
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA luminescence-based assay for monitoring changes in alpha-synuclein aggregation in living cells†
Travis J. Nelson‡
a,
Tiffany Truong‡§
a,
BaoLong Truonga,
Camden V. Bilyeua,
Jia Zhaoa and
Cliff I. Stains *abcde
*abcde
aDepartment of Chemistry, University of Nebraska – Lincoln, Lincoln, NE 68588, USA. E-mail: cstains@virginia.edu
bNebraska Center for Integrated Biomolecular Communication, University of Nebraska – Lincoln, Lincoln, NE 68588, USA
cCancer Genes and Molecular Regulation Program, Fred & Pamela Buffet Cancer Center, University of Nebraska Medical Center, Omaha, NE 68198, USA
dDepartment of Chemistry, University of Virginia, Charlottesville, VA 22904, USA
eUniversity of Virginia Cancer Center, University of Virginia, Charlottesville, VA 22904, USA
First published on 28th April 2020
Abstract
Parkinson's disease is characterized by the accumulation of protein aggregates in the brain, termed Lewy bodies. Lewy bodies are predominantly composed of α-synuclein and mutations that increase the aggregation potential of α-synuclein have been associated with early on-set disease. Assays capable of reporting on the solubility of α-synuclein in living cells could provide a means to interrogate the influence of mutations on aggregation as well as identify small molecules capable of modulating the aggregation of α-synuclein. Herein, we repurpose our previously reported self-assembling NanoLuc luciferase fragments to engineer a platform for detecting α-synuclein solubility in living cells. This new assay is capable of reporting on changes in α-synuclein solubility caused by disease-relevant mutations as well as inhibitors of aggregation. In the long term, this new assay platform provides a means to investigate the influence of mutations on α-synuclein solubility as well as identify potential tool compounds capable of modulating α-synuclein aggregation.
Parkinson's disease (PD) is a major neurodegenerative disease, affecting an estimated 572 out of every 100
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people in the US over the age of 45,1 and is characterized by the formation of protein aggregates termed Lewy bodies (LBs). LBs consist primarily of α-synuclein2,3 and display a cross-β structure.4 Evidence indicates that LBs can be propagated in patients in a mechanism reminiscent of prion protein propagation.5 Importantly, mutations that increase the aggregation potential of α-synuclein (αSYN) have been associated with more aggressive disease phenotypes.6 Thus, methods for assessing αSYN solubility in living cells could ultimately provide insights into mutant αSYN aggregation potential as well as aid in the identification of compounds capable of modulating αSYN aggregation.7,8
000 people in the US over the age of 45,1 and is characterized by the formation of protein aggregates termed Lewy bodies (LBs). LBs consist primarily of α-synuclein2,3 and display a cross-β structure.4 Evidence indicates that LBs can be propagated in patients in a mechanism reminiscent of prion protein propagation.5 Importantly, mutations that increase the aggregation potential of α-synuclein (αSYN) have been associated with more aggressive disease phenotypes.6 Thus, methods for assessing αSYN solubility in living cells could ultimately provide insights into mutant αSYN aggregation potential as well as aid in the identification of compounds capable of modulating αSYN aggregation.7,8
