
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhodium-catalyzed phosphorylation reaction of water-soluble disulfides using hypodiphosphoric acid tetraalkyl esters in water†
Mieko Arisawa *,
Kohei Fukumoto and
Masahiko Yamaguchi
*,
Kohei Fukumoto and
Masahiko Yamaguchi
Department of Organic Chemistry, Graduate School of Pharmaceutical Sciences, Tohoku University, Aoba, Sendai, 980-8578, Japan. E-mail: arisawa@m.tohoku.ac.jp
First published on 6th April 2020
Abstract
RhCl3 catalyzed the exchange reaction of disulfides and hypodiphosphoric acid tetraalkyl esters in water under homogeneous conditions, which indicated the hypodiphosphoric acid tetraalkyl esters to be novel and efficient phosphorylation reagents in water. The reaction was used in the phosphorylation of unprotected glutathione disulfide.
Chemical modification of biomacromolecules such as peptides and proteins is critical to tuning their biological activity. Such modification should be conducted in water, because of the solubility of such biomacromolecules. The reaction should also proceed selectively in the presence of diverse heteroatom functional groups without requiring the use of protecting groups. Transition-metal-catalysis can be an effective method for chemical modification, although the availability of such reactions in homogeneous water has been limited. The click reaction has been used for such purposes, which, however, requires the introduction of an alkyne moiety into the biomacromolecules.1a–c Direct chemical modification of peptides and proteins is preferable, and reactions at tyrosine, tryptophan, and histidine residues have been examined.2a–d Disulfide RS–SR is an important sulfur functional group contained in cysteine residues and is involved in the construction of tertiary structures of proteins, which can exhibit chemical reactivities different from those of nitrogen and oxygen functional groups. Derivatization of disulfides is an interesting subject for the modification of the structure and function of biomacromolecules. Conventional methods for protein disulfide modification generally involve two-step procedures of reduction to thiol RS–H followed by and alkylation to form sulfides RS–R′.3a–e One-step procedures involving direct cleavage of the S–S bond in disulfides and transfer of the organothio group to other organic molecules are attractive for such modifications because of their simplicity. In addition, disulfides are more stable and easier to handle than thiols. Previously, we developed the S–S bond exchange reaction of glutathione and cystine in homogeneous water,4 which was applicable to chemical modification of insulin.5 The disulfide exchange reaction is in the equilibrium state; accordingly, it is necessary to use one of the substrates in large excess to effectively obtain the products.
It has also been described that a rhodium complex can cleave and exchange the P–P bond of tetraalkyldiphosphine disulfides with the S–S bond of disulfides in organic solvents.6 This reaction using an equimolar amount of the substrates provides quantitative amount of phosphinothioates, because of the energetically favorable nature of the reaction. It was then considered that the phosphorylation could be used for the effective modification of peptide disulfides in water, provided that an appropriate water-soluble phosphorylation reagent could be developed. Described herein is the rhodium-catalyzed phosphorylation of water-soluble disulfides using hypodiphosphoric acid tetraalkyl esters in water (Scheme 1). RhCl3 catalytically cleaved and exchanged S–S and P–P bonds, and the method was successfully applied to the phosphorylation of glutathione in water. The hypodiphosphoric acid tetraalkyl esters have not been used as phosphorylation reagents, and their high reactivity in the presence of rhodium catalyst is shown herein.
