure determination
The single-crystal structures of Pb3Ba3Zn6(BO3)8 and α-BaZn2(BO3)2 were determined by single-crystal XRD techniques at room temperature. The colourless crystals with approximate dimensions of 0.11 × 0.09 × 0.08 mm3 for Pb3Ba3Zn6(BO3)8 and 0.11 × 0.08 × 0.05 mm3 for α-BaZn2(BO3)2 were selected for the structure analysis on a Bruker SMART APEX II CCD diffractometer, which was used to collect data using monochromatic Mo-Kα radiation (λ = 0.71073 Å) at 296(2) K. The data were integrated with the SAINT program and calculated on the SHELXTL crystallographic software package.40 The structures were solved by SHELXS-97 and refined on F2 by a full-matrix least-squares method.41 The structural symmetries were checked with PLATON.42 The crystal data and structure refinement information are summarized in Table 1. The atomic coordinates equivalent isotropic displacement parameters, bond valence sum, and selected bond lengths are summarized in Tables S1–S4,† respectively.| Empirical formula | Pb3Ba3Zn6(BO3)8 | α-BaZn2(BO3)2 |
|---|---|---|
| a R 1 = ∑||Fo| − |Fc||/∑|Fo| and wR2 = [∑w(Fo2 − Fc2)2/∑wFo4]1/2 for Fo2 > 2σ(Fo2). | ||
| Formula weight | 1896.29 | 385.70 |
| Temperature | 296(2) K | |
| Wavelength | 0.71073 Å | |
| Crystal system | Monoclinic | |
| Space group | C2/c (15) | P21/n(14) |
| Unit cell dimensions | a = 26.55(3) Å | a = 10.216(3) Å |
| b = 4.973(5) Å | b = 4.9556(13) Å | |
| c = 18.588(19) Å | c = 12.161(3) Å | |
| β = 106.587(12)° | β = 111.638(13)° | |
| Volume | 2352(4) Å3 | 573.3(3) Å3 |
| Z | 4 | 4 |
| Density (g cm−3) | 5.356 | 4.468 |
| Absorption coefficient (mm−1) | 32.424 | 15.054 |
| F(000) | 3304 | 696 |
| Theta range for data collection | 3.14 to 25.00° | 2.24 to 27.50° |
| Limiting indices | −23 ≤ h ≤ 31, −5 ≤ k ≤ 5, −22 ≤ l ≤ 16 | −13 ≤ h ≤ 12, −6 ≤ k ≤ 6, −15 ≤ l ≤ 15 |
| Reflections collected/unique | 5525/2044 [R(int) = 0.0575] | 7671/1317 [R(int) = 0.0381] |
| Completeness to theta | 99.4% | 99.6% |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 2044/6/182 | 1317/0/101 |
| Goodness-of-fit on F2 | 1.025 | 1.042 |
| Final R indices [Fo2 > 2σ(Fo2)]a | R 1 = 0.0492 | R 1 = 0.0222 |
| wR 2 = 0.1364 | wR 2 = 0.0461 | |
| R indices (all data)a | R 1 = 0.0588 | R 1 = 0.0312 |
| wR 2 = 0.1438 | wR 2 = 0.0485 | |
| Largest diff. peak and hole | 4.804 and −3.337 e Å−3 | 1.010 and −0.694 e Å−3 |
Thermal properties
The thermal analysis of Pb3Ba3Zn6(BO3)8 was carried out on a NETZSCH STA 449C thermal analyzer instrument in an atmosphere of flowing N2. The measurement temperature ranged from 25 °C to 1200 °C with a heating rate of 5 °C min−1.Spectroscopic properties
The Fourier transform infrared spectroscopy spectrum of Pb3Ba3Zn6(BO3)8 was measured on a Shimadzu IRAffinity-1 Fourier transform IR spectrometer ranging from 400 to 4000 cm−1.The 3700DUV Shimadzu spectrophotometer was used to measure the diffuse reflectance spectrum of Pb3Ba3Zn6(BO3)8 at room temperature over the range from 190 to 2500 nm. The Kubelka–Munk function was applied to transform the reflectance spectrum into absorbance spectrum.