Classic methods for detection of protein aggregates rely on small molecule-based optical probes (such as thioflavin T and Congo red).9–11 While these methods remain the gold standards for in vitro experiments, recent efforts have focused on the development of approaches to assess protein aggregation within living systems.12–16 Our lab has developed a cell-based, bioluminescent assay system that relies on the rapid reassembly of fragmented NanoLuc luciferase (Nluc),17 an engineered luciferase that utilizes coelenterazine or analogues thereof as substrates.18–22 In this system, proteins of interest (POIs) are fused to the N-terminus of residues 1–65 of Nluc (termed N65). Reassembly with the complementary Nluc fragment, consisting of residues 66–171 (termed 66C), leads to reassembly of active Nluc and production of a luminescent signal. Changes in the solubility of the POI influences the amount of N65 available for reassembly, making luminescence from this assay system proportional to the solubility of the POI (Fig. 1). We have previously shown that this assay platform is capable of reporting the influence of mutations as well as small molecules on the solubility of amyloid-beta (Alzheimer's), huntingtin (Huntington's), and amylin (type 2 diabetes).23,24 Herein, we re-engineer this system to report on the solubility of αSYN in living cells. We demonstrate the ability to detect the influence of disease-relevant mutations and small molecules on the solubility of αSYN. In the long term, this new assay platform could be used to study the influence of mutations on aggregation in living cells as well as identify tool compounds capable of modulating aggregation.25–28
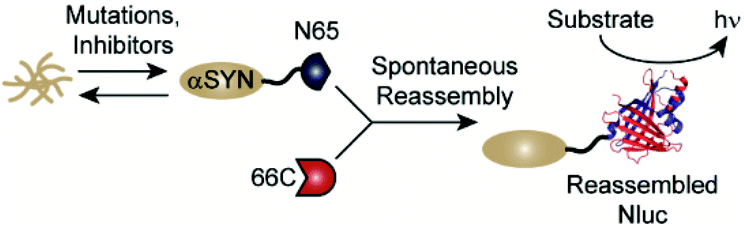 | ||
| Fig. 1 A split-Nluc assay system for interrogating changes in αSYN solubility. αSYN is covalently fused to the N-terminus of N65. Mutations to the αSYN sequence or treatment with inhibitors alters the amount of αSYN-N65 available for reassembly with 66C. Light production from reassembled Nluc (PDB: 5B0U) can be used as a proxy for soluble αSYN. | ||
In order to engineer a split-Nluc system for detection of αSYN solubility, we fused wild-type (WT)-αSYN to the N-terminus of N65. We then asked if this new assay platform was capable of reporting on the influence of mutations on αSYN solubility. Specifically, we chose the A30P6,29–31 and A53T32 mutations that have been shown to increase oligomerization of αSYN. In addition, we chose to investigate the influence of the G51D mutation that has been shown to decrease the rate of aggregation of αSYN.33 Accordingly, we performed site-directed mutagenesis to generate these mutants (Fig. 2a and Table S1†). Bacterial cells were transformed with each N65 fusion along with 66C.17 Expression was induced with IPTG, cultures were normalized for cell density, and luminescence from reassembled Nluc was determined. We observed 89% and 61% decreases in the luminescence of cells expressing the A30P and A53T mutants relative to WT-αSYN (Fig. 2b), consistent with previous reports.6,29–32 Interestingly, we note that previous work indicates that both A30P and A53T have a higher propensity to form oligomers in vitro as well as in cell-based assays.32,34 In order to further probe the ability of our system to detect αSYN oligomerization, we turned our attention to the disease-associated H50Q and E46K mutants that have been shown to display similar oligomer formation compared to WT-αSYN in vitro and in mammalian cells.34–36 As expected, the H50Q and E46K mutants display indistinguishable luminescent intensities in the assay compared to WT-αSYN (Fig. S1†). In addition, we assessed the influence of the synthetic A30P/A76P mutant, which has been shown to increase oligomerization.34,37 The A30P/A76P mutant displayed a significant 77% reduction in luminescence relative to WT-αSYN (Fig. S1†). Consequently, the split-Nluc system may be reporting on differences in the oligomer formation of these mutants relative to WT-αSYN, this observation is being further investigated by our laboratory. Conversely, the G51D mutant displayed a reproducible 12% increase in luminescence, in-line with previous observations of reduced aggregation kinetics of this mutant.33 Regardless of the specific identity of the aggregate species that inhibits reassembly of the split-Nluc assay system, this platform is clearly capable of reporting on established trends in αSYN aggregation as a result of mutations.