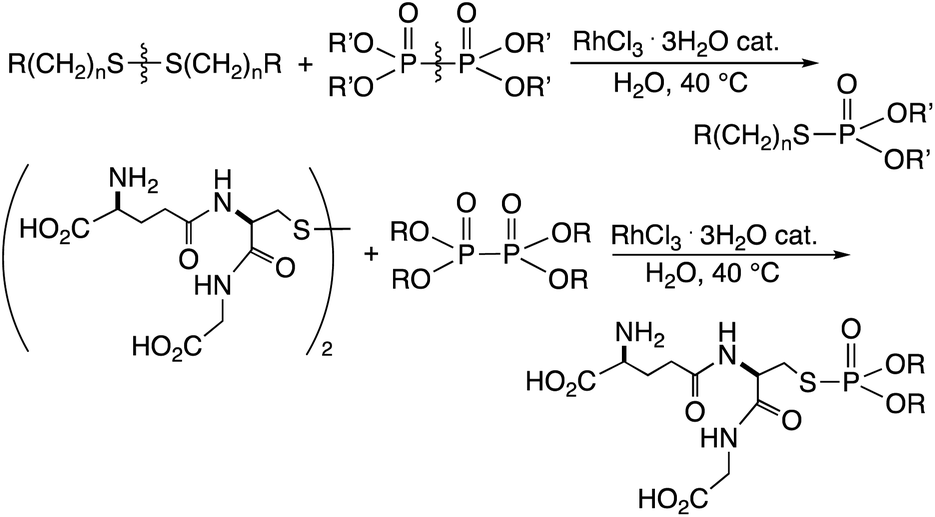 | ||
| Scheme 1 Rhodium-catalyzed phosphorylation reaction of water-soluble disulfides using hypodiphosphoric acid tetraalkyl esters in water. | ||
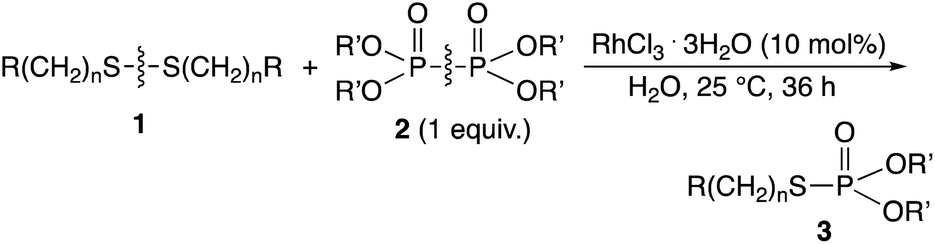
Diphosphine and diphosphine disulfides with aryl and alkyl groups that we previously studied are water-insoluble.6,7a–c In contrast, hypodiphosphoric acid tetraalkyl esters containing methyl, ethyl, butyl, and cyclic alkyl groups were found to be water-soluble and could be used as phosphorylation reagents in water in the presence of a rhodium catalyst. The hypodiphosphoric acid tetraalkyl esters were synthesized by copper-catalyzed dehydrogenative couplings of diethyl phosphonate.8 A reaction of hypodiphosphoric acid tetraalkyl esters involving P–P bond cleavage was reported, in which hypodiphosphoric acid tetraethyl ester 2a thermally isomerized to diphosphoric(III, V) acid tetraethyl ester at 190–200 °C.9 We here employed the water-solubled hypodiphosphoric acid tetraalkyl esters for the phosphorylation reaction of disulfides in water. There have been no reports of direct phosphorylation of organodisulfides using hypodiphosphoric acid tetraalkyl esters. The thiophosphorylation reaction by P–S bond formation has generally been conducted in organic solvents.10a–d,11a–c,12a–c Phosphorylation reactions of cysteine peptides at SH groups and thiophosphorylation to α,β-unsaturated carbonyl units of peptides in water were reported.13a–c
Rhodium-catalyzed phosphorylation of di(hydroxyalkyl)disulfides using hypodiphosphoric acid tetraalkyl esters as reagents was examined in a homogeneous water solution. When di(3-hydroxypropyl)disulfide 1a was treated with 2a (1 equiv.) in the presence of RhCl3·3H2O (10 mol%) in water at 25 °C for 36 h, O,O-diethyl S-(3-hydroxypropyl)phosphorothioate 3aa (52%) was obtained, which was accompanied by diethyl phosphate 4 (20%) and phosphorous acid diethyl ester 5 (20%) and the recovery of 1a (40%) (Scheme 2). 2a was not recovered, and the formation of 4 and 5 revealed the competitive reaction of 2a with water. Increasing the amount of 2a from 1 to 3 equiv. did not change the yield of 3aa (51%). The yield of 3aa was improved to 66% using 25 mol% of the catalyst. It was noted that these mixtures provided pH 3. Reactions in phosphate buffer (pH 7.4) and aqueous ammonia (pH 10.0) gave 3aa in 50%, 49% yields, respectively. The formation of 3aa was not much affected by pH. When the reaction was conducted at 40 °C for 20 h and at reflux for 3 h, the yield of 3aa decreased to 28% and 38%, respectively. No reaction occurred in the absence of RhCl3·3H2O. Other metal complexes such as [Rh(OAc)2]2 (7%), [Rh(cod)2]+BF4− (5%), and PdCl2·2NaCl·3H2O (5%) were ineffective even when 40 mol% of the catalyst at reflux for 3 h was used.