Theoretical calculations
The band structures and density of states (DOS) of Pb3Ba3Zn6(BO3)8 and α-BaZn2(BO3)2 were calculated by using the plane-wave pseudo-potential based on the density functional theory with the CASTEP program.43 The generalized gradient-approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional was chosen to describe the exchange–correlation potential. Under the norm-conserving pseudopotential (NCP), the following orbital electrons were treated as valence electrons: Pb 5d106s26p2, Ba 5s25p66s2, Zn 3d104s2, B 2s22p1, O 2s22p4.44–46 The energy cut-off was set at 910 eV for Pb3Ba3Zn6(BO3)8 and 830 eV for α-BaZn2(BO3)2. The Monkhorst–Pack k-point sampling was set at 3 × 3 × 1 for Pb3Ba3Zn6(BO3)8 and 3 × 5 × 2 for α-BaZn2(BO3)2 in the Brillouin zone. The other calculation parameters and convergent criteria were set by the default values of CASTEP.Results and discussion
Structure of Pb3Ba3Zn6(BO3)8
Pb3Ba3Zn6(BO3)8 crystallizes in a monoclinic crystal system with a centrosymmetric space group of C2/c (no. 15). In the asymmetric unit, there are two unique Pb atoms, two unique Ba atoms, three unique Zn atoms, four unique B atoms and twelve unique O atoms. In the structure, the B atoms are three-coordinated, with B–O bond lengths ranging from 1.30(3) to 1.42(3) Å. However, the Zn atoms are four-coordinated, with the Zn–O bond lengths ranging from 1.903(12) to 2.031(12) Å. The bond valence sum (BVS) values of Pb (1.994–2.062), Ba (2.055–2.153), Zn (1.979–2.105), B (2.951–3.093), and O (1.818–2.171) were in good agreement with reasonable values.In the structure, the ZnO4 tetrahedra have two different connection modes, the isolated ZnO4 tetrahedra and the 1D [Zn4O12]∞ chain (Fig. 2(a)). The [Zn4O12]∞ chains share vertices with isolated BO3 triangles to form the [Zn4B2O15]∞ chains (Fig. 2(b)), which are further connected by the isolated ZnO4 tetrahedra and BO3 triangles to form the three-dimensional (3D) framework (Fig. 2(c)). The Ba and Pb atoms are filled in the space created by the network. Notably in the structure, there are three types of tunnels along the b axis: A-type, B-type and C-type. For the A-tunnel, as shown in Fig. S1(b),† we can notice that it is a super-large tunnel with 40 members, which is very rare in the borate compounds reported so far. The length of this super-tunnel has a large value of 20.607 Å (determined by the distance of Zn(2)–Zn(2)) and the width is 4.467 Å (determined by the distance of