DOI:
10.1039/D0RA00935K
(Paper)
RSC Adv., 2020,
10, 10897-10903
Biomimetic synthesis of galantamine via laccase/TEMPO mediated oxidative coupling†
Received
31st January 2020
, Accepted 5th March 2020
First published on 17th March 2020
Abstract
Laccase-mediated intramolecular oxidative radical coupling of N-formyl-2-bromo-O-methylnorbelladine afforded a novel and isolable spirocyclohexadienonic intermediate of galantamine. High yield and conversion of substrate were obtained in the presence of the redox mediator 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO). This laccase procedure, with an overall yield of 34%, represents a scalable and environmentally friendly alternative to previously reported syntheses of galantamine based on the use of potassium ferricyanide as an unspecific radical coupling reagent.
Introduction
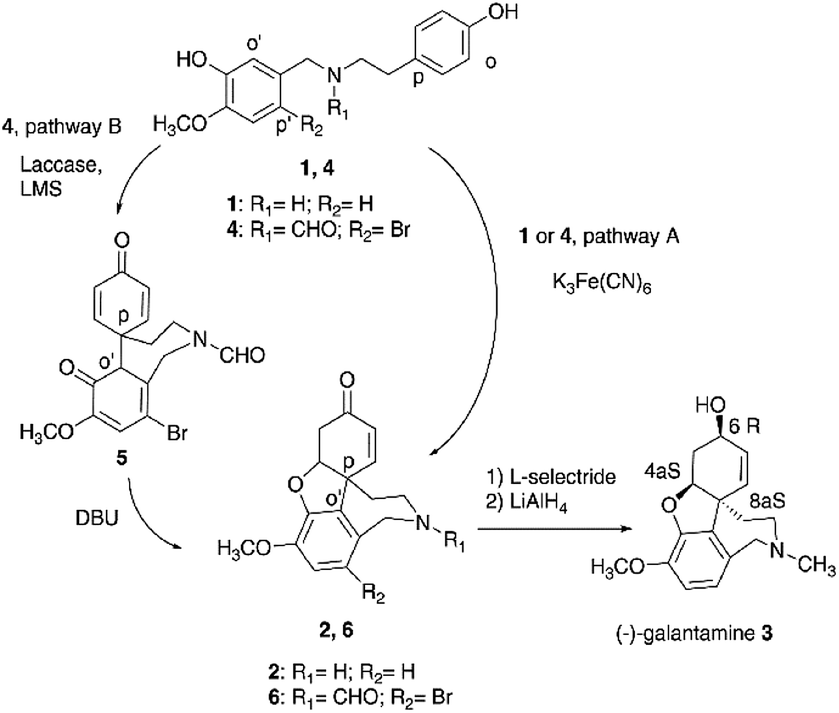
The oxidative radical coupling is a reaction of pivotal relevance in the biosynthesis of polycyclic natural substances, including the synthesis of bioactive aromatic alkaloids narwedine 2 and galantamine 3 from O-methylnorbelladine 1 (Fig. 1).1,2 Galantamine, isolated from the Caucasian snow-drop and from the bulbs of different species of Amarillidaceae family, is the active principle contained in Remynil® and Razadyne®, two drugs that act as allosteric ligands (APL) of human nicotinic acetylcholine receptors.3–5 In particular, compound 3 controls the neuronal release of different neurotransmitters such as glutamate, γ-aminobutyric acid (GABA) and bioactive monoamines in the therapy of the Alzheimer's disease.3 The biosynthetic pathway of galantamine proceeds by regioselective para–ortho (p–o′) intramolecular radical coupling and phenolic addition in compound 1 to yield 2, followed by reduction and alkylation.4 At the industrial scale, the intramolecular radical coupling is realized in very low yield (1.4%) by use of potassium ferricyanide K3Fe(CN)6, due to the occurrence of undesired side-reactions involving less hindered aromatic positions of the substrate (pathway A, Fig. 1).6 K3Fe(CN)6 is characterized by acute toxicity, skin corrosion, and serious eye and respiratory tract irritation, as well as by unfavorable environmental impact.7 The role of K3Fe(CN)6 in the cyclization of compound 1 has not been reported in detail, even if the formation of phenoxyl radicals by iron-mediated one-electron abstraction process is expected as operative process.8 Synthetic alternative to the use of K3Fe(CN)6 involves the cyclization of nitrogen protected N-formyl-2-bromo-O-methylnorbelladine 4 to yield N-formyl-1-bromo-narwedine 6 (54% yield) under similar experimental conditions (pathway A, Fig. 1).5,6,9 Laccase (EC 1.10.3.2) is the simplest multi-copper oxidoreductase in nature (E0 = 0.5 to 0.8 V vs. normal hydrogen electrode NHE) able to catalyze the oxidation of phenols and other organic substrates with concomitant reduction of molecular dioxygen (O2) to water (H2O).10–12 Several examples of the use of laccase in the oxidative radical coupling are reported.13 The industrial interest in this enzyme is related to the high value of its catalytic constant, large substrate promiscuity, and high thermal resistance, associated to the use of air as primary oxidant.14–16 In order to develop a novel and environmentally friendly synthesis of galantamine 3 as alternative to the K3Fe(CN)6 process, we report here that laccase (Trametes versicolor) efficiently catalyzes the one-pot para–ortho (p–o′) oxidative radical coupling of 4 to a novel and isolable spirocyclohexadienonic intermediate of narwedine 6 and galantamine 3, namely 9-bromo-7-methoxy-4′,6-dioxo-3,4,5a,6-tetrahydrospiro-[benzo[c]azepine-5,1′-cyclohexane]-2′,5′-diene-2(1H)-carbaldehyde 5 (pathway B, Fig. 1). The effect of low molecular weight redox-mediators (laccase mediator system, LMS), including 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)diammonium salt (ABTS) and 1-hydroxybenzotriazole (HOBt), in the selectivity and yield of the cyclization was evaluated. These compounds can enhance the efficacy of the radical process by transferring the oxidative redox potential from the active site of the enzyme to the bulk of the solution.17–22 The laccase/TEMPO mediated intramolecular radical coupling of 4 to 5, followed by simple alkaline catalyzed intramolecular enolic cycloaddition and canonical reduction procedure afforded galantamine 3 in a total yield higher than that previously reported for K3Fe(CN)6 under optimal experimental conditions.
