
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTitanium phosphonate oxo-alkoxide “clusters”: solution stability and facile hydrolytic transformation into nano titania†
Fredric G. Svenssona,
Geoffrey Danielb,
Cheuk-Wai Tai c,
Gulaim A. Seisenbaevaa and
Vadim G. Kessler*a
c,
Gulaim A. Seisenbaevaa and
Vadim G. Kessler*a
aDepartment of Molecular Sciences, Swedish University of Agricultural Sciences, Box 7015, 750 07 Uppsala, Sweden. E-mail: vadim.kessler@slu.se
bDepartment of Biomaterials and Technology/Wood Science, Swedish University of Agricultural Sciences, Box 7008, 75007 Uppsala, Sweden
cDepartment of Materials and Environmental Chemistry, Stockholm University, 106 91 Stockholm, Sweden
First published on 14th February 2020
Abstract
Titanium (oxo-) alkoxide phosphonate complexes were synthesized using different titanium precursors and tert-butylphosphonic acid (tBPA) as molecular models for interaction between phosphonates and titania surfaces and to investigate the solution stability of these species. Reflux of titanium(IV) ethoxide or titanium(IV)(diisopropoxide)bis(2,4-pentadionate) with tert-butylphosphonic acid in toluene–ethanol mixture or acetone yielded seven titanium alkoxide phosphonate complexes; [Ti5(μ3-O)(μ2-O)(μ-HOEt)2(μ-OEt)3(μ2-OEt)(μ3-tBPA)3(μ3-HtBPA)(μ2-tBPA)2(μ2-HtBPA)]·3EtOH, 1, [Ti4O(μ-OEt)5(μ2-OEt)7(μ3-tBPA)], 2, [Ti4(μ2-O)2(μ-OEt)2(μ-HOEt)2(μ2-tPBA)2(μ2-HtPBA)6]·4EtOH, 3, [Ti4(μ2-O)2(μ-OEt)2(μ-HOEt)2(μ2-tPBA)2(μ2-HtPBA)6]·2EtOH, 4, [Ti6(μ2-O)(μ3-O)2(μ2-OEt)5(μ-OEt)6(μ3-tBPA)3(μ3-HtBPA)], 5, [Ti4(μ-iOPr)4(acac)4(μ2-tBPA)4], 6 and [Ti5(μ4-O)(μ2-O)3(μ2-OEt)4(μ-OEt)6(μ-HOEt)(μ3-tBPA)]2, 7. The binding mode of tBPA to the titanium oxo-core were either double or triple bridging or a combination of the two. No monodentate or chelating coordination was observed. 31P NMR spectrometry of dissolved single crystals indicates that 1 and 5 retain their solid-state structures in solution, the latter even on moderate heating, while 6 and 7 dissolved into several other forms. The complexes were found to be sensitive towards hydrolysis, proceeding in a topotactic fashion with densification of the material into plates and lamellae resulting finally in “core–shell” nanoparticles with a crystalline core (anatase) and an amorphous outer shell upon contact with water at room temperature as observed by HRTEM and AFM analyses. 31P NMR data supported degradation after addition of water to solutions of the complexes. Hydrolysis under different conditions affords complex oxide structures of different morphologies.
Introduction
Titanium coordination compounds are of growing interest for technological applications. They are used as precursors for organic–inorganic hybrid materials and metal organic frameworks, utilizing organic ligands containing polymerizable groups for further modification of the materials.1–3 Monodisperse crystalline titania (anatase) nanoparticles can be obtained from titanium coordination compounds by shift in solution equilibrium4 or upon hydrolysis and thermolysis.5 Titanium coordination complexes also find use as catalysts, particularly for olefin polymerization.6,7 The discovery of photocatalytic activity of titania by Honda and Fujishima8 in the 1970's has resulted in a number of applications, such as self-cleaning glasses, wastewater remediation and hydrogen gas production.9,10 Much work has been done on modifying titanium alkoxides with carboxylic acids,11,12 β-diketonate13,14 and phenols;15 leaving the field of titanium phosphonate complexes less explored. Recently however, there has been an increased interest in titanium phosphonates and the literature now contains quite a rich flora of titanium alkoxide complexes with phosphonate ligands. Sizes range from dinuclear complexes16 to nano-size complexes [Ti26O26(OEt)39(PhenylPO3)6]Br.17 Because of the high affinity of the phosphonate group to metal oxide surfaces, including titania,18,19 it provides an attractive anchor group for functionalizing inorganic nanoparticles with organic ligands for applications such as hybrid nanoadsorbents,20 organic–inorganic hybrid materials,21 mesoporous titanium phosphonate structures for catalysis22,23 and for biological applications. Phospholipids are a major constituent of the cell membrane. Le and co-workers have shown that some phospholipids interact quite strongly with commercial titania nanoparticles by electrostatic interactions, with the interaction being pH dependent.24 Thus, coating of implants with titania helps the attachment of new cells to implant surfaces and are of interest for regenerative medicine.25,26 Common phosphonic acids for modification of titanium alkoxides are tert-butylphosphonic acid (tBPA) and phenylphosphonic acid (PPA). The phosphonate group is interesting as a ligand as it has up to three oxygen atoms available for coordination to metal atoms and can be mono-, bi- or tridentate coordinating. Bi- and tridentate coordination is referred to as bridging.17,18 In most cases of bi- and tridentate coordination of phosphonate oxygen to titanium, each oxygen binds to different titanium centers. While chelates of phosphonate to titanium center are also known,27 they do not appear to be common, probably because of straining bond angles. Because of the high reactivity of modified titanium alkoxo-complexes,28 these compounds readily hydrolyze into titania5,28 in the presence of moisture or even traces of water in solvents. The photocatalytic activity of titanium (oxo-) complexes and titanium phosphonate complexes has been reported in several recent papers,29–32 in which liquid aqueous medium was applied. However, the authors never investigated the fate of the titanium oxo-complexes in water, using any suitable characterization techniques for analysis of possible transformations such as atomic force microscopy or electron microscopy. A plausible supposition could be made that the studies of “single molecule photocatalysis” could in reality refer to titania nanoparticles resulting from hydrolysis of the applied “clusters”. As the ligands of the complexes are transferred to the surfaces of the formed metal oxide nanoparticles, there will be a difference in photocatalytic activity of the “dissolved clusters”. Different coatings on titania nanoparticles have been shown to influence the catalytic activity.33 Coordination complexes form as the most thermodynamically stable species under the current conditions.34 If some conditions are changed (e.g. temperature, solvent or introduction of new ligands), a new thermodynamically more stable species may form. Likewise, a solid-state structure may exist in equilibrium with other forms in solution,16,35,36 making solution characterization (e.g. NMR) of coordination complexes necessary. Recent work by Nyman and co-workers involved use of small angle X-ray scattering to characterize coordination complexes in solution.37,38 Most titanium phosphonates reported in the literature so far were synthesized under basic conditions (<1 equivalent of phosphonic acid).39–41 In a previous publication we thoroughly investigated the solution stability of two titanium oxo-alkoxides modified with phenylphosphonate ligands using multiple NMR techniques.42 In this work we synthesized a series of titanium (oxo-) alkoxides modified with tert-butylphosphonic acid, using 0.85, 1, 2 and 4 equivalents of phosphonic acid, to obtain formally basic, neutral and acidic reaction conditions. We extended solution stability studies to also include investigation of their hydrolytic stability. Seven different titanium alkoxide phosphonate structures were obtained by reacting titanium(IV) ethoxide or titanium(IV) diisopropoxide-bis-2,4-pentadionate, Ti(acac)2iOPr2, with tert-butylphosphonic acid on reflux in different solvent systems. The stability (in CDCl3 or acetone-d6) of selected complexes were investigated using 31P nuclear magnetic resonance (NMR) spectrometry. In our observations for a collection of phenoxide,5 carboxylate43 and selected phosphonate titanium alkoxide derivatives (this work) we were not able to observe any pronounced stability to hydrolysis in aqueous medium. General mechanisms demonstrated earlier43 for hydrolysis in boiling water appear to be even the case for selected phosphonate complexes investigated in this work.Experimental section
Chemicals
Titanium(IV) ethoxide (Aldrich), titanium(IV)(diisopropoxide)bis(2,4-pentadionate) (Alfa Aesar) and tert-butylphosphonic acid (Sigma-Aldrich) were used as received. Acetone (Merck) was dried over molecular sieves (4 Å), 99.7% ethanol (Solveco) was refluxed and distilled over calcium metal and toluene (PA, Merck) was either used as received (“wet”) refluxed and distilled over lithium aluminium hydride (anhydrous). All synthetic procedures were carried out in a glovebox under nitrogen atmosphere and refluxed at ambient atmosphere.Analyses
Single-crystal X-ray diffraction data were recorded with a Bruker D8 SMART APEX II CCD diffractometer (graphite monochromator) using λ(Mo-Kα) = 0.71073 Å radiation. The structures were solved by direct methods. Metal atom coordinates were obtained from the initial solutions and other non-hydrogen atoms by Fourier synthesis. Details of data collection and refinement are summarized in ESI Table S1.† For FTIR, a Perkin–Elmer FTIR spectrometer Spectrum-100 was used. Crystals were milled in paraffin oil and their spectra recorded by a total of 8–16 scans in 400–4000 cm−1 range. For NMR, a Bruker Avance III 600 MHz Cryoprobe spectrometer and a Bruker Avance III 600 MHz SmartProbe spectrometer were used for recording of all spectra (at 30 °C), using Bruker Topspin version 2.1 software. NMR spectra for 1, 5 and 7 were recorded in anhydrous CDCl3 and 6 in anhydrous acetone-d6. Atomic force microscopy (AFM) analyses were performed with a Bruker FastScan with ScanAsyst. A Hitachi TM-1000 scanning electron microscope, equipped with an Oxford Instruments EDS system, was used for SEM imaging. For TEM analyses, JEOL JEM-2100F TEM (200 kV operating voltage), Philips CM-20 Super Twin microscope (200 kV operation voltage), Philips CM12 (120 kV operation voltage) and FEI Tecnai (200 kV operation voltage) were used.[Ti5(μ3-O)(μ2-O)(μ-HOEt)2(μ-OEt)3(μ2-OEt)(μ3-tBPA)3(μ3-HtBPA)(μ2-tBPA)2(μ2-HtBPA)]·3EtOH, 1, tert-butylphosphonic acid (tBPA), 0.48 mmol (2 eq.) was dissolved in a mixture of 0.3 mL “wet” toluene and 4 mL anhydrous ethanol. Titanium(IV) ethoxide, 0.24 mmol, was then added, giving a clear colourless solution. After reflux for 2 hours, the reaction mixture was concentrated by rotary evaporation to ∼1 mL. The RM became very pale yellow. After storage at −18 °C for a few days, colourless crystals suitable for single crystal X-ray diffraction were obtained. Compound 1 crystallized in monoclinic space group C2/c. 1 was obtained in ∼80% yield. 13C NMR δ ppm: 129.04, 128.25, 58.53, 25.34, 25.06, 24.88, 18.40. 31P NMR δ ppm: 33.96, 26.57, 24.92, 24.44, 19.79, 18.05, 16.24. IR, cm−1: 1140 (b), 1052 (s), 918 (s), 833 (s), 727 (s), 669 (s), 620 (b). Only major signals are reported.