B(4)–B(4)). Also, two four-coordinated Pb(1) atoms and two six-coordinated Ba(1) atoms are located in this sort of tunnel (Fig. S2†). For the B-tunnels, as shown in Fig. S1(c),† there are 12 members with the length of 6.795 Å and width of 4.311 Å. Also, one six-coordinated Ba(2) atom dominates this tunnel (Fig. S2†). Apart from these, there is a small C-type tunnel with 8 members in Fig. S1(d);† the cross-sectional dimensions of this tunnel are 4.874 Å × 3.582 Å. Also, one eight-coordinated Pb(2) atom is located in this tunnel (Fig. S2†). The larger A and B tunnels suggest that the Pb2+ and Ba2+ cations can be replaced by other cations, which provides an idea for the synthesis of new compounds. Besides, the large tunnels in the frameworks could make Pb3Ba3Zn6(BO3)8 a promising functional material for application in catalysts, ion-exchangers and molecular sieves.
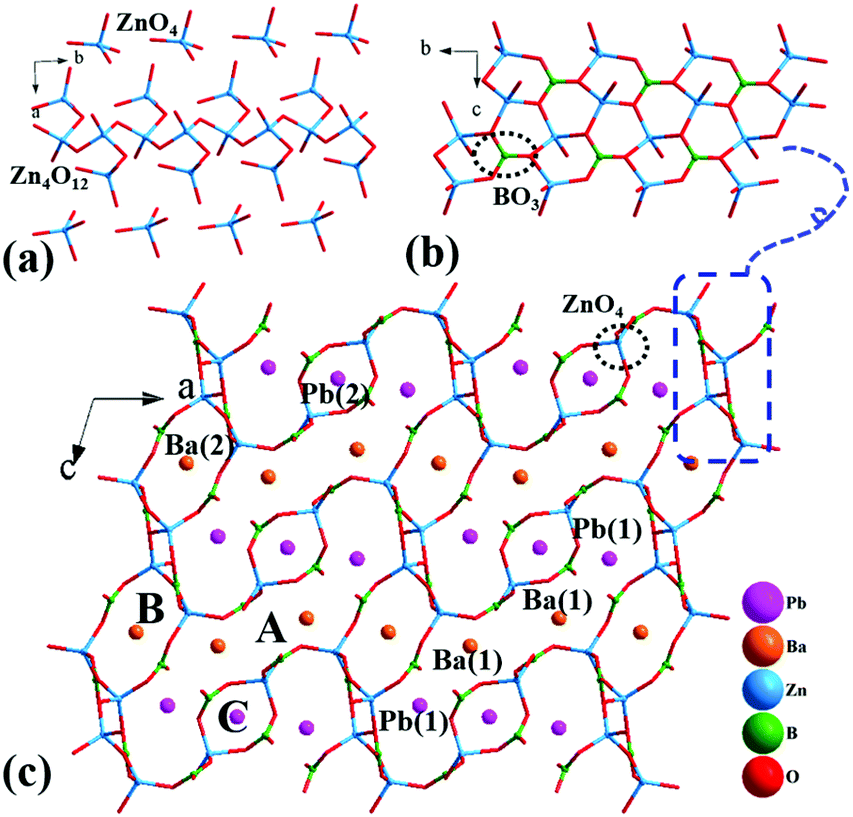 | ||
| Fig. 2 Crystal structure of Pb3Ba3Zn6(BO3)8: (a) the isolated ZnO4 tetrahedra and [Zn4O12]∞ chain; (b) the [Zn4B2O15]∞ chain; (c) crystal structure viewed along the b-axis. All the Pb–O and Ba–O bonds are removed for clarity. | ||
Structure of α-BaZn2(BO3)2
α-BaZn2(BO3)2 crystallizes in a monoclinic symmetry, with the space group P21/n (no. 14). In the asymmetric unit, the Ba, Zn, B and O atoms occupy one, two, two and six crystallographically unique sites, respectively. In the structure, the bond lengths of Ba–O, Zn–O and B–O range from 2.651(3) to 3.328(3) Å, 1.898(3) to 2.060(3) Å and 1.361(6) to 1.395(6) Å, respectively. The calculated total bond valences (Ba, 1.917; Zn, 1.999 and 2.084; B, 2.944 and 2.964; O, 1.908 to 2.068) indicate that the Ba, Zn, B and O atoms are in the oxidation states of +2, +2, +3 and −2, respectively.The Zn(1)O4 tetrahedra and the B(1)O3 triangles alternately connect with each other to form a [ZnBO4] chain (Fig. 3(a)), and two Zn(2)O4 tetrahedra connect with each other to form Zn2O6 dipolymers, which are further linked by B(2)O3 triangle to form a [Zn2B2O8] chain (Fig. 3(b)). Then, the [ZnBO4] chain and [Zn2B2O8] chain alternately link by sharing corner O atoms to form [Zn2B2O6] 2D layers (Fig. 3(c)), and the Ba atoms reside in the space between the two layers (Fig. 3(d)). Similar to Pb3Ba3Zn6(BO3)8, the structure of α-BaZn2(BO3)2 contains two different types of Zn–O units: an isolated Zn2O6 dipolymer and 1D [Zn2O7] chain (Fig. S3†), which has not been reported so far, to the best of our knowledge.