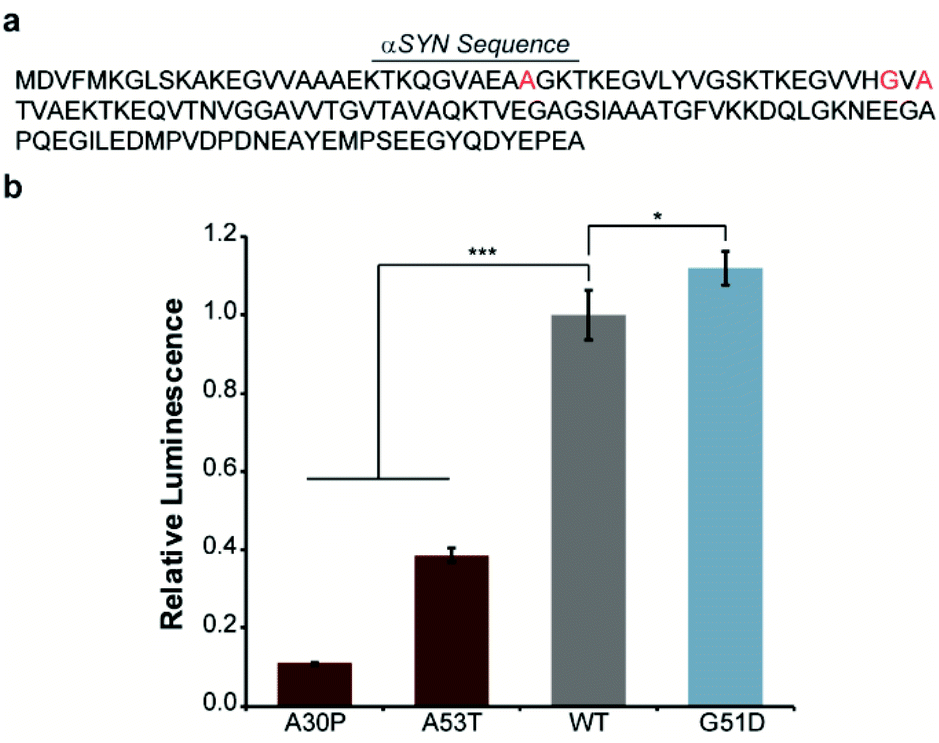 | ||
| Fig. 2 Disease-relevant mutations influence αSYN solubility. (a) The amino acid sequence of αSYN is shown with mutation sites indicated in red. (b) Luminescence from bacterial cells expressing the indicated mutant or wild-type (WT) αSYN fused to N65 in the presence of 66C. Error bars represent the standard deviation of two (A30P) or three (A53T, WT, and G51D) biological replicates assayed in triplicate. * indicates a p-value of <0.05 and *** indicates a p-value of <0.001. | ||
Next, we asked whether this re-engineered system was capable of reporting on the influence of small molecules on αSYN solubility. For validation purposes, we chose molecules that have been previously shown to disrupt αSYN aggregation both in vitro and in cells, namely EGCG38,39 and D-mannitol (Fig. 3a and b).40,41 Accordingly, bacteria expressing WT-αSYN-N65 and 66C were grown in the presence of these inhibitors, normalized to cell density, and assayed for luminescence. We observed a reproducible 88% increase in luminescence from cells cultured in the presence of 1 μM EGCG (Fig. 3c). Increasing the concentration of EGCG to 10 μM did not result in significantly increased luminescent signal (Fig. S2†) compared to 1 μM EGCG (Fig. 3c), indicating that EGCG was saturating under these experimental conditions. Likewise, we observed an 88% increase in luminescence from cells treated with 0.5 M D-mannitol (Fig. 3d). In agreement with previous literature, increasing the concentration of D-mannitol to 1 M resulted in inhibition of cell growth and a decrease in inhibitory activity as assessed by luminescence (Fig. S3†). This data supports the observation of a reversed dose-dependent response for D-mannitol.40 Taken together, these experiments demonstrate the ability of the split-Nluc system to report upon changes in αSYN solubility in response to known inhibitors of αSYN aggregation.
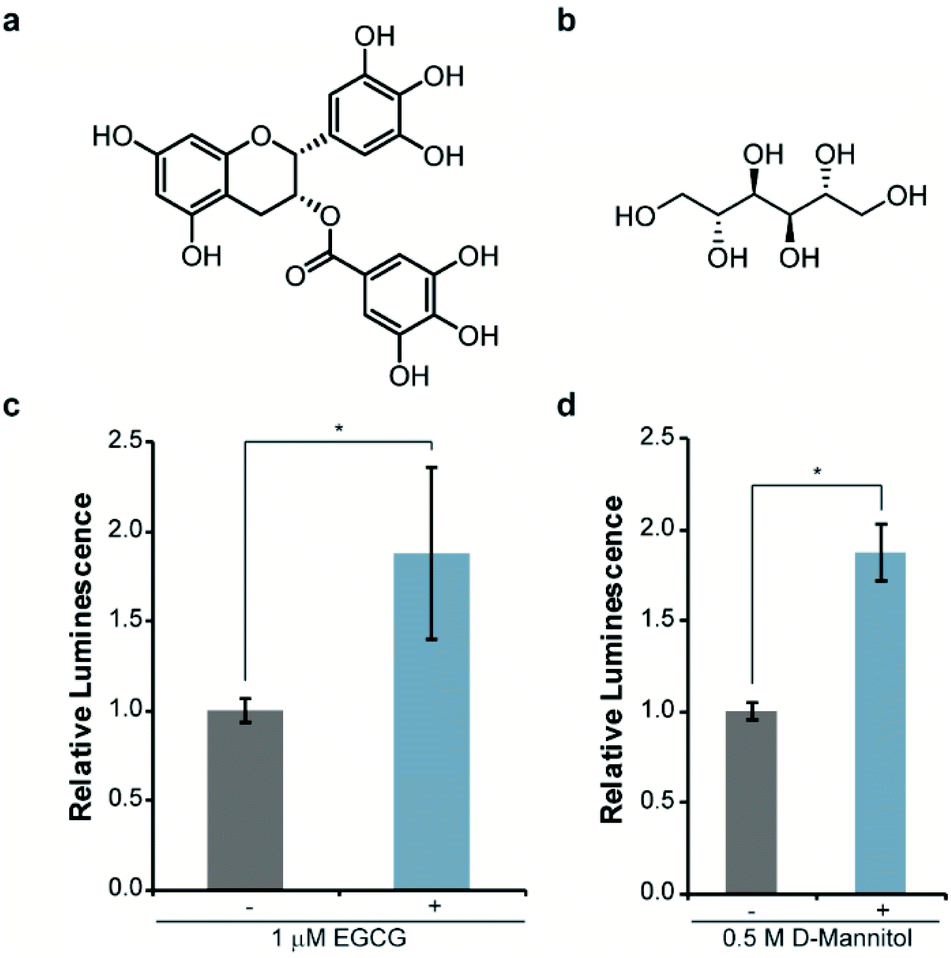 | ||