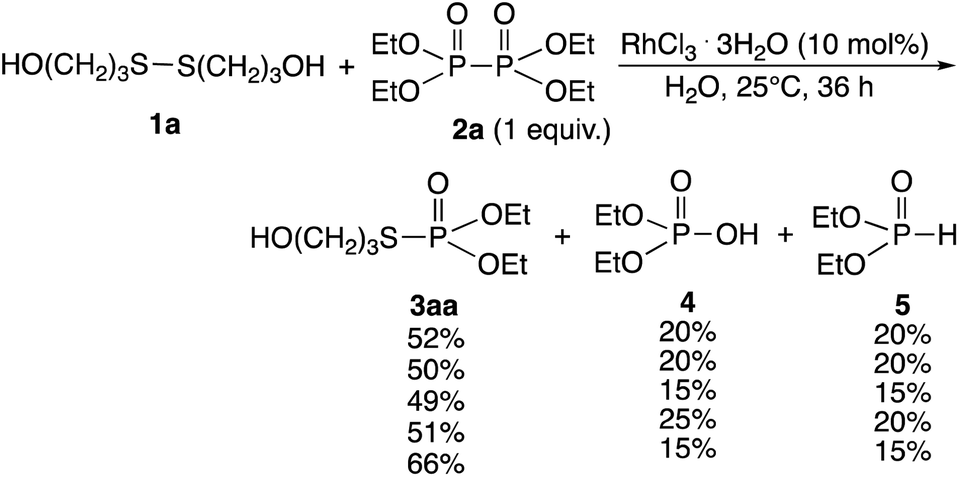 | ||
| Scheme 2 Rhodium-catalyzed phosphorylation reaction of disulfide 1a using hypodiphosphoric acid tetraethyl esters 2a. aUnder phosphate buffer (pH 7.4). bUnder aqueous ammonia (pH 10.0). cUse of 2a (3 equiv.). dUse of RhCl3 (25 mol%). | ||
It was determined that 4 and 5 were formed by the reaction of 2a and water in the presence of RhCl3·3H2O. When 2a was reacted in excess water in the presence of RhCl3·3H2O (10 mol%) at 25 °C for 36 h, 4 (20%) and 5 (20%) were obtained (Scheme 3). The hydrolytic reaction rate was considerably lower than the phosphorylation reaction rates of 1a and 2a. Neither 4 nor 5 was formed in the absence of RhCl3·3H2O. 3aa was not formed by the reaction of 4 or 5 with 1a, which confirmed that 3aa was directly formed from 2a.