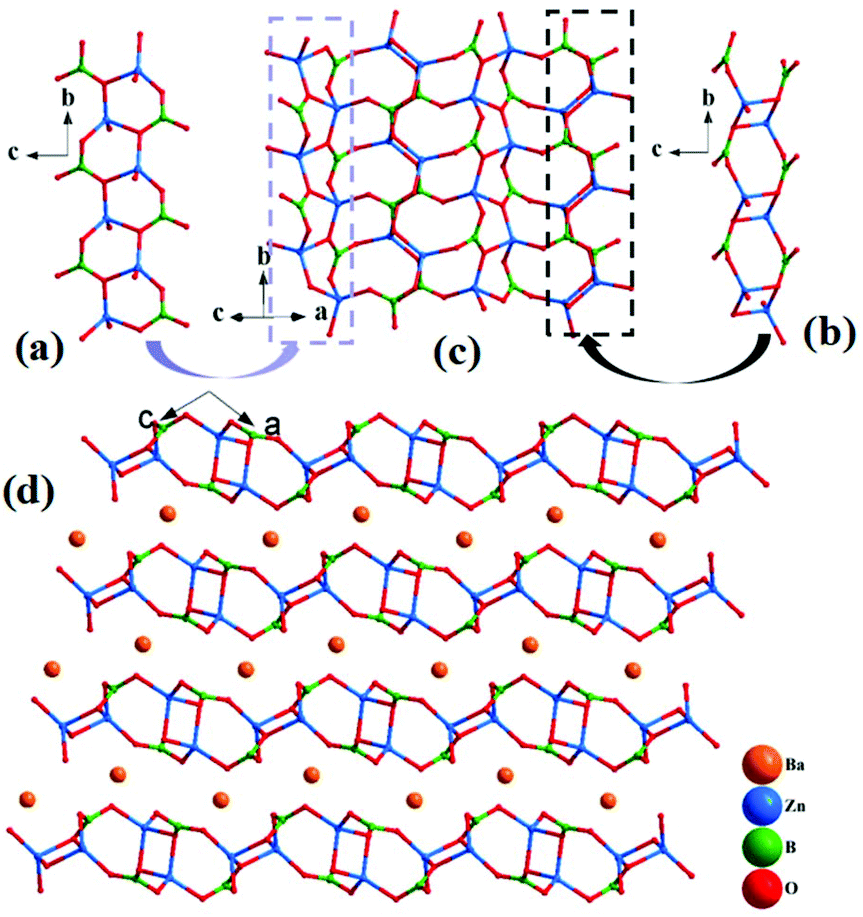 | ||
| Fig. 3 (a) [ZnBO4] chain; (b) [Zn2B2O8] chain; (c) the [Zn2B2O6]∞ layer; (d) crystal structure viewed along the b-axis; all the Ba–O bonds are removed for clarity. | ||
On the contrary, β-BaZn2(BO3)2 possesses a three-dimensional framework consisting of ZnO4 tetrahedra and BO3 triangles. As shown in Fig. S4(a),† four ZnO4 tetrahedra connect with each other by sharing corner O atoms to form Zn4O13 multimers, which further link with each other to form Zn4O12 chains. Then, the Zn4O12 chains connect with the isolated BO3 triangles to form a 3D framework structure. The Ba atoms are filled in the space created by the network, Fig. S4(c).†
Structure comparison
We summarize the anhydrous zincoborates in Table S5† and note that most of these compounds contain one type of Zn–O units, such as isolated ZnOn (n = 4, 5, 6) polyhedra, or they connect with each other to construct 0D multimer, 1D chains, 2D layers and 3D networks by sharing corners, edges or faces. Besides, the O atoms in ZnOn polyhedra can be replaced by other atoms and can form various units, such as the Zn2O6F unit in KCdZnBO3F, Zn3O12X unit in Zn3B7O13X (X = Cl, Br) and Zn4O12Se unit in Zn8(BO2)12Se2.47–51 It is worth noting that there are only five compounds possessing two different types of Zn–O units: isolated Zn2O7 and Zn2O6 in Ba2KZn3(B3O6)(B6O13),33 isolated Zn2O7 and Zn3O10 in Ba4K2Zn5(B3O6)3(B9O19)34 and isolated ZnO4 and Zn2O6 in α-, β- and γ-Pb2Ba4Zn4B14O31.38 In these five compounds, two types of Zn–O units are all 0D (Fig. S5†). However, for Pb3Ba3Zn6(BO3)8 and α-BaZn2(BO3)2, the two types of Zn–O units are 0D and 1D (Fig. S3†), which are reported for the first time in zincoborates.Thermal properties
The DSC-TG curves for Pb3Ba3Zn6(BO3)8 are shown in Fig. 4a. The TG result showed that Pb3Ba3Zn6(BO3)8 exhibited no obvious weight loss up to 1000 °C. The DSC curve for Pb3Ba3Zn6(BO3)8 exhibited one sharp and one mild endothermic peak at about 678 and 790 °C, respectively. To further confirm the thermal property, firstly, the polycrystalline sample of Pb3Ba3Zn6(BO3)8 (5 g) was