[Ti4(μ4-O)(μ-OEt)5(μ2-OEt)7(μ3-tBPA)], 2, was synthesized from a synthetic procedure analogous to that for compound 1 but with anhydrous toluene more diluted with ethanol. Small colorless crystals were obtained after a few days. 2 crystallized in triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Compound 2 was obtained in ∼80% yield. IR, cm−1: 1138 (b), 1101 (s), 1036 (s), 1022 (s), 979 (m), 834 (s), 718 (s), 597 (b), 545 (m). Only major signals are reported.
. Compound 2 was obtained in ∼80% yield. IR, cm−1: 1138 (b), 1101 (s), 1036 (s), 1022 (s), 979 (m), 834 (s), 718 (s), 597 (b), 545 (m). Only major signals are reported.
[Ti4(μ2-O)2(μ-OEt)2(μ-HOEt)2(μ2-tPBA)2(μ2-HtPBA)6]·4EtOH, 3 and 4, [Ti4(μ2-O)2(μ-OEt)2(μ-HOEt)2(μ2-tPBA)2(μ2-HtPBA)6]·2EtOH. 0.95 mmol (4 eq.) tBPA was added to 0.24 mmol titanium(IV) ethoxide in 0.4 mL freshly distilled dry toluene plus 4 mL ethanol and refluxed for 2 hours. The RM was then concentrated to ∼1.5 mL and stored at −18 °C. After a few days, small, colorless crystals suitable for single crystal X-ray diffraction were obtained. 3 and 4 co-crystallized in the same flask and were distinguished by appearance. 3 and 4 crystallized in the monoclinic space groups P2(1)/n and P2(1)/c, respectively. 3 and 4 was obtained in ∼70% yield. This protocol could also yield either 1 or 5 as major product when the solvents (especially EtOH) had not been distilled immediately before synthesis. 31P NMR δ ppm: 37.64, 22.34. IR, cm−1: 3324 (b), 2335 (s), 1133 (s), 1050 (s), 945 (m), 832 (s), 659 (s), 504 (s). Only major signals are reported.
[Ti6(μ2-O)(μ3-O)2(μ2-OEt)5(μ-OEt)6(tBPA)3(HtBPA)], 5, was synthesized by refluxing titanium(IV) ethoxide with 2 eq. of tBPA in a mixture of 0.4 mL “wet” toluene and 4 mL ethanol. It crystallized in the triclinic space group P. 5 was obtained in ∼80% yield. 31P NMR δ ppm: 35.31, 27.42, 23.51 and 20.98 ppm. 1H NMR δ ppm: 6.22 (s), 4.48 (m), 3.75 (q, J = 6.99, 7.06 Hz), 1.49 (m), 1.37 (m), 1.19 (d, J = 3.66 Hz) 1.15 (d, J = 4.65 Hz). IR, cm−1: 1063 (s), 1011 (s), 978 (s), 833 (s), 766 (b), 612 (b), 572 (b). Only major signals are reported.
[Ti4(μ-iOPr)4(acac)4(tBPA)4], 6, 0.17 mmol (0.85 eq.) tBPA was added to 0.20 mmol of Ti(acac)2(iOPr)2 in 5 mL anhydrous acetone (the solution turned deep orange) and refluxed for 3 hours. The RM was concentrated to ∼1 mL and then stored at −18 °C. Orange crystals of 6 suitable for single X-ray diffraction were obtained within a few days. Compound 6 crystallized in the monoclinic space group C2/c. 6 was obtained in ∼60% yield. 31P NMR δ ppm: 27.03, 27.00, 26.08, 25.95, 25.72, 25.14, 24.58, 23.29, 21.19, 20.12, 16.81. IR, cm−1: 1592 (m), 1027 (s), 1109 (m), 983 (m), 850 (s), 834 (s), 665 (s), 613 (m). Only major signals are reported.
[Ti5(μ4-O)(μ2-O)3(μ2-OEt)4(μ-OEt)6(μ-HOEt)(μ3-tBPA)]2, 7, 0.24 mmol tBPA was added to 0.24 mmol titanium(IV) ethoxide in 0.3 mL toluene plus 3 mL ethanol and refluxed for 0.50 h. The RM was stored at 4 °C. After a week, massive precipitation of colorless crystals had occurred. 7 crystallized in the triclinic space group P. Compound 7 was obtained in ∼80% yield. 31P NMR δ ppm: 35.23, 34.68, 34.00, 33.59, 33.06, 32.95. IR, cm−1: 1136 (s), 1024 (s), 976 (s), 922 (s), 833 (w). Only major signals are reported.
Results and discussion
A variety of different structures were obtained by reacting titanium(IV) ethoxide with tert-butylphosphonic acid using different solvents and Ti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :tBPA ratios. An overview is given in the Scheme 1. Compounds 1, 2 and 5 where obtained by refluxing titanium(IV) ethoxide with two eq. of tBPA in an ethanol:toluene mixture. By using four eq. of tBPA, compounds 3 and 4 where afforded in a mixture with each other. To obtain 3 and 4 without contamination of 1 or 5 the solvents needed to be distilled immediately prior to synthesis. Correspondingly, rigours exclusion of water was necessary to obtain compound 2 without contamination of 1 or 5. Apparently, the Ti:tBPA ratio is not the sole determinant factor of which compound that forms but also trace amounts of water and contents of the solvents.
:tBPA ratios. An overview is given in the Scheme 1. Compounds 1, 2 and 5 where obtained by refluxing titanium(IV) ethoxide with two eq. of tBPA in an ethanol:toluene mixture. By using four eq. of tBPA, compounds 3 and 4 where afforded in a mixture with each other. To obtain 3 and 4 without contamination of 1 or 5 the solvents needed to be distilled immediately prior to synthesis. Correspondingly, rigours exclusion of water was necessary to obtain compound 2 without contamination of 1 or 5. Apparently, the Ti:tBPA ratio is not the sole determinant factor of which compound that forms but also trace amounts of water and contents of the solvents.
 | ||
| Scheme 1 Synthetic procedures employed for isolation of complexes described in this work. | ||
Molecular structures
Detailed crystallographic information for compounds 1 to 7 are given in ESI Table 1.† The phosphonate group has several potential binding modes; monodentate, bidentate (bridging or chelating) or tridentate coordination. For the compounds obtained at 0.85, 1 or 2 eq. tBPA, bi-, tridentate or a combination of the two are observed. It might have been anticipated to observe monodentate coordination in phosphonate for compounds 3 and 4, obtained from acidic syntheses (4 eq. tBPA), but instead all phosphonates had a bidentate coordination mode. This indicates good affinity of the phosphonate oxygen to titania even under acidic conditions and that phosphonate groups could be used as stable anchor groups for low-pH applications. All phosphonate groups in compounds 2, 5, 6 and 7 (2, 2, 0.85 and 1 eq. tBPA, respectively) had tridentate coordination. In 1, there is a mixture of bi- and tridentatly coordinating phosphonates.It is important to note that the structures obtained on reflux are apparently the same as those obtained on solvothermal synthesis as testified in particular by the structure of 2 (see below) that is formed on both types of synthesis.20 Its pentanuclear core has been observed in numerous products of solvothermal synthesis. In general, the structure of a modified (oxo) alkoxide demonstrates a dense packing of cations and ligands. Solvothermal synthesis as well as prolonged reflux serve simply to generate oxo-ligands via condensation (side) reactions.