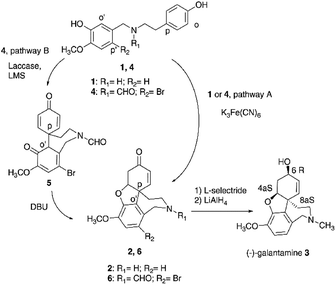 |
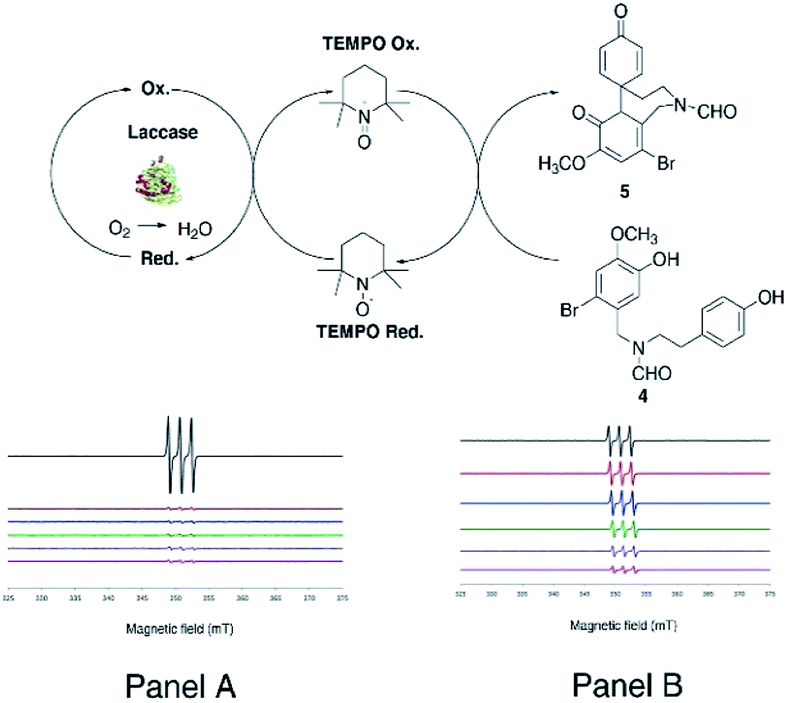
| | Fig. 1 Synthesis of galantamine 3. Pathway A: synthesis of galantamine 3 from O-methylnorbelladine 1 or N-formyl-2-bromo-O-methylnorbelladine 4 by use of K3Fe(CN)6. Pathway B: laccase mediated synthesis of galantamine 3 from N-formyl-2-bromo-O-methylnorbelladine 4. LMS: laccase mediator system. DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene. | |
Results and discussion
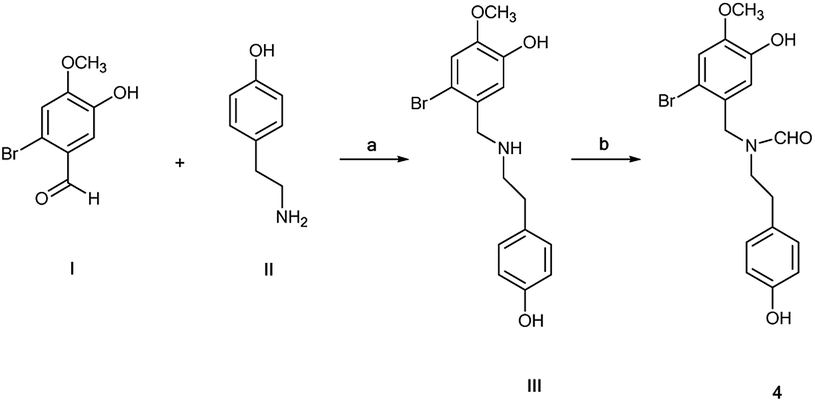
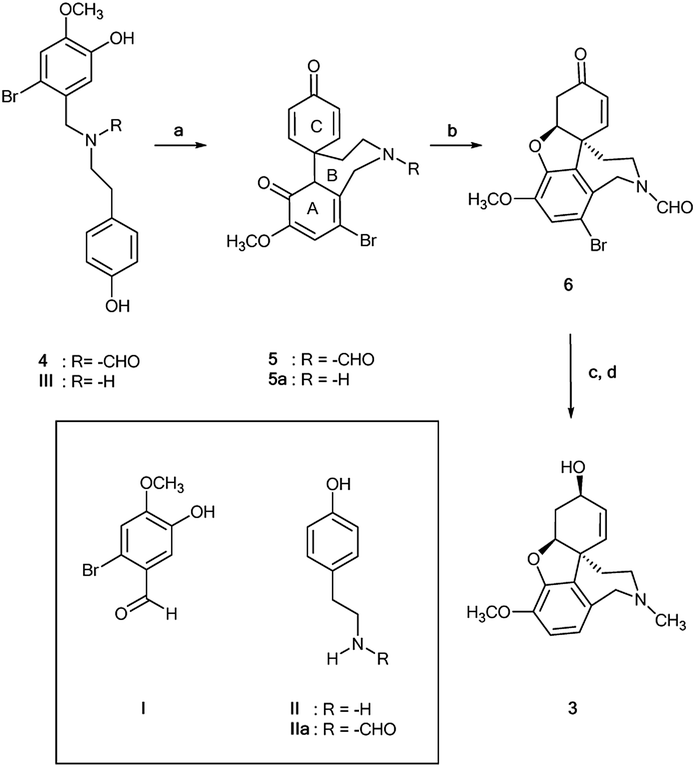
N-Formyl-2-bromo-O-methylnorbelladine 4 was prepared as described in Scheme 1. This synthetic pathway simplifies the conventional procedure.9 Briefly, commercially available 2-bromo-5-hydroxy-4-methoxybenzaldehyde I and 4-hydroxy-phenylethylamine (tyramine) II were coupled with NaBH4 in MeOH at 25 °C to yield III in 95% yield. Compound III was successively reacted with ethyl formate (HCOOC2H5) in DMF at 80 °C to afford desired 4 in 92% yield. The oxidative radical coupling was performed by addition of laccase from Trametes versicolor (1000 U mmol−1) to compound 4 (380 mg, 1.0 mmol) in 1,4-dioxane (5 mL) and sodium acetate buffer (20 mL; 0.5 M; pH 4.5) mixture under gentle stirring at 25 °C for 3.0 h in air. 1,4-Dioxane was used to circumvent the problem of the low solubility of 4 in the buffer. Under these experimental conditions, the spirocyclohexadienonic derivative 5 was isolated in low yield (18%), besides higher amounts of the compound I, N-formyl-tyramine IIa, and unreacted substrate (Scheme 2; Table 1, entry 1). In accordance with data previously reported, compounds I and IIa were probably obtained by the undesired laccase mediated oxidation of the benzylic position in 4 followed by elimination of water molecule and hydrolysis of the corresponding enamonium ion intermediate.23
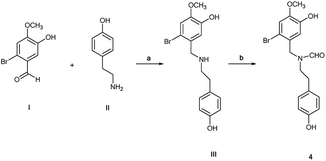 |
| | Scheme 1 Simplified synthesis of N-formyl-2-bromo-O-methylnorbelladine 4 from 2-bromo-5-hydroxy-4-methoxybenzaldehyde I and tyramine II; (a) NaBH4, MeOH, 3 h, 95%; (b) HCOOC2H5, DMF, 80 °C, 24 h, 92%. | |
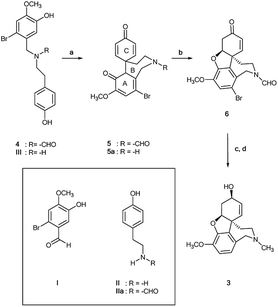 |
| | Scheme 2 Biomimetic laccase-mediated synthesis of galantamine 3 starting from N-formyl-2-bromo-O-methylnorbelladine 4. Compounds I, II and IIa are probably due to the undesired laccase catalyzed benzylic oxidation of 4; (a) laccase Tv, mediator, O2, 1,4-dioxane/sodium acetate buffer; (b) DBU, CH2Cl2, 91%; (c) L-selectride, THF, −78 °C; (d) LiAlH4, THF, r.t., 61%. | |
Table 1 Laccase-mediated oxidative radical coupling of N-formyl-2-bromo-O-methylnorbelladine 4 and 2-bromo-O-methylnorbelladine III to spirocylohexadienonic derivatives 5 and 5aa
| Entry |
Substrate |
Laccase (U mmol−1) |
Mediator (eq.) |
Conversion (%) |
Product(s) |
Yield (%) |
| The reaction was performed treating the substrate in 1,4-dioxane and sodium acetate buffer with laccase in the presence or absence of the redox mediator under gentle stirring at 25 °C for 3.0 h in an air atmosphere. The experiment was conducted in triplicate. 0.3 equivalents of mediator with respect to substrate. 0.6 equivalents of mediator with respect to substrate. The reaction was performed with 0.6 equivalents of mediator at 50 °C. The reaction was performed with 0.6 equivalents of mediator in acetonitrile at 25 °C. |
| 1 |
4 |
1000 |
— |
80 |
5(I)[IIa] |
18(31)[27] |
| 2 |
4 |
100 |
ABTSb |
76 |
5(I)[IIa] |
19(27)[27] |
| 3 |
4 |
100 |
TEMPOb |
81 |
5(I)[IIa] |
21(30)[28] |
| 4 |
4 |
100 |
HOBtb |
73 |
(I)[IIa] |
(27)[27] |
| 5 |
4 |
100 |
TEMPOc |
83 |
5(I)[IIa] |
38(20)[19] |
| 6 |
4 |
100 |
TEMPOd |
71 |
5(I)[IIa] |
30(22)[18] |
| 7 |
4 |
100 |
TEMPOe |
68 |
5(I)[IIa] |
25(23)[16] |
| 8 |
4 |
1000 |
TEMPOc |
>98 |
5(I)[IIa] |
70(16)[13] |
| 9 |
III |
1000 |
TEMPOc |
80 |
5a(I)[II] |
8(37)[33] |
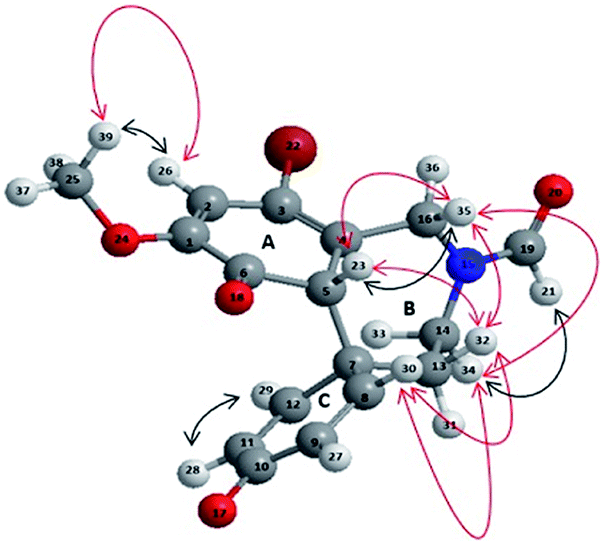