placed in a platinum crucible and heated to 750 °C and held at this temperature for 3 days. The powder XRD patterns demonstrated that the phase mainly was β-BaZn2(BO3)2 (Fig. S6†). Secondly, the samples of Pb3Ba3Zn6(BO3)8 were heated to 1000 °C followed by cooling to room temperature at a rate of 5 °C per hour. Then the residue was mechanically separated from the crucible and some block crystal was obtained. Also, the crystal was determined by single-crystal XRD techniques, and the result showed that it was α-BaZn2(BO3)2. The powder XRD patterns of the residue were in agreement with the calculated pattern of α-BaZn2(BO3)2, as shown in Fig. 4b, which proved the existence of α-BaZn2(BO3)2. The above experiments show that Pb3Ba3Zn6(BO3)8 melts incongruently, and decomposes into β-BaZn2(BO3)2 at around 750 °C.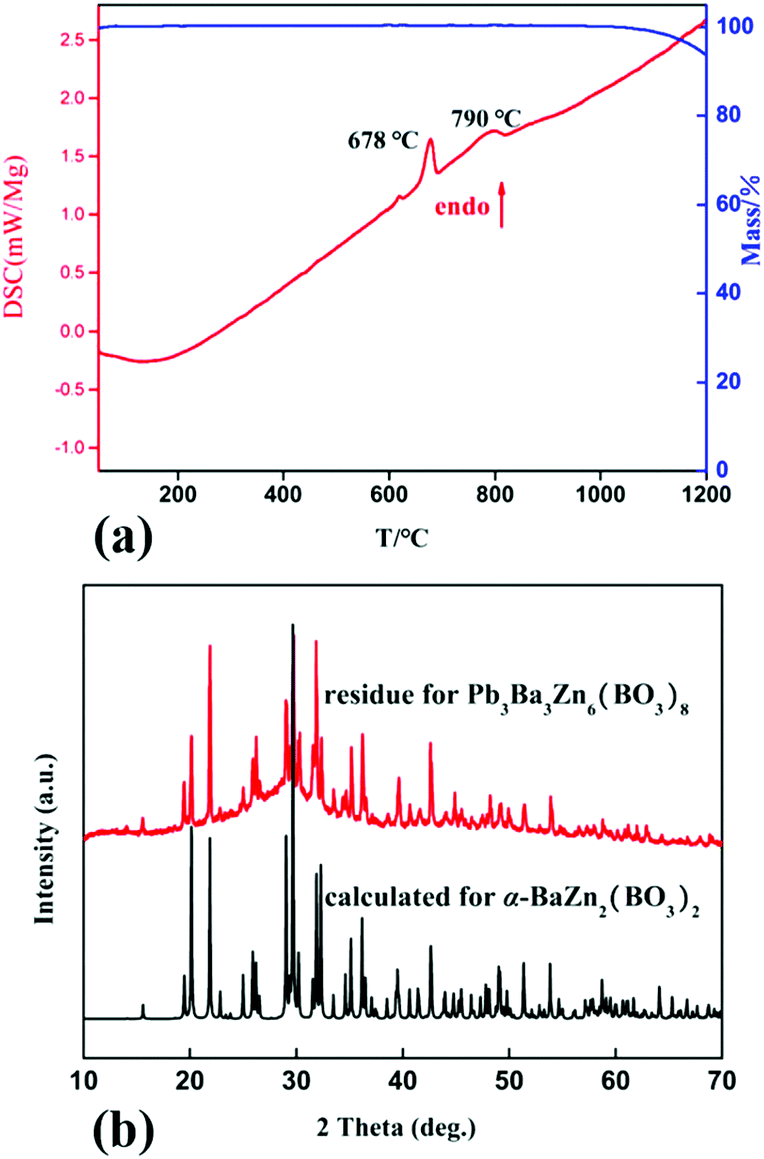 | ||
| Fig. 4 (a) TG/DSC curves for Pb3Ba3Zn6(BO3)8; (b) powder XRD patterns of the residue. | ||
Spectroscopic properties
The IR spectrum is shown in Fig. 5(a). The peak at 1645 cm−1 is a characteristic peak of H–O–H bending motion,52 which may be caused by the moisture-absorbing KBr. The peaks at 1440 and 890 cm−1 belong to the BO3 asymmetric and symmetric stretching vibrations, respectively.52 The peaks at 1135 and 450 cm−1 are attributed to the stretching and bending of the Zn4–O, respectively.31 The weak peaks at 730 and 620 cm−1 belong to the out-of-plane bending of BO3 groups.52 Also, the absorption peak at 570 cm−1 corresponds to the bending of BO3 groups.52 These results confirm the existence of BO3 triangles and ZnO4 tetrahedra in the structure of Pb3Ba3Zn6(BO3)8.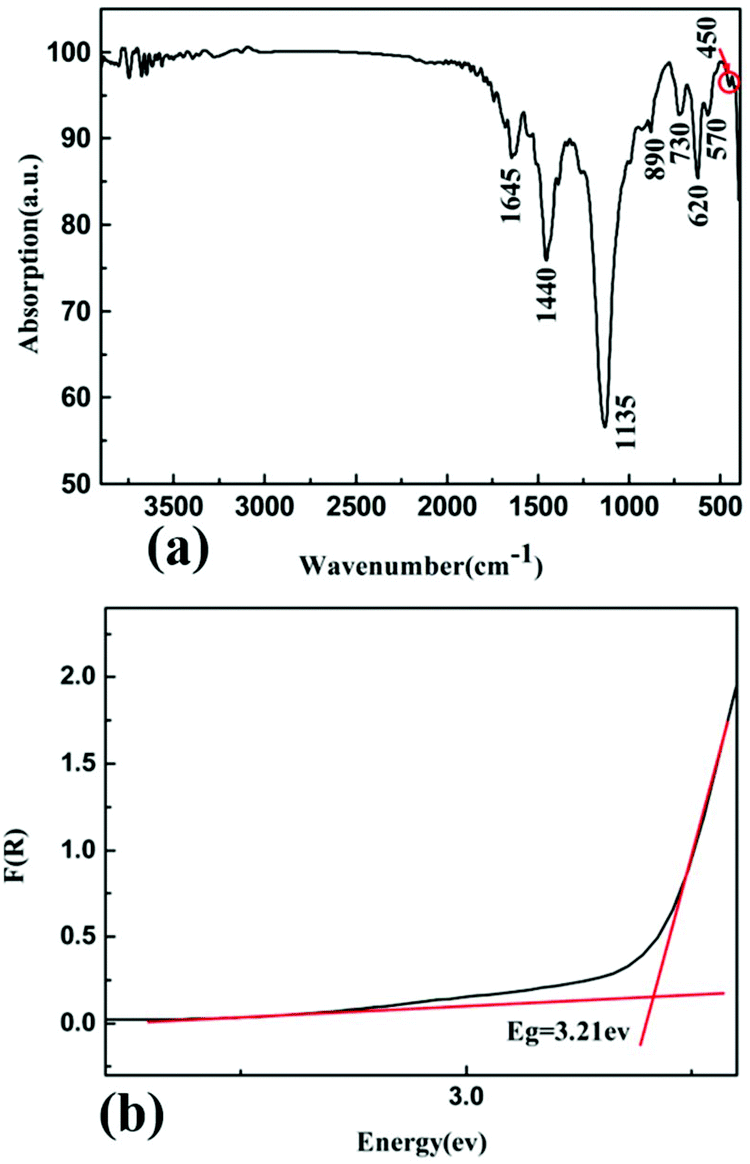 | ||
| Fig. 5 (a) The infrared spectrum of Pb3Ba3Zn6(BO3)8; (b) the UV-vis-NIR diffuse reflectance spectrum of Pb3Ba3Zn6(BO3)8. | ||
The UV-vis-NIR diffuse reflectance spectrum of Pb3Ba3Zn6(BO3)8 was measured from 190 to 2600 nm. On the basis of the diffuse-reflectance spectrum, it was converted to absorbance based on the Kubelka–Munk function and is shown in Fig. 5(b). The experimental energy gap of Pb3Ba3Zn6(BO3)8 was 3.21 eV.