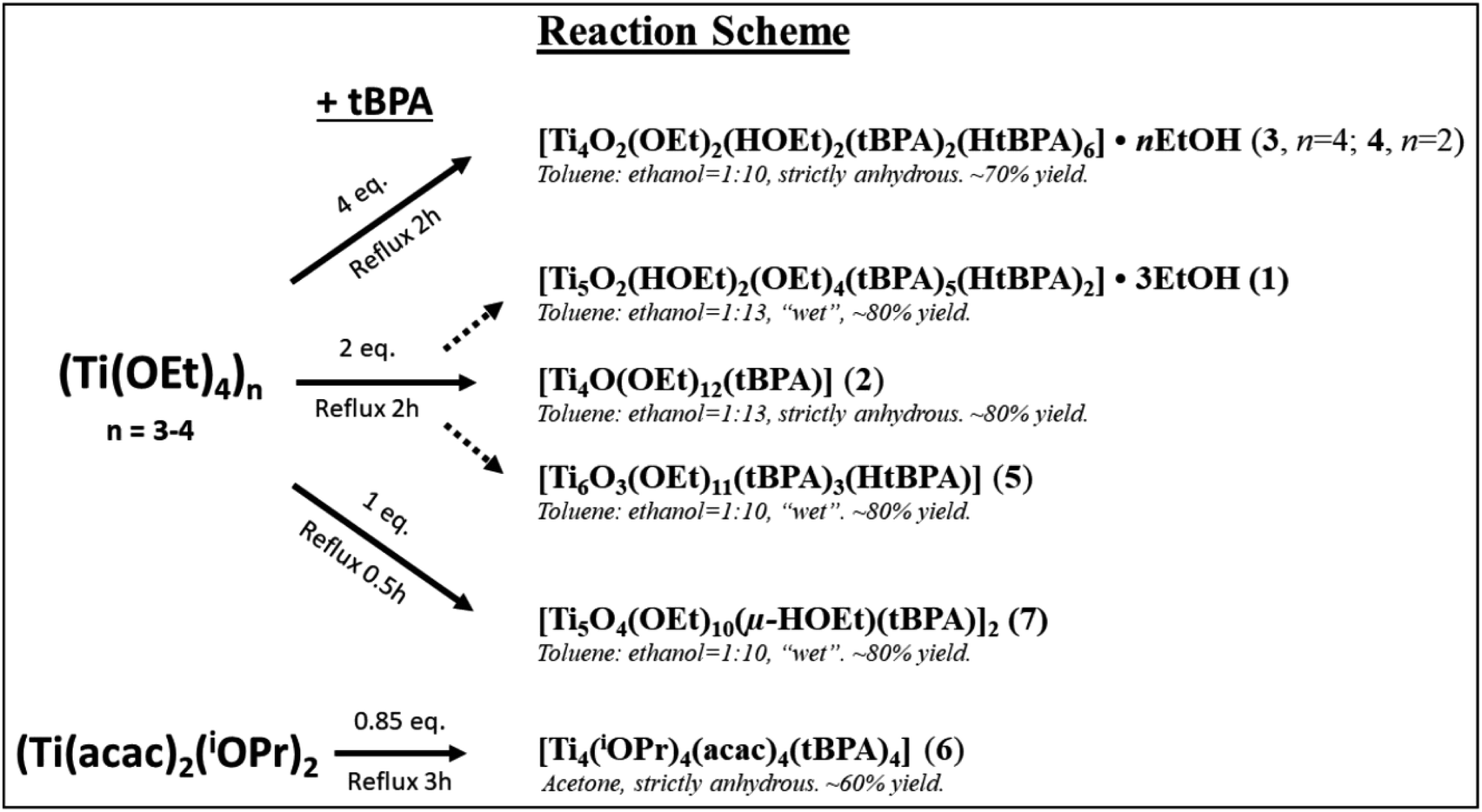
Compound 1, [Ti5(μ3-O)(μ2-O)(μ-HOEt)2(μ-OEt)3(μ2-OEt)(μ3-tBPA)3(μ3-HtBPA)(μ2-tBPA)2(μ2-HtBPA)]·3EtOH, crystallized in the monoclinic space group C2/c (Fig. 1). It consists of a pentanuclear oxo-core where all five titanium atoms are octahedrally coordinated by oxygen atoms. The degree of condensation (O/Ti) is 0.40. Ti1, Ti2 and Ti4 are connected by a μ3-oxygen (O5), forming a Ti3O subunit. Ti3 and Ti5 connect via a μ3-oxygen (O11). The two Ti3O subunits are connected via five phosphonate groups (P1 to P5). The average Ti–O distance in 1 is 1.963 Å, with the μ3-O connecting Ti1, Ti2 and Ti4 having a significantly longer average bond, 2.276 Å. The (P)O–Ti bonds comes in two populations, one shorter than on average 1.921 Å and one longer at 1.962 Å. The structure also contains one bridging and five terminal ethoxy groups. Four of the phosphonate groups are tridentatly coordinating and three are bidentatly coordinating. Four hydrogen atoms are needed to achieve a neutral complex. Based on Ti–O bond lengths from the crystallographic data, two terminal ethoxide ligands (O16, 2.133 Å, and O24, 1.889 Å) and two phosphonate oxygen atoms (O12 and O30) are likely protonated sites. It is noteworthy that in this structure the alkoxide groups and protonated phosphonic acid molecules are coordinated by/“adsorbed” on the titanium oxide core are present simultaneously. This indicates that optimization of molecular geometry can potentially dominate over acid–base interactions in this class of compounds (see bond lengths for (P)O–Ti and P–O in ESI Table S2†).
 | ||
| Fig. 1 Molecular structure of compound 1. Titanium atoms are displayed as green, oxygen as red, phosphorus as magenta and carbon atoms are grey. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (o): Ti1–O6 1.750(5), Ti1–O3 1.912(4), Ti1–O2 1.925(5), Ti2–O10 1.750(5), Ti2–O7 1.896(5), Ti2–O9 1.914(5), Ti3–O11 1.807(5), Ti3–O15 1.925(5), Ti3–O14 1.938(5), Ti4–O17 1.744(6), Ti4–O19 1.914(5), Ti4–O18 1.916(5), Ti5–O24 1.899(5), Ti5–O23 1.936(5), Ti5–O22 1.961(5), Ti3–O11–Ti5 137.6(2), Ti1–O4–Ti2 112.1(2). | ||
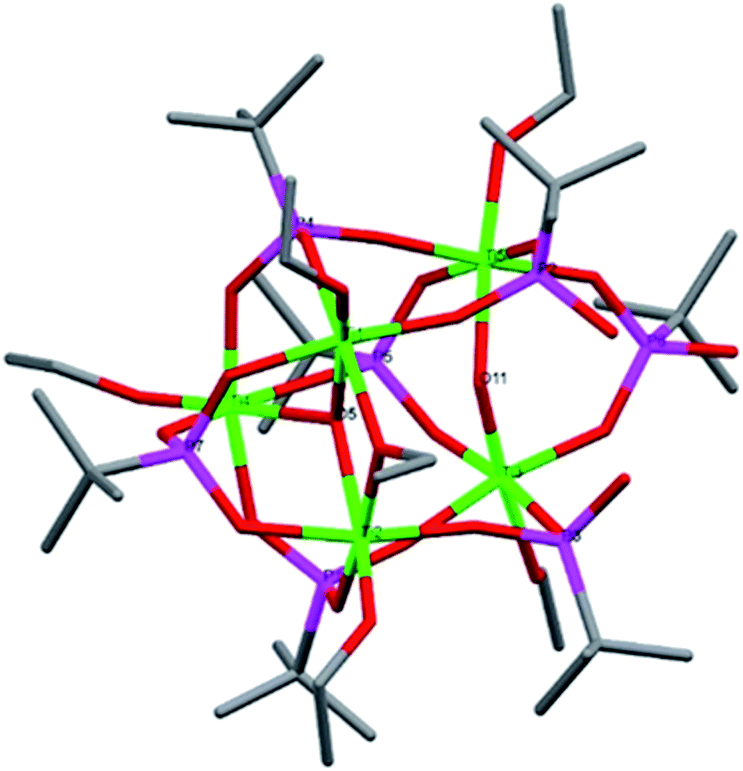
Compound 2, [Ti4(μ4-O)(μ-OEt)5(μ2-OEt)7(μ3-tBPA)], crystallized in the triclinic space group P (Fig. 2). 2 is structurally similar to a complex previously reported by us.20 It suffers from disorder at the Ti4 position. Compound 2 is a tetranuclear (Ti4O) titanium oxo-complex with one tridentatly bonding tBPA ligand coordinating Ti2, Ti4 and Ti4/Ti4a. It features one μ4-O bridge (O1), connecting the four titanium atoms. The degree of condensation (O/Ti) is 0.25. The average Ti–O bond distance is 1.905 Å while for (P)O–Ti it is 1.968 Å (see bond lengths for (P)O–Ti and P–O in ESI Table S3†).
 | ||
| Fig. 2 Molecular structure of compound 2. Green it titanium, red is oxygen, magenta is phosphorous and grey is carbon. Hydrogen atoms have been omitted for clarity. The two dark green atoms are alternate positions for the disordered Ti4. Selected bond lengths (Å) and angles (o): Ti1–O6B 1.776(12), Ti1–O4 1.797(8), Ti1–O5 1.826(10), Ti2–O8 1.777(5), Ti2–O10 1.915(6), Ti2–O2 1.961(5), Ti3–O13 1.750(5), Ti3–O12 1.831(5), Ti3–O3 1.968(5), Ti4–O14 1.788(9), Ti4–O16 1.791(8), Ti4–O5A 2.004(10), Ti4A–O14A 1.738(13), Ti4A–O16A 1.781(12), Ti4A O15B 1.932(14), Ti1–O5A–Ti4 105.8(4), Ti2–O7–Ti3 104.1(2). | ||
Compound 3, [Ti4(μ2-O)2(μ-OEt)2(μ-HOEt)2(μ2-tPBA)2(μ2-HtPBA)6]·4EtOH, is a tetranuclear centrosymmetric titanium alkoxide phosphonate complex crystallizing in monoclinic space group P2(1)/n (Fig. 3). It contains four octahedrally coordinated titanium atoms, with one terminal ethanol group on each titanium. The core has a distorted cubic motif if both Ti and P atoms are considered. Two μ2-O bridges, O2 and O2a, connects Ti1–Ti2 and Ti1a–Ti2a, respectively. The complex contains eight tBPA ligands, all of them coordinate bidentatly to two different titanium atoms. The complex has four solvating ethanol molecules. The average Ti–O bond length is 1.956 Å and 1.944 Å for the (P)O–Ti bonds (see bond lengths for (P)O–Ti and P–O in ESI Table S4†). The Ti1–O2–Ti2 distances are unsymmetrical, with bond lengths of 1.682 Å and 2.014 Å for Ti1–O2 and Ti2–O2, respectively. The degree of condensation (O/Ti) is 0.50. To achieve a neutral complex, eight hydrogens are needed. For the asymmetric unit, one hydrogen is found on a terminal ethanol ligand, having a substantially elongated Ti–O bond (O5, 2.188 Å), while the other three are located at the tBPA ligands (O3, O9 and O13).