Compound 5 was characterized by the presence of a tricyclic condensed system, containing the azepine ring (B) and two hexadienonic rings (A and C, respectively), as a mixture (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) of two rotamers do to the steric hindrance of the N-formyl protecting group.9 The structure of 5 was unambiguously assigned by different 1D-NMR (1H-NMR, 13C-NMR and distortionless enhancement by polarization transfer analysis DEPT-135), and 2D-NMR (homonuclear 1H–1H correlated spectroscopy COSY, homonuclear 1H–1H overhauser spectroscopy NOESY and 1H–DEPT-135 heteronuclear single-quantum coherence HSQC) analyses (for detailed 1D and 2D NMR spectra see ESI, S2–S25;† representative 1H-NMR, 13C-NMR HSQC, DEPT-135, 1H–1H COSY and 1H–1H NOESY data for compound 5 are reported in ESI, Table S1†). The prevalence of the keto-tautomer in ring A (with respect to the possible phenolic alternative) was confirmed by the couple of signals at 6.41 ppm and 6.44 ppm (double singlet) in the 1H-NMR analysis (unchangeable after D2O treatment), corresponding to H-23 proton (Fig. 2), and by the signals at 186 ppm and 181 ppm in the 13C-NMR analysis, corresponding to C-6 and C-10 quaternary carbons, respectively (in accordance with the proposed structure these signals disappeared in the DEPT-135 experiment). The COSY analysis clearly showed the interaction between H-23 and H-35, while the para–ortho regioselectivity in the azepine ring B was further confirmed by the NOESY correlation between H-32 and both H-23 and H-35, as well as by the short-range heteronuclear correlation (HSQC) involving these hydrogen atoms and the respective connected carbons. In order to increase the yield of compound 5, the oxidative radical coupling was repeated in the presence of three laccase redox mediators characterized by different reaction mechanisms, namely ABTS (E0 = 0.5 V), TEMPO (E0 = 0.75 V), and HOBt (E0 = 1.13 V). The electron-transfer (ET) process is operative in the case of ABTS, while oxoammonium ion mediated one-electron transfer mechanism, and radical H-abstraction (HAT), are effective processes in the case of TEMPO and HOBt, respectively.24–28
:1 ratio) of two rotamers do to the steric hindrance of the N-formyl protecting group.9 The structure of 5 was unambiguously assigned by different 1D-NMR (1H-NMR, 13C-NMR and distortionless enhancement by polarization transfer analysis DEPT-135), and 2D-NMR (homonuclear 1H–1H correlated spectroscopy COSY, homonuclear 1H–1H overhauser spectroscopy NOESY and 1H–DEPT-135 heteronuclear single-quantum coherence HSQC) analyses (for detailed 1D and 2D NMR spectra see ESI, S2–S25;† representative 1H-NMR, 13C-NMR HSQC, DEPT-135, 1H–1H COSY and 1H–1H NOESY data for compound 5 are reported in ESI, Table S1†). The prevalence of the keto-tautomer in ring A (with respect to the possible phenolic alternative) was confirmed by the couple of signals at 6.41 ppm and 6.44 ppm (double singlet) in the 1H-NMR analysis (unchangeable after D2O treatment), corresponding to H-23 proton (Fig. 2), and by the signals at 186 ppm and 181 ppm in the 13C-NMR analysis, corresponding to C-6 and C-10 quaternary carbons, respectively (in accordance with the proposed structure these signals disappeared in the DEPT-135 experiment). The COSY analysis clearly showed the interaction between H-23 and H-35, while the para–ortho regioselectivity in the azepine ring B was further confirmed by the NOESY correlation between H-32 and both H-23 and H-35, as well as by the short-range heteronuclear correlation (HSQC) involving these hydrogen atoms and the respective connected carbons. In order to increase the yield of compound 5, the oxidative radical coupling was repeated in the presence of three laccase redox mediators characterized by different reaction mechanisms, namely ABTS (E0 = 0.5 V), TEMPO (E0 = 0.75 V), and HOBt (E0 = 1.13 V). The electron-transfer (ET) process is operative in the case of ABTS, while oxoammonium ion mediated one-electron transfer mechanism, and radical H-abstraction (HAT), are effective processes in the case of TEMPO and HOBt, respectively.24–28
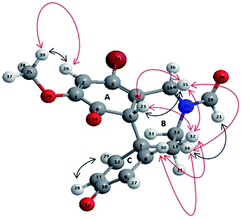 |
| | Fig. 2 3D structure of compound 5 representing COSY (black arrows) and NOESY (red arrows) interactions. | |