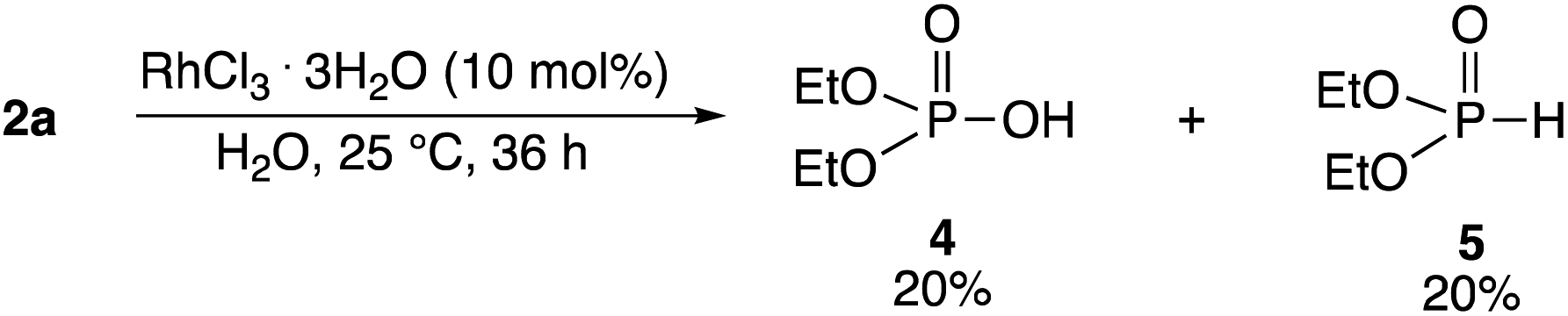 | ||
| Scheme 3 Formation of diethyl phosphate 4 and phosphorous acid diethyl ester 5 from 2a in water. | ||
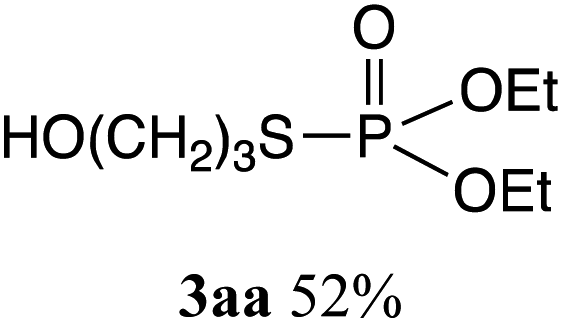
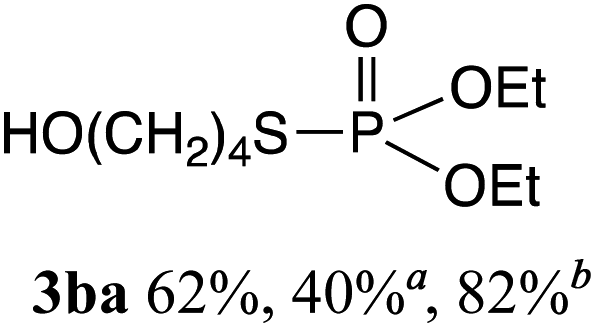
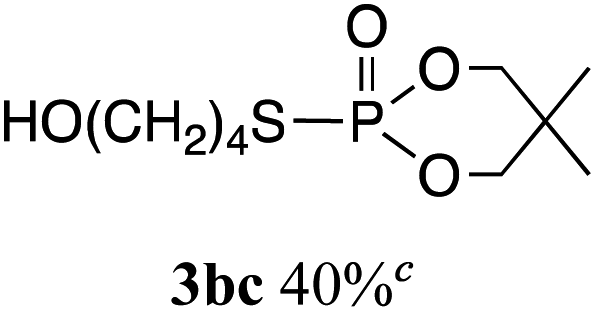
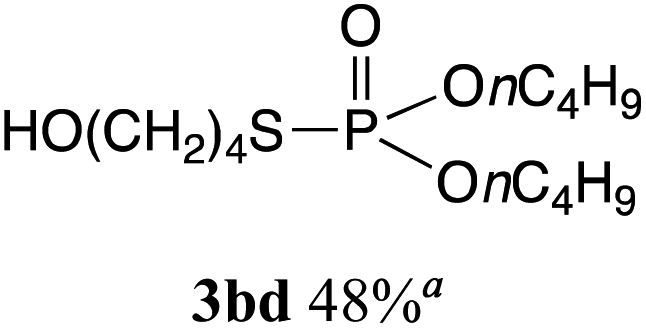
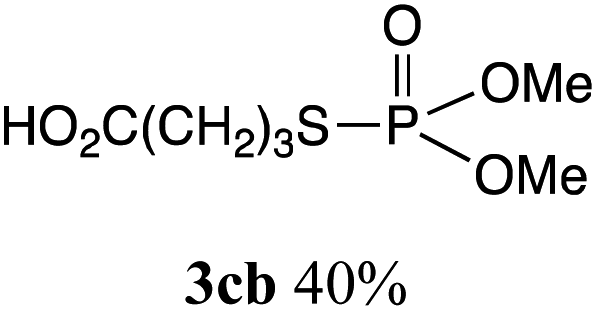
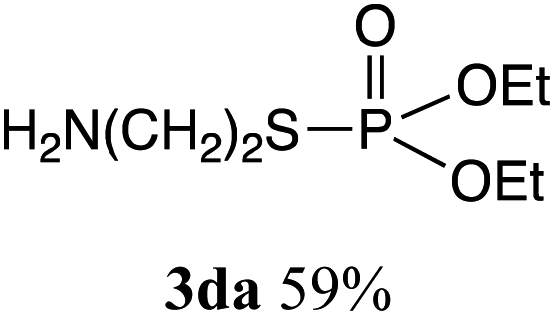
The phosphorylation of water-soluble disulfides was examined (Table 1). The reaction of di(4-hydroxybutyl)disulfides 1b and 2a proceeded in water at 25 °C for 36 h, and 3ba was obtained in 62% yield. The reaction of hypodiphosphoric acid tetramethyl esters 2b and 1b gave 3bb in 52% yield. The yields of 3ba (40%) and 3bb (46%) decreased when the reaction was conducted at 40 °C for 20 h. The reaction of 1b with hypodiphosphoric acid tetrabutyl ester 2c and 5,5,5′,5′-tetramethyl-bis-(1,3,2)-dioxaphosphinane 2,2′-dioxide 2d gave the corresponding products 3bc (40%) and 3bd (48%) at elevated temperatures of 40 °C and 70 °C, respectively. The reaction did not proceed at 25 °C because of the lower solubility of 2c and 2d in water: 2c was not soluble in water at 25 °C but became soluble at 70 °C. Analogously, the reactions of bis(3-carboxybutyl)disulfide 1c with 2a and 2b in water gave the phosphorylated products 3ca and 3cb in 48% and 40%, respectively. The reaction of bis(2-aminoethyl)disulfide 1d and 2a also proceeded to give the corresponding product 3da (59%).