Theoretical calculations
To gain further insight into the electronic structures and optical properties of the title compounds, the band structures and DOS were also calculated. As shown in Fig. 6(a) and (c), Pb3Ba3Zn6(BO3)8 and α-BaZn2(BO3)2 are indirect band gap compounds with energy values of 3.43 and 3.39 eV, and for Pb3Ba3Zn6(BO3)8 the calculated value is close to the experimental result.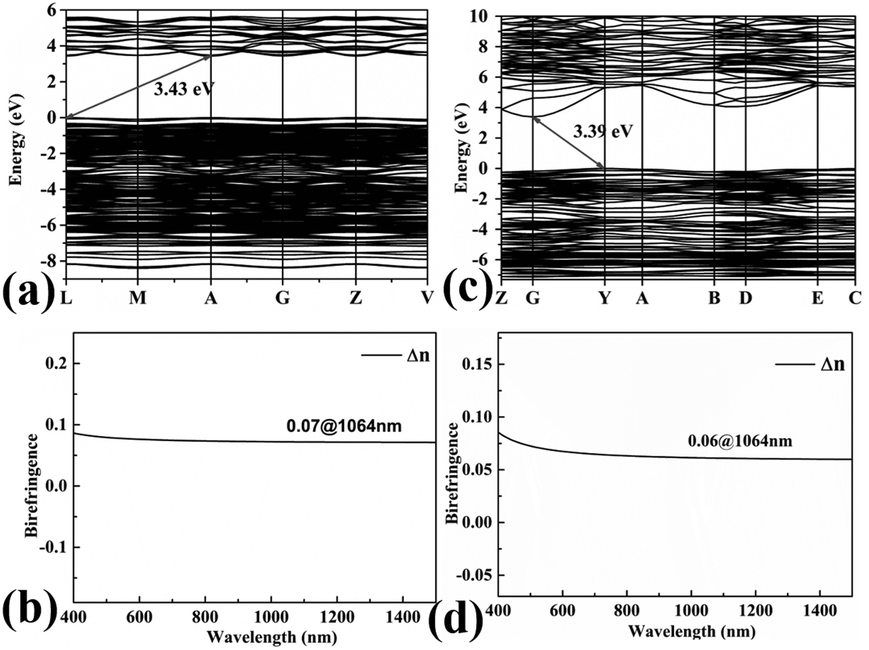 | ||
| Fig. 6 The calculated band structures and calculated birefrigence (Δn) of Pb3Ba3Zn6(BO3)8 (a) and (b), and α-BaZn2(BO3)2 (c) and (d). | ||
The birefringences of Pb3Ba3Zn6(BO3)8 and α-BaZn2(BO3)2 were also calculated using CASTEP. As shown in Fig. 6(b) and (d), the birefringences at 1064 nm were 0.07 and 0.06 for Pb3Ba3Zn6(BO3)8 and α-BaZn2(BO3)2, respectively.
The DOS of Pb3Ba3Zn6(BO3)8 and α-BaZn2(BO3)2 are presented in Fig. 7(a) and (b). For Pb3Ba3Zn6(BO3)8, near the Fermi level region, the top of the VBs is mainly occupied by the O-2p, B-2p and Zn-3d orbitals. Also, the bottom of the CBs is mainly occupied by the Pb-6p states, which suggests that the B–O and Zn–O groups make the main contribution to the band gap. While for α-BaZn2(BO3)2, the top of the VBs is mainly contributed by O-2p, B-2p and Zn-3d states and the bottom of the CBs is mainly contributed by Zn-4s and Ba-4d states. This means that the B–O and Zn–O groups control the band gap of α-BaZn2(BO3)2. From Fig. 7, we can see that the d orbital of Zn atoms in Pb3Ba3Zn6(BO3)8 is farther from the Fermi level compared with α-BaZn2(BO3)2. It was indicated that the d–p hybridization in Pb3Ba3Zn6(BO3)8 has little effect on the band gap.53,54 This is why the band gap of Pb3Ba3Zn6(BO3)8 (3.43 eV) is a little larger than that of α-BaZn2(BO3)2 (3.39 eV).
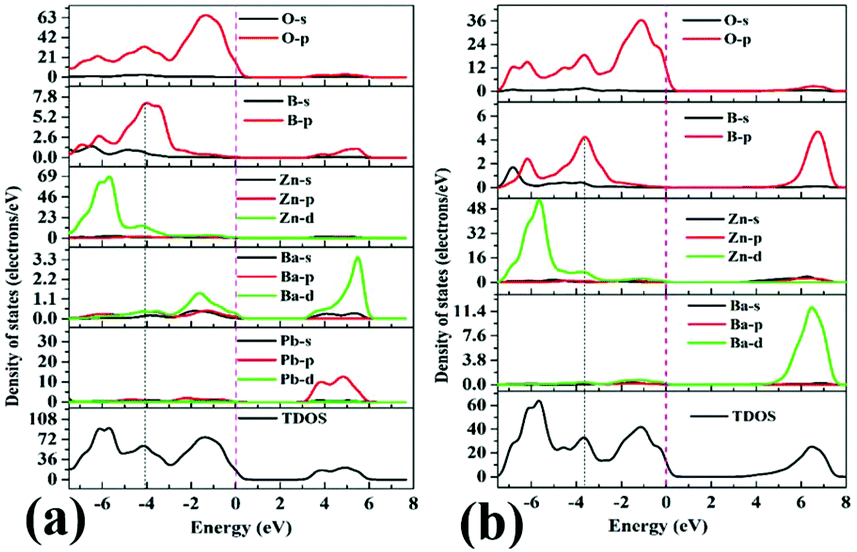 | ||
| Fig. 7 The total and partial densities ofstates of Pb3Ba3Zn6(BO3)8 (a), and α-BaZn2(BO3)2 (b). | ||
Conclusions
Two new zincoborates, namely, Pb3Ba3Zn6(BO3)8 and α-BaZn2(BO3)2, were synthesized and structurally characterized. The structure of Pb3Ba3Zn6(BO3)8 features a 3D framework consisting of BO3 triangles, ZnO4 tetrahedra and [Zn4O15]∞ chains. The structure of α-BaZn2(BO3)2 consists of [Zn2B2O6] layers formed by [ZnBO4] chains and [Zn2B2O8] chains. Pb3Ba3Zn6(BO3)8 and α-BaZn2(BO3)2 both contain 0D and 1D Zn–O units, which is reported for the first time in zincoborates, as far as we know. The TG/DSC curves indicated that Pb3Ba3Zn6(BO3)8 melts incongruently. Also, the powder XRD pattern of the residue of Pb3Ba3Zn6(BO3)8 proves the existence of α-BaZn2(BO3)2. The infrared spectrum confirms the existence of BO3 triangles and ZnO4 tetrahedra in Pb3Ba3Zn6(BO3)8. Also, the experimental band gap of Pb3Ba3Zn6(BO3)8 was 3.21 eV, which coincides with the calculated value. The first principles studies show that Pb3Ba3Zn6(BO3)8 and α-BaZn2(BO3)2 both are indirect band gap compounds with the band gaps of 3.43 and 3.39 eV, respectively. Those are primarily determined by the B–O and Zn–O groups for Pb3Ba3Zn6(BO3)8 and α-BaZn2(BO3)2, respectively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by National Natural Science Foundation of China (Grant No. 51972230 and 61975096); Research and Free Exploration Project of Shenzhen City (Grant No. JCYJ20180305164316517); Natural Science Foundation of Shandong Province (Grant No. ZR2017MF031).Notes and references
- M. Luo, Y. X. Song, F. Liang, N. Ye and Z. S. Lin, Inorg. Chem. Front., 2018, 5, 916 RSC
.
