DOI:
10.1039/D0RA00563K
(Paper)
RSC Adv., 2020,
10, 8949-8957
Synthesis of lutein esters using a novel biocatalyst of Candida antarctica lipase B covalently immobilized on functionalized graphitic carbon nitride nanosheets†
Received
18th January 2020
, Accepted 22nd February 2020
First published on 2nd March 2020
Abstract
Lutein scavenges free radicals and inhibits vision damage caused by photo oxidation, while decomposing easily with light and heat. Its stability and bioavailability can be tremendously improved by lutein ester synthesis. However, green and efficient esterification preparation methods are urgently needed. In this study, which used functionalized graphitic carbon nitride nanosheets (g-C3N4-Ns) as the immobilized carrier, a novel biocatalyst was designed and prepared to accommodate Candida antarctica lipase B (CALB), considerably enhancing the performance. It was characterized by TEM, XRD, FTIR, XPS, TGA, and BET to demonstrate successful preparation and then applied to catalyze esterification between lutein and succinate anhydride in dimethyl formamide (DMF) solvent resulting in a conversion rate up to 92% at 50 °C in 60 h, 34% more than free CALB under the same conditions. We believe this is the highest esterification rate in lutein esters synthesis and it has great potential to facilitate eco-friendly and efficient preparation.
1. Introduction
Exhibiting the activity of provitamin A and the advantages of anti-oxidation, lutein, together with β-carotene, zeaxanthin, lycopene, β-cryptoxanthin, and α-carotene are the six carotenoids present in human serum,1,2 which improve intercellular gap junction communication and boost the immune system.3 Especially, lutein has been recognized as a key supplement in the human diet4 for its capacity to scavenge reactive oxygen (such as the free ROO˙, HO˙, HOCl) and nitrogen species, and delay age-related macular degeneration.5 Unfortunately, mammals, including humans, cannot synthesize lutein themselves and must rely on diet for ingestion.3 Furthermore, due to its high degree of unsaturation, lutein is easily decomposed under light and heat conditions.6 To improve its stability and bioavailability,7 esterifying the hydroxyl groups at both ends of lutein by different anhydrides to form lutein diester is considered to be an ingenious strategy, in addition, the original lutein can be endowed with different biological activities and physiological functions.2,3,8
Traditionally, the reported approaches for lutein ester preparation are chemical catalysis,8 solvent extraction,9 and supercritical carbon dioxide extraction10 from marigolds, microalgae,11 or other plants. However, these methods have several non-negligible shortcomings such as complicated processes, solvent pollution, and environmental contamination. Compared to the methods mentioned above, as one new potential method, esterification by lipase shows many advantages such as high efficiency, selectivity, specificity, mild catalytic media, and environmental friendliness.12 Unluckily, like protein, lipase is high cost, sensitive to the reaction condition, and easily inactivated.13 Immobilizing lipase on various carriers in different ways, can promise the function of improving the thermal stability and reusability, leading to lower cost of lipase application.14,15 However, very few papers have described the enzymatic synthesis of lutein esters. Wang16 and Hou17 tried to catalyze the synthesis of lutein palmitate by Novozyme 435 and lipase-Pluronic F-127. Nevertheless, in their studies, toluene, methyl tert-butyl ether (MTBE), or methyl isobutyl ketone (MIBK) were used as the solvent for lutein esterification, which are not suitable for food applications. Therefore, the development of new immobilized lipases for efficient and green esterification of lutein esters is urgently needed.
As an essential participator in the immobilization process, supporting material deeply affects lipase activity. Two-dimensional nanomaterials such as graphene oxide18 and carbon nanosheets19,20 have gained extensive attention21 due to their large surface area, abundant active sites, and good biocompatibility. As a superior material, 2D carbon nitride nanosheets exhibit excellent performance in many fields. Among the seven phases of carbon nitride, graphitic carbon nitride is the most stable formation and possesses outstanding advantages.22 Recently, our group first synthesized g-C3N4-Ns based immobilized Candida rugosa lipase (CRL) and considered it as a highly promising material;23 however, since the surface area of g-C3N4-Ns (74.374 m2 g−1) and the protein amount of the immobilized lipase (44.76 mg g−1) were not particularly satisfactory, its further application could not be realized.
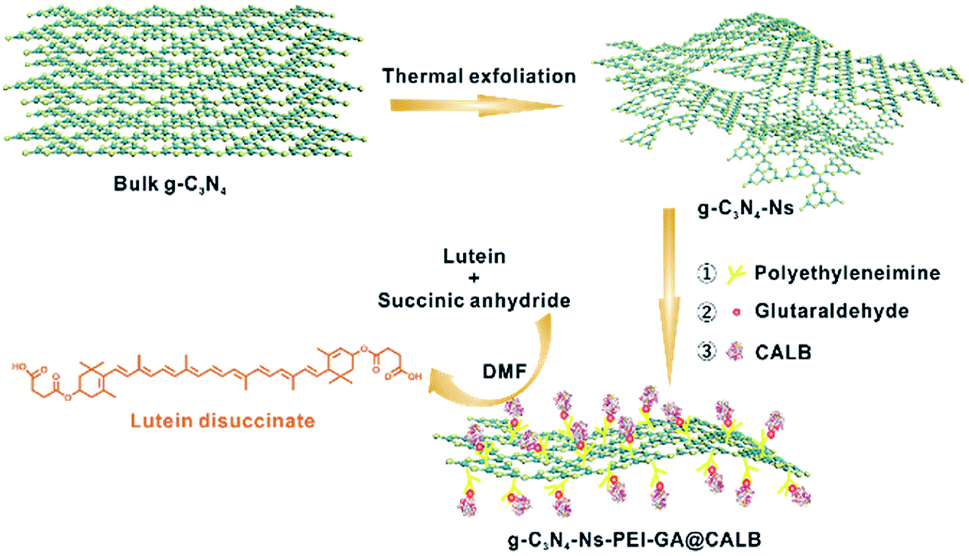
In this work, we prepared a new bioreactor that utilized polyethyleneimine (PEI)-modified g-C3N4-Ns as the carrier and anchored CALB on g-C3N4-Ns with the assistance of glutaraldehyde (GA). The whole procedure is shown in Scheme 1 and the design concept can be explained as follows: (1) an easy, low-cost thermal polymerization method was chosen to prepare bulk graphitic carbon nitride (bulk g-C3N4) with a stable formation and good biocompatibility. The van der Waals forces between the two-dimensional planes of the bulk g-C3N4 were destroyed by thermal action stripping it into single layers of graphitic carbon nitride nanosheets and obtaining a large surface area that aided the attachment of CALB; (2) to aid water dispersibility of g-C3N4-Ns, branched PEI was selected as the amino modification to adorn the g-C3N4-Ns surface for the rich –NH2 group. In addition, positively charged branched PEI could easily be anchored on the negatively charged graphite carbon nitride nanosheets via electrostatic adherence, and sufficient amino groups on the surface of g-C3N4-Ns-PEI provided numerous binding sites for CALB; (3) as a bridge, GA made it possible for the oxygen-containing groups (such as carboxyl, hydroxyl, and carbonyl) on CALB to covalently connect to the amino groups on the graphite carbon nitride nanosheets, effectively preventing enzyme leakage during immobilization and esterification; (4) with good solubility for lutein and succinic anhydride, non-polluting DMF was selected as the reaction solvent, which induced catalytic efficiency improvement during esterification. Simultaneously, g-C3N4-Ns-PEI@CALB was well dispersed in DMF, which is beneficial to mass transformation. According to the results, the maximum conversion rate of lutein disuccinate was up to 92% within 60 h. To the best of our knowledge, this is the first study about immobilizing CALB on g-C3N4-Ns lutein ester synthesis, and we obtained the highest esterification rate in the literature.