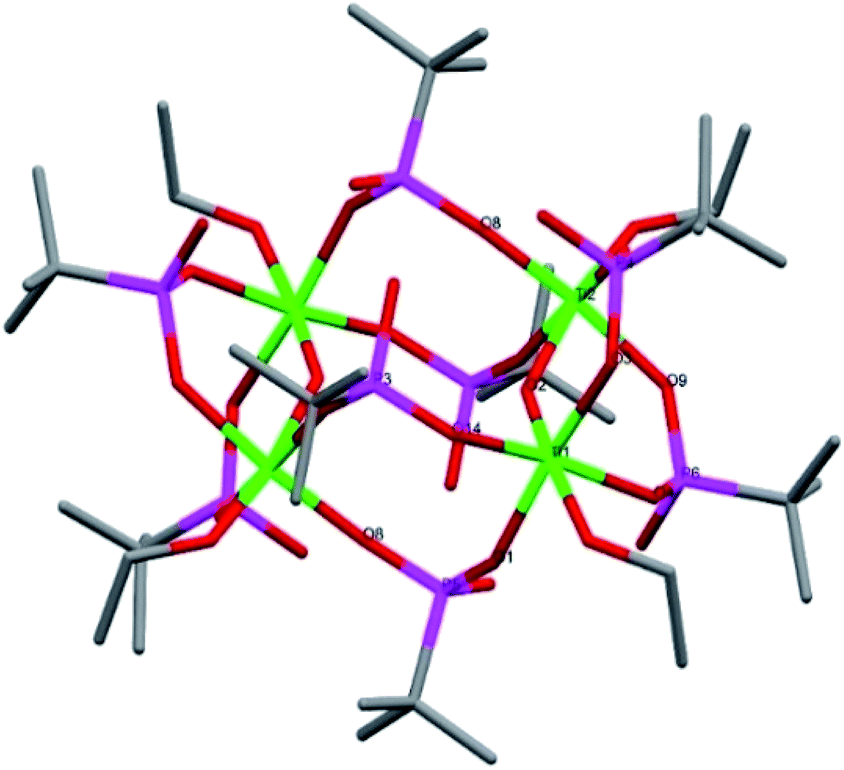 | ||
| Fig. 3 Molecular structure of compound 3. Green it titanium, red is oxygen, magenta is phosphorous and grey is carbon. The solvating ethanol molecules and hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (o): Ti1–O2 1.682(4), Ti1–O1 1.959(4), Ti1–O14 1.986(4), Ti1–O4 2.001(4), Ti2–O6 1.767(4), Ti2–O8 1.902(4), Ti2–O7 1.984(4), Ti2–O9 2.007(4) Ti1–O2–Ti2 142.79(19), P5–O1–Ti1 139.4(2). | ||
Compound 4, [Ti4(μ2-O)2(μ-OEt)2(μ-HOEt)2(μ2-tPBA)2(μ2-HtPBA)6]·2EtOH, is chemically identical to compound 3, but crystallizes in the closely related monoclinic space group P2(1)/c and it contains only two solvating ethanol molecules. It co-crystallized with 3 and was distinguished by appearance. The average Ti–O bond length is 1.961 Å, and for (P)O–Ti is 1.977 Å (see bond lengths for (P)O–Ti and P–O in ESI Table S5†). The Ti1–O1–Ti2 bond lengths are 2.011 Å and 1.745 Å, respectively. To achieve a neutral complex, eight hydrogen atoms are needed. For the asymmetric unit, one terminal ethoxide ligand with substantially elongated Ti–O bond (O8, 2.144 Å) is protonated (an ethanol molecule), as are three coordinating phosphonate oxygen atoms (O3, O5 and O6).
Compound 5, [Ti6(μ2-O)(μ3-O)2(μ2-OEt)5(μ-OEt)6(μ3-tBPA)3(μ3-HtBPA)], is a hexanuclear titanium oxo-alkoxide phosphonate complex crystallizing in the triclinic space group P (Fig. 4). The degree of condensation (O/Ti) is 0.50. All six titanium atoms have an octahedral coordination environment. There are two μ3-oxygens (O1 and O15) connecting three titanium atoms each (Ti1–Ti3 and Ti4–Ti6, respectively), forming two Ti3O-subunits. There is one μ2-O (O11) and three phosphonate groups (P7, P9 and P10) connecting the two subunits. The structure contains five bridging ethoxide groups and six terminal ethoxide groups. The average Ti–O bond length is 1.960 Å, and for (P)O–Ti bonds 1.964 Å (see bond lengths for (P)O–Ti and P–O in ESI Table S6†). The two μ3-oxygens, O1 and O15, bond to titanium with average bond length of 1.996 Å and 2.135 Å, respectively. One phosphonate oxygen atom (O13, 2.037 Å) is protonated to give a neutral complex.
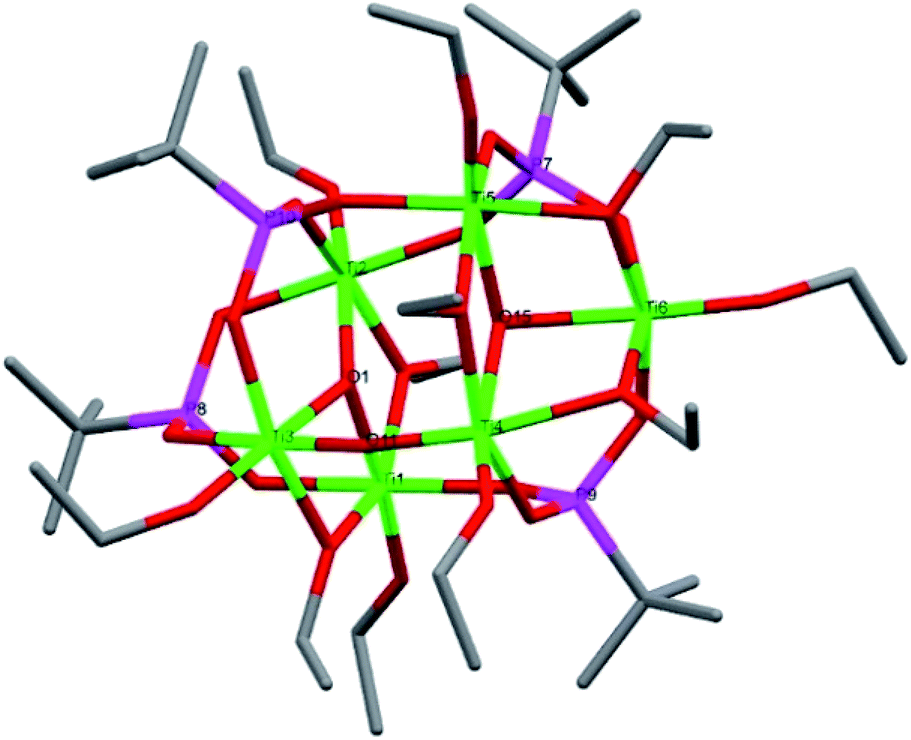 | ||
| Fig. 4 Molecular structure of compound 5. Green it titanium, red is oxygen, magenta is phosphorous and grey is carbon. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and bond angles (o): Ti1–O6 1.797(3), Ti1–O4 1.934(3), Ti1–O2 1.979(3), Ti2–O10 1.784(3), Ti2–O8 1.947(3), Ti2–O7 1.965(3), Ti3–O14 1.788(3), Ti3–O11 1.892(3), Ti3–O12 1.950(3), Ti4–O11 1.742(3), Ti4–O20 1.782(3), Ti4–O17 1.990(3), Ti5–O22 1.954(3), Ti5–O16 1.967(3), Ti5–O18 2.010(3), Ti6–O26 1.744(3), Ti6–O19 1.926(3), Ti6 O25 1.934(3), Ti2–O1–Ti3 139.19(12), Ti4–O15–Ti5 102.42(10), Ti6–O15–Ti5 98.79(10). | ||
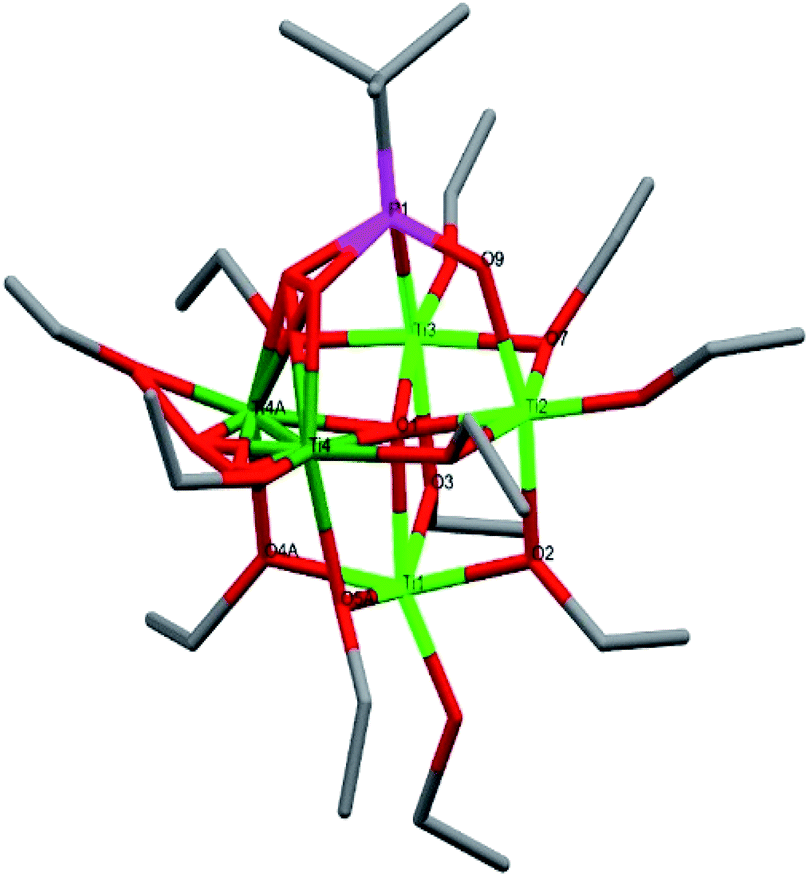
Compound 6, [Ti4(μ-iOPr)4(acac)4(μ3-tBPA)4], is a centrosymmetric tetranuclear titanium phosphonate-acetylacetonate alkoxide complex crystallizing in the monoclinic space group C2/c (Fig. 5). The titanium oxo-core forms a distorted cube. All phosphonate oxygen atoms coordinate to three different titanium atoms. Each titanium atom has a terminal isopropoxide group. The average Ti–O bonds in 6 is 1.935 Å. The (P)O–Ti average bond length is 1.936 Å, which is shorter than for the acac ligands of 2.005 Å (see bond lengths for (P)O–Ti and P–O in ESI Table S7†). An analogous structure having phenylphosphonic acid ligands instead of tert-butylphosphonic acid was recently reported.44 In the Ti(acac)2iOPr2 precursor there are two acac and two iOPr ligands per titanium but in compound 6 there are only one of each ligand per titanium. The acac-ligand is chelating and generally considered to be strongly bound to Ti(IV) centers. Its removal by interaction with an acidic phosphonic acid reagent with preservation of isopropoxide in the coordination sphere is unexpected. This may be due to a Meerwein–Ponndorf–Verley (MPV) reduction between a iOPr ligand and an acetone solvent molecule, followed by an aldol condensation between an acetone and the reduced acac ligand with the subsequent release of Me2C(O)CH2C(O)CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(OH)CH3. MPV reduction with solvent condensation was previously observed for another titanium alkoxide complex in acetone.5
C(OH)CH3. MPV reduction with solvent condensation was previously observed for another titanium alkoxide complex in acetone.5
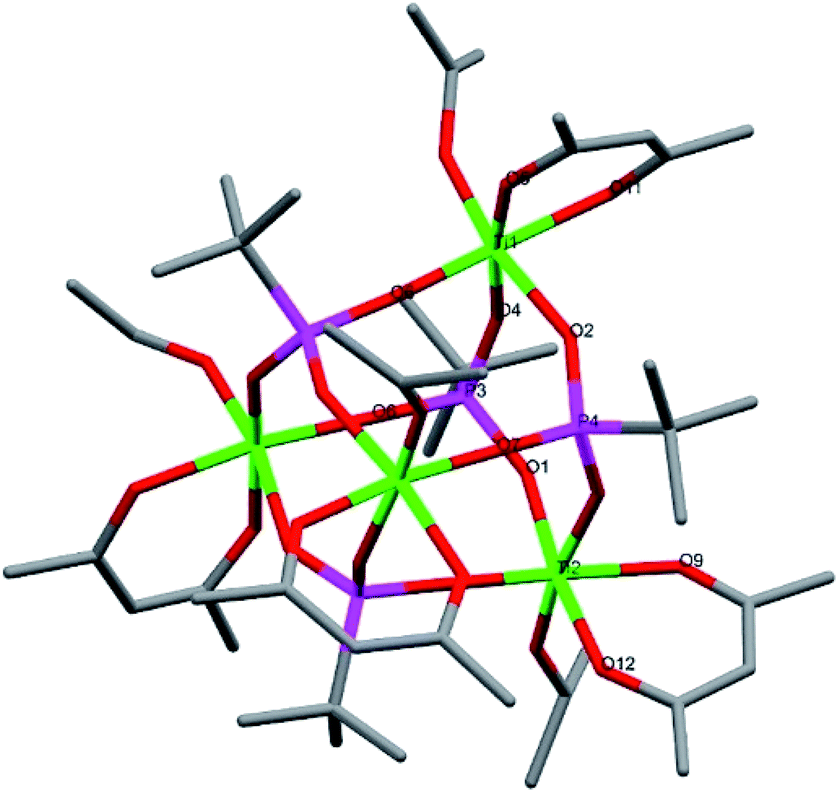 | ||
| Fig. 5 Molecular structure of compound 6. Green it titanium, red is oxygen, magenta is phosphorous and grey is carbon. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (o): Ti1–O3 1.790(4), Ti1–O6 1.890(4), Ti1–O4 1.914(4), Ti1–O5 1.995(4), Ti2–O10 1.801(5), Ti2–O7 1.882(4), Ti2–O1 1.949(4), Ti2–O12 1.978(4), P4–O2–Ti1 147.5(3), P3–O1–Ti2 146.7(3). | ||
Compound 7, [Ti5(μ4-O)(μ2-O)3(μ2-OEt)4(μ-OEt)6(μ-HOEt)(μ3-tBPA)]2, is a dimeric structure with two Ti5O4 cores linked by two μ2-OEt bridges. 7 crystallized in the triclinic space group P (Fig. 6). The asymmetric unit of 7 has a pentanuclear oxo-core (Ti5O4) with six terminal and five bridging ethoxide molecules and one tridentatly coordinating tBPA. Ti1, Ti3, Ti4 and Ti5 are connected via a μ4-oxygen (O1) and Ti2 connects to Ti1, Ti3 and Ti5 via O15, O11 and O14, respectively. The average Ti–O bond length was 1.960 Å and 1.995 Å for the (P)O–Ti (see bond lengths for (P)O–Ti and P–O in ESI Table S8†). The average Ti–O length for the three μ2-oxygens was 1.851 Å and 2.034 Å for the μ3-oxygen. For the asymmetric unit, one hydrogen atoms is needed for a neutral complex. This hydrogen atom is attached to a terminal ethoxide ligand with a substantially elongated Ti–O bond (O18B, 2.271 Å).
 | ||
| Fig. 6 Molecular structure of compound 7. Green it titanium, red is oxygen, magenta is phosphorous and grey is carbon. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (o): Ti1–O7 1.769(5), Ti1–O15 1.838(4), Ti1–O8 1.976(4), Ti2–O11 1.846(4), Ti2–O14 1.848(5), Ti2–O15 1.859(5), Ti3–O13 1.788(5), Ti3–O11 1.796(4), Ti3–O4 1.979(5), Ti4–O40 1.762(5), Ti4–O9 1.945(6), Ti4–O2 1.965(5), Ti5–O6 1.793(5), Ti5–O14 1.807(4), Ti5–O5 1.998(5), Ti4–O2–Ti5 105.3(2), Ti3–O11–Ti2 133.7(2), Ti5–O14–Ti2 125.8(2). | ||
