
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective catalytic degradation of a lignin model compound into phenol over transition metal sulfates†
Min-ya Wu,
Jian-tao Lin,
Zhuang-qin Xu,
Tian-ci Hua,
Yuan-cai Lv,
Yi-fan Liu,
Rui-han Pei,
Qiong Wu and
Ming-hua Liu *
*
Fujian Provincial Engineering Research Center of Rural Waste Recycling Technology, College of Environment & Resources, Fuzhou University, No. 2 Xueyuan Road, Shangjie Town, Minhou County, Fuzhou, Fujian 350116, China. E-mail: mhliu2000@fzu.edu.cn
First published on 16th January 2020
Abstract
Transition metal salts were employed as the catalysts to improve the selective degradation of the α-O-4 lignin model compound (benzyl phenyl ether (BPE)) in the solvothermal system. The results concluded that most of the transition metal salts could enhance BPE degradation. Among which, NiSO4·6H2O exhibited the highest performance on BPE degradation (90.8%) for 5 h and phenol selectivity (53%) for 4 h at 200 °C. In addition, the GC-MS analysis indicated that the intermediates during BPE degradation included a series of aromatic compounds, such as phenol, benzyl methyl ether and benzyl alcohol. Furthermore, the mechanisms for BPE degradation and phenol selectivity in the NiSO4·6H2O system involved the synergetic effects between the acid catalysis and coordination catalysis, which caused the effective and selective cleavage of the C–O bonds.
Introduction
As a natural renewable aromatic polymer, lignin can be depolymerized into small molecule compounds as high value-added chemicals under certain conditions.1 Lignin has a complex three-dimensional spatial structure, composing of three phenyl-propanoid monomers with a large amount of C–O and C–C bonds.2,3 Meanwhile, it contains methoxyl, phenolic hydroxyl, alcohol hydroxyl and other characteristic functional groups. However, due to its unique complexity, lignin is prevented from further conversion into value-added chemicals.4,5 Consequently, most of lignin is only used as a waste product or low quality fuel.The C–O ether bonds are the most important linkages in the structure of lignin.6 The most inter-unit linkages in lignin are β-O-5, α-O-4 and 4-O-5 ether bonds,7 in which the α-O-4 is the most vibrant due to the lowest bond dissociation energy of the aliphatic C–O bond.8,9 At present, lignin related model compounds, such as α-O-4 benzyl phenyl ether (BPE), have been investigated to disclose selective degradation of lignin into small aromatics because of the structural complex and variable of lignin. So far, a great number of homogenous and heterogeneous catalysts have been applied extensively for cleaving the C–O ether bonds by preserving the aromatic ring functionality.10–12 For example, Paone et al.13 achieved the efficient cleavage of the C–O bonds in benzyl phenyl ether (BPE) under catalytic transfer hydrogenation conditions using 2-propanol as the H-donor and Pd/Fe3O4 as catalyst. Roberts et al.14 studied the influence of alkali carbonates on BPE degradation efficiency reactions between 270 °C and 370 °C and they found that toluene and 2,4-benzyl phenol were primary productions by formation of a cation-BPE adduct.
However, most lignin depolymerization processes still suffered obvious disadvantage of low conversion and the degradation products were uncontrollable. The single catalyst could not meet the dual needs of further improving the degradation efficiency and achieving higher product selectivity. Therefore, the method of multiple catalysts combined to degrade lignin has been widely investigated. Recently, transition metal salts have been more widely used as primary catalysts or synergistic catalysts due to their more reactive chemical properties in biomass conversion. Shu et al.15 proposed to use CrCl3 cooperated with Pd/C to depolymerize lignin, and achieved highly controllable product distribution. Tribulová et al.16 found that low molecular products, such as monosaccharides, were formed when transition metal sulfates were used to hydrolyze cellulose, which improved the cellulose sensitivity to oxidation. Zakzeski et al.17 studied the oxidative degradation of lignin in ionic liquids with varies transition metal salts as co-catalysts. The results showed the positive impact of the metal salts on the catalytic degradation of lignin. Besides, the catalytic activity of metal salts could be enhance remarkably by the ionic liquid under mild conditions.
In addition, the effect of solvent for cleaving C–O bonds of aryl ethers were also concerned.18–20 Methanol has obvious solvent thermal effect and benign hydrogen supply, which can produce H+ and other free radicals under mild conditions. Besides, it is cheap and bio-renewable. Importantly, it can stabilize the monocyclic radicals generated in the degradation process, resulting in generating more monomer aromatic groups and reducing the role of polymerization.21,22
In this study, in order to investigate the effect of transition metal sulfates on catalytic cleavage of C–O bond and products selectivity, and confirm the possibility of catalytic degradation of lignin as primary catalyst or cooperate with others. The transition metal sulfates were employed as homogenous catalyst to degrade BPE in methanol. The conditions such as temperature and time for BPE degradation and phenol selectivity were studied. In addition, the intermediates during BPE degradation were detected and the synergistic mechanism for the selective cleavage of the C–O bonds were proposed. This research would be a beneficial reference for the future efficient and product-controlled depolymerization of lignin by using the cooperation effect of multiple catalysts.