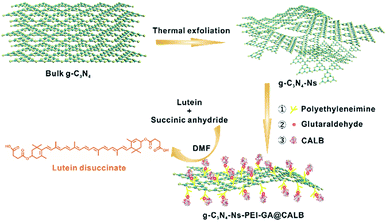 |
| | Scheme 1 Schematic representation of the formation of g-C3N4-Ns-PEI-GA@CALB. | |
2. Experimental
2.1. Materials
Candida antarctica lipase B (CALB, liquid, enzyme activity 5000 U mL−1) was supplied by Beijing Gaoruisen Biological Technology Co., Ltd (Beijing, China). Lutein (75%) and succinic anhydride were purchased from Aladdin (Shanghai, China). Dicyandiamide (DCDA, 99%) and p-nitrophenylpalmitate (p-NPP, Aldrich, 99%) were obtained from Sigma-Aldrich (St. Louis, USA). Chromatographically pure methyl alcohol, acetonitrile, and acetic acid were purchased from Merck Biotechnology Co., Ltd (Shanghai, China). The BCA Protein Assay Kit was obtained from Shanghai Yuanye Biotechnology Co., Ltd (Shanghai, China). GA solution (25%), dimethyl sulfoxide (DMSO), chloroform, methyl tert-butyl ether, acetone, and DMF were supplied by Sinopharm Chemical Reagent Co., Ltd (Shanghai, China).
2.2. Preparation of g-C3N4-Ns
The preparation of g-C3N4-Ns is based on the existing literature with a slight adjustment to the procedure.24 First, 3 g of dicyandiamide was weighed and placed in a 30 mL alumina crucible, capped, and left a seam. Second, the alumina crucible was heated to 550 °C at 2.3 °C min−1 and held at 550 °C for 4 h in a tube furnace. This process was performed in air atmosphere and cooled to room temperature (25 °C) naturally. The hard yellow product was bulk g-C3N4, which was ground into powder by using an agate mortar. Third, 1.5 g bulk g-C3N4 powder was evenly spread on a 50 mL porcelain boat and heated to 500 °C at 5 °C min−1 in a tube furnace, and kept for 2 h in air followed by cooling naturally. Finally, the light yellow powders were collected and preserved in a dry environment.
2.3. Preparation of g-C3N4-Ns-PEI
The method for the preparation of functionalized g-C3N4-Ns by PEI was based on the literature and had been appropriately modified.25 One gram of obtained g-C3N4-Ns was dispersed in 20 mL phosphate buffer (PBS) (0.1 M, pH 7), stirred, and sonicated for 10 min, and 10 g of PEI was dissolved in 10 mL PBS buffer (0.1 M, pH 7) by stirring. Successively, the dissolved PEI was added drop-wise to the prepared PBS buffer, containing g-C3N4-Ns. After stirring for 48 h at room temperature, the mixture was centrifuged and washed by deionized water several times. Finally, the sediment was dried in a 60 °C oven and then preserved for use.
2.4. Covalently immobilized CALB on g-C3N4-Ns-PEI (g-C3N4-Ns-PEI-GA@CALB)
The immobilization of CALB on g-C3N4-Ns-PEI was described in the previous report.23 There were two steps to immobilize CALB on g-C3N4-Ns-PEI. First, 200 mg of g-C3N4-Ns-PEI was well dispersed in 6.34 mL PBS buffer (0.1 M, pH 7 or other pH value according to the experimental request) and then 2 mL 25% GA was added. The mixture was incubated in a shaker with a rotating speed of 160 rpm and temperature of 55 °C. After agitating for 12 h, the sample was separated by vacuum filtration to remove the PBS and redundant GA and washed by deionized water several times. Next, the solid was dried in the oven at 60 °C. Subsequently, 18 mL PBS and 6 mL CALB liquid were first mixed by a shaker for 5 min, then 200 mg of the prepared g-C3N4-Ns-PEI-GA was added. The reaction liquid was gently vibrated at 160 rpm and in 37 °C for 8 h. Next, the material was centrifuged and washed by deionized water three times, followed by freeze-drying in a vacuum freeze drier. Finally, the immobilized lipase g-C3N4-Ns-PEI-GA@CALB was prepared for use. The protein loading content of g-C3N4-Ns-PEI-GA@CALB was determined by BCA Protein Assay Kit, taking bovine serum albumin as the standard. Additionally, the protein loading content of g-C3N4-Ns-PEI-GA@CALB was calculated using the subtraction method of the initial protein content and the supernatant protein content after immobilization.
2.5. Characterization of prepared materials
To observe the morphologies of the series materials at different steps of the experiment, different methods were used for characterization, including UV-vis absorption spectroscopy (DU 800 UV-vis spectrometer), scanning electron microscopy (SEM, ZEISS MERLIN Compact), transmission electron microscopy (TEM, Tecnai G2 F30, FEI), Fourier-transform infrared spectroscopy (FT-IR, Bruker Tensor 27 FT-IR spectrometer Thermoelectron), X-ray diffraction (XRD, Bruker D8 Advance), X-ray photoelectron spectroscopy (XPS, ESCALAB 250Xi), thermogravimetric analysis (TGA, TAQ50), and Brunauer–Emmett–Teller (BET) analysis (ASAP2020).
2.6. Enzymatic activity assay and thermal stability of g-C3N4-Ns-PEI-GA@CALB
The activities of CALB and g-C3N4-Ns-PEI-GA@CALB were evaluated by hydrolyzing p-NPP in PBS at 37 °C for 5 min, which was conducted in a table concentrator at 160 rpm. The concentration of the hydrolysis product p-NP (p-nitrophenol) was measured by UV spectrophotometer at 410 nm. The relative activities were defined by presuming the maximum absorbance value of the free or immobilized lipase catalysis under optimal conditions as 100%. Thirty milligrams g-C3N4-Ns-PEI-GA@CALB (or 30 μL CALB) were mixed with 1 mL PBS (0.1 M, pH 7.0), and then 1 mL 0.5% p-nitrophenylpalmitate ethanol solution (w/v) was added. The mixture was incubated at 37 °C for 5 min at 160 rpm in a shaker and was terminated by adding 2 mL of Na2CO3 (25 g L−1) solution. Subsequently, 3 mL of the supernatant was obtained by centrifuging the reaction solution. Then the supernate was suitably diluted and detected on an ultraviolet-visible spectrophotometer at 410 nm. Thermal stabilities of CALB and g-C3N4-Ns-PEI-GA@CALB were studied by measuring the residual activities of the lipases after incubation at different temperatures for 1 h.
2.7. Synthesis of different lutein esters
In the typical experiment, 140 mg lutein was first dissolved in 1 mL DMF by sonication in a hermetic round-bottom flask and 500 mg succinic anhydride was added to the solution. After shaking for several minutes, 100 mg immobilized lipase g-C3N4-Ns-PEI-GA@CALB was added. To protect the lutein from oxidization, nitrogen was injected into the area above the liquid to expel the air. Next, the flask was shaken at 45 °C and 160 rpm in a horizontal shaker for a certain time.
2.8. Analysis of the conversion rate of lutein disuccinate
The esterification rates of lutein monosuccinate and lutein disuccinate were detected by High Performance Liquid Chromatography (HPLC) analysis. One hundred microliters of terminal solution of esterification reaction were taken out and mixed with 5 mL chromatographically pure methyl alcohol. The samples could be analyzed by HPLC after filtration through a 0.22 μm membrane. The HPLC analysis was conducted using a YMC-Carotenoid S-5 μm (250 × 4.6 mm I.D.). Mobile phases A and B were methyl alcohol (0.05% acetic acid) and acetonitrile (0.05% acetic acid), respectively. The conditions of HPLC analysis were as follows: the elution condition was binary high-pressure elution, flow velocity was 1 mL min−1, column temperature was 40 °C, injection volume was 20 μL, and the absorbance was detected at 450 nm. Finally, the esterification rates of lutein disuccinate and lutein monosuccinate were calculated by the area normalization method.
2.9. Reusability of g-C3N4-Ns-PEI-GA@CALB
The reusability of g-C3N4-Ns-PEI-GA@CALB was studied by adding 10 mg of g-C3N4-Ns-PEI-GA@CALB to 1 mL DMF solution (containing 140 mg lutein and 500 mg succinic anhydride) and reacting the mixture for 24 h at 50 °C in the protection of nitrogen. Subsequently, g-C3N4-Ns-PEI-GA@CALB was regained by centrifuging the previous reaction solution and discarding the supernatant, and was used for the next cycle. The relative esterification rates were defined by presuming the first cycle as 100%.
3. Results and discussion
3.1. Synthesis of the g-C3N4-Ns-PEI-GA@CALB
The synthesis procedure is illustrated in Scheme 1. First, the carrier material g-C3N4-Ns was prepared by a simple “top-down” method, thermal oxidation etching of bulk g-C3N4 in the air, obtaining 2D-nanosheets with a high surface area of 118.858 m2 g−1. Then, the surface of g-C3N4 was modified by branched PEI via electrostatic interaction to create abundant functional groups (–NH2). Next, a majority of CALB was immobilized by the covalent connection via GA, while some was anchored by PEI and g-C3N4 via electrostatic interactions or physical absorptions, respectively. Finally, to esterify lutein, the two substrates and g-C3N4-Ns-PEI-GA@CALB were well dispersed in the DMF solvent. The large surface of g-C3N4-Ns could provide sufficient space for substrate access to CALB and ensured the catalytic performance.