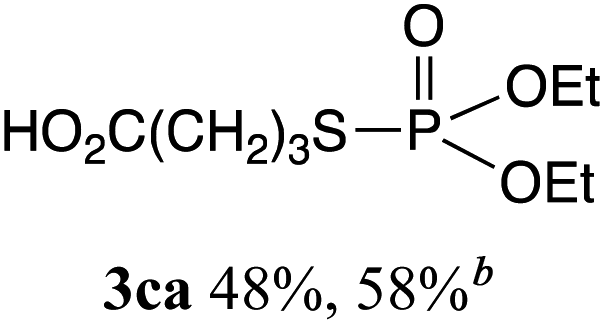
Water-soluble diselenides 6a–6c was reacted with 2a at 25 °C for 36 h in water, and the phosphorylated 7aa–7ac with the P–Se bonds were obtained in good yields (Scheme 4). The cleavage and exchange of S–S/Se–Se bonds and P–P bonds effectively using the rhodium complex provided P–S/P–Se bonds.
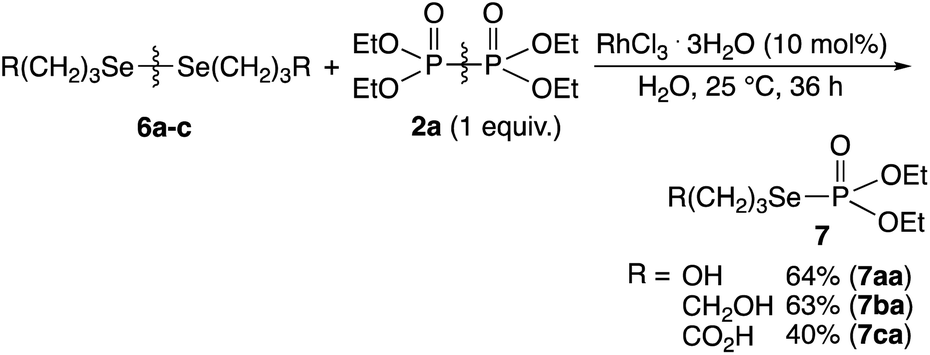 | ||
| Scheme 4 Rhodium-catalyzed phosphorylation reaction of water-soluble diselenides using hypodiphosphoric acid tetraethyl ester 2a. | ||
The reaction was applied to unprotected glutathione disulfide 8 (Table 2). When 8 and 2a (1 equiv.) were reacted in water at 40 °C for 36 h in the presence of RhCl3·3H2O (10 mol%), carboxymethyl N-[N-L-γ-glutamyl-S-(dimethoxyphosphinyl)-L-cysteinyl]glycine 9a was obtained in 77% yield; 9a was isolated by reversed-phase chromatography. When the reaction was conducted at concentrations of 0.125 M, 0.25 M, and 0.5 M at 25 °C in the presence of RhCl3·3H2O (20 mol%), 9a was obtained in 28%, 48%, and 55% yields, respectively. The yield of 9a improved with increasing concentration. The rhodium catalyst is essential in the phosphorylation reaction of 8. The reaction of 8 and the hypodiphosphoric acid tetramethyl ester 2b also gave the glutathione derivative 9b in 66% yield. The reaction of 5,5,5′,5′-tetramethyl-bis-(1,3,2)-dioxaphosphinane 2,2′-dioxide 2c proceeded at an elevated temperature of 70 °C to give the product 9c at 64% yield. Phosphorylation of glutathione (reduced form) in water did not form 9a. In addition, reactions of 2a with methionine, tyrosine, histidine, and tryptophan under rhodium-catalyzed condition did not proceed. 