Experimental section
Degradation of benzyl phenyl ether
The degradation of BPE was conducted 0.3000 g BPE, 1.4 mmol transition metal salts, and 20 mL methanol were separately poured into a 50 mL reactor equipped with PPL coating in a glove box (LM1000S, Dellix Industry Co. Ltd, China). N2 was kept in the headspace of the test tube before sealing the Teflon coated rubber cap with aluminium crimp. The autoclave was heated to given temperature (rate of 7 °C min−1). When the reaction was finished, the mixture was cooled down to room temperature during 30 min using flowing water. At selected time intervals, 1 mL of liquid sample was taken out for analysis. After filtered through 0.22 μm glass fiber filters (Tianjin Branch billion Lung Experimental Equipment Co., Ltd, China), the residual BPE concentration and the generated phenol concentration were monitored. Used catalyst was washed thrice with methanol. Then it was dried at 40 °C until a constant weight. The system without catalyst was set the control. All experiments were conducted in triplicates.Analytical methods
The BPE concentration was monitored by high-performance liquid chromatography (HPLC, Agilent, USA), which equipped with an UV detector set at 254 nm; and column temperature 40 °C and 80% methanol (v/v), 20% ultra water (v/v) was used as mobile phase at a flow rate of 1.0 mL min−1. The products during BPE degradation process were determined by gas chromatography coupled to concentration spectrometry (GC-MS, Agilent 7890B + 5977, USA), which equipped with a HP-5ms column (30 m × 0.25 mm × 0.25 μm), and ion source was EI. The oven temperature was programmed as 50 °C hold 2 min, and then ramped up to 280 °C with 10 °C min−1 and hold for another 5 min. The injector kept at 300 °C in spit mode (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with helium as the carrier gas. The column flow rate was 1 mL min−1. The residual concentration of the reactant BPE as well as the products were determined by calibrating the peak areas with products solutions of known concentrations of these compounds. In addition, the recovered catalyst was characterized by X-ray diffraction (XRD, Miniflex 600, Rigaku Co., Japan) spectrum for recovered catalyst was obtained using Cu Kα radiation at 40 kV and 40 mA.
:1) with helium as the carrier gas. The column flow rate was 1 mL min−1. The residual concentration of the reactant BPE as well as the products were determined by calibrating the peak areas with products solutions of known concentrations of these compounds. In addition, the recovered catalyst was characterized by X-ray diffraction (XRD, Miniflex 600, Rigaku Co., Japan) spectrum for recovered catalyst was obtained using Cu Kα radiation at 40 kV and 40 mA.
The primary products from this process were divided into gaseous fraction, volatile products, nonvolatile products and residual solid. The weight of the gaseous fraction was negligible. The yield and selectivity of degradation products were evaluated by the aromatic ring balance and according to the following equations (eqn (1) and (2)) based on the HPLC and GC-MS results.
| BPE degradation efficiency = ((1 − n/n0)) × 100% | (1) |
| Degradation products selectivity = (nn/(2(n0 − n))) × 100% | (2) |
Results and discussion
Benzyl phenyl ether degradation with different catalysts