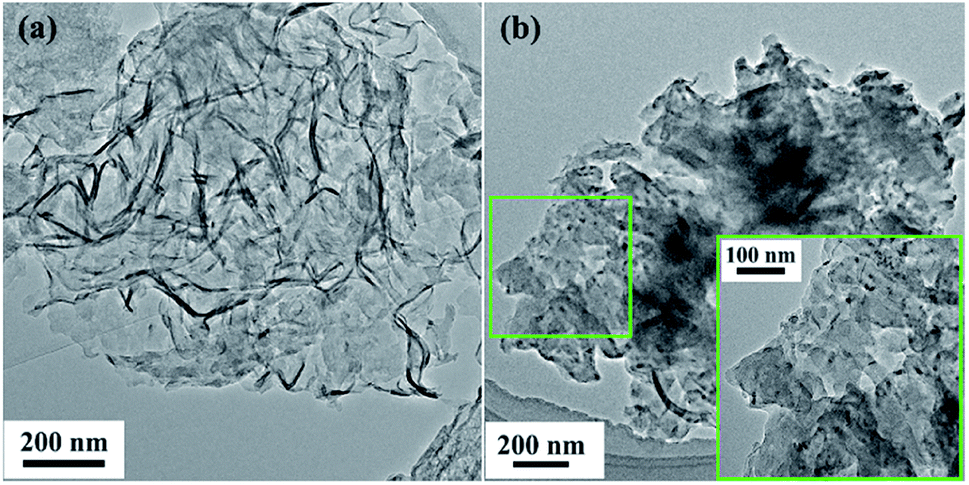
3.1.1. Characterization of g-C3N4-Ns-PEI-GA@CALB. The morphology of prepared materials was investigated by transmission electron microscopy (TEM) and the TEM images of g-C3N4-Ns and g-C3N4-Ns-PEI-GA@CALB are exhibited in Fig. 1a and b, respectively. After thermal stripping of the bulk g-C3N4, which was formed by thermal polymerization of dicyandiamide, we retained the 2D g-C3N4-Ns, which displayed soft silk-like nano-sized lamellar structures with folds spread on the flat surface. However, after immobilizing the CALB on g-C3N4-Ns, many small dark dots appeared on the surface of the silk in the high-resolution picture of Fig. 1b, revealing that CALB was successfully connected with g-C3N4-Ns. This conclusion was consistent with the preparation of a new biocatalyst C3N4-Ns@CRL in our previous report.23
 |
| | Fig. 1 TEM images of g-C3N4-Ns (a) and g-C3N4-Ns-PEI-GA@CALB (b). | |
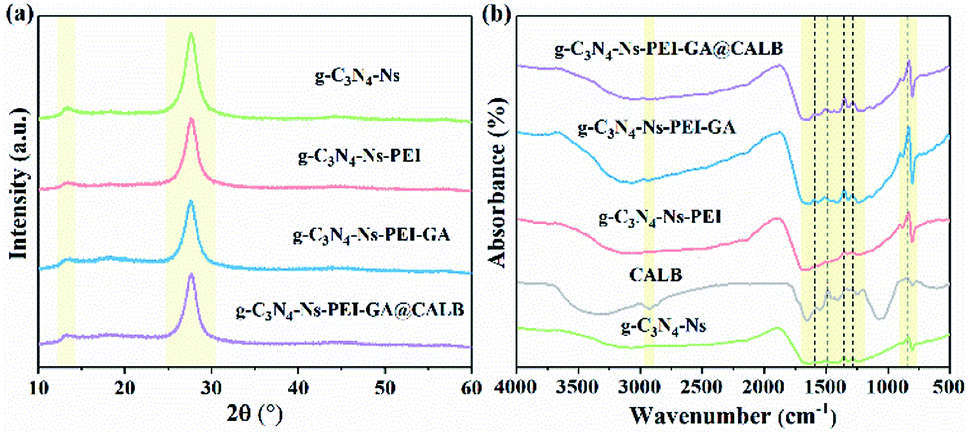
To investigate the lattice structures of the materials prepared in the process of immobilization, XRD analysis was involved. As shown in Fig. 2a, at 13° and 27°, the similar characteristic diffraction angles can be observed in the four samples. The weak low-angle reflection (peak 13°) stem results from the repeat units tri-s-triazine in the parallel planes lattice, and the distinct peak at 27° can be attributed to the interlayer diffraction of the layered structures of g-C3N4-Ns. A slight decrease is observed in peak intensity at 13° and 27° after PEI adsorption, which should be attributed to the lower crystallinity of g-C3N4-Ns caused by PEI. Moreover, there is no marked difference in peak strength of the four sample curves, which indicates that g-C3N4-Ns invariably maintains the original crystal structure during PEI modification, GA connection, and CALB immobilization.
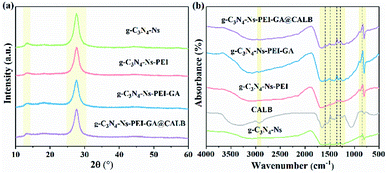 |
| | Fig. 2 XDR pattern (a) and FT-IR spectra (b) of g-C3N4-Ns, g-C3N4-Ns-PEI, g-C3N4-Ns-PEI-GA and g-C3N4-Ns-PEI-GA@CALB. | |
In addition, we conducted FT-IR spectroscopy to further verify the functional groups and chemical bonds on g-C3N4-Ns related products, and the result is shown in Fig. 2b. First, the peaks between 900 cm−1 and 1800 cm−1 are likely attributed to the stretching and vibration of the C–N skeleton. The details suggest that the peaks at 1500–1300 cm−1 are caused by shear vibration and in-plane flexural vibration of N–H in the aromatic structure. However, the peaks in the range of 1300–800 cm−1 can be ascribed to the stretching and vibration of trigonal C–N(–C)–C and bridging C–NH–C units. Second, the two peaks around 820 cm−1 in the graph originated from the out-of-plane bending vibration of the H atom on the heptazine ring. This evidence further proves that PEI, GA, and CALB attach to g-C3N4-Ns successfully. Moreover, comparing the curves of free CALB and g-C3N4-Ns-PEI-GA@CALB, the N–H peak of free CALB at 3000 cm−1, weakened after immobilization, which can be explained by the successful formation of amide bonds between CALB and GA.