Infrared spectrometry
All compounds were analysed by Fourier transform infrared spectrometry (FTIR). Crystals milled in a dry nitrogen atmosphere were analyzed in paraffin oil to avoid hydrolysis. The fingerprint area for all compounds is complex, giving overlapping signals. Compound 1 showed absorption at 832 cm−1 and 1140 cm−1, assigned to Ti–O–Ti and PO vibrations, respectively. For compound 2, an absorption at 1022 cm−1 is assigned to the P–O–Ti bond and a sharp signal at 980 cm−1 is assigned as a P–O vibration. A signal at 834 cm−1 that corresponds to Ti–O absorption could also be found. With compound 3 + 4, the absorption at 1133 cm−1 is assigned to PO. The absorption at 832 cm−1 corresponds to a Ti–O bond vibration. A broad signal at 3324 cm−1 indicated the presence of OH-groups. The FTIR spectrum of compound 5 contains absorptions at 833 cm−1, 1023 cm−1, which are assigned to P–O–Ti and Ti–O–Ti vibrations, respectively. A vibration at 3329 cm−1 indicated OH-groups. Compound 6 absorbs at 834 cm−1, 982 cm−1 and 1026 cm−1, which are assigned to Ti–O–Ti, P–O and P–O–Ti, respectively. Compound 7 showed absorptions at 833 cm−1 (Ti–O–Ti), 976 cm−1 (P–O), 1024 cm−1 (P–O–Ti) and 1136 cm−1 (PO). FTIR signals are summarized in Table 1.
Hydrolysis studies
In our previous work with phenoxide and carboxylate containing titanium alkoxides we have demonstrated their facile hydrolysis in contact with water.5,43 We wanted to continue our investigation on phosphonate-containing titanium alkoxide complexes, which have recently been reported as hydrolytically stable and tested in aquatic media for UV-driven water-splitting.29,30,32 Titanium alkoxides are often modified to contain chelating (and bridging) ligands to stabilize the colloid oxide phase.45 This modification, however, does not necessarily result in hydrolytic stability of the complexes in solutions with high water content or in pure aqueous solvent, which also has been pointed out by others.46 It should be mentioned that apparent stabilization against hydrolysis has been proved for carboxylate metal–organic frameworks (MOFs), caused by thermodynamic gain due to stability of a continuous 3D crystal structure.47 In a previous publication43 we reported the facile production of highly porous, diatom-like structures by hydrolysis of penicillamine modified titanium complexes at elevated temperatures. We hypothesized that also phosphonate modified titanium complexes can be used to obtain complex titania structures upon hydrolysis. Thus, hydrolysis of 7 was performed under different conditions (see ESI† for details). When 7 (i.e. its dissolved forms, see the NMR description below) in toluene was injected quickly via syringe into a heated dilute suspension of fibrillar nanocellulose without stirring, well-defined smooth spheres was obtained (ESI Fig. S2†). Fibrillar nanocellulose was added as a Pickering emulsifier as it has a pronounced ability to stabilize oil droplets injected to aqueous media.48,49 After heating to 500 °C, to improve crystallinity, the structures in Fig. 7a were obtained. When the same solution was injected into a water/acetone mixture at room temperature, corrugated spheres as shown in Fig. 7b were obtained instead. Corrugation of the surfaces increased with increased v% acetone, the structure in Fig. 7b was obtained with 20 v% acetone. The observed pattern at the surface may be explained by an instant mixture of solvents at the toluene:water interface upon injection, followed by rapid simultaneous spinodal decomposition and precursor hydrolysis. The morphology of the material obtained in Fig. 7a (and ESI Fig. S3†) is very similar to those obtained by Collins and co-workers upon hydrolysis of unmodified titanium alkoxides in aqueous ammonia solutions.50 When injecting the titanium phosphonate precursor via quick ejection from a small-diameter syringe, small droplets are formed. Immediate hydrolysis of the titanium alkoxide phosphonates in water will retain the original spherical shape as observed in Fig. 7b and ESI Fig. S2.† According to energy-dispersive X-ray spectrometry (EDS) analyses, phosphonates are removed during the heat treatment (Fig. 7a) but are retained at the surface of the corrugated spheres, which were not subjected to any heat treatment (Fig. 7b). As was discerned from the crystallographic data, the tBPA can coordinate to titanium even when partially protonated. Accordingly they can evaporate during heating and thus no phosphorous was detected during elemental analysis of the heat-treated sample (Fig. 7a). Compound 7 has a Ti:P ratio of 5:1 while the surface of the hydrolysis product has a Ti:P ratio of 3:1 according to EDS analysis, showing thus increased P content.
 | ||
| Fig. 7 Hydrolysis products of phosphonate modified titanium oxo-alkoxides. (a) Hydrolysis in a CNF-suspension at 90 °C with subsequent heating afforded a hierarchical porous material while (b) hydrolysis at room temperature in a 20 v% aqueous solution with acetone afforded spheres with highly corrugated surfaces. EDS spectrum for each structure are shown below. Even though there might exist equilibrium products with higher P content in solution, there will likely be a ligand transfer to the surface during condensation of the titanium-oxo cores into nanoparticles. The total surface area decreases and ligands will concentrate on the surface of nanoparticles and on the outer layer of the sample, leading to increase in the phosphor content measured by EDS. In the case of compound 4, hydrolysed in MilliQ-water (Fig. S11†), the Ti:P ratio changes from 1:2 in the complex to 1:1 in the hydrolysis product. This suggest loss of the ligand to the solvent. | ||
Having obtained complex macroscale hydrolysis products, we were interested to study the nanoscale products of decomposition of the complexes. To study the influence of chelating ligands on the hydrolytic stability of the titanium phosphonate complexes, compound 6 was synthesized. Its titanium precursor, Ti(acac)2iOPr2, has two acac ligands per titanium, of which one is replaced by a tBPA ligand during the assembly of the complex.