Catalysts played a vital role in the BPE degradation.14 To study the effects of different transition metal on the BPE degradation, 11 transition metal compounds were chosen as the catalyst and the results were shown in Fig. 1 and Table S1.† As presented in Fig. 1a, in the control system, BPE degradation efficiency was rather low (3.6%) in the absence of the catalysts, suggesting that the possible effect of methanol on BPE degradation was negligible. Whereas, the rapid decline of BPE was observed in various transition metal sulfates systems, suggesting that transition metal sulfates could effectively accelerate catalytic degradation of BPE. As reported in previous studies, the Lewis acid catalyst could effectively degrade the lignin and model compounds through the cleavage of C–O linkages.23–25 Herein, the hydrogen ion concentration (c(H+)) detection results (Fig. 1c) showed that these systems with different transition metal sulfates maintained more hydrogen ion than that of control during BPE degradation, which kept systems more acidic and confirmed the essence of Lewis acid catalyst. Thus, as Lewis acids, the transition metal sulfates could enhance the BPE degradation. In order to confirm this, the catalytic activities of traditional proton acids as H2SO4 was investigated. More than 90% degradation efficiency and less than 20% of phenol selectivity could be achieved. It demonstrated clearly that lignin model compound could be efficiently degraded through acid catalysis of metal sulfates. Meanwhile, the product distribution were significantly depended on other element.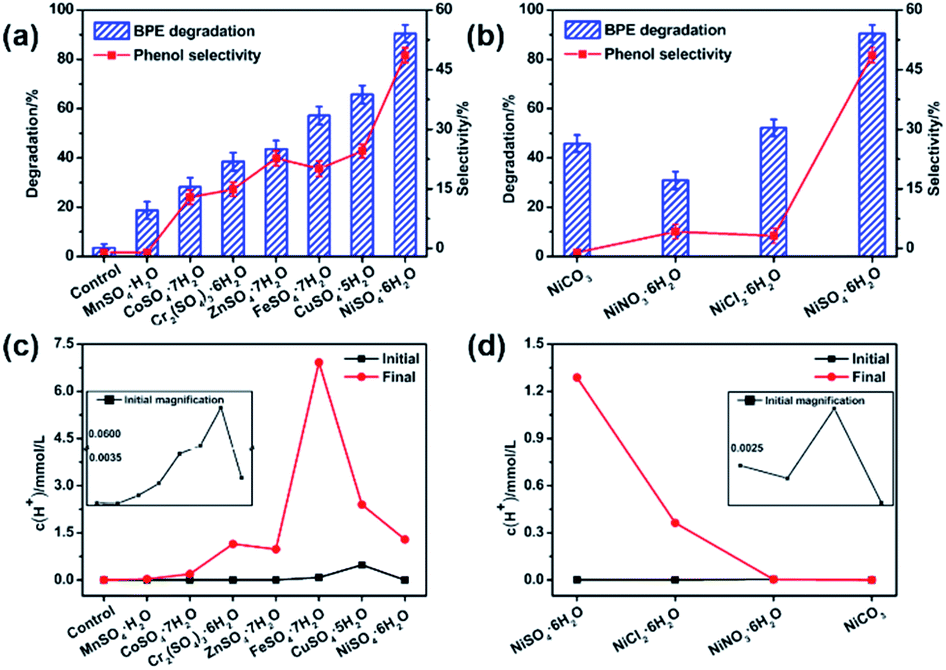 | ||
| Fig. 1 (a and b) Effect of transition metal salts on BPE degradation efficiency and phenol selectivity. Condition: 300 mg BPE, 1.4 mmol transition metal salts, 20 mL methanol, 200 °C, 5 h. (c and d) Hydrogen ion concentration (c(H+)) of catalyst systems. Condition: 1.4 mmol transition metal salts, 20 mL methanol, 200 °C, 5 h. | ||
As shown in Fig. 1a, these transition metal sulfates showed rather different performance on BPE degradation and phenol selectivity. The maximum degradation efficiency (90.4%) occurred to NiSO4·6H2O system, following CuSO4·5H2O (65.7%), FeSO4·7H2O (57.2%), ZnSO4·7H2O (43.6%), Cr2(SO4)3·6H2O (38.5%), CoSO4·7H2O (28.3%), and the minimum degradation efficiency was observed in MnSO4·H2O system (18.7%). Meanwhile, the similar results happened to the phenol selectivity, suggesting that NiSO4·6H2O exhibited the best performance on BPE degradation and phenol selectivity. After treatment (200 °C, 5 h), the hydrogen ion concentration (Table S1†) in these systems without BPE were as follows: FeSO4·7H2O (6.92 mmol L−1) > CuSO4·5H2O (2.40 mol L−1) > NiSO4·6H2O (1.29 mmol L−1) > Cr2(SO4)3·6H2O (1.15 mmol L−1) > ZnSO4·7H2O (0.98 mmol L−1) > CoSO4·7H2O (0.19 mmol L−1) > MnSO4·H2O (0.03 mmol L−1). According to the results in Fig. 1a and c, it could be found that the systems with higher hydrogen ion concentration exhibited better performance. However, it seemed no significant positive correlation between the BPE degradation efficiency and final hydrogen ion concentration, which meant that besides the hydrogen ion concentration, the transition metal cations might play a key role in enhancing BPE degradation and phenol production. As previously reported by others, the difference among these system were probably attributed to the strengths of the substrate–catalyst interactions in presence of different metal centers.26,27 According to Paul et al.'s investigation, during the pyrolysis of cellulose by metal salts, the mechanism mainly involved both acid and ionic catalysis.28 In addition, they also found that the acidity or alkalinity of the metal salt was less important than the ionic catalysis. Considering the highest electronegativity of Ni atom among these transition metal, it could be inferred that NiSO4·6H2O could produce more coordination points besides acidic centers and increase the reaction activities through the substrate–catalyst interactions during the process. Furthermore, the effect of anion on BPE degradation was further investigated, NiCl2·6H2O, NiNO3·6H2O and NiCO3 were also investigated with NiSO4·6H2O as catalyst. As displayed in Fig. 1b, both BPE degradation efficiency