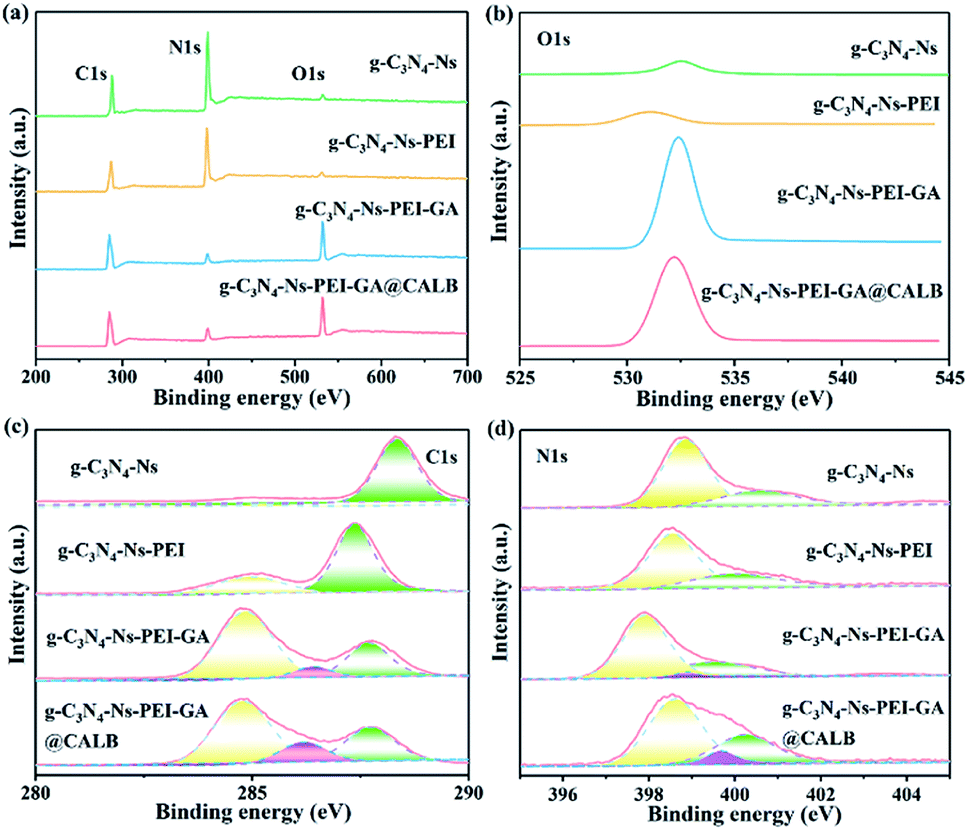
The composition and content of elements, as well as chemical bonds of the materials were studied by XPS. As shown in Fig. 3a, the elements of C1s (287.0 eV), N1s (399.0 eV), and O1s (532.0 eV) are all observed. However, distinct differences can be noticed for each element in the characteristics and content. In Fig. 3b, the O1s peaks of g-C3N4-Ns and g-C3N4-Ns-PEI at 532.51 eV and 531.04 eV, respectively, are very small and negligible; however, after the addition of GA, the intensity rapidly increases resulting from the oxygen on GA, which shows that GA successfully links with g-C3N4-Ns-PEI. Furthermore, the O1s peak of g-C3N4-Ns-PEI-GA@CALB increases slightly compared with the non-enzyme version, and these inconspicuous changes may be caused by small quantities of oxygen-containing functional groups in CALB. In Fig. 3c, after PEI addition, the C1s peak of g-C3N4-Ns-PEI separates into two peaks at 284.99 eV and 287.35 eV, respectively. The new peak at 284.99 eV is ascribed to the C–C bonds introduced by PEI. Moreover, compared with the g-C3N4-Ns, the peak (288.34 eV, sp2 (N)2–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) is shifted to weak binding-energy regions (287.35 eV) due to the electrostatic interaction between protonated PEI and negative g-C3N4-Ns. Subsequently, after the introduction of GA, a new peak located at 286.41 eV appeared, which is assigned to sp2 CO in GA and CO–NH formed by the aldimine condensation between PEI amino and GA aldehyde groups. Similarly, the deconvoluted peaks of g-C3N4-Ns-PEI-GA@CALB are displayed at 284.76 eV, 286.19 eV, and 287.71 eV, corresponding to C–C, CO, or CO–NH and (N)2–CN, respectively. These results disclose that PEI, GA, and CALB are all anchored on the g-C3N4-Ns surface solidly. The N1s peak of g-C3N4-Ns in Fig. 3d can be separated into two peaks at 398.85 eV and 400.57 eV, which represent the sp2 hybridization of C–NC or N–(C)3 bonds in tri-s-triazine rings and the very small amino existing in (C)2–NH bonds. After the g-C3N4-Ns modified by PEI, the peak of (C)2–NH shifted to weaker binding energy by 0.59 eV, indicating that the electrostatic interaction between PEI and g-C3N4-Ns improves the electron distribution density outside the nitrogen nucleus and the atomic nucleus weakens the electron binding force and results in the decreased energy intensity. In addition, compared with g-C3N4-Ns-PEI, g-C3N4-Ns-PEI-GA and g-C3N4-Ns-PEI-GA@CALB showed new peaks at 398.88 eV and 399.67 eV, respectively, indicating the formation of NH–CO bonds, by the reaction between GA and PEI as well as CALB, respectively. Meanwhile, due to the formation of new amide bonds, the intensity of C–NC and N–(C)3 in g-C3N4-Ns-PEI-GA shifted to the weak binding-energy area by 0.93 eV compared with that of g-C3N4-Ns-PEI.
N) is shifted to weak binding-energy regions (287.35 eV) due to the electrostatic interaction between protonated PEI and negative g-C3N4-Ns. Subsequently, after the introduction of GA, a new peak located at 286.41 eV appeared, which is assigned to sp2 CO in GA and CO–NH formed by the aldimine condensation between PEI amino and GA aldehyde groups. Similarly, the deconvoluted peaks of g-C3N4-Ns-PEI-GA@CALB are displayed at 284.76 eV, 286.19 eV, and 287.71 eV, corresponding to C–C, CO, or CO–NH and (N)2–CN, respectively. These results disclose that PEI, GA, and CALB are all anchored on the g-C3N4-Ns surface solidly. The N1s peak of g-C3N4-Ns in Fig. 3d can be separated into two peaks at 398.85 eV and 400.57 eV, which represent the sp2 hybridization of C–NC or N–(C)3 bonds in tri-s-triazine rings and the very small amino existing in (C)2–NH bonds. After the g-C3N4-Ns modified by PEI, the peak of (C)2–NH shifted to weaker binding energy by 0.59 eV, indicating that the electrostatic interaction between PEI and g-C3N4-Ns improves the electron distribution density outside the nitrogen nucleus and the atomic nucleus weakens the electron binding force and results in the decreased energy intensity. In addition, compared with g-C3N4-Ns-PEI, g-C3N4-Ns-PEI-GA and g-C3N4-Ns-PEI-GA@CALB showed new peaks at 398.88 eV and 399.67 eV, respectively, indicating the formation of NH–CO bonds, by the reaction between GA and PEI as well as CALB, respectively. Meanwhile, due to the formation of new amide bonds, the intensity of C–NC and N–(C)3 in g-C3N4-Ns-PEI-GA shifted to the weak binding-energy area by 0.93 eV compared with that of g-C3N4-Ns-PEI.
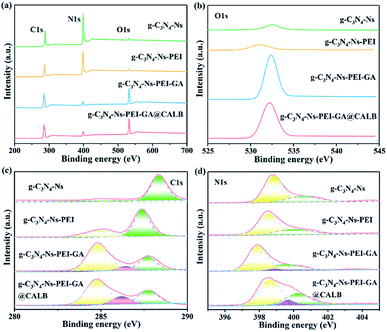 |
| | Fig. 3 XPS spectra (a), high-resolution O1s (b), high-resolution C1s (c) and high-resolution N1s (d) of g-C3N4-Ns, g-C3N4-Ns-PEI, g-C3N4-Ns-PEI-GA and g-C3N4-Ns-PEI-GA@CALB. | |
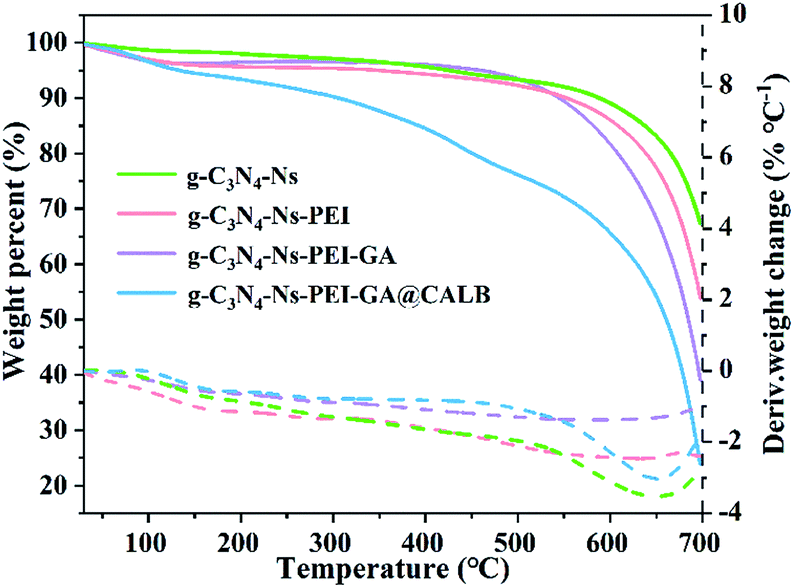
With the help of TGA, we observed the weight loss of different samples during temperature increase in nitrogen atmosphere. In Fig. 4, when the temperature reaches 650 °C, the curve of g-C3N4-Ns decreases, indicating g-C3N4-Ns begins to degrade at this point, yet the critical temperatures of g-C3N4-Ns-PEI and g-C3N4-Ns-PEI-GA decomposition are 600 °C and 500 °C, respectively, demonstrating that the PEI and GA adhere to g-C3N4-Ns successfully. The curve of g-C3N4-Ns-PEI-GA@CALB continually decreases between 100 °C and 700 °C and has three evident loss weight steps at 100 °C, 450 °C, and 650 °C, which is due to the evaporation of water, dissociation of CALB, and pyrolysis of g-C3N4-Ns. The results also proved that CALB is successfully connected to g-C3N4-Ns-PEI-GA.