Crystals of compounds 1 and 6 (representing phosphonate modified titanium alkoxide complexes without and with chelating ligands, respectively) were immersed in MilliQ-water at room temperature. A white residue formed at the bottom. This powder was analysed by high resolution atomic force microscopy (AFM). The AFM analyses (Fig. 8a and b, and ESI Fig. S5a and b†) revealed homogenous particles of about 20 nm in size. Formation of titanium (oxo-) alkoxide species is the first step in the sol–gel process, occurring via simultaneous hydrolysis–condensation reactions.28 They then coalesce into nuclei in the form of nanoparticles. The group of Kanaev has studied the formation of titania nanoparticles from hydrolysis of titanium alkoxides in solution. They found an almost immediate emergence of “clusters” upon reaction between titanium(IV) isopropoxide and water. The size of these “clusters” were dependent on the organic solvent used, being 5.2 nm and 3.8 nm for isopropanol and n-propanol, respectively, illustrating hydrolytic transformation into nuclei of oxide phases in contact with water.51 The transformation from the oxo-alkoxide complex into the oxide phase is facilitated by the acidity of the replacing ligands. Fornasieri and co-workers reported the substitution of ethoxide ligands in the Ti16O16(OEt)32 complex were more facile for protic ligands with lower pKα-values and lesser steric hindrance.52 Addition of 1 and 6 to MilliQ-water lead to slightly acidic suspensions (pH ∼ 4 for both). Hence, replacement of organic ligands by H3O+ should occur via proton-assisted nucleophilic substitution, with the subsequent release of the protonated organic ligands (iPrOH, Hacac, H2tBPA) and formation of titanium dioxide. HRTEM analyses of hydrolyzed 1 and 6 reveal highly crystalline titania (anatase) cores of between 2 nm to 4 nm in diameter (Fig. 10a and b). The lattice fringe distance is 0.2 nm, which corresponds to the 2 0 0 plane of anatase. As seen in Fig. 10b, a crystal fragment of 6 transforms topotactically into a titania structure by the numerous nucleation sites. Larger crystal fragments of 1 display clear lamellar structure typical for intermediate state in the topotactic reactions preceding nucleation (Fig. 11). This is a result of the hydrolysis–condensation reactions on contact with water. The formation of more condensed species results first in a densification and contraction of the original structure, forming “sheets” of the amorphous oxo-alkoxide aggregates. Further densification on hydrolysis results in breakdown of sheets and formation of oxide (anatase) nanoparticles – nuclei of the oxide phase with pronounced crystal structure surrounded by disordered amorphous matrix with lower density (Fig. 9).53 While modification of titanium alkoxides with chelating ligands have been reported to increase hydrolytic stability in organic solvents, this is less likely to be the case in pure aqueous solvent. Therefore, the residues of modified titanium alkoxide complexes intended for aqueous catalysis should be carefully investigated by high-resolution microscopy (TEM, AFM).
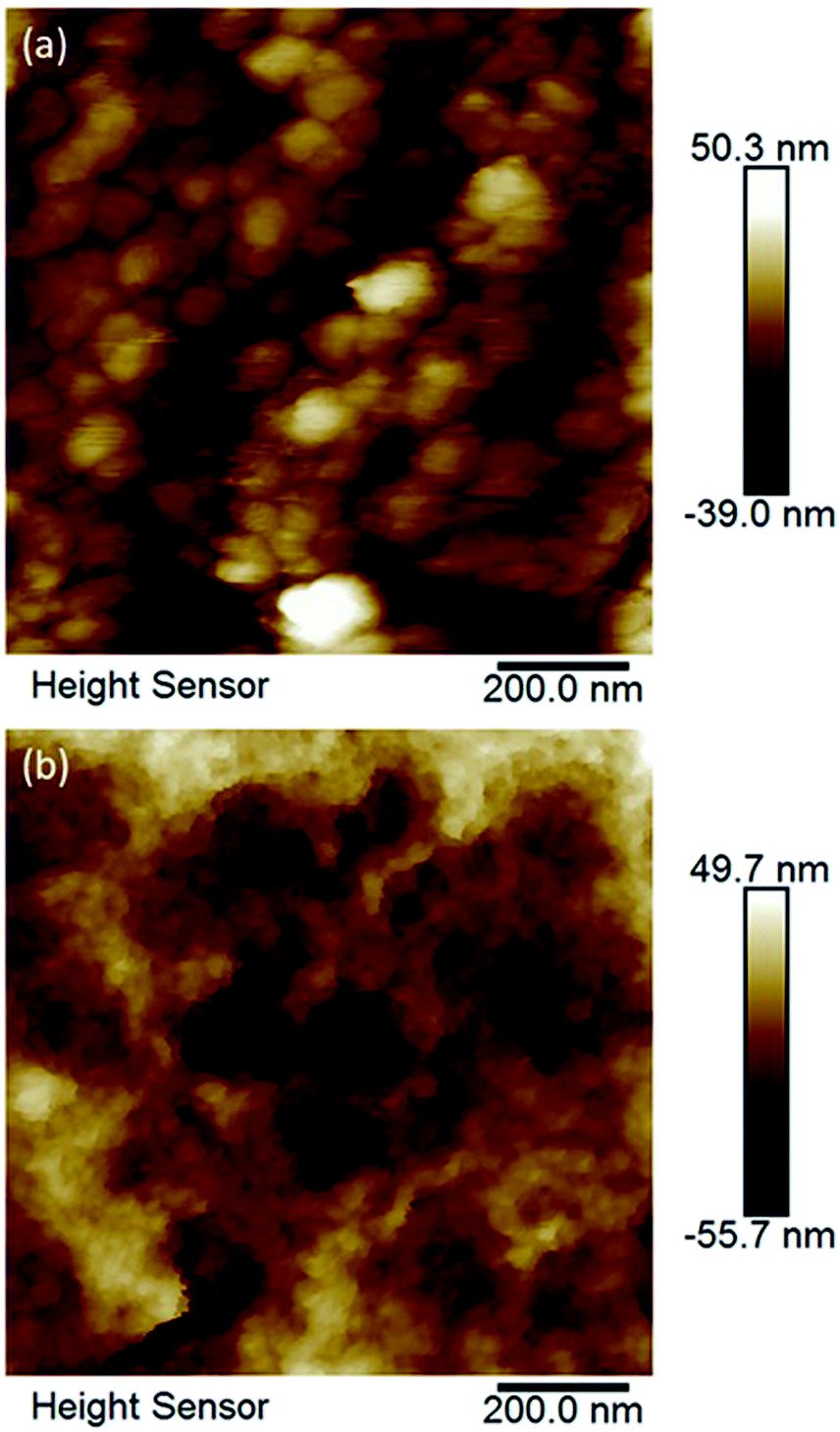 | ||
| Fig. 8 AFM micrographs of (a) hydrolysed compound 1 (no chelating ligands), and (b) hydrolysed compound 6 (one chelating ligand per titanium atom). Both compounds yielded rather uniform titania nanoparticles which apparently are aggregates of primary anatase nuclei according to TEM. | ||
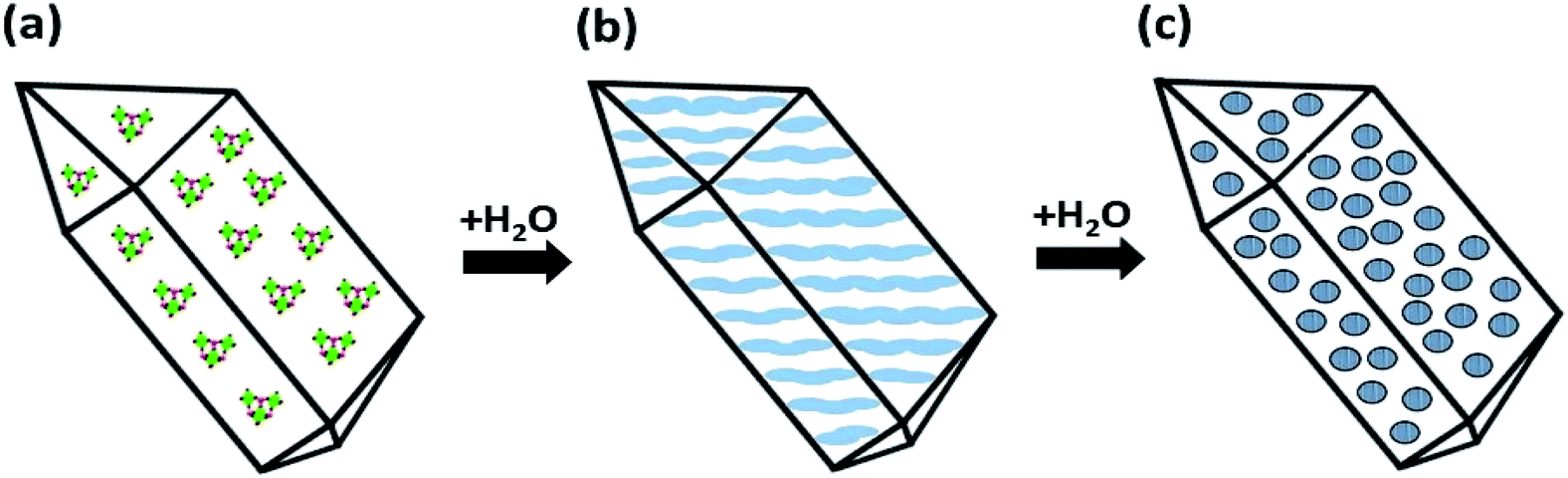 | ||
| Fig. 9 Transformation of an (oxo) alkoxide crystal to crystalline anatase particles on topotactic hydrolysis in water as proposed earlier in ref. 53. (a) Addition of water to the molecular precursor crystal (a) causes hydrolysis and contraction of the crystal structure, leading to densification with formation of the amorphous lamellar structures (b). Randomly scattered crystalline (anatase) nuclei emerge in the disordering amorphous phase upon further hydrolysis (c, see also Fig. 10b). | ||
Phosphorus NMR
The 31P NMR spectra of phosphorus containing coordination compounds are complex and the shifts depend on several factors, including bond angles, in addition to the chemical surrounding,54 making it difficult to assign shifts to particular phosphorous atoms in more complex phosphonate compounds. As this study focuses on solution stability of the complexes, only the compounds that could be obtained as pure product are presented (31P NMR spectrum of compound 4 is shown in ESI Fig. S1e†). Single crystals of compounds 1, 5, 6 and 7 were dissolved in anhydrous CDCl3 (1, 5 and 7) or anhydrous acetone-d6 (6) and 31P NMR spectra were recorded (Fig. 12). NMR spectra of 2 have been reported earlier in ref. 20. The crystallographic isomers 3 and 4 turned always to be contaminated by 5, which made identification of the signals not fully conclusive. Compound 1 showed seven high intensity, distinct phosphorous signals (33.95, 26.57, 24.92, 24.44, 19.79, 18.05 and 16.24 ppm), which were assigned to the seven different phosphorous atoms in 1. This indicates that the structure of 1 observed in the solid-state is preserved as the major species in solution under applied conditions. The four down-field signals (24.44, 19.79, 18.05 and 16.24 ppm) are considerably broadened compared to the three up-field signals. The former signals may be assigned to the four tridentate phosphorus atoms, where the broadening is caused by faster relaxation compared to the three bidentate phosphorous atoms. For compound 5 there are four distinct phosphorous signals (35.31, 27.44, 23.53 and 21.01 ppm) again showing that the solid-state molecular structure of 5 appears to be preserved as the most stable species in solution. | ||