| Fig. 3 Monitoring changes in αSYN solubility upon treatment with known inhibitors of aggregation. The structures of EGCG (a) and D-mannitol (b) are shown. Luminescence from bacterial cells expressing WT-αSYN-N65 and 66C in the absence of presence of the indicated concentration of EGCG (c) or D-mannitol (d). Error bars represent the standard deviation of three (EGCG) or two (D-mannitol) biological replicates assayed in triplicate. * indicates a p-value of <0.05. | ||
Conclusions
We have re-engineered our split-Nluc assay system to provide a platform for the analysis of αSYN solubility in cells. This method provides a luminescent readout of αSYN solubility and is capable of reporting on the influence of mutations as well as small molecules on the aggregation of αSYN. We note that previous work indicates that αSYN is translocated to the periplasm of E. coli.42 Ongoing work in our lab is aimed at determining whether the split-Nluc system described herein detects periplasmic and/or cytosolic populations of αSYN. Nonetheless, the ability to detect previously reported changes in αSYN solubility in response to six different point mutations suggests that the system is reporting on changes in solubility. Moreover, treatment with two small molecule inhibitors known to increase the solubility of αSYN clearly results in an increase in luminescence of the assay system. Thus, while we cannot rule out changes in translocation influencing luminescence in all cases, the effects observed herein are attributed primarily to previously reported changes in αSYN solubility. In the long term, we envision that this approach could be utilized to probe fundamental aspects of αSYN folding as well as identify tool compounds capable modulating the aggregation of αSYN within cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the NIH (R35GM119751) and the University of Virginia. The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.References
- C. Marras, J. C. Beck, J. H. Bower, E. Roberts, B. Ritz, G. W. Ross, R. D. Abbott, R. Savica, S. K. Van Den Eeden, A. W. Willis, C. M. Tanner and P. G. Parkinson's Foundation, npj Parkinson's Dis., 2018, 4, 21 CrossRef CAS PubMed
.