As a general procedure, compound 4 (380 mg, 1.0 mmol) dissolved in 1,4-dioxane (5 mL) and sodium acetate buffer (20 mL, 0.5 M, pH 4.5) mixture was treated with laccase (100 U mmol−1 or 1000 U mmol−1) in the presence of the appropriate redox mediator (from 0.3 to 0.6 equivalents with respect to substrate) under gentle stirring at 25 °C for 3.0 h in air. Irrespective from the experimental conditions, laccase (100 U mmol−1) in the presence of ABTS or TEMPO (0.3 equivalents) afforded 5 in a yield similar to that previously obtained with 1000 U mmol−1 of laccase alone (Table 1, entries 2 and 3, respectively, versus entry 1). Conversely, degradative and over-oxidation side-processes prevailed when the reaction was performed in the presence of HOBt (Table 1, entry 4). Since the work-up of the laccase/ABTS cyclization produced an emulsion of difficult treatment, the oxidation was further optimized with TEMPO in a larger amount (0.6 equivalents) as selected redox mediator. In this latter case, compound 5 was obtained in appreciable yield (38%) (Table 1, entry 5). The increase of the reaction temperature (50 °C), as well as the use of acetonitrile (CH3CN) as an alternative solvent did not improve the yield of 5 (Table 1, entries 6 and 7, respectively). Better results were finally obtained with 1000 U mmol−1 of laccase and 0.6 equivalents of TEMPO, in which case compound 5 was isolated in 70% yield (Table 1, entry 8). The EPR spectra confirmed the occurrence of a catalytic cycle during the oxidation of 4 with laccase and TEMPO (Fig. 3) (for detailed EPR spectra see ESI, S26 and S27†). Measurements were performed in situ by treatment of compound 4 (0.3 mmol) with laccase (0.14 mM) using two concentrations of TEMPO (0.6 mM and 6 μM) in 1,4-dioxane/buffer medium (1.2 mL, 1,4-dioxane:buffer 1:4 ratio) The EPR signal of TEMPO decreased rapidly in the absence of compound 4, as a consequence of the formation of the corresponding oxoammonium ion (Fig. 3, Panel A). On the other hand, the EPR signal decreased slowly in the presence of compound 4 due to the expected regeneration of the native radical species by laccase (Fig. 3, Panel B). In Table 2 are reported the quantitative EPR data recorded for the cyclization at different reaction times and at the two indicated concentration values of TEMPO, both in the absence (Table 2 entries 1 and 3) and in the presence of compound 4 (Table 2, entries 2 and 4).
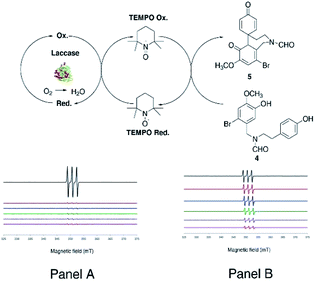 |
| | Fig. 3 Redox cycle of the transformation of 4 into 5 performed by laccase and mediated by TEMPO. EPR signal of TEMPO in absence (Panel A) and in the presence (Panel B) of the substrate (entry 2 and 4 in Table 2). | |
Table 2 EPR analysis and spin density quantitation (spins per mm3)a of TEMPO during the oxidation of compound 4 with laccase
| Entry |
t = 0 min |
t = 30 min |
t = 60 min |
t = 120 min |
t = 180 min |
t = 240 min |
| Error value ± 0.1. Spin quantitation in the presence of TEMPO (0.6 mM) and laccase (0.14 mM). Spin quantitation in the presence of TEMPO (0.6 mM), laccase (0.14 mM) and compound 4 (0.25 mM). Spin quantitation in the presence of TEMPO (6 μM) and laccase (0.14 mM). Spin quantitation in the presence of TEMPO (6 μM), laccase (0.14 mM) and compound 4 (0.25 mM). |
| 1b |
1.5 × 1014 |
1 × 1014 |
0.7 × 1014 |
0.7 × 1014 |
0.3 × 1014 |
0.8 × 1014 |
| 2c |
3 × 1014 |
2.3 × 1014 |
2 × 1014 |
1.4 × 1014 |
1.3 × 1014 |
1 × 1014 |
| 3d |
8 × 1015 |
4 × 1014 |
2 × 1014 |
0.9 × 1014 |
1.3 × 1014 |
1.2 × 1014 |
| 4e |
1.6 × 1015 |
1.5 × 1015 |
1.2 × 1015 |
0.9 × 1015 |
0.9 × 1015 |
0.6 × 1015 |
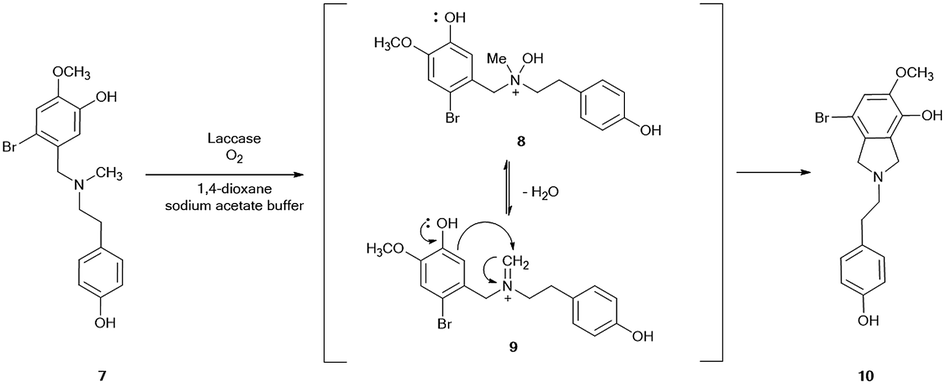
The role of the nitrogen-protecting group in the efficacy of the reaction was then evaluated by analyzing the oxidation of unprotected 2-bromo-O-methylnorbelladine III under optimal experimental conditions (Scheme 2). In this latter case, the corresponding tricyclic intermediate 5a was isolated in very low yield besides the compound I and tyramine II (Table 1, entry 9), confirming the beneficial role of the formyl protecting group in avoiding undesired oxidative side-processes.23 Moreover, the same procedure applied to N-methyl-2-bromo-O-methylnorbelladine 7, bearing an alkyl electron donating group instead of the electron withdrawing formyl moiety, afforded the isoindoline derivative 10 as the only recovered product in 76% yield besides the unreacted