| Fig. 10 TEM micrographs of hydrolysis products of 1 and 6. (a) Anatase particles from the hydrolysis of 1 are visible as dark spots. The insert shows a magnified particle. The lattice fringe distance is 0.2 nm. (b) A crystal fragment of 6. The dark spots are anatase nuclei of about 2 nm in diameter. The inset shows a magnified particle. The lattice fringe spacing of the particle is 0.2 nm. | ||
 | ||
| Fig. 11 A fragment of crystal 6 after immersion into water. The lamellar structure originates from hydrolysis–condensation reactions of 6 with water that results first in cracking of the original crystal into plates with subsequent formation of layers of densified titanium oxo-alkoxide material. | ||
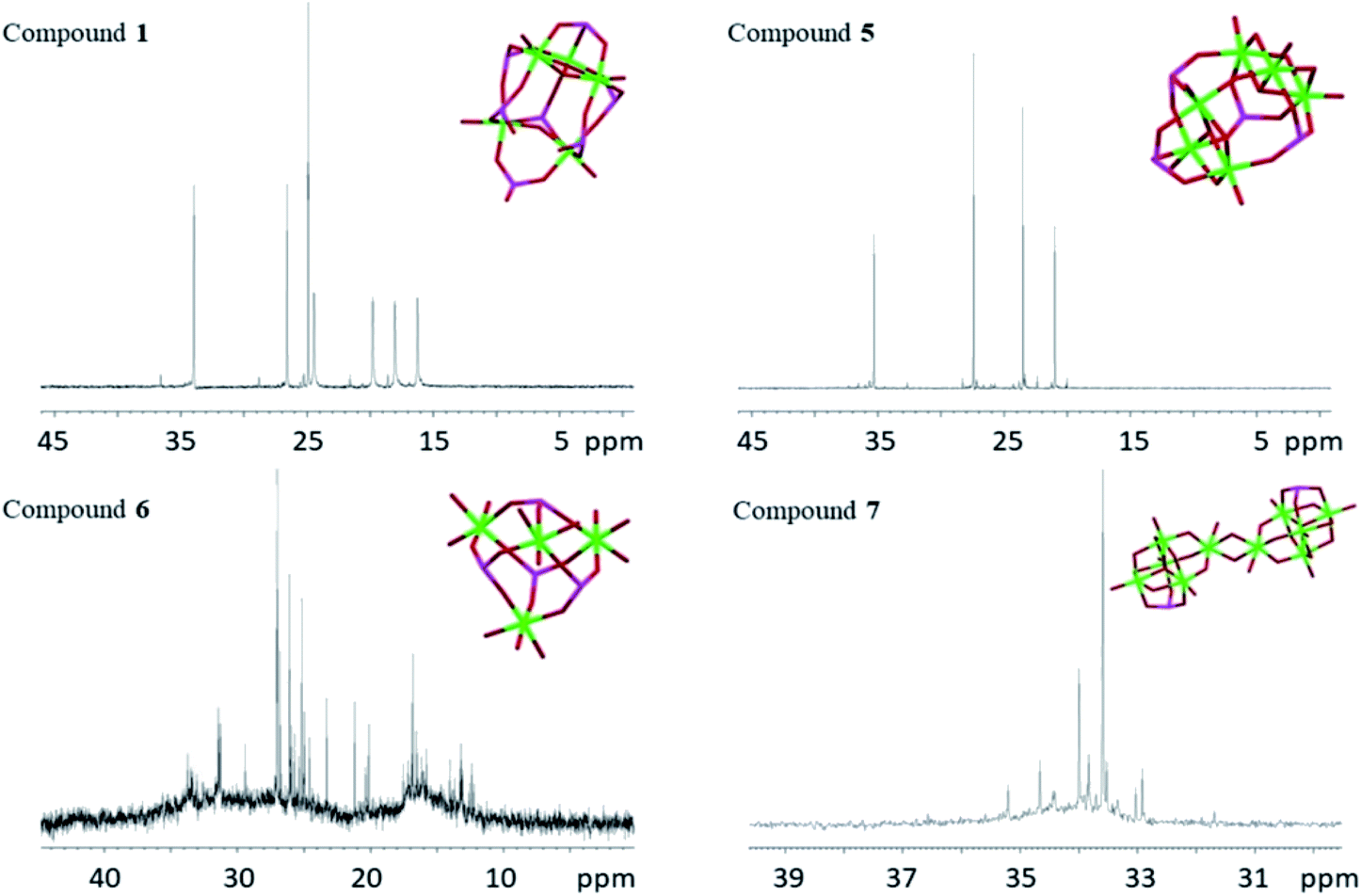 | ||
| Fig. 12 31P NMR spectra of compounds 1, 5, 6 and 7. The titanium oxo-cores with phosphorus atoms are inserted in their respective spectrum. Green is titanium, red is oxygen and magenta is phosphorus. | ||
Compound 6, however, revealed a complex set of several lower intensity peaks in its phosphorous spectrum. Obviously, the solid-state molecular structure of 6 is not representative for the solution, but probably exists in equilibrium with other species. For 7, two major signals (33.59 and 34.00 ppm), along with several weaker peaks, were recorded. The two major signals indicate presence of at least two major species in solution. Temperature variable 31P NMR spectra were recorded between 5 °C to 50 °C for compound 5 in CDCl3 (ESI Fig. S1c†). The original signals are retained at all temperatures, although intensity might decrease a little with increasing temperature. Some additional signals appear and gain increased intensities with increasing temperature, which might be indicative of degradation or dissociative equilibrium products of 5. After completing the temperature series, an aliquot of 30 μL of MilliQ-water was added to the NMR tube (the sample became turbid after shaking, see ESI Fig. S1d†) and a new spectrum was recorded. This time, no phosphorus signals could be detected, which indicated hydrolysis of the complex (ESI Fig. S1c†). Equilibria between different species of modified titanium alkoxo-complexes and possibility of existence for multiple forms in solution have also been pointed out previously.16,35,36 The term “cluster”, now used in a very broad and non-specific context, was originally coined for molecules with well-defined stable metal–metal bonds.55 As a consequence of the presence of such strong metal–metal bonds in clusters, these kind of compounds often show rather high stability, and the core-structure can be retained under quite harsh conditions.56 Metal coordination compound cores consisting of a M–O–M framework, without metal–metal bonds, are less rigid and the nuclearity and geometric structure of the core may be changed even under rather benign conditions.57,58
Conclusions
Titanium(IV) alkoxides undergo facile reaction with tert-butylphosphonic acid to produce oligonuclear heteroleptic complexes. The spheroidal molecules of complexes easily form crystals which precipitates from a toluene:ethanol solution. These structures can serve as plausible models for surface interactions between phosphonate ligands and titania. The coordination was found to always be doubly or triply bridging indicating strong interaction with titania and propose the phosphonate group to be an attractive anchoring group for titania, even at low pH. Hydrolytic and solution stabilities were investigated by a combination of techniques, i.e. EM, AFM and NMR. The complexes both in solution and in solid state underwent rapid hydrolysis in contact with water, forming crystalline anatase nuclei. This is in line with our earlier observations for carboxylate and phenoxide complexes. By controlling the hydrolysis conditions complex structured oxide materials could be obtained.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The support from the Swedish Research Council (Vetenskapsrådet) (grant 2014-3938) is gratefully acknowledged. We thank Dr Peter Agback for assistance with NMR measurements.References
- L. Rozes and C. Sanchez, Chem. Soc. Rev., 2011, 40, 1006–1030 RSC.
- M. Carraro and S. Gross, Materials, 2014, 7, 3956–3989 CrossRef CAS PubMed.