and phenol generation for NiCl2·6H2O, NiNO3·6H2O and NiCO3 were inferior to those for NiSO4·6H2O. It can be explained by two reasons. On one hand, as depicted in Fig. 1d, the hydrogen ion concentration value of NiCO3 system was near zero, which resulting in superior BPE degradation efficiency but inferior phenol selectivity. According to Roberts, et al.'s investigation, alkali carbonates can promote the ether bonds cleaved by forming the cation-BPE adduct.14 However, due to the lower protons concentration in the system, the generated phenol would react with benzyl alcohol and form molecular compounds, causing a decline of the phenol yield.14 On the other hand, the anion could significant affect the reactivity of the catalyst by charging the density on the metal center.29 During the cellulose decomposition, SO42− in Lewis acid was found to be an excellent hydrogen bonding acceptor and nucleophilic reagent.30 Hence, the higher phenols selectivity and BPE degradation was mainly contributed to the synergic effect between Ni2+ and SO42−. Based on the above results, NiSO4·6H2O was an excellent catalyst for BPE degradation.
Effect of reaction temperature on benzyl phenyl ether degradation
During lignin decomposition, temperature was one of the most important factors and it played a vital position in activating the catalyst and fracturing the C–O and C–C bonds.10 Herein, the BPE degradation and phenol generation at varied reaction temperature (180–220 °C) were monitored and the results were shown in Fig. 2. It was noticed that BPE degradation process was highly dependent on temperature. The degradation efficiency dramatically increased as the rise of temperature before 200 °C (from 19.6 to 93.5%). Subsequently, BPE degradation efficiency kept steady when the reaction temperature was over 200 °C. Similarly, the phenol selectivity also exhibited rapid increasing (from 180 °C to 200 °C). Once the temperature was over 200 °C, the phenol selectivity significant decline (from 48.7 to 44.4%). All the results suggested that the phenol selectivity was more sensitive to temperature than BPE degradation efficiency. According to previous reports, much higher temperature partly caused the carbonization of organic compounds and accelerated the condensation reaction among degradation products, which resulted in the decline of the phenol selectivity.31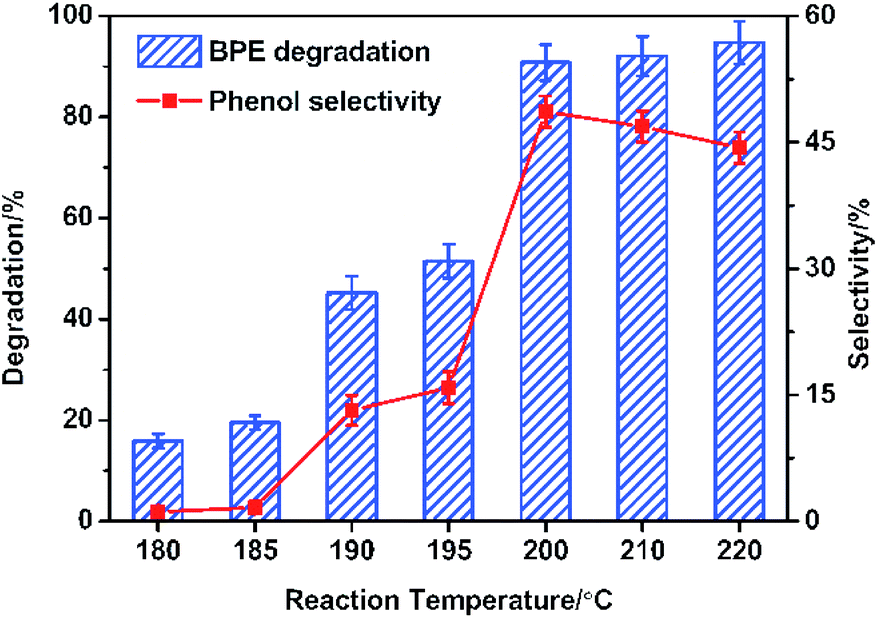 | ||
| Fig. 2 Effect of reaction temperature on BPE degradation efficiency and phenol selectivity. Condition: 300 mg BPE, 1.4 mmol NiSO4·6H2O, 20 mL methanol, 5 h. | ||
Benzyl phenyl ether, hydrogen ion concentration and catalyst variation under different time
To study the effect of the reaction time on BPE degradation and phenol selectivity, BPE degradation and phenol generation with various time were monitored under 200 °C (Fig. 3). As seen in Fig. 3a, when the reaction time prolonged from 0.5 h to 5 h, BPE degradation efficiency gradually increased from 5.6% to 90.8%. However, the phenol selectivity varied significantly with prolonged reaction time. It increased from 16.3% for 0.5 h to 53.1% for 4 h, but dropped to 48.7% for 5 h, which meant that the phenol might participate in the subsequent reaction. According to Long et al.'s study, long reaction time was beneficial to BPE degradation but harmful to the phenol production.31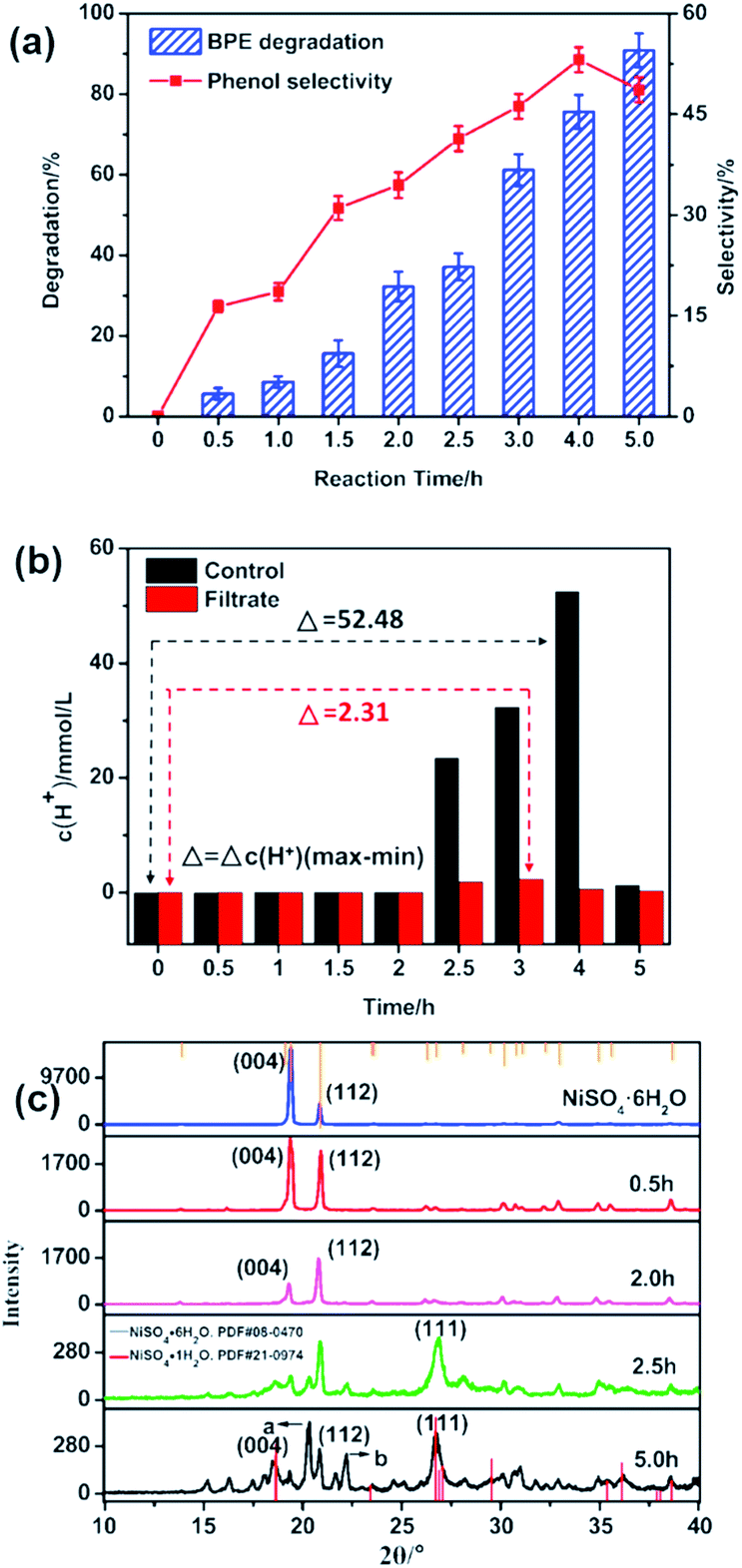 | ||
| Fig. 3 (a) Effect of reaction time on BPE degradation; (b) the hydrogen ion concentration during the process. (c) X-ray diffraction pattern for recovered catalysts. Condition: 300 mg BPE, 1.4 mmol NiSO4·6H2O, 20 mL methanol, 200 °C. | ||