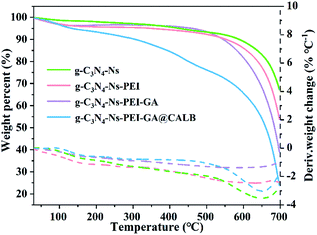 |
| | Fig. 4 TGA analysis of g-C3N4-Ns, g-C3N4-Ns-PEI, g-C3N4-Ns-PEI-GA and g-C3N4-Ns-PEI-GA@CALB. | |
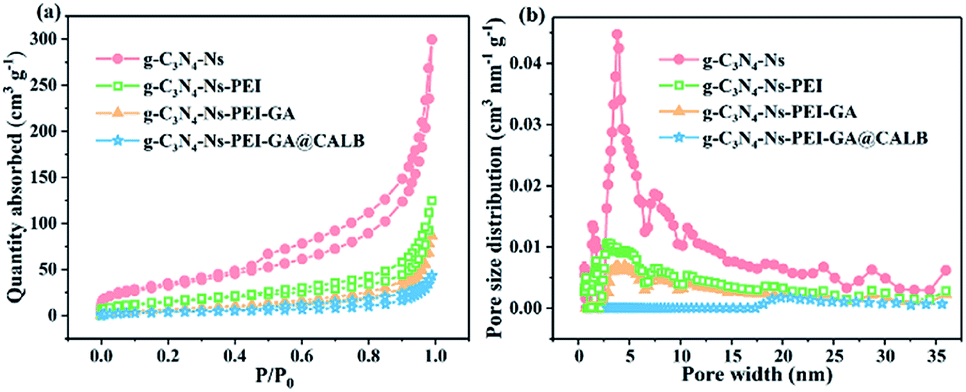
According to the BET gas sorptometry measurement in Fig. 5a, the prepared g-C3N4-Ns has a large surface area of 118.858 m2 g−1, whereas the surface areas of g-C3N4-Ns-PEI, g-C3N4-Ns-PEI-GA, and g-C3N4-Ns-PEI-GA @CALB are 54.005 m2 g−1, 26.947 m2 g−1, and 14.163 m2 g−1, respectively, exhibiting a significant decrease compared with the pure g-C3N4-Ns (ESI, Table S1†). Due to the large amount of the PEI polymers, which are attached to the g-C3N4-Ns covering the triazine rings, the surface area of g-C3N4-Ns decreases sharply. Subsequently, the covalent connection between GA and CALB leads to a further decrease in g-C3N4-Ns-PEI surface area. As shown in Fig. 5b, it is evident that g-C3N4-Ns has a mixed pore distribution with the size of 3.794 nm, which may originate from the defects and holes in the layers. Meanwhile, g-C3N4-Ns-PEI, g-C3N4-Ns-PEI-GA, and g-C3N4-Ns-PEI-GA@CALB have pores of 3.058 nm, 3.060 nm, and 2.897 nm, proving that g-C3N4-Ns maintains its intact layer structure. In addition, compared with the g-C3N4-Ns (0.444 cm3 g−1), the pore volume of g-C3N4-Ns-PEI-GA@CALB (0.178 cm3 g−1) reduces by 0.266 cm3 g−1. The reason may be that PEI, GA, and CALB attach or migrate to the edges of the pores on g-C3N4-Ns, leading to decreased pore volume.
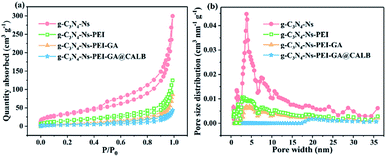 |
| | Fig. 5 Nitrogen adsorption–desorption isotherms (a) and pore size distribution (b) of g-C3N4-Ns, g-C3N4-Ns-PEI, g-C3N4-Ns-PEI-GA and g-C3N4-Ns-PEI-GA@CALB. | |
3.2. Factors in the immobilization of g-C3N4-Ns-PEI-GA@CALB
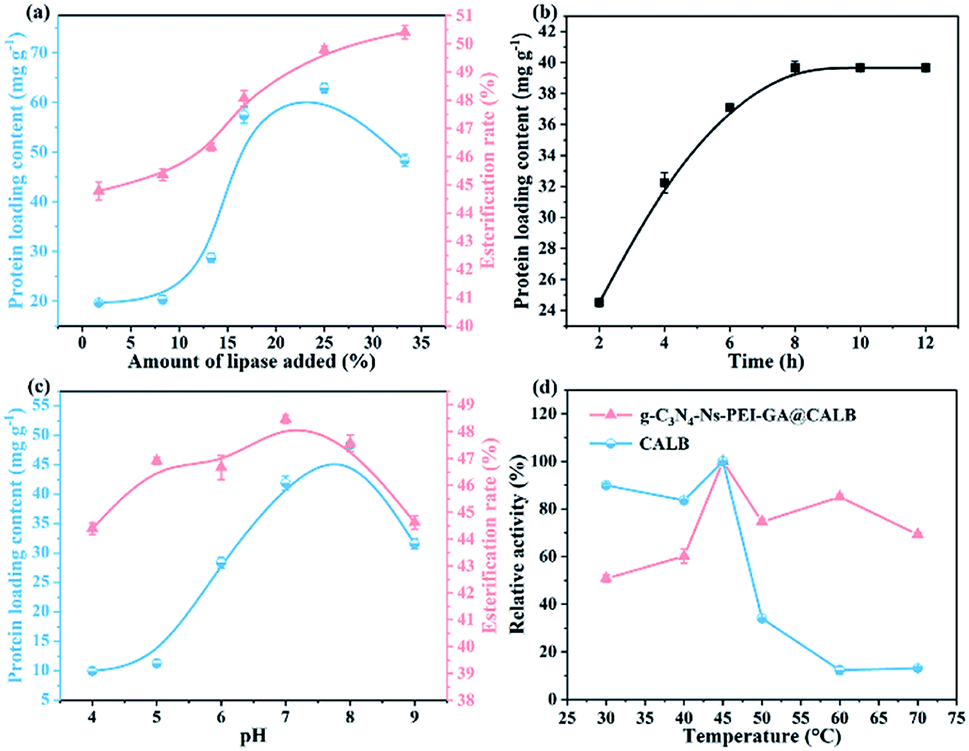
3.2.1. Effect of the lipase concentration on protein loading content and esterification rate. As is understood, the initial lipase concentration affects the amount of protein adhered to the materials and the activity. From Fig. 6a, with the increase in initial lipase concentration in the range of 2–25%, the protein loading content of g-C3N4-Ns-PEI-GA@CALB also increases. However, the amount decreases when the lipase concentration reaches 25%. The decline could be explained by that the aggregation of excessive CALB instead of distribution as a monolayer at the surface of g-C3N4-Ns in the immobilization, which is unfavorable to the attachment of CALB on the g-C3N4-Ns surface. Meanwhile, the esterification rate increases with lipase concentration, rapidly between 2% and 16% and slowly between 16% and 34%. It can be inferred that high loading of CALB has the potential to form lipase dimers and the active center will be covered, and combined with that the lipases of the underlying layer cannot interact with substrates, resulting in the low activity and slowly increased esterification rate.26 Therefore, 25% is considered the optimum initial concentration of lipase. As shown in Fig. 6b, the protein loading content achieved the maximum value in 8 h, then entered the plateau period, which indicates that the binding sites on the surface of g-C3N4-Ns are saturated.