2a and RhCl3·3H2O can likely phosphorylate disulfides in peptides without being affected by hydroxy, carboxy, amino, sulfide, phenol, thiol, imidazolyl, and indolyl groups.
|
||
|---|---|---|
| Entry | R | Yield |
| a Reaction concentration: 0.5 M. Reaction time: 24 h.b Use of RhCl3 3H2O (20 mol%). Reaction time: 24 h.c Reaction temperature: 25 °C.d Reaction concentration: 0.125 M.e Reaction concentration: 0.5 M.f Without RhCl3·3H2O.g Reaction temperature: 70 °C. | ||
| 1 | CH3CH2 (2a) | 77%, 64%a, 72%b, 28%c,d, 48%b,c, 55%b,e, n.d.f (9a) |
| 2 | CH3 (2b) | 66% (9b) |
| 3 | 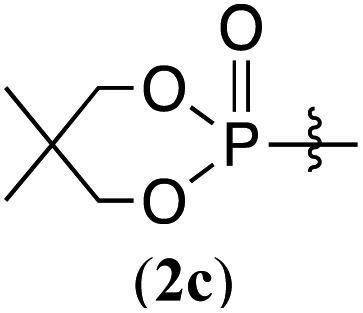 |
64%g (9c) |

A similar reaction mechanism for rhodium-catalyzed phosphorylation in organic solvents may be considered involving a rhodium(I) phosphonate intermediate, which is formed from hypodiphosphoric acid tetraalkyl esters and RhCl3 (Fig. 1).7a,b Oxidative addition of hypodiphosphoric acid tetraalkyl esters to rhodium(I) phosphonate provides a rhodium(III) triphosphonate intermediate, which undergoes an organothio exchange reaction with the disulfide R′SSR′ forming the R′S–Rh(III)–P(O)(OR)2 complex and a thiophosphorylated product. Another thiophosphorylated product may be liberated by reductive elimination with the regeneration of the rhodium(I) phosphonate.