In order to further explore the catalytic mechanism of NiSO4·6H2O, the hydrogen ion concentration during BPE degradation was closely inspected at different reaction times (Fig. 3b). In the control system, the hydrogen ion concentration showed a relatively stable fluctuation between 0–2 h. Afterwards, when the reaction time prolonged from 2 h to 2.5 h, the hydrogen ion concentration increased to 23.40 mmol L−1 dramatically, and maintained increasing until the reaction time was extended to 4 h, but rose to 1.29 mmol L−1 at 5 h. Meanwhile, the similar results happened to the BPE degradation system. In both systems, it was observed that the hydrogen ion concentration evidently increased during the process, which demonstrated the essence of NiSO4·6H2O as a Lewis acid catalyst.23–25 Nevertheless, the difference of hydrogen ion concentration (Δc(H+)) during the whole process in both systems was rather different. In the filtrate, the change of hydrogen ion concentration was lower than that in control system, which indicated that hydrogen ions participated in the degradation process of BPE. According to previous studies, the H+ played a very important role in BPE degradation.13
In order to further explore the mechanism of NiSO4·6H2O for BPE degradation, the recovered catalysts were characterized by XRD. As displayed in Fig. 3c, during 0–2 h, no new diffraction peaks turned up, but the intensity of the (004) and (112) crystal faces diffraction peaks of NiSO4·6H2O significantly weakened, indicating that NiSO4·6H2O participated in BPE degradation process, but no significant change in the structure occurred to NiSO4·6H2O. Then, when the reaction time prolonged to 2.5 h, the intensity of the diffraction peaks of NiSO4·6H2O were weaker and a new diffraction peak at 26.7° appeared. After confirmed by the PDF card (no. 21-0974), the new diffraction peak was assigned to the (111) crystal face diffraction peak of NiSO4·H2O, suggesting that NiSO4·6H2O lost crystal water during the BPE degradation. Similarly, the crystal water loss process of NiSO4·6H2O was also confirmed in the previous reports.32 Besides, the corresponding hydrogen ion concentration showed a significant increasing within 2.0 to 2.5 h, indicating that the dehydration reaction of NiSO4·6H2O in methanol caused a significant increasing in the concentration of hydrogen ions in the system. Besides, there were two unknown diffraction peaks as a and b occurred at 5 h, which were speculated to be NiSO4·xH2O (0 ≤ x < 1), which meant catalyst continued to lose water after 2.5 h. According to Huang et al.'s study, NiSO4·6H2O could achieve dehydration by co-pyrolysis with methanol and the structure had been changed, which further promoted the release of protons by methanol and formed an acidic environment in the BPE degradation system.33 Therefore, the dehydrated catalyst played an important role in the acid-catalyzed degradation of BPE.
Identification of the intermediates and the pathway of benzyl phenyl ether during the process
Furthermore, the variation of the degradation products was detected and the results were displayed in Fig. S1 and Table S2.† The corresponding spectra of MS were attach in appendix (Fig. S2†). During BPE degradation, approximately five types of possible products were identified through the National Institute of Standards and Technology (NIST11) library. Peak 4 was BPE (RT = 17.235 min), with molecular ion at 184 m/z and major fragment ions at 91 m/z. The peaks (1–3) with retention time at 9.810, 7.048 and 7.888 min were the primary productions of BPE degradation, which involved phenol, benzyl methyl ether, benzyl alcohol, respectively. The peak 5 (RT = 18.800 min) with molecular ion at 184 m/z was confirmed as 2-benzyl phenol, which was formed through secondary reaction between the intermediates.Based on the GC-MS, the yield and selectivity of specific compounds (phenol, benzyl methyl ether and benzyl alcohol) with time were illustrated in Fig. 4 and Table S3.