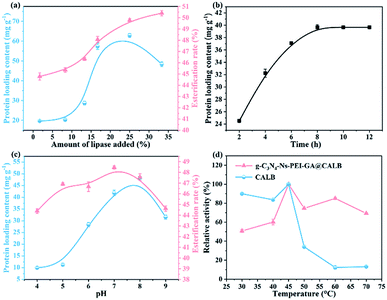 |
| | Fig. 6 Effect of the initial enzyme concentration on the protein loading content and the esterification rate of lutein disuccinate (a) and on the immobilization time (b); effect of pH (c) on protein content and esterification rate and temperature (d) on the activity of free and immobilized lipase. | |
3.2.2. Effect of pH and temperature on the activity of lipase. From Fig. 6c we draw the conclusion that the immobilized protein reaches the maximum value at the pH = 7.0, implying that the neutral environment is conducive to the fixation of CALB on g-C3N4-Ns-PEI-GA, which can be explained by electric charge distribution. When the pH value is higher than the isoelectric point, the net charge of protein is negative; on the contrary, the protein is positively charged when the pH is below the isoelectric point. Therefore, CALB can be absorbed on carriers via electrostatic adsorption due to the negative charge of CALB (pI = 6) and positively charged PEI (pI = 8) on the surface, which acts as an auxiliary mean of chemical covalently immobilized CALB. Furthermore, compared to free CALB, the conversion rate of lutein disuccinate for immobilized CALB remains relatively high between pH 5.0 and 8.0, indicating that g-C3N4-Ns-PEI-GA@CALB can maintain high activity under a wide range of pH condition and has good acid–base tolerance. The carrier material g-C3N4 shields and protects CALB from the deformation and destruction caused by acid or alkali in the PBS.27 Furthermore, the thermal stability of free CALB and g-C3N4-Ns-PEI-GA@CALB are contrasted in Fig. 6d. In the curves, both reach the maximum activity at the optimal temperature of 45 °C. At the higher temperature range, the relative residual activity of free CALB decreases to 12% on account of the conformation of the enzyme damages. Conversely, CALB covalently binds with g-C3N4-Ns@PEI showing excellent heat-resistance, with relative residual activity of approximately 69% even at 70 °C. This phenomenon may be explained by the covalent bonds formed when immobilization reduces the conformational flexibility of enzyme molecules, which effectively restricts the distortion or damage during temperature elevation.28
3.3. Factors in the esterification of lutein and succinic anhydride
3.3.1. Effect of different lipase on the lutein disuccinate conversion rate. The unique conformation of active centre endows lipase strict selectivity for substrates and reaction systems.29 In order to figure out the most suitable lipase for the synthesis of lutein disuccinate, we selected four different lipases including CALB, CRL, g-C3N4-Ns-PEI-GA@CALB and Novozyme 435 for the 48 h reaction. The results are shown in Table 1, the esterification rate of lutein disuccinate catalyzed by g-C3N4-Ns-PEI-GA@CALB is the highest (62.2%) compared with other lipases. Even for commercial Novozyme 435, the catalytic performance is not good. Because most of Novozyme 435 float on the solvent and cannot fully interact with the substrate.
Table 1 Lipase screening for synthesis lutein disuccinate
| Lipase |
Conversion rate (%) |
| CALB |
21.7 |
| CRL |
5.6 |
| g-C3N4-Ns-PEI-GA@CALB |
62.2 |
| Novozyme 435 |
25.3 |
3.3.2. Effect of different organic solvents on the lutein disuccinate conversion rate. On account of the solubility of lutein and succinic anhydride, as well as CALB activity expression, an optimal medium is indispensable to provide a moderate environment for esterification. Considering the solubility of lutein and succinic anhydride, we selected nine solvents (ESI, Table S2†) to monitor the activity of CALB during esterification in 48 h. In the presence of CALB, no significant reaction occurs between lutein and succinic anhydride when hexyl alcohol and N-butyl alcohol are involved. On the contrary, the conversion rate of lutein disuccinate reaches the maximum of 64.32% when DMF is used as the solvent, followed by chloroform (56.46%) and toluene (47.72%). In conclusion, DMF is the best candidate for esterification and activity expression of CALB. The results reveal that as a less toxic and polluting solvent, DMF is more suitable for enzyme reaction of edible substrates16,17 and can be employed to replace the commonly used polluted and toxic reagents for lutein esterification.
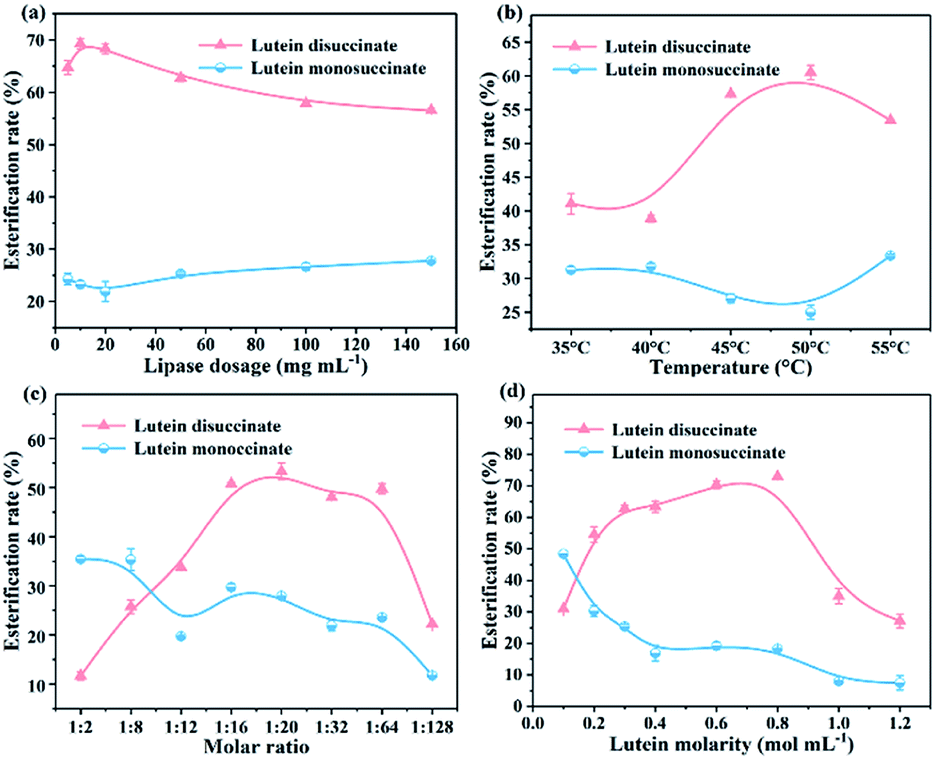
3.3.3. Effect of the immobilized lipase dosage and temperature on esterification rate. The effect of immobilized lipase dosage on the esterification rate of lutein monosuccinate and lutein disuccinate is illustrated in Fig. 7a. As shown in the curves, the esterification rate of lutein disuccinate reaches 69% when the concentration of g-C3N4-Ns-PEI-GA@CALB is 10 mg mL−1 and reduces gradually with increased immobilized lipase dosage. Nevertheless, when the dosage of CALB exceeds 10 mg mL−1, the superfluous solid could not maintain intimate contact with the solvent, which affects the stable reaction system. Meanwhile, the overdosed biocatalyst may cause the agglomeration and precipitation of intermediate monosuccinate resulting in the inaccessibility of g-C3N4-Ns-PEI-GA@CALB.30 Furthermore, the effect of the esterification temperature on the activity of g-C3N4-Ns-PEI-GA@CALB is displayed in Fig. 7b. As expected, the esterification rate increases with temperature from 35 to 50 °C and reaches its maximum. This is because the relative higher temperature elevates the esterification rate by increasing the solubility of reactants while enhancing the frequency of effective collisions and affinity between the substrates and CALB molecules.31 However, when the temperature is above 50 °C, a slight decrease in the esterification rate occurs due to the decomposition of lutein and the irreversible deactivation of CALB at the high temperature.