substrate (Scheme 3). In line with data previously reported for the oxidation of aromatic benzylamines with inorganic copper salts, compound 10 was probably obtained by formation of the corresponding quaternary amine N-oxide 8 (due to the activation of the nitrogen atom by the methyl substituent), followed by elimination of water to iminium ion 9 and successive phenolic intramolecular addition (Scheme 3).29 The activity of isoindoline derivatives as acetylcholine esterase (AChE) inhibitors against Alzheimer's disease has beenreported.30 Galantamine 3 was finally obtained from 5 by a two steps procedure (Scheme 2), including the synthesis of N-formyl-1-bromo-narwedine 6 by treatment with 1,8-diazabicyclo[5.4.0]undec-7-ene DBU (91%), followed by conventional reduction with both L-selectride and lithium aluminum hydride (61%).7,31,32
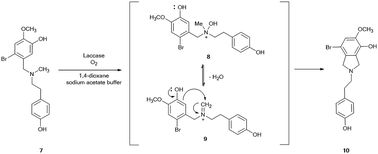 |
| | Scheme 3 Proposed mechanism for the laccase-mediated synthesis of isoindoline 10 starting from N-methyl-2-bromo-O-methylnorbelladine 7. | |
Conclusions
In conclusion, a novel and biomimetic synthesis of galantamine was developed under environmentally friendly conditions by laccase/TEMPO mediated oxidative radical coupling of N-formyl-2-bromo-O-methylnorbelladine 4 to spirocyclohexadienonic derivative 5. In the optimal experimental conditions (1000 U mmol−1 of laccase and 0.6 equivalents of TEMPO) the N-formyl-1-bromo-narwedine 6 was obtained in a total yield (65%) higher than that previously reported for the K3Fe(CN)6 procedure (54%).6 Moreover, the total yield for galantamine 3 was 34%. Further studies are ongoing to evaluate the use of supported laccase in the synthesis of compound 5, as well as, the laccase mediated synthesis of novel bioactive isoindoline derivatives starting from N-alkyl-narwedine derivatives.
Experimental
Materials, methods and experimental section
Reagents and laccase from Trametes versicolor were obtained from commercial suppliers (Sigma-Aldrich Srl, Milan, Italy). Chemical reactions were monitored using thin layer chromatography on precoated aluminium silica gel Merck 60 F254 plates and an UV lamp was used for visualization. Merck silica gel 60 (230–400 mesh) was used for flash chromatography. All products were dried in high vacuum (10–3 mbar). 1H NMR, 13C NMR, DEPT-135, COSY, NOESY and HSQC spectra were recorded on a Bruker Avance DRX400 (400 MHz/100 MHz) spectrometer. Chemical shifts for protons are reported in parts per million (δ scale) and internally referenced to the CD3OD, CDCl3, DMSO-d6 or (CD3)2O, signal at δ 3.33 ppm, 7.28 ppm, 2.50 ppm and 3.31 ppm respectively. Coupling constants (J) are reported in Hz. Multiplicities are reported in the conventional form: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad. Mass spectra (MS) data were obtained using an Agilent 1100 LC/MSD VL system (G1946C) with a 0.4 mL min−1 flow rate using a binary solvent system of 95:5/methyl alcohol:water.
Enzyme activity assay
The enzyme activity was assayed by using 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)diammonium salt (ABTS) procedure. ABTS (5.0 mM), sodium acetate buffer (2.0 mL, pH 5.0), and the enzyme solution (200 μL) were used as standard solution. The formation of the cation radical was detected by measuring the increase of absorbance at 420 nm (ε420 = 36000 M−1 cm−1). One unit of laccase activity has been defined as the amount of enzyme that catalyzed the oxidation of 1.0 μmol of ABTS in 200 μL reaction mixture at 25 °C during 1.0 min.
EPR analysis
CW (Continuous wave) X-band (9 GHz) EPR spectra were recorded at room temperature with a Bruker E580 Elexsys Series using the Bruker ER4122SHQE cavity. The 1 mm diameter quartz capillaries were filled in with the samples and analyzed with the EPR spectrometer. The spin quantitation was carried out against an internal reference (Bruker) of irradiated solid alanine (3 mm length, 5 mm diameter) sealed under N2 atmosphere, and containing a total of 2.05 × 10−7 ± 10% spins, using the SpinCounting program provided in the Xepr software (Bruker). Experimental conditions for spectra acquisition were: ν = 9.86 GHz, modulation 0.1 mT, microwave power 2 mW.