† As shown in Fig. 4, with the prolonging of the reaction time (0–5 h), the yields of phenol, benzyl methyl ether and benzyl alcohol increased and reached maximum (1.410, 0.905 and 0.330 mmol, respectively) at 5 h, which was consistent with the results of GC-MS chromatogram (Fig. S1†). Instead, the selectivity of the specific compounds was rather different. For phenol, the selectivity increased before 4 h, but declined at 5 h. The selectivity of benzyl methyl ether began to decrease at 2.5 h. All the results indicated that during BPE degradation, the intermediates also participated in some side reaction, resulting in the decline of the benzyl methyl ether and phenol selectivity, which was in accordance with the results in Fig. 3.
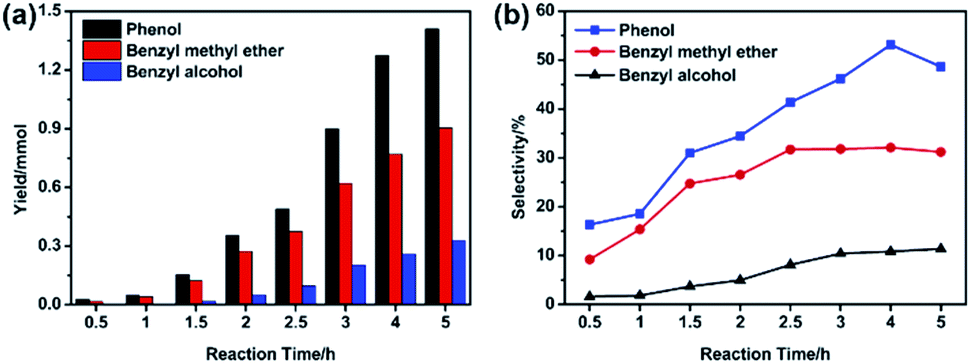 | ||
| Fig. 4 The yield (a) and selectivity (b) of degradation products. Condition: 300 mg BPE, 1.4 mmol NiSO4·6H2O, 20 mL methanol, 200 °C. | ||
 | ||
| Fig. 5 The pathway of BPE degradation. Condition: 300 mg BPE, 1.4 mmol NiSO4·6H2O, 200 °C. | ||
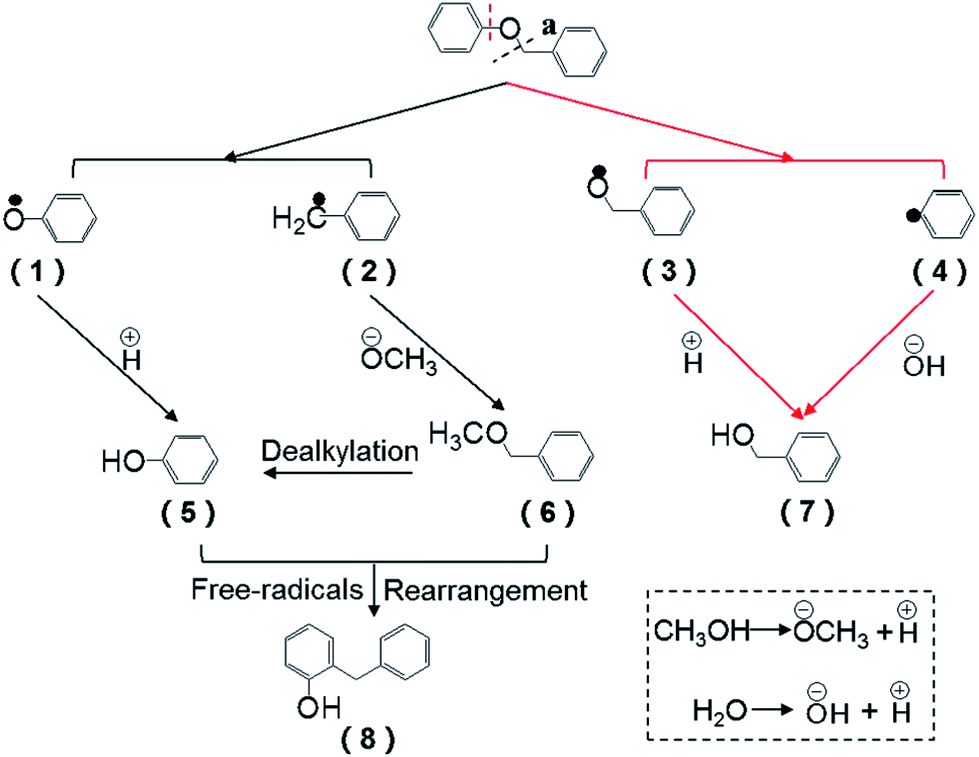
Based on the intermediates, the pathway of BPE during the reaction system was proposed. During the synergistic effect of catalyst and heat, methanol and the lost crystal water was transformed into the methoxyl group, hydroxyl group and hydrogen ions, respectively, while the decomposition of BPE via the cleavage of C–O bonds involved two ways. In route a (Fig. 5), BPE was cleaved to phenoxy radicals (1) and benzyl radicals (2). Afterwards, the phenoxy radicals (1) combined with the hydrogen ions preferentially and the benzyl radicals (2) bonded to the methoxyl group, leading to the formation of phenol (5) and benzyl methyl ether (6). In route b, BPE was broken into benzoxy radicals (3) and phenyl radicals (4). Then the benzyl radicals (3) combined with the hydrogen ions to form benzyl alcohol and phenyl radicals (4) bonded to hydroxyl group to generate phenol. According to related research, Caromatic–O bond dissociation energy was much higher than Calkyl–O.10 Thus, it could be concluded that route a was the main pathway for BPE decomposition. Meanwhile, the formation dealkylation of benzyl methyl ether (6) was ascribed into the rearrangement reaction between the phenol (5) and intermediates, which was similar to Yang et al.'s findings.34 In addition, benzyl methyl ether (6) were prone to rearrangement reaction with phenol and formed a new C–C bond, resulting the presence of 2-benzyl phenol (8).
 | ||
| Fig. 6 Catalytic mechanism of NiSO4·6H2O. Condition: 300 mg BPE, 1.4 mmol NiSO4·6H2O, 20 mL methanol, 200 °C, 5 h. | ||
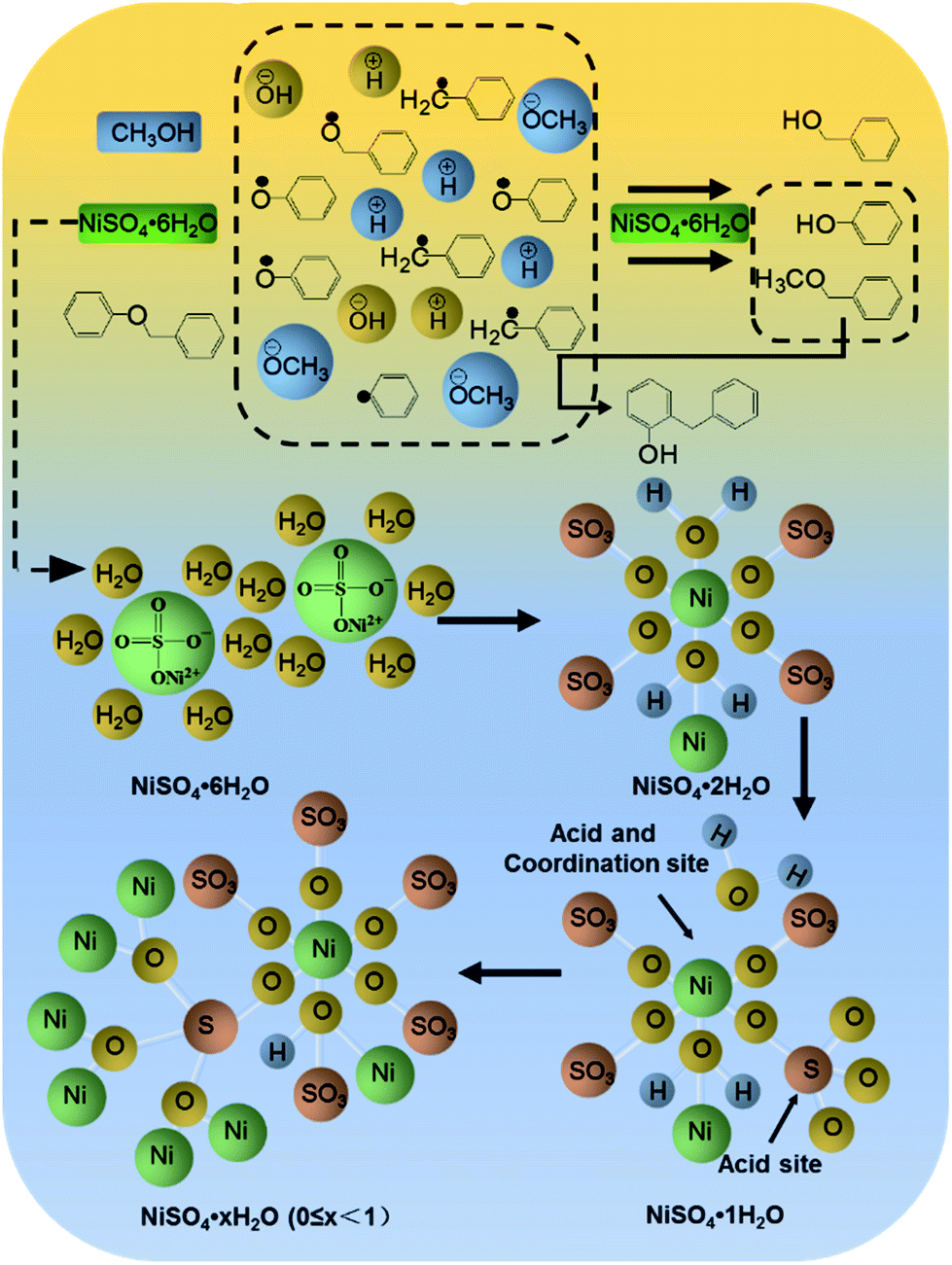
Proposed catalytic mechanism
Based on the degradation pathway and the structural analysis of the catalyst, the main degradation mechanism for BPE was proposed in Fig. 6. First, NiSO4·6H2O maintained acid condition during the BPE degradation as acid catalyst (Fig. 3b). During the co-pyrolysis process of NiSO4·6H2O and methanol, NiSO4·6H2O gradually lost water (Fig. 3c). Meanwhile, the sp3d2 orbital hybridization occurred at nickel atom, and lead to the catalyst formed an octahedral metastable transition state (Fig. 6).32 In this structure, an empty track on the nickel atoms tended to form Lewis acid site besides that of S atom. Hence, NiSO4·6H2O had more than one acidic site, which was beneficial to the breakage of ether bonds at acid condition. Simultaneously, the SO42−, which acted as an excellent hydrogen bond donor and nucleophile, synergized with Ni2+ to promote the cleavage of C–O bonds. This mechanism accorded well with most acid catalytic lignin depolymerization process.35,36In addition, NiSO4·6H2O had the potential as coordination catalysis, which exhibited important influence on not only BPE degradation