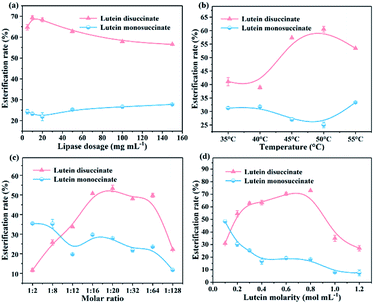 |
| | Fig. 7 Effect of lipase dosage (a) and temperature (b) on the esterification; effect of molar ratio (c) and lutein molarity (d) on esterification rate. | |
3.3.4. Effect of lutein molarity and molar ratio on esterification rate. Among other important factors, the effect of concentration and substrate molar ratio were studied. To explore the optimal molar ratio of lutein and succinic anhydride, the ratios from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 to 1:128 were designed to perform the esterification and the results are shown in Fig. 7c. During this period, the rate of esterification reaches the maximum around 1:16, and is relatively stable in the range of 1:16–1:20, then rapidly decreases above 1:32. According to the “Ping-Pong bi-bi” catalytic mechanism,32 CALB first forms the first transition state with substrate succinic anhydride which as the acyl donor, and then the hydroxyl group at one end of lutein acts as nucleophile to deacylate the acylase intermediate to form a second transition state. Therefore, moderate excessive succinic anhydride could promote the formation and accumulation of the first transitional states in the CALB catalysis, and meanwhile, accelerate entering the second transition state. When the succinic anhydride is overdosed, the decreased esterification rate may be attributed to the superfluous and insoluble succinic anhydride in DMF, forming an obstruction between the dissolved substrates and lipases.33 In Fig. 7d, the esterification rate increases with the addition of substrate and the optimum lutein concentration is 0.7 mol mL−1. This may explain why the full interaction of CALB and substrate is easily achieved with the appropriately substrate concentration. For higher substrate concentration (>0.8 mol mL−1), the esterification rate significantly declines to 27% when lutein molarity reaches 1.2 mol mL−1. Therefore, appropriate concentration of substrates can promote the reactions, and higher substrate concentration may induce the augmentation of the media viscosity, lowering catalytic efficiency at high cost.34
:2 to 1:128 were designed to perform the esterification and the results are shown in Fig. 7c. During this period, the rate of esterification reaches the maximum around 1:16, and is relatively stable in the range of 1:16–1:20, then rapidly decreases above 1:32. According to the “Ping-Pong bi-bi” catalytic mechanism,32 CALB first forms the first transition state with substrate succinic anhydride which as the acyl donor, and then the hydroxyl group at one end of lutein acts as nucleophile to deacylate the acylase intermediate to form a second transition state. Therefore, moderate excessive succinic anhydride could promote the formation and accumulation of the first transitional states in the CALB catalysis, and meanwhile, accelerate entering the second transition state. When the succinic anhydride is overdosed, the decreased esterification rate may be attributed to the superfluous and insoluble succinic anhydride in DMF, forming an obstruction between the dissolved substrates and lipases.33 In Fig. 7d, the esterification rate increases with the addition of substrate and the optimum lutein concentration is 0.7 mol mL−1. This may explain why the full interaction of CALB and substrate is easily achieved with the appropriately substrate concentration. For higher substrate concentration (>0.8 mol mL−1), the esterification rate significantly declines to 27% when lutein molarity reaches 1.2 mol mL−1. Therefore, appropriate concentration of substrates can promote the reactions, and higher substrate concentration may induce the augmentation of the media viscosity, lowering catalytic efficiency at high cost.34
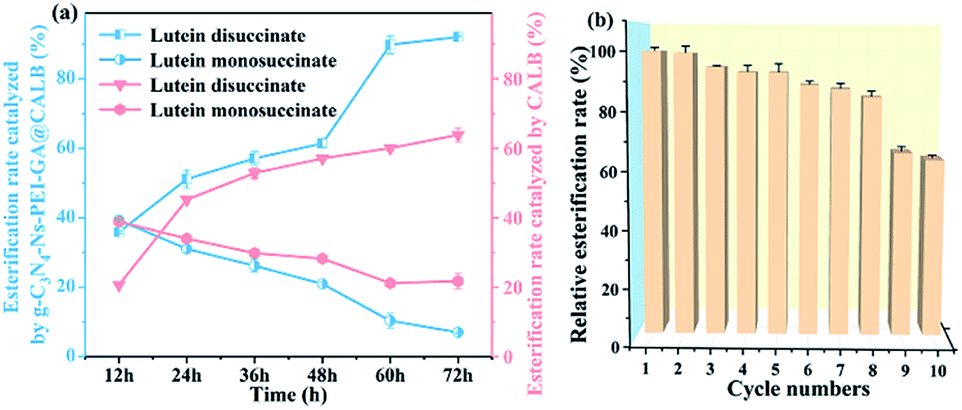
3.3.5. Time course of free and immobilized lipase catalyzed esterification reactions. The time course could explain the reaction process and mechanism, and liquid chromatography was used to analyze samples with different reaction time. As shown in Fig. 8a, with the extended reaction time, the content of lutein disuccinate and lutein monosuccinate gradually increases and decreases, respectively. In consequence, we can infer that lutein monosuccinate is formed first and then transforms to lutein disuccinate by esterification. The CALB has strong stereo-selectivity due to the “pocket” structure of its active site that has very limited space available compared with other lipases. Lutein is composed of a conjugated long chain of eighteen carbon atoms with two ionone rings at its ends, which may sterically hinder its hydroxyl access to the active CALB pocket. Longer esterification time is required to strengthen the interaction between the substrate and CALB. In our enzymatic reaction, the esterification rate of lutein disuccinate catalyzed by our immobilized lipase is up to 92% within 60 h while by free lipase is merely 60%, indicating the great performance of g-C3N4-Ns-PEI-GA@CALB in lutein esterification. Immobilized CALB on the g-C3N4-Ns-PEI-GA can improve thermal stability, solvent tolerance, and conformational stability of the active center. However, free CALB tends to be inactivated when it is directly exposed to solvent DMF at high temperature for a long time. The results are also consistent with the viewpoints mentioned above.
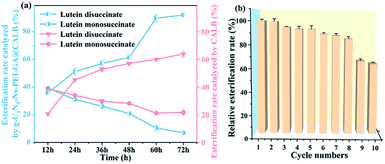 |
| | Fig. 8 The time course of the esterification reaction (a); recycling study of g-C3N4-Ns-PEI-GA@CALB and the first cycle assign 100% (b). | |
3.3.6. Reusability of the immobilized lipase. As shown in the Fig. 8b, the esterification rate decreases slightly with the cycle times, and it is worth noting that the conversion rate remains at 84% after eight cycles, which may benefit from the role of g-C3N4-Ns. The CALB is firmly bound to carbon nitride by covalent attachment, which effectively reduces the lipase leakage and ensures that g-C3N4-Ns-PEI-GA@CALB maintains high catalytic activity during every cycles. After the ninth cycle, the esterification rate decreases to 65%, which may be attributed to the mechanical loss of g-C3N4-Ns-PEI-GA@CALB in the centrifugation and irreversible partial inactivation of CALB when catalyzes for several cycles at high temperatures.
4. Conclusions
In summary, we prepared g-C3N4-Ns with a large surface area of 118.858 m2 g−1 and g-C3N4-Ns-PEI-GA@CALB with a high protein loading content of 60 mg g−1. The immobilized lipase exhibited excellent catalysis ability in esterifying lutein and succinic anhydride. The optimal esterification conditions were as follows: molarity of lutein, 0.7 mol mL−1; molar ratio of lutein and succinic anhydride, 1:16; dosage of g-C3N4-Ns-PEI-GA@CALB, 10 mg mL−1; and temperature, 50 °C. Under the optimum condition, the esterification rate could up to 92%. In addition, compared to free CALB, the thermo tolerance of g-C3N4-Ns-PEI-GA@CALB was greatly enhanced, in which the lipase activity of the former was 85% of initial activity while the latter remained only 12% after incubation at 60 °C for 1 h. Meanwhile, g-C3N4-Ns-PEI-GA@CALB presented excellent reusability in the catalyzing esterification reaction; for 8 runs the relative esterification rate was 84%. These results inspire us to explore more 2D nanomaterials in the design of the carrier materials and synthesize new structural lipids with different functions by esterification.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to the National Science Foundation of China (no. 31872896 and 31501423), the Young Elite Scientists Sponsorship Program by CAST (2017QNRC001), the Agricultural Science and Technology Innovation Project of Chinese Academy of Agricultural Sciences (CAAS-ASTIP-2013-OCRI) for financial support.
References
- H. Fu, B. Xie, G. Fan, S. Ma, X. Zhu and S. Pan, Effect of esterification with fatty acid of β-cryptoxanthin on its thermal stability and antioxidant activity by chemiluminescence method, Food Chem., 2010, 122, 602–609 CrossRef.
- D. J. Li, J. F. Song, A. Q. Xu and C. Q. Liu, Optimization of the ultrasound-assisted synthesis of lutein disuccinate using uniform design, Ultrason. Sonochem., 2014, 21, 98–103 CrossRef PubMed.
- A. Pérez-Gálvez and M. I. Mínguez-Mosquera, Esterification of xanthophylls and its effect on chemical behavior and bioavailability of carotenoids in the human, Nutr. Res., 2005, 25, 631–640 CrossRef.