- Q. Xia, L. Liao, D. Cheng, D. M. Duong, M. Gearing, J. J. Lah, A. I. Levey and J. Peng, Front. Biosci., 2008, 13, 3850–3856 CrossRef CAS PubMed
- M. G. Spillantini, M. L. Schmidt, V. M. Lee, J. Q. Trojanowski, R. Jakes and M. Goedert, Nature, 1997, 388, 839–840 CrossRef CAS PubMed
- M. D. Tuttle, G. Comellas, A. J. Nieuwkoop, D. J. Covell, D. A. Berthold, K. D. Kloepper, J. M. Courtney, J. K. Kim, A. M. Barclay, A. Kendall, W. Wan, G. Stubbs, C. D. Schwieters, V. M. Lee, J. M. George and C. M. Rienstra, Nat. Struct. Mol. Biol., 2016, 23, 409–415 CrossRef CAS PubMed
- K. Araki, N. Yagi, K. Aoyama, C. J. Choong, H. Hayakawa, H. Fujimura, Y. Nagai, Y. Goto and H. Mochizuki, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 17963–17969 CrossRef CAS PubMed
- L. Narhi, S. J. Wood, S. Steavenson, Y. Jiang, G. M. Wu, D. Anafi, S. A. Kaufman, F. Martin, K. Sitney, P. Denis, J. C. Louis, J. Wypych, A. L. Biere and M. Citron, J. Biol. Chem., 1999, 274, 9843–9846 CrossRef CAS PubMed
- J. A. Kritzer, S. Hamamichi, J. M. McCaffery, S. Santagata, T. A. Naumann, K. A. Caldwell, G. A. Caldwell and S. Lindquist, Nat. Chem. Biol., 2009, 5, 655–663 CrossRef CAS PubMed
- T. F. Outeiro and S. Lindquist, Science, 2003, 302, 1772–1775 CrossRef CAS PubMed
- M. Biancalana and S. Koide, Biochim. Biophys. Acta, 2010, 1804, 1405–1412 CrossRef CAS PubMed
- H. Puchtler and F. Sweat, J. Histochem. Cytochem., 1965, 13, 693–694 CrossRef CAS PubMed
- H. Puchtler, F. Sweat and M. Levine, J. Histochem. Cytochem., 1962, 10, 355–364 CrossRef CAS
- K. L. Maxwell, A. K. Mittermaier, J. D. Forman-Kay and A. R. Davidson, Protein Sci., 1999, 8, 1908–1911 CrossRef CAS PubMed
- G. S. Waldo, B. M. Standish, J. Berendzen and T. C. Terwilliger, Nat. Biotechnol., 1999, 17, 691–695 CrossRef CAS PubMed
- W. C. Wigley, R. D. Stidham, N. M. Smith, J. F. Hunt and P. J. Thomas, Nat. Biotechnol., 2001, 19, 131–136 CrossRef CAS PubMed
- Y. Liu, M. Fares, N. P. Dunham, Z. Gao, K. Miao, X. Jiang, S. S. Bollinger, A. K. Boal and X. Zhang, Angew. Chem., Int. Ed., 2017, 56, 8672–8676 CrossRef CAS PubMed
- K. H. Jung, S. F. Kim, Y. Liu and X. Zhang, ChemBioChem, 2019, 20, 1078–1087 CrossRef CAS PubMed
- T. J. Nelson, J. Zhao and C. I. Stains, Methods Enzymol., 2019, 622, 55–66 Search PubMed
- A. S. Dixon, M. K. Schwinn, M. P. Hall, K. Zimmerman, P. Otto, T. H. Lubben, B. L. Butler, B. F. Binkowski, T. Machleidt, T. A. Kirkland, M. G. Wood, C. T. Eggers, L. P. Encell and K. V. Wood, ACS Chem. Biol., 2016, 11, 400–408 CrossRef CAS PubMed
- M. P. Hall, J. Unch, B. F. Binkowski, M. P. Valley, B. L. Butler, M. G. Wood, P. Otto, K. Zimmerman, G. Vidugiris, T. Machleidt, M. B. Robers, H. A. Benink, C. T. Eggers, M. R. Slater, P. L. Meisenheimer, D. H. Klaubert, F. Fan, L. P. Encell and K. V. Wood, ACS Chem. Biol., 2012, 7, 1848–1857 CrossRef CAS PubMed
- O. Shimomura, T. Masugi, F. H. Johnson and Y. Haneda, Biochemistry, 1978, 17, 994–998 CrossRef CAS PubMed
- Y. Tomabechi, T. Hosoya, H. Ehara, S. I. Sekine, M. Shirouzu and S. Inouye, Biochem. Biophys. Res. Commun., 2016, 470, 88–93 CrossRef CAS PubMed
- H. W. Yeh, O. Karmach, A. Ji, D. Carter, M. M. Martins-Green and H. W. Ai, Nat. Methods, 2017, 14, 971–974 CrossRef CAS PubMed