efficiency but also the selectivity of productions. On the one hand, the sp3d2 orbital hybridization occurred at nickel atom also formed coordination site, which were apt to form stable complexes with the oxygen atoms on α-O-4 bonds (Fig. 6). The stable complexes could effectively reduce the activation energy of ether bond and then resulted in the fracture of C–O bonds in the BPE, resulting in high BPE degradation efficiency. Hence, BPE was depolymerized into phenoxy radicals and benzyl radicals in methanol (Fig. 5) through the acid catalysis and coordination catalysis of NiSO4·6H2O. On the other hand, Ni atom could weaken the O–H bond in methanol through coordination, which promoted the release of protons by methanol and accelerated hydrogenation reactions.14 Since the hydrogen absorption capacity of phenoxy radicals was better than that of benzyl radicals, the proton preferentially bonded with phenoxy group to form phenol, leading to the increasing of phenol.21 Meanwhile, the benzyl radicals combined with methoxyl group to formed benzyl methyl ether. Afterwards, with prolonged reaction time, the O atoms on the methoxyl group of benzyl methyl ether exhibited partial positive charge due to the coordination between Ni and O, which greatly enhanced the electrophilicity and reactivity of the methyl.37 Meanwhile, as an excellent nucleophile, the SO42− could attack the methyl, caused the separation of the methyl from methoxyl group. Afterwards, the initial position of the methyl was replaced by protons and then formed phenol. Accordingly, as the reaction time prolonged, the difference between the yields of phenol and benzyl methyl ether increased (Fig. 4).
Besides, according to previous report, due to the attack of Ni atom, the lost crystal water was broken away and formed protons and a small amount of hydroxyl14 and then BPE could be hydrolyzed to form phenol and benzyl alcohol10,38(Fig. 5). In He et al.'s investigation, they found that the cleavage of the α-O-4 bond in water could occur via parallel hydrolysis and form phenol and benzyl alcohol.11
During the reaction, due to the instability of carbocation at benzyl radicals, benzyl methyl ether were prone to rearrangement reaction with phenol and formed a new C–C bond, resulting the decline of the phenol, benzyl methyl ether, intermediates and the presence of some new products, such as 2-benzyl phenol (Fig. 5).39
Conclusions
The transition metal sulfates could be used as catalysts to effectively decompose the lignin model compounds. Among which, NiSO4·6H2O exhibited excellent performance on the degradation of benzyl phenyl ether (BPE) and the phenol selectivity. Due to the synergetic effects between the acid catalysis mechanism and the coordination catalysis mechanism, NiSO4·6H2O effectively and selectively clove the C–O bonds in BPE and formed a series of aromatic compounds, such as phenol, benzyl methyl ether and benzyl alcohol. Consequently, this study would provide theoretical and experimental basis for the decomposition of lignin by metal salt base catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the Production and Study Project of Cooperation in Universities and Colleges in Fujian Province (No. 2019H6008) and Natural Science Foundation of China (No. 21577018).Notes and references
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337(6095), 695–699 CrossRef CAS.
- J. Zakzeski, A. L. Jongerius, P. C. Bruijnincx and B. M. Weckhuysen, ChemSusChem, 2012, 5(8), 1602–1609 CrossRef CAS.
- V. K. Ponnusamy, D. D. Nguyen, J. Dharmaraja, S. Shobana, J. R. Banu, R. G. Saratale, S. W. Chang and G. Kumar, Bioresour. Technol., 2019, 271, 462–472 CrossRef CAS.
- P. Azadi, O. R. Inderwildi, R. Farnood and D. A. King, Renewable Sustainable Energy Rev., 2013, 21(5), 506–523 CrossRef CAS.