- A. Robert, B. Sandra, C. Reinhold and S. Andeas, Determination of carotenoid stereoisomers in commercial dietary supplements by high-performance liquid chromatography, J. Agric. Food Chem., 2004, 52, 6086–6090 CrossRef PubMed.
- E. Rodrigues, L. R. Mariutti and A. Z. Mercadante, Scavenging capacity of marine carotenoids against reactive oxygen and nitrogen species in a membrane-mimicking system, Mar. Drugs, 2012, 10, 1784–1798 CrossRef CAS.
- A. Robert, S. Andeas and C. Reinhold, Effects of heating and illumination on trans–cis isomerization and degradation of carotene and lutein in isolated spinach chloroplasts, J. Agric. Food Chem., 2005, 53, 9512–9518 CrossRef.
- A. Subagio, H. Wakaki and N. Morita, Stability of lutein and its myristate esters, Biosci., Biotechnol., Biochem., 1999, 63, 1784–1786 CrossRef CAS.
- T. K. Dey, I. Maiti, S. Chakraborty, M. Ghosh and P. Dhar, Enzymatic synthesis of lipophilic lutein-PUFA esters and assessment of their stabilization potential in EPA-DHA rich fish oil matrix, J. Food Sci. Technol., 2019, 56, 2345–2354 CrossRef CAS.
- C. M. Cerón, C. Inmaculada, F. J. Sánchez, G. F. Acién, M. Emilio and M. J. Fernández-Sevilla, Recovery of lutein from Microalgae biomass: development of a process for scenedesmus almeriensis biomass, J. Agric. Food Chem., 2008, 56, 11761–11766 CrossRef.
- Y. Gong, S. Plander, H. Xu, B. Simandi and Y. Gao, Supercritical CO2 extraction of oleoresin from marigold (Tagetes erecta L.) flowers and determination of its antioxidant components with online HPLC-ABTS+ assay, J. Sci. Food Agric., 2011, 91, 2875–2881 CrossRef CAS.
- J. H. Lin, D. J. Lee and J. S. Chang, Lutein production from biomass: marigold flowers versus microalgae, Bioresour. Technol., 2015, 184, 421–428 CrossRef CAS.
- S. A. Ansari and Q. Husain, Potential applications of enzymes immobilized on/in nano materials: A review, Biotechnol. Adv., 2012, 30, 512–523 CrossRef CAS.
- S. Rohit, C. Yusuf, C. Uttam and Banerjee, Production, purification, characterization, and applications of lipases, Biotechnol. Adv., 2001, 19, 627–662 CrossRef.
- D. N. Tran and K. J. Balkus, Perspective of recent progress in immobilization of enzymes, ACS Catal., 2011, 1, 956–968 CrossRef.
- S. Zhang, Q. Deng, Y. Li, M. Zheng, C. Wan, C. Zheng, H. Tang, F. Huang and J. Shi, Novel amphiphilic polyvinylpyrrolidone functionalized silicone particles as carrier for low-cost lipase immobilization, R. Soc. Open Sci., 2018, 5, 172368 CrossRef PubMed.
- R. Wang, M. Hou, Y. Zhang, J. Ge and Z. Liu, Enzymatic synthesis of lutein dipalmitate in organic solvents, Catal. Lett., 2015, 145, 995–999 CrossRef.
- M. Hou, R. Wang, X. Wu, Y. Zhang, J. Ge and Z. Liu, Synthesis of lutein esters by using a reusable lipase-Pluronic conjugate as the catalyst, Catal. Lett., 2015, 145, 1825–1829 CrossRef.
- M. Mathesh, B. Luan, T. O. Akanbi, J. K. Weber, J. Liu, C. J. Barrow, R. Zhou and W. Yang, Opening Lids: modulation of lipase Immobilization by graphene oxides, ACS Catal., 2016, 6, 4760–4768 CrossRef.
- Z. Wang, K. Wang, Y. Wang, S. Wang, Z. Chen, J. Chen and J. Fu, Large-scale fabrication of N-doped porous carbon nanosheets for dye adsorption and supercapacitor applications, Nanoscale, 2019, 11, 8785–8797 RSC.
- S. Zhang, Q. Deng, H. Shangguan, C. Zheng, J. Shi, F. Huang and B. Tang, Design and preparation of carbon nitride based amphiphilic Janus N-doped carbon/MoS2 nanosheets for interfacial enzyme nanoreactor, ACS Appl. Mater. Interfaces, 2020 DOI:10.1021/acsami.9b18735.
- T. R. B. Ramakrishna, T. D. Nalder, W. Yang, S. N. Marshall and C. J. Barrow, Controlling enzyme function through immobilisation on graphene, graphene derivatives and other two dimensional nanomaterials, J. Mater. Chem. B, 2018, 6, 3200–3218 RSC.
- L. Chen and J. Song, Tailored graphitic carbon nitride nanostructures: synthesis, modification, and sensing applications, Adv. Funct. Mater., 2017, 27, 1702695 CrossRef.
- Y. Li, Z. Ruan, M. Zheng, Q. Deng, S. Zhang, C. Zheng, H. Tang, F. Huang and J. Shi, Candida rugosa lipase covalently immobilized on facilely-synthesized carbon nitride nanosheets as a novel biocatalyst, RSC Adv., 2018, 8, 14229–14236 RSC.
- P. Niu, L. Zhang, G. Liu and H.-M. Cheng, Graphene-like carbon nitride nanosheets for improved photocatalytic activities, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef.
- H. Cai, X. An, J. Cui, J. Li, S. Wen, K. Li, M. Shen, L. Zheng, G. Zhang and X. Shi, Facile hydrothermal synthesis and surface functionalization of polyethyleneimine-coated iron oxide nanoparticles for biomedical applications, ACS Appl. Mater. Interfaces, 2013, 5, 1722–1731 CrossRef.
- S. Zhang, J. Shi, Q. Deng, M. Zheng, C. Wan, C. Zheng, Y. Li and F. Huang, Preparation of carriers based on ZnO nanoparticles decorated on graphene oxide (GO) nanosheets for efficient Immobilization of lipase from Candida rugose, Molecules, 2017, 22, 1205 CrossRef.
- D. Yu, X. Zhang, D. Zou, T. Wang, T. Liu, L. Wang, W. Elfalleh and L. Jiang, Immobilized CALB catalyzed transesterification of soybean oil and phytosterol, Food Biophys., 2018, 13, 208–215 CrossRef.
- D. Spinelli, E. Fatarella, A. Di Michele and R. Pogni, Immobilization of fungal (Trametes versicolor) laccase onto Amberlite IR-120 H beads: optimization and characterization, Process Biochem., 2013, 48, 218–223 CrossRef.
- J. Uppenberg, N. Ohrner, M. Norin, K. Hult and T. A. Jones, Crystallographic and molecular-modeling studies of lipase B from Candida antarctica reveal a stereospecific pocket for secondary alcohols, Biochemistry, 1996, 34, 16838–16851 CrossRef.
- S. Šabeder, M. Habulin and Ž. Knez, Lipase-catalyzed synthesis of fatty acid fructose esters, J. Food Eng., 2006, 77, 880–886 CrossRef.
- A. M. Gumel, M. S. Annuar, T. Heidelberg and Y. Chisti, Thermo-kinetics of lipase-catalyzed synthesis of 6-O-glucosyldecanoate, Bioresour. Technol., 2011, 102, 8727–8732 CrossRef.
- G. D. Yadav and P. S. Lathi, Kinetics and mechanism of synthesis of butyl isobutyrate over immobilised lipases, Biochem. Eng. J., 2003, 16, 245–252 CrossRef.
- Y. Ning, F. Chen, X. Xu, Y. Jin, Z. Wang, K. Yang and Y. Jia, Biosynthesis of neokestose laurate catalyzed by Candida antarctica lipase B and its antimicrobial activity against food pathogenic and spoilage bacteria, J. Agric. Food Chem., 2017, 65, 11092–11099 CrossRef CAS.
- J. Kim, M. Y. Chung, H. D. Choi, I. W. Choi and B. H. Kim, Enzymatic synthesis of structured Monogalactosyldiacylglycerols enriched in pinolenic acid, J. Agric. Food Chem., 2018, 66, 8079–8085 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SEM images, the surface area, pore width, pore volume of materials and other supplementary data. See DOI: 10.1039/d0ra00563k |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Jie Shi
a,
Jie Shi