Procedure for the synthesis of compound III
Tyramine (II) (0.45 mmol, 1.05 eq.) was added to a solution of 2-bromo-5-hydroxy-4-methoxybenzaldehyde (I) (0.43 mmol, 1 eq.) in MeOH (2 mL) and the mixture was stirred for 2 h at room temperature. After this period, NaBH4 (0.47 mmol, 1.1 eq.) was added to the reaction mixture and it was stirred for 10 min at 0 °C and 3 h at room temperature. The reaction mixture was then filtered over Celite® and the filtrate was evaporated under reduced pressure. The residue was purified by silica gel gradient column chromatography (CHCl3/MeOH/NH4OH 10:1:0.1) to afford III (yield = 95%). 1H-NMR ((CD3)2CO, 400 MHz): δ = 7.07 (s, 1H, ArH), 7.05 (d, 2H, J = 2 Hz, 2 × ArH), 7.02 (s, 1H, ArH), 6.76, 6.75 (dd, 2H, J = 8.4, 2 Hz, 2 × ArH), 3.85 (s, 3H, OCH3), 3.75 (s, 2H, ArCH2N), 2.83–2.79 (m, 2H, NCH2CH2Ar), 2.74, 2.71 (dd, 2H, J = 14.8, 6.8 Hz, NCH2CH2Ar) ppm. 13C-NMR ((CD3)2CO, 100 MHz) δ = 35.2, 50.6, 52.4, 55.7, 111.4, 115.0, 115.4, 116.7, 129.5, 130.9, 132.0, 146.0, 147.1155.5 ppm. DEPT-135 ((CD3)2CO, 100 MHz) δ = 35.2 (CH2), 50.6 (CH2), 52.4 (CH2), 55.7 (CH3), 115.0 (CH), 115.4 (CH), 116.7 (CH), 129.5 (CH) ppm. MS (ESI): m/z for [C16H18BrNO3]−: 350.07.
Procedure for the synthesis of compound 4
Ethyl formate (2.8 mL) was added to a solution of III (0.35 mmol, 1.0 eq.) in DMF (1 mL) and the mixture was stirred for 24 h at 80 °C. After the reaction, ethyl formate was removed under reduced pressure and ethyl acetate was added to the reaction mixture. The organic layer was washed with an aqueous solution of LiCl (3% aq.) and then with brine and dried over sodium sulfate. The organic layer was evaporated under reduced pressure. The residue was purified by silica gel gradient column chromatography (CHCl3/MeOH 15:1) to afford 4 (yield = 92%).1H-NMR ((CD3)2CO, 400 MHz): showed the presence of two rotamers. δ = 8.32, 8.01 (s, 1H, CHO), 7.16, 7.13 (s, 1H, ArH), 7.06–7.02 (m, 4H, 4 × ArH), 6.86, 6.85 (s, 1H, ArH), 6.79–6.75 (m, 4H, 4 × ArH), 4.56, 4.41 (s, 2H; ArCH2N), 3.88, 3.87 (s, 3H, OCH3), 3.44 (t, 2H, J = 14.4, 7.2 Hz, NCH2CH2Ar), 3.36–3.32 (m, 2H, NCH2CH2Ar), 2.78 (t, 2H, J = 14.4, 7.2 Hz, NCH2CH2Ar), 2.68–2.64 (m, 2H, NCH2CH2Ar) ppm. 13C-NMR ((CD3)2CO, 100 MHz): showed the presence of two rotamers. δ = 32.4, 34.0, 43.7, 44.1, 48.4, 50.5, 55.7, 55.8, 111.4, 111.8, 115.2, 115.3, 115.5, 115.9, 116.1, 116.7, 128.1, 128.7, 129.1, 129.6, 129.7, 129.8, 146.5, 146.6, 147.8, 148.1, 155.8, 156.0, 162.6, 162.7 ppm. DEPT-135 ((CD3)2CO, 100 MHz) δ = 32.3 (CH2), 34.0 (CH2), 43.7 (CH2), 44.0 (CH2), 48.4 (CH2), 50.7 (CH2), 55.7 (CH3), 115.2 (CH), 115.3 (CH), 115.4 (CH), 115.9 (CH), 116.1 (CH), 116.7 (CH), 129.6 (CH), 129.8 (CH), 162.6 (CH), 162.7 (CH) ppm. MS (ESI): m/z for [C17H18BrNO4]−: 378.02.
Procedure for the synthesis of compound 5
Laccase (1000 U) was added to a stirred solution of compound 4 (1 mmol, 1 eq.) in 1,4-dioxane (5 mL) and acetate buffer, pH 4.5, 0.5 M (20 mL). TEMPO (0.6 mmol, 0.6 eq.) was added immediately. The solution was stirred for 3 h under O2 atmosphere. After this period the aqueous solution was extracted with ethyl acetate (3 × 50 mL). The organic layer was washed with brine, dried over sodium sulfate and evaporated under reduced pressure. The residue was purified by silica gel gradient column chromatography (CHCl3/MeOH 20:1) to afford 5 (yield = 70%). 1H-NMR (CDCl3, 400 MHz): showed the presence of two rotamers. δ = 8.18, 7.90 (s, 1H, CHO), 7.05, 6.98 (d, 4H, J = 7.6 Hz, 4 × CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO), 6.80, 6.74 (d, 4H, J = 8 Hz, 4 × CHCHCO), 6.44, 6.41 (s, 1H, CHCO), 5.96, 5.93 (s, 1H, CHCOCH3), 4.35, 4.10 (s, 2H, CH2N), 3.85 (s, 6H, 2 × OCH3), 3.49 (t, 4H, J = 13.6, 6.8 Hz, 2 × NCH2CH2), 2.79 (t, 4H, J = 12.8, 6.4 Hz, 2 × NCH2CH2) ppm. 13C-NMR (CDCl3, 100 MHz): showed the presence of two rotamers. δ = 32.6, 34.3, 40.7, 45.1, 46.0, 49.8, 56.4, 56.5, 107.5, 107.7, 115.7, 115.9, 128.8, 129.9, 130.5, 131.2, 143.3, 155.0, 158.9, 163.3, 181.6, 186.9 ppm. DEPT-135 (CDCl3, 100 MHz) δ = 32.6 (CH2), 34.3 (CH2), 40.7 (CH2), 45.1 (CH2), 46.0 (CH2), 49.8 (CH2), 56.4 (CH3), 56.5 (CH3), 107.5 (CH), 107.7 (CH), 115.6 (CH), 115.9 (CH), 129.9 (CH), 130.4 (CH), 131.1 (CH), 163.3 (CH) ppm. LR-MS (ESI): m/z for [C17H16BrNO4]−: 376.03.