- C. A. Gasser, G. Hommes and F. X. Corvini, Appl. Microbiol. Biotechnol., 2012, 95(5), 1115–1134 CrossRef CAS PubMed.
- K. Barta, G. R. Warner, E. S. Beach and P. T. Anastas, Green Chem., 2013, 16(1), 191–196 RSC.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115(21), 11559–11624 CrossRef CAS PubMed.
- E. Dorrestijn, L. J. J. Laarhoven and I. W. C. E. Mulder, J. Anal. Appl. Pyrolysis, 2000, 54(1–2), 153–192 CrossRef CAS.
- M. Stoneham, Contemp. Phys., 2009, 50(3), 494 Search PubMed.
- J. He, L. Lu, Z. Chen, D. Mei and J. A. Lercher, J. Catal., 2014, 311(3), 41–51 CrossRef CAS.
- J. He, Z. Chen, D. Mei, J. A. Lercher and A. Johannes, J. Catal., 2014, 309(6), 280–290 CrossRef CAS.
- J. Ji, H. Guo, C. Li, Z. Qi, B. Zhang, T. Dai, M. Jiang, C. Ren, A. Wang and T. Zhang, ChemCatChem, 2018, 10(2), 415–421 CrossRef CAS.
- E. Paone, C. Espro, R. Pietropaolo and F. Mauriello, Catal. Sci. Technol., 2016, 6(22), 7937–7941 RSC.
- V. Roberts, S. Fendt, A. A. Lemonidou, X. Li and J. A. Lercher, Appl. Catal., B, 2010, 95(1), 71–77 CrossRef CAS.
- R. Shu, J. Long, Z. Yuan, Z. Qi, T. Wang, C. Wang and L. Ma, Bioresour. Technol., 2015, 179, 84–90 CrossRef CAS.
- T. Tribulová, F. Kačík, D. V. Evtuguin, I. Čabalová and J. Ďurkovič, Cellulose, 2019, 26(4), 2625–2638 CrossRef.
- J. Zakzeski, A. L. Jongerius and B. M. Weckhuysen, Green Chem., 2010, 12(7), 1225–1236 RSC.
- M. Rashidi, J. N. Beltramini and D. Martin, J. Incl. Phenom. Macrocycl. Chem., 2019, 94(3–4), 297–305 CrossRef CAS.
- R. Gao, Y. Li, H. Kim, J. K. Mobley and J. Ralph, ChemSusChem, 2018, 11(13), 2045–2050 CrossRef CAS.
- X. X. Besse, Y. Schuurman and N. Guilhaume, Appl. Catal., B, 2017, 209, 265–272 CrossRef CAS.
- X. Jiang, Q. Lu, X. Dong, C. Chen and C. Dong, Chin. J. Biotechnol., 2015, 31(10), 1512 Search PubMed.
- D. Shen, J. Wei and S. Gu, J. Anal. Appl. Pyrolysis, 2017, 127, 176–182 CrossRef CAS.
- M. M. Hepditch and R. W. Thring, Can. J. Chem. Eng., 2000, 78(1), 226–231 CrossRef CAS.
- L. Yang, Y. Li and P. E. Savage, Ind. Eng. Chem. Res., 2014, 53(7), 2633–2639 CrossRef CAS.
- G. L. Huppert, B. C. Wu, S. H. Townsend, S. C. Paspek and M. T. Klein, Ind. Eng. Chem. Res., 1989, 28(2), 161–165 CrossRef CAS.
- N. A. Rebacz and P. E. Savage, Ind. Eng. Chem. Res., 2010, 49(2), 535–540 CrossRef CAS.
- S. Kobayashi, S. Nagayama and T. Busujima, J. Am. Chem. Soc., 1998, 29(52), 1522–2667 Search PubMed.
- P. T. Williams and P. A. Horne, Renew. Energy, 1994, 4(1), 1–13 CrossRef CAS.
- G. Bartoli, R. Dalpozzo, A. D. Nino, L. Maiuolo, M. Nardi, A. Procopio and A. Tagarelli, Eur. J. Org. Chem., 2004,(10), 2176–2180 CrossRef CAS.
- C. J. Jiang, Cellul. Chem. Technol., 2014, 48(1–2), 75–78 CAS.
- J. X. Long, W. Y. Lou, L. F. Wang, B. L. Yin and X. H. Li, Chem. Eng. Sci., 2015, 122, 24–33 CrossRef CAS.
- H. Hattori, S. Miyashita and K. Tanabe, Bull. Chem. Soc. Jpn., 1971, 44(4), 893–895 CrossRef CAS.
- H. M. Huang, Acta Chim. Sin., 1983, 41(12), 1081–1086 CAS.
- B. Yang, H. Lin, K. Miao, P. Zhu, L. Liang, K. Sun, H. Zhang, J. Fan, V. Meunier and Y. Li, Angew. Chem., Int. Ed. Engl., 2016, 55(34), 9881–9885 CrossRef CAS PubMed.
- J. Long, Z. Qi, T. Wang, X. Zhang, X. Ying and L. Ma, Bioresour. Technol., 2014, 154(1), 10–17 CrossRef CAS PubMed.
- S. Jia, B. J. Cox, X. Guo, Z. C. Zhang and J. G. Ekerdt, ChemSusChem, 2010, 3(9), 1078–1084 CrossRef CAS PubMed.
- J. Hu, D. Shen, S. Wu, H. Zhang and R. Xiao, RSC Adv., 2015, 5(55), 43972–43977 RSC.
- C. Yokoyama, K. Nishi and S. Takahashi, J. Jpn. Pet. Inst., 1997, 40(6), 465–473 CrossRef CAS.
- N. Yan, C. Zhao, P. J. Dyson, C. Wang, L. T. Liu and Y. Kou, ChemSusChem, 2010, 1(7), 626–629 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: GC-MS and MS spectra. See DOI: 10.1039/c9ra09706f |
| This journal is © The Royal Society of Chemistry 2020 |