- U. Schubert, Chem. Soc. Rev., 2011, 40, 575–582 RSC.
- G. A. Seisenbaeva, G. Daniel, J. M. Nedelec and V. G. Kessler, Nanoscale, 2013, 5, 3330–3336 RSC.
- F. G. Svensson, G. A. Seisenbaeva and V. G. Kessler, Eur. J. Inorg. Chem., 2017, 4117–4122 CrossRef CAS.
- S. Ishii, J. Saito, M. Mitani, J. Mohri, N. Matsukawa, Y. Tohi, S. Matsui, N. Kashiwa and T. Fujita, J. Mol. Catal. A: Chem., 2002, 179, 11–16 CrossRef CAS.
- M. Mitani, J. Mohri, Y. Yoshida, J. Saito, S. Ishii, K. Tsuru, S. Matsui, R. Furuyama, T. Nakano, H. Tanaka, S. Kojoh, T. Matsugi, N. Kashiwa and T. Fujita, J. Am. Chem. Soc., 2002, 124, 3327–3336 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- K. Nakata and A. Fujishima, J. Photochem. Photobiol., C, 2012, 13, 169–189 CrossRef CAS.
- D. Y. C. Leung, X. L. Fu, C. F. Wang, M. Ni, M. K. H. Leung, X. X. Wang and X. Z. Fu, ChemSusChem, 2010, 3, 681–694 CrossRef CAS PubMed.
- N. Steunou, F. Robert, K. Boubekeur, F. Ribot and C. Sanchez, Inorg. Chim. Acta, 1998, 279, 144–151 CrossRef CAS.
- S. Doeuff, Y. Dromzee, F. Taulelle and C. Sanchez, Inorg. Chem., 1989, 28, 4439–4445 CrossRef CAS.
- S. Verma, S. Rani, S. Kumar and M. A. M. Khan, Ceram. Int., 2018, 44, 1653–1661 CrossRef CAS.
- K. Siwinska-Stefanska, J. Zdarta, D. Paukszta and T. Jesionowski, J. Sol-Gel Sci. Technol., 2015, 75, 264–278 CrossRef CAS.
- K. Gigant, A. Rammal and M. Henry, J. Am. Chem. Soc., 2001, 123, 11632–11637 CrossRef CAS PubMed.
- R. J. Errington, J. Ridland, K. J. Willett, W. Clegg, R. A. Coxall and S. L. Heath, J. Organomet. Chem., 1998, 550, 473–476 CrossRef CAS.
- Y. Chen, E. Trzop, J. D. Sokolow and P. Coppens, Chem.–Eur. J., 2013, 19, 16651–16655 CrossRef CAS PubMed.
- M. Nilsing, S. Lunell, P. Persson and L. Ojamae, Surf. Sci., 2005, 582, 49–60 CrossRef CAS.
- M. Nilsing, P. Persson and L. Ojamae, Chem. Phys. Lett., 2005, 415, 375–380 CrossRef CAS.
- G. A. Seisenbaeva, I. V. Melnyk, N. Hedin, Y. Chen, P. Eriksson, E. Trzop, Y. L. Zub and V. G. Kessler, RSC Adv., 2015, 5, 24575–24585 RSC.
- P. H. Mutin, G. Guerrero and A. Vioux, J. Mater. Chem., 2005, 15, 3761–3768 RSC.
- Y. P. Zhu, T. Y. Ma, T. Z. Ren, J. Li, G. H. Du and Z. Y. Yuan, Appl. Catal., B, 2014, 156, 44–52 CrossRef.
- T. Y. Ma, X. Z. Lin, X. J. Zhang and Z. Y. Yuan, New J. Chem., 2010, 34, 1209–1216 RSC.
- Q. C. Le, M. H. Ropers, H. Terrisse and B. Humbert, Colloids Surf., B, 2014, 123, 150–157 CrossRef CAS PubMed.
- E. A. Abou Neel, W. Chrzanowski and J. C. Knowles, Mater. Sci. Eng., C, 2014, 35, 307–313 CrossRef CAS PubMed.
- J. S. Shah, P. K. C. Venkatsurya, W. W. Thein-Han, R. D. K. Misra, T. C. Pesacreta, M. C. Somani and L. P. Karjalainen, Mater. Sci. Eng., C, 2011, 31, 458–471 CrossRef CAS.
- M. Mehring, G. Guerrero, F. Dahan, P. H. Mutin and A. Vioux, Inorg. Chem., 2000, 39, 3325–3332 CrossRef CAS PubMed.
- V. G. Kessler, G. I. Spijksma, G. A. Seisenbaeva, S. Hakansson, D. H. A. Blank and H. J. M. Bouwmeester, J. Sol-Gel Sci. Technol., 2006, 40, 163–179 CrossRef CAS.
- J. X. Liu, M. Y. Gao, W. H. Fang, L. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2016, 55, 5160–5165 CrossRef CAS PubMed.
- W. H. Fang, L. Zhang and J. Zhang, Dalton Trans., 2017, 46, 803–807 RSC.
- K. Z. Su, M. Y. Wu, Y. X. Tan, W. J. Wang, D. Q. Yuan and M. C. Hong, Chem. Commun., 2017, 53, 9598–9601 RSC.
- H. T. Lv, H. M. Li, G. D. Zou, Y. Cui, Y. Huang and Y. Fan, Dalton Trans., 2018, 47, 8158–8163 RSC.
- M. E. Carlotti, E. Ugazio, S. Sapino, I. Fenoglio, G. Greco and B. Fubini, Free Radical Res., 2009, 43, 312–322 CrossRef CAS PubMed.
- G. A. Lawrence, Introduction to Coordination Chemistry, John Wiley & Sons, 2010 Search PubMed.
- C. G. Lugmair and T. D. Tilley, Inorg. Chem., 1998, 37, 1821–1826 CrossRef CAS.
- D. Chakraborty, V. Chandrasekhar, M. Bhattacharjee, R. Kratzner, H. W. Roesky, M. Noltemeyer and H. G. Schmidt, Inorg. Chem., 2000, 39, 23–26 CrossRef CAS PubMed.
- R. E. Ruther, B. M. Baker, J. H. Son, W. H. Casey and M. Nyman, Inorg. Chem., 2014, 53, 4234–4242 CrossRef CAS PubMed.
- N. A. Vanagas, J. N. Wacker, C. L. Rom, E. N. Glass, I. Colliard, Y. S. Qiao, J. A. Bertke, E. Van Keuren, E. J. Schelter, M. Nyman and K. E. Knope, Inorg. Chem., 2018, 57, 7259–7269 CrossRef CAS PubMed.
- M. Czakler, C. Artner and U. Schubert, Monatsh. Chem., 2015, 146, 1249–1256 CrossRef CAS PubMed.
- K. Sharma, R. Antony, A. C. Kalita, S. K. Gupta, P. Davis and R. Murugavel, Inorg. Chem., 2017, 56, 12848–12858 CrossRef CAS PubMed.
- M. Czakler, C. Artner and U. Schubert, Eur. J. Inorg. Chem., 2013, 2013, 5790–5796 CrossRef CAS.
- F. G. Svensson and V. G. Kessler, Polyhedron, 2020, 178, 114276 CrossRef.
- G. A. Seisenbaeva, M. P. Moloney, R. Tekoriute, A. Hardy-Dessources, J. M. Nedelec, Y. K. Gun'ko and V. G. Kessler, Langmuir, 2010, 26, 9809–9817 CrossRef CAS PubMed.
- R. Hayami, T. Sagawa, S. Tsukada, K. Yamamoto and T. Gunji, Polyhedron, 2018, 147, 1–8 CrossRef CAS.
- The Sol-Gel Handbook, ed. D. Levy and M. Zayat, Wiley-VHC, 2015 Search PubMed.
- W. Luo, J. L. Hou, D. H. Zou, L. N. Cui, Q. Y. Zhu and J. Dai, New J. Chem., 2018, 42, 11629–11634 RSC.
- N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS PubMed.
- L. Bai, S. Q. Huan, W. C. Xiang and O. J. Rojas, Green Chem., 2018, 20, 1571–1582 RSC.
- S. Varanasi, L. Henzel, L. Mendoza, R. Prathapan, W. Batchelor, R. Tabor and G. Garnier, Front. Chem., 2018, 6, 409 CrossRef PubMed.
- A. Collins, D. Carriazo, S. A. Davis and S. Mann, Chem. Commun., 2004, 568–569, 10.1039/b315018f.
- K. Cheng, K. Chhor and A. Kanaev, Chem. Phys. Lett., 2017, 672, 119–123 CrossRef.
- G. Fornasieri, L. Rozes, S. Le Calve, B. Alonso, D. Massiot, M. N. Rager, M. Evain, K. Boubekeur and C. Sanchez, J. Am. Chem. Soc., 2005, 127, 4869–4878 CrossRef CAS PubMed.
- G. A. Seisenbaeva, G. Daniel, J. M. Nedelec, Y. K. Gun'ko and V. G. Kessler, J. Mater. Chem., 2012, 22, 20374–20380 RSC.
- W. Gao, L. Dickinson, C. Grozinger, F. G. Morin and L. Reven, Langmuir, 1996, 12, 6429–6435 CrossRef CAS.
- F. A. Cotton, Inorg. Chem., 1964, 3, 1217–1220 CrossRef CAS.
- M. N. Sokolov, P. A. Abramov, A. L. Gushchin, I. V. Kallnina, D. Y. Naumov, A. V. Virovets, E. V. Peresypkina, C. Vicent, R. Llusar and V. P. Fedin, Inorg. Chem., 2005, 44, 8116–8124 CrossRef CAS PubMed.
- T. A. Bazhenova, N. V. Kovaleva, G. V. Shilov, G. N. Petrova and D. A. Kuznetsov, Eur. J. Inorg. Chem., 2016, 5215–5221, DOI:10.1002/ejic.201600804.
- A. Radtke, P. Piszczek, T. Muziol and A. Wojtczak, Inorg. Chem., 2014, 53, 10803–10810 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full details of X-ray structure solution and refinement. CCDC 1872957–1872963 (for compounds 1–7 respectively). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra10691j |
| This journal is © The Royal Society of Chemistry 2020 |