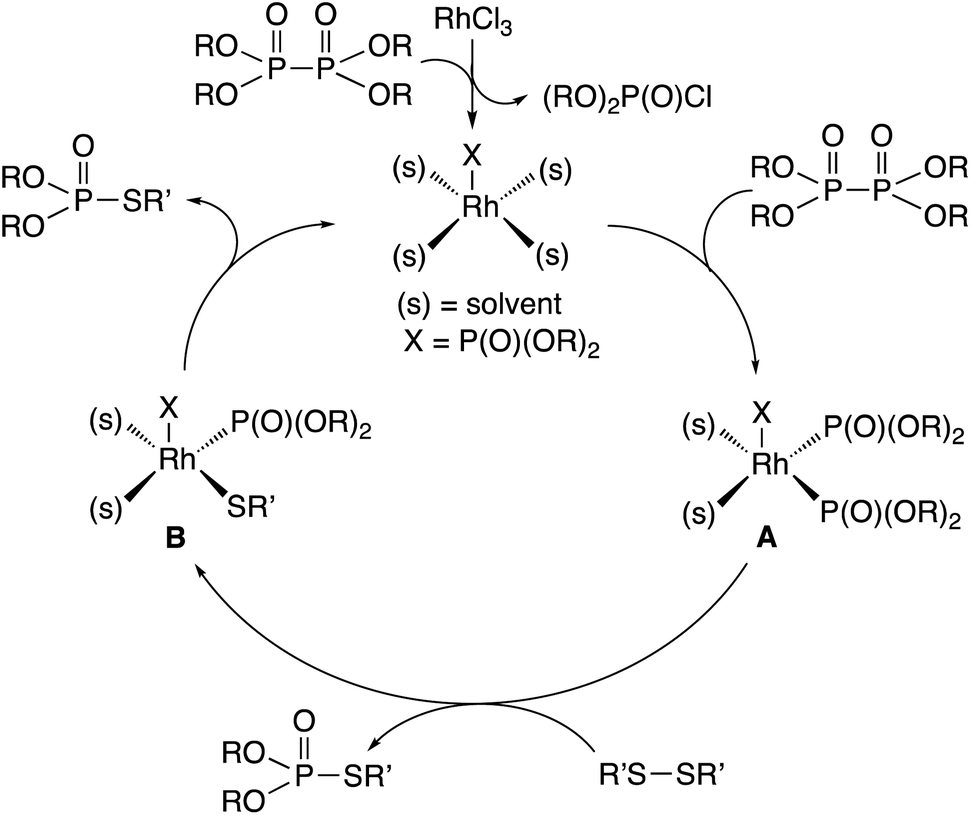 | ||
| Fig. 1 Proposed reaction mechanism. | ||
Conclusions
In summary, RhCl3 catalyzed the phosphorylation reaction of water-soluble disulfides, including unprotected glutathione disulfide, with hypodiphosphoric acid tetraalkyl esters in water under homogeneous conditions. We have shown that RhCl3 effectively activates disulfides in water without being affected by presence of the amino and carboxy groups. This method may be applicable to complex proteins. This reaction is of additional interest because phosphorylation of serine and tyrosine is a critical process for signal transduction in biological cells, and cysteine phosphorylation can exhibit interestingly biological phenomena.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Platform Project for Supporting Drug Discovery and Life Science Research from AMED under Grant number JP18am0101100, JSPS KAKENHI Grant number 17K19112, and Tohoku University Center for Gender Equality Promotion (TUMUG).Notes and references
- For examples, (a) S. Li, L. Wang, F. Yu, Z. Zhu, D. Shobaki, H. Chen, M. Wang, J. Wang, G. Qin, U. J. Erasquin, L. Ren, Y. Wang and C. Cai, Chem. Sci., 2017, 8, 2107 RSC; (b) J. Martell and E. Weerapana, Molecules, 2014, 19, 1378 CrossRef PubMed; (c) S. I. Presolski, V. P. Hong and M. G. Finn, Curr. Protoc. Chem. Biol., 2011, 3, 153 CrossRef PubMed.
- For examples, (a) J. Ohata, M. B. Minus, M. E. Abernathy and Z. T. Ball, J. Am. Chem. Soc., 2016, 138, 7472 CrossRef CAS PubMed; (b) S. D. Tilley and M. B. Francis, J. Am. Chem. Soc., 2006, 128, 1080 CrossRef CAS PubMed; (c) J. M. Antos and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 10256 CrossRef CAS PubMed; (d) Review: E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef CAS PubMed.
- For examples, (a) G. D'Alessio, M. C. Maloni and A. Parente, Biochemistry, 1975, 14, 1116 CrossRef PubMed; (b) D. L. Sondack and A. Light, J. Biol. Chem., 1971, 246, 1630 CAS; (c) P. A. Price, W. H. Stein and S. Moore, Disulfide bridged modification of proteins via reduction, J. Biol. Chem., 1969, 244, 929 CAS; (d) Review: S. L. Kuan, T. Wang and T. Weil, Chem.–Eur. J., 2016, 22, 17112 CrossRef CAS PubMed; (e) S. Brocchini, A. Godwin, S. Balan, J. Choi, M. Zloh and S. Shaunak, Adv. Drug Delivery Rev., 2008, 60, 3 CrossRef CAS PubMed.