CHCO), 6.80, 6.74 (d, 4H, J = 8 Hz, 4 × CHCHCO), 6.44, 6.41 (s, 1H, CHCO), 5.96, 5.93 (s, 1H, CHCOCH3), 4.35, 4.10 (s, 2H, CH2N), 3.85 (s, 6H, 2 × OCH3), 3.49 (t, 4H, J = 13.6, 6.8 Hz, 2 × NCH2CH2), 2.79 (t, 4H, J = 12.8, 6.4 Hz, 2 × NCH2CH2) ppm. 13C-NMR (CDCl3, 100 MHz): showed the presence of two rotamers. δ = 32.6, 34.3, 40.7, 45.1, 46.0, 49.8, 56.4, 56.5, 107.5, 107.7, 115.7, 115.9, 128.8, 129.9, 130.5, 131.2, 143.3, 155.0, 158.9, 163.3, 181.6, 186.9 ppm. DEPT-135 (CDCl3, 100 MHz) δ = 32.6 (CH2), 34.3 (CH2), 40.7 (CH2), 45.1 (CH2), 46.0 (CH2), 49.8 (CH2), 56.4 (CH3), 56.5 (CH3), 107.5 (CH), 107.7 (CH), 115.6 (CH), 115.9 (CH), 129.9 (CH), 130.4 (CH), 131.1 (CH), 163.3 (CH) ppm. LR-MS (ESI): m/z for [C17H16BrNO4]−: 376.03.
Procedure for the synthesis of compound 6
A mixture of 5 (1 mmol, 1.0 eq.) and DBU (1.8 mmol, 1.8 eq.) in dichloromethane (20 mL) was stirred at 0 °C until no started material was detected by TLC. The mixture was then poured into HCl (0.5 M, 40 mL) and extracted with dichloromethane (3 × 40 mL). The organic layer was then dried over sodium sulfate and evaporated under reduced pressure. The residue was purified by silica gel gradient column chromatography (CHCl3/MeOH 40:1) to afford 6 (yield = 91%).1H-NMR (DMSO-d6, 400 MHz): showed the presence of two rotamers. δ = 8.13, 8.11 (s, 1H, CHO), 7.26–7.23, 7.21–7.18 (dd, 1H, J = 10.4, 2 Hz, COCHCH), 7.13, 7.09 (s, 1H, ArH), 5.99–5.96 (s, 1H, COCHCH), 5.44–5.40, 5.03–4.99, 4.76–4.72 (d, 2H, J = 16.2, ArCH2N), 4.30–4.26 (d, 1H, J = 15.6 Hz, CHOAr), 4.03–4.00, 3.72–3.65, 3.14–3.08, 2.83–2.79, 1.99–1.92 (m, 4H, NCH2CH2), 3.83 (s, 3H, OCH3) 2.31–2.28, 2.39–2.20 (d, 2H, J = 14.0 Hz, CHCH2CO). 13C-NMR (DMSO-d6, 100 MHz): showed the presence of two rotamers. δ = 34.3, 37.6, 45.6, 45.7, 49.8, 50.0, 51.1, 56.6, 87.8, 112.1, 113.2, 116.6, 116.7, 127.1, 128.3, 128.4, 131.7, 131.9, 144.5, 144.6, 144.8, 145.0, 147.3, 147.5, 162.9, 195.1 ppm. MS (ESI): m/z for [C17H16BrNO4]+: 378.03.
Procedure for the synthesis of compound 3
A solution of L-selectride (2.35 mmol, 2.35 eq.) in THF (2.35 mL, 1.0 M solution) was added to a solution of 6 (1 mmol, 1.0 eq.) in THF (20 mL) and the mixture was stirred for 6 h at −78 °C. After this period, a suspension of LiAlH4 (2.35 mmol, 2.35 eq.) in THF (6 mL) was added to the reaction mixture and it was stirred for 12 h at room temperature. After this time, an aqueous solution of sodium sulfate was added to the reaction mixture that was then extracted with chloroform. The organic layer was dried over sodium sulfate and evaporated under reduced pressure. The residue was dissolved in methanol and 47% aq. HBr in methanol was added and the solution was stirred for 1 h at less than 30 °C. Galantamine 3 was then isolated by filtration, washed with MeOH, and dried under reduced pressure (yield = 61%). 1H-NMR (DMSO-d6, 400 MHz): δ = 9.80 (br, 1H), 6.85 (d, 1H, J = 8.0 Hz), 6.81 (d, 1H, J = 8.4 Hz), 6.17 (d, 1H, J = 10 Hz), 5.91–5.89 (dd, 1H, J = 4, 14 Hz), 4.80 (d, 1H, J = 13.2 Hz), 4.60 (s, 1H), 4.50 (s, 1H), 4.35 (bs, 1H), 4.10 (s, 1H), 3.76 (m, 4H), 3.51 (d, 1H), 2,85 (bs, 1H) 2.50–2.10 (m, 3H), 1.90 (bs, 1H) ppm. 13C-NMR (DMSO-d6, 100 MHz): 31.4, 46.8, 55.3, 56.1, 59.9, 86.8, 112.4, 122.6, 125.7, 130.4, 133.5, 145.4, 146.9 ppm. MS (ESI): m/z for [C17H20BrNO3]+: 366.08.
Procedure for the synthesis of compound 7
Formaldehyde (0.22 mmol, 1.05 eq.) was added to a solution of III (0.21 mmol, 1 eq.) in MeOH (2 mL) and the mixture was stirred for 3 h. After this period, NaBH4 (0.23 mmol, 1.1 eq.) was added to the reaction mixture and stirred for 10 min at 0 °C and for 3 h at room temperature. The reaction mixture was then filtered over Celite® and the filtrate was evaporated under reduced pressure. The residue was purified by silica gel gradient column chromatography (CHCl3:MeOH 10:1) to afford 7 (yield = 96%).1H-NMR (CDCl3, 400 MHz): 7.07 (s, 1H, ArH), 7.05 (d, 2H, J = 8 Hz, ArH), 7.00 (s, 1H, ArH), 6.77 (d, 2H, J = 8 Hz), 3.88 (s, 3H, OCH3), 3.60 (s, 2H, ArCH2N), 2.81–2.78 (m, 2H, ArCH2CH2N), 2.71–2.68 (m, 2H, ArCH2CH2N), 2.34 (s, 3H, NCH3) ppm. 13C-NMR (CDCl3, 100 MHz): 32.5, 41.7, 50.8, 59.3, 60.4, 113.8, 114.9, 115.3, 116.9, 129.8, 145.1, 146.4, 153.9 ppm. MS (ESI): m/z for [C17H20BrNO3]−: 364.02.