- C. Wu, L. H. Li, J. L. Song, G. Yang, M. G. Humphreye and C. Zhang, Inorg. Chem. Front., 2017, 4, 692 RSC
- Z. Z. Zhang, Y. Wang, H. Li, Z. H. Yang and S. L. Pan, Inorg. Chem. Front., 2019, 6, 546 RSC
- M. Zhang, D. H. An, C. Hu, X. L. Chen, Z. H. Yang and S. L. Pan, J. Am. Chem. Soc., 2019, 141, 3258 CrossRef CAS
- X. L. Chen, B. B. Zhang, F. F. Zhang, Y. Wang, M. Zhang, Z. H. Yang, K. R. Poeppelmeier and S. L. Pan, J. Am. Chem. Soc., 2018, 140, 16311 CrossRef CAS
- M. Ma, D. N. Liu, S. Hao, R. M. Kong, G. Du, A. M. Asiri, Y. D. Yao and X. P. Sun, Inorg. Chem. Front., 2017, 4, 840 RSC
- J. L. C. Rowsell, N. J. Taylor and L. F. Nazar, J. Am. Chem. Soc., 2002, 124, 6522 CrossRef CAS
- Z. Z. Zhang, Y. Wang, B. B. Zhang, Z. H. Yang and S. L. Pan, Angew. Chem., Int. Ed., 2018, 57, 6577 CrossRef CAS
- J. M. Doux, L. Leguay, A. Le Gal La Salle, O. Joubert and E. Quarez, Solid State Ionics, 2018, 324, 260 CrossRef CAS
- W. Y. Geng, X. F. Zhou, J. Y. Ding and Y. H. Wang, J. Mater. Chem. C, 2019, 7, 1982 RSC
- Z. Jia and M. J. Xia, New J. Chem., 2017, 41, 12416 RSC
- M. Mutailipu, X. Su, M. Zhang, Z. H. Yang and S. L. Pan, Inorg. Chem. Front., 2017, 4, 281 RSC
- H. W. Yu, H. P. Wu, S. L. Pan, Z. H. Yang, X. L. Hou, X. Su, Q. Jing, K. R. Poeppelmeier and J. M. Rondinelli, J. Am. Chem. Soc., 2014, 136, 1264 CrossRef CAS
- T. Baiheti, S. J. Han, A. Tudi, Z. H. Yang, H. H. Yu and S. L. Pan, Inorg. Chem. Front., 2019, 6, 1461 RSC
- S. F. Jin, G. M. Cai, W. Y. Wang, M. He, S. C. Wang and X. L. Chen, Angew. Chem., Int. Ed., 2010, 49, 4967 CrossRef CAS
- W. J. Yao, X. X. Jiang, H. W. Huang, T. Xu, X. S. Wang, Z. S. Lin and C. T. Chen, Inorg. Chem., 2013, 52, 8291 CrossRef CAS
- M. Luo, F. Liang, Y. X. Song, D. Zhao, N. Ye and Z. S. Lin, J. Am. Chem. Soc., 2018, 140, 6814 CrossRef CAS
- S. G. Jantz, M. Dialer, L. Bayarjargal, B. Winkler, L. van Wüllen, F. Pielnhofer, J. Brgoch, R. Weihrich and H. A. Höppe, Adv. Opt. Mater., 2018, 6, 1800497 CrossRef
- T. Pilz, H. Nuss and M. Jansen, J. Solid State Chem., 2012, 186, 104 CrossRef CAS
- H. P. Wu, H. W. Yu, Q. Bian, Z. H. Yang, S. J. Han and S. L. Pan, Inorg. Chem., 2014, 53, 12686 CrossRef CAS
- G. Q. Shi, Y. Wang, F. F. Zhang, B. B. Zhang, Z. H. Yang, X. L. Hou, S. L. Pan and K. R. Poeppelmeier, J. Am. Chem. Soc., 2017, 139, 10645 CrossRef CAS
- Y. Wang, B. B. Zhang, Z. H. Yang and S. L. Pan, Angew. Chem., Int. Ed., 2018, 57, 2150 CrossRef CAS
- X. F. Wang, Y. Wang, B. B. Zhang, F. F. Zhang, Z. H. Yang and S. L. Pan, Angew. Chem., Int. Ed., 2017, 56, 14119 CrossRef CAS
- Z. Z. Zhang, Y. Wang, B. B. Zhang, Z. H. Yang and S. L. Pan, Chem. – Eur. J., 2018, 24, 11267 CrossRef CAS
- Z. Z. Zhang, Y. Wang, B. B. Zhang, Z. H. Yang and S. L. Pan, Inorg. Chem., 2018, 57, 4820 CrossRef CAS
- M. Mutailipu, M. Zhang, B. B. Zhang, L. Y. Wang, Z. H. Yang, X. Zhou and S. L. Pan, Angew. Chem., Int. Ed., 2018, 57, 6095 CrossRef CAS
- G. Q. Shi, F. F. Zhang, B. B. Zhang, D. W. Hou, X. L. Chen, Z. H. Yang and S. L. Pan, Inorg. Chem., 2016, 56, 344 CrossRef
- G. P. Han, G. Q. Shi, Y. Wang, B. B. Zhang, S. J. Han, F. F. Zhang, Z. H. Yang and S. L. Pan, Chem. – Eur. J., 2018, 24, 4497 CrossRef CAS