- M. Arisawa, A. Suwa and M. Yamaguchi, J. Organomet. Chem., 2006, 691, 1159 CrossRef CAS.
- M. Arisawa, M. Kuwajima, A. Suwa and M. Yamaguchi, Heterocycles, 2010, 80, 1239 CrossRef CAS.
- M. Arisawa, T. Ono and M. Yamaguchi, Tetrahedron Lett., 2005, 46, 5669 CrossRef CAS.
- For examples, (a) M. Arisawa, T. Tazawa, W. Ichinose, H. Kobayashi and M. Yamaguchi, Adv. Synth. Catal., 2018, 360, 3488 CrossRef CAS; (b) M. Arisawa, T. Yamada, S. Tanii, Y. Kawada, H. Hashimoto and M. Yamaguchi, Chem. Commun., 2016, 52, 13580 RSC; (c) M. Arisawa and M. Yamaguchi, Tetrahedron Lett., 2010, 51, 4840 CrossRef CAS.
- Y. Zhou, S. Yin, Y. Gao, Y. Zhao, M. Goto and L.-B. Han, Angew. Chem., Int. Ed., 2010, 49, 6852 CrossRef CAS PubMed.
- J. Michalski, W. Stec and A. Zwierzak, Chem. Ind., 1965, 8, 347 Search PubMed.
- For examples of nucleophilic substitution, (a) C. M. Timperley, S. A. Saunders, J. Szpalek and M. J. Waters, J. Fluorine Chem., 2003, 119, 161 CrossRef CAS; (b) P.-Y. Renard, H. Schwebel, P. Vayron, L. Josien, A. Valleix and C. Mioskowski, Chem.–Eur. J., 2002, 8, 2910 CrossRef CAS PubMed; (c) T.-L. Au-Yeung, K.-Y. Chan, W.-K. Chan, R. K. Haynes, I. D. Williams and L. L. Yeung, Tetrahedron Lett., 2001, 42, 453 CrossRef CAS; (d) R. G. Harvey, H. I. Jacobson and E. V. Jensen, J. Am. Chem. Soc., 1963, 85, 1623 CrossRef CAS.
- For examples of oxidative coupling of thiols and phosphonates, (a) H. Huang, J. Ash and J. Y. Kang, Org. Biomol. Chem., 2018, 16, 4236 RSC; (b) S. Song, Y. Zhang, A. Yeerlan, B. Zhu, J. Liu and N. Jiao, Angew. Chem., Int. Ed., 2017, 56, 2487 CrossRef CAS PubMed; (c) Y. Moon, Y. Moon, H. Choi and S. Hong, Green Chem., 2017, 19, 1005 RSC.
- Using phosphorothioates, (a) D. J. Jones, E. M. O'Leary and T. P. O'Sullivan, Tetrahedron Lett., 2018, 59, 4279 CrossRef CAS; (b) X.-Y. Chen, M. Pu and H.-G. Cheng, Angew. Chem., Int. Ed., 2019, 58, 11395 CrossRef CAS PubMed; (c) X. Zhang, Z. Shi, C. Shao, J. Zhao, D. Wang, G. Zhang and L. Li, Eur. J. Org. Chem., 2017, 1884 CrossRef CAS.
- (a) M. J. Piggott and P. V. Attwood, Amino Acids, 2017, 49, 1309 CrossRef CAS PubMed; (b) J. Bertran-Vicente, M. Penkert, O. Nieto-Garcia, J.-M. Jeckelmann, P. Schmieder, E. Krause and C. P. R. Hackenderger, Nat. Commun., 2016, 7, 12703 CrossRef CAS PubMed; (c) A. K. Buchowiecka, Amino Acids, 2019, 51, 1365 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02377a |
| This journal is © The Royal Society of Chemistry 2020 |