Procedure for the synthesis of compound 10
Laccase (1000 U) was added to a stirred solution of compound 7 (1 mmol, 1 eq.) in 1,4-dioxane (5 mL) and acetate buffer, pH 4.5, 0.5 M (20 mL), and the mixture was stirred for 6 h under O2 atmosphere. After this period the aqueous solution was extracted with ethyl acetate (3 × 50 mL). The organic layer was washed with brine, dried over sodium sulfate and evaporated under reduced pressure. The residue was purified by silica gel gradient column chromatography (CHCl3/MeOH 15:1) to afford 10 (yield = 76%).1H-NMR (CDCl3, 400 MHz): 7.04 (d, 2H, J = 8.4 Hz, ArH), 6.99 (s, 1H, ArH), 6.77 (d, 2H, J = 8.4 Hz, ArH), 3.87 (s, 3H, OCH3), 3.75 (s, 2H, ArCH2N), 2.83 (br s, 4H, NCH2CH2Ar), 2.42 (s, 2H, ArCH2N) ppm. 13C-NMR (CDCl3, 100 MHz): 154.3, 146.2, 134.6, 131.0, 128.2, 125.8, 122.6, 116.7, 115.3, 108.8, 64.5, 58.4, 56.0, 53.2, 35.6 ppm. 35.6, 53.2, 56.0, 58.4, 64.5, 108.8, 115.3, 116.7, 122.6, 125.8, 128.2, 131.0, 134.6, 146.2, 157.3 MS (ESI): m/z for [C17H18BrNO3]−: 362.08.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by funds from: MIUR Ministero dell'Istruzione, dell'Università della Ricerca Italiano, project PRIN 2017, ORIGINALE CHEMIAE in Antiviral Strategy – Origin and Modernization of Multi-Component Chemistry as a Source of Innovative Broad Spectrum Antiviral Strategy, cod. 2017BMK8JR (L. B. and R. S.).
Notes and references
- A. I. Scott, Q. Rev., 1965, 19, 1 RSC
 .
. - D. H. Barton, R. Kirby, J. B. Taylor and G. M. Thomas, J. Chem. Soc., 1963, 4545–4558 RSC .
- M. Villarroya, A. G. García, J. Marco-Contelles and M. G. López, Expert Opin. Invest. Drugs, 2007, 16, 1987–1988 CrossRef CAS PubMed .
- B. Janssen and B. Schäfer, ChemTexts, 2017, 3, 7 CrossRef .
- J. Marco-Contelles, M. d. C. Carreiras, C. Rodríguez, M. Villarroya and A. G. García, Chem. Rev., 2006, 106, 116–133 CrossRef CAS PubMed .
- B. Küenburg, L. Czollner, J. Fröhlich and U. Jordis, Org. Process Res. Dev., 1999, 3, 425–431 CrossRef .
- M. Node, S. Kodama, Y. Hamashima, T. Katoh, K. Nishide and T. Kajimoto, Chem. Pharm. Bull., 2006, 54, 1662–1679 CrossRef CAS PubMed .
- K. B. Wiberg, H. Maltz and M. Okano, Inorg. Chem., 1968, 7, 830–831 CrossRef CAS .
- J. Szewczyk, A. H. Lewin and F. I. Carroll, J. Heterocycl. Chem., 1988, 25, 1809–1811 CrossRef CAS .
- A. Messerschmidt, Multi-Copper Oxidases, World Scientific Publishing Co. Pte. Ltd, Singapore, 1997 Search PubMed .
- L. Jönsson, K. Sjöström, I. Häggström and P. O. Nyman, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1995, 1251, 210–215 CrossRef .
- M. Mogharabi and M. A. Faramarzi, Adv. Synth. Catal., 2014, 356, 897–927 CrossRef CAS .
- T. Kudanga, G. S. Nyanhongo, G. M. Guebitz and S. Burton, Enzyme Microb. Technol., 2011, 48, 195–208 CrossRef CAS PubMed .
- N. Santhanam, J. M. Vivanco, S. R. Decker and K. F. Reardon, Trends Biotechnol., 2011, 29, 480–489 CrossRef CAS PubMed .
- F. Xu, Ind. Biotechnol., 2005, 1, 38 CrossRef CAS .
- S. Riva, Trends Biotechnol., 2006, 24, 219–266 CrossRef CAS PubMed .
- M. Fabbrini, C. Galli and P. Gentili, J. Mol. Catal. B: Enzym., 2002, 16, 231–240 CrossRef CAS .
- V. A. Golubev, Y. N. Kozlov, A. N. Petrov and A. P. Purmal, Catalysis of Redox Processes by Nitroxyl Radicals, Springer, Heidelberg, Berlin, 1992 Search PubMed .
- V. V. Abalyaeva and O. N. Efimov, Russ. J. Electrochem., 2002, 38, 1093–1097 CrossRef CAS .
- R. Bourbonnais, D. Leech and M. G. Paice, Biochim. Biophys. Acta, Gen. Subj., 1998, 1379, 381–390 CrossRef CAS .
- K. Li, F. Xu and K. L. Eriksson, Appl. Environ. Microbiol., 1999, 65, 2654 CrossRef CAS PubMed .
- O. V. Morozova, G. P. Shumakovich, S. V. Shleev and Y. I. Yaropolov, Appl. Biochem. Microbiol., 2007, 43, 523–535 CrossRef CAS .
- P. Galletti, F. Funiciello, R. Soldati and D. Giacomini, Adv. Synth. Catal., 2015, 357, 1840–1848 CrossRef CAS .
- R. Bourbonnais, M. G. Paice, B. Freiermuth, E. Bodie and S. Borneman, Appl. Environ. Microbiol., 1997, 63, 4627 CrossRef CAS PubMed .
- A. Marjasvaara, J. Jänis and P. J. Vainiotalo, Mass Spectrom., 2008, 43, 470–477 CrossRef CAS PubMed .
- B. Branchi, C. Galli and P. Gentili, Org. Biomol. Chem., 2005, 3, 2604–2614 RSC .
- P. Baiocco, A. M. Barreca, M. Fabbrini, C. Galli and P. Gentili, Org. Biomol. Chem., 2003, 1, 191–197 RSC .
- C. Galli and P. Gentili, J. Phys. Org. Chem., 2004, 17, 973–977 CrossRef CAS .
- G. Rousselet, P. Capdevielle and M. Maumy, Tetrahedron Lett., 1995, 36, 4999–5002 CrossRef CAS .
- E. Andrade-Jorge, L. A. Sánchez-Labastida, M. A. Soriano-Ursúa, J. A. Guevara-Salazar and J. G. Trujillo-Ferrara, Med. Chem. Res., 2018, 27, 2187–2198 CrossRef CAS .
- M. C. Carreño and A. Urbano, Synlett, 2005, 1, 1–25 Search PubMed .
- J. Szewczyk, J. W. Wilson, A. H. Lewin and F. I. Carroll, J. Heterocycl. Chem., 1995, 32, 195–199 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00935k |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a