- Z. Wang, M. Zhang, X. Su, S. L. Pan, Z. H. Yang, H. Zhang and L. Liu, Chem. – Eur. J., 2015, 21, 1414 CrossRef CAS
- X. B. Pan, H. P. Wu, M. Wen, B. B. Zhang, Z. H. Yang and S. L. Pan, Inorg. Chem. Front., 2018, 5, 2501 RSC
- H. P. Wu, X. Su, S. J. Han, Z. H. Yang and S. L. Pan, Inorg. Chem., 2016, 55, 4806 CrossRef CAS
- Y. N. Chen, M. Zhang, C. Hu, Z. H. Yang and S. L. Pan, Inorg. Chem. Front., 2018, 5, 1787 RSC
- S. Busche and K. Bluhm, Z. Naturforsch., B: J. Chem. Sci., 1996, 51, 319 CAS
- X. A. Chen, Y. J. Chen, L. Wu, X. A. Chang and W. Q. Xiao, Solid State Sci., 2014, 27, 47 CrossRef CAS
- H. W. Yu, H. P. Wu, S. L. Pan, B. B. Zhang, L. Y. Dong, S. J. Han and Z. H. Yang, New J. Chem., 2014, 38, 285 RSC
- G. S. Yang, P. F. Gong, Z. S. Lin and N. Ye, Chem. Mater., 2016, 28, 9122 CrossRef CAS
- Q. Huang, L. J. Liu, X. Y. Wang, R. K. Li and C. T. Chen, Inorg. Chem., 2016, 55, 12496 CrossRef CAS
- H. W. Yu, H. P. Wu, Q. Jing, Z. H. Yang, P. S. Halasyamani and S. L. Pan, Chem. Mater., 2015, 27, 4779 CrossRef CAS
- R. W. Smith and D. A. Keszler, J. Solid State Chem., 1992, 100, 325 CrossRef CAS
-
SAINT, version 7.60A, Bruker Analytical X-ray Instruments, Inc., Madison, WI, 2008 Search PubMed
-
G. M. Sheldrick, SHELXS-97, Program for X-ray Crystal Structure Refinement, University of Gottingen, Germany, 1997 Search PubMed
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Rrfson and M. C. Payne, Z. Kristallogr., 2005, 220, 567 CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS
- A. M. Rappe, K. M. Rabe, E. Kaxiras and J. D. Joannopoulos, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 1227 CrossRef
- J. S. Lin, A. Qteish, M. C. Payne and V. Heine, Phys. Rev. B, 1993, 47, 4174 CrossRef
- R. K. Li and P. Chen, Inorg. Chem., 2010, 49, 1561 CrossRef CAS
- F. Zhang, Z. W. Jiao, D. G. Shen, G. Q. Shen and X. Q. Wang, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2010, 66, i1 CrossRef CAS PubMed
- S. Y. Mao, M. E. Mendoza-Alvarez, W. Depmeier, F. Kubel and H. Schmid, Ferroelectrics, 1991, 115, 91 CrossRef CAS
- J. Campa-Molina, S. Ulloa-Godinez, A. Barrera, L. Bucio and J. Mata, J. Phys.: Condens. Matter, 2006, 18, 4827 CrossRef CAS
- K. L. Moran, T. E. Gier, W. T. A. Harrison, G. D. Stucky, H. Eckert, K. Eichele and R. E. Wasylishen, J. Am. Chem. Soc., 1993, 115, 10553 CrossRef CAS
- J. Li, S. P. Xia and S. Y. Gao, Spectrochim. Acta, Part A, 1995, 51, 519 CrossRef
- B. C. Shih, Y. Xue and P. H. Zhang, Phys. Rev. Lett., 2010, 105, 146401 CrossRef
- C. A. Ataide, R. R. Pela, M. Marques and L. K. Teles, Phys. Rev. B: Condens. Matter Mater. Phys., 2017, 95, 045126 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: CIF file; check cif; atomic coordinates and equivalent isotropic displacement parametersand selected bond distances and angles tables for Pb3Ba3Zn6(BO3)8 and α-BaZn2(BO3)2; table of the basic information of the existing anhydrous-zincoborates. CCDC 1949368 (Pb3Ba3Zn6(BO3)8) and 1949369 (α-BaZn2(BO3)2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qi01122f |
| This journal is © the Partner Organisations 2020 |