- J. Zhao, T. J. Nelson, Q. Vu, T. Truong and C. I. Stains, ACS Chem. Biol., 2016, 11, 132–138 CrossRef CAS PubMed
- J. Zhao, Q. Vu and C. I. Stains, Mol. BioSyst., 2016, 12, 2984–2987 RSC
- W. Kim and M. H. Hecht, J. Mol. Biol., 2008, 377, 565–574 CrossRef CAS PubMed
- A. F. McKoy, J. Chen, T. Schupbach and M. H. Hecht, J. Biol. Chem., 2012, 287, 38992–39000 CrossRef CAS PubMed
- C. I. Stains, K. Mondal and I. Ghosh, ChemMedChem, 2007, 2, 1674–1692 CrossRef CAS PubMed
- C. Wurth, N. K. Guimard and M. H. Hecht, J. Mol. Biol., 2002, 319, 1279–1290 CrossRef CAS PubMed
- H. A. Lashuel, B. M. Petre, J. Wall, M. Simon, R. J. Nowak, T. Walz and P. T. Lansbury, Jr, J. Mol. Biol., 2002, 322, 1089–1102 CrossRef CAS PubMed
- J. Li, V. N. Uversky and A. L. Fink, Biochemistry, 2001, 40, 11604–11613 CrossRef CAS PubMed
- J. Li, V. N. Uversky and A. L. Fink, Neurotoxicology, 2002, 23, 553–567 CrossRef CAS PubMed
- K. A. Conway, S. J. Lee, J. C. Rochet, T. T. Ding, R. E. Williamson and P. T. Lansbury, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 571–576 CrossRef CAS PubMed
- M. B. Fares, N. Ait-Bouziad, I. Dikiy, M. K. Mbefo, A. Jovicic, A. Kiely, J. L. Holton, S. J. Lee, A. D. Gitler, D. Eliezer and H. A. Lashuel, Hum. Mol. Genet., 2014, 23, 4491–4509 CrossRef CAS PubMed
- D. F. Lazaro, E. F. Rodrigues, R. Langohr, H. Shahpasandzadeh, T. Ribeiro, P. Guerreiro, E. Gerhardt, K. Krohnert, J. Klucken, M. D. Pereira, B. Popova, N. Kruse, B. Mollenhauer, S. O. Rizzoli, G. H. Braus, K. M. Danzer and T. F. Outeiro, PLoS Genet., 2014, 10, e1004741 CrossRef PubMed
- D. Ghosh, M. Mondal, G. M. Mohite, P. K. Singh, P. Ranjan, A. Anoop, S. Ghosh, N. N. Jha, A. Kumar and S. K. Maji, Biochemistry, 2013, 52, 6925–6927 CrossRef CAS PubMed
- R. A. Fredenburg, C. Rospigliosi, R. K. Meray, J. C. Kessler, H. A. Lashuel, D. Eliezer and P. T. Lansbury, Jr, Biochemistry, 2007, 46, 7107–7118 CrossRef CAS PubMed
- D. P. Karpinar, M. B. Balija, S. Kugler, F. Opazo, N. Rezaei-Ghaleh, N. Wender, H. Y. Kim, G. Taschenberger, B. H. Falkenburger, H. Heise, A. Kumar, D. Riedel, L. Fichtner, A. Voigt, G. H. Braus, K. Giller, S. Becker, A. Herzig, M. Baldus, H. Jackle, S. Eimer, J. B. Schulz, C. Griesinger and M. Zweckstetter, EMBO J., 2009, 28, 3256–3268 CrossRef CAS PubMed
- J. Bieschke, J. Russ, R. P. Friedrich, D. E. Ehrnhoefer, H. Wobst, K. Neugebauer and E. E. Wanker, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7710–7715 CrossRef CAS PubMed
- D. E. Ehrnhoefer, J. Bieschke, A. Boeddrich, M. Herbst, L. Masino, R. Lurz, S. Engemann, A. Pastore and E. E. Wanker, Nat. Struct. Mol. Biol., 2008, 15, 558–566 CrossRef CAS PubMed
- R. Shaltiel-Karyo, M. Frenkel-Pinter, E. Rockenstein, C. Patrick, M. Levy-Sakin, A. Schiller, N. Egoz-Matia, E. Masliah, D. Segal and E. Gazit, J. Biol. Chem., 2013, 288, 17579–17588 CrossRef CAS PubMed
- A. Paul, B. D. Zhang, S. Mohapatra, G. Li, Y. M. Li, E. Gazit and D. Segal, Front. Mol. Biosci., 2019, 6, 16 CrossRef CAS PubMed
- G. Ren, X. Wang, S. Hao, H. Hu and C. C. Wang, J. Bacteriol., 2007, 189, 2777–2786 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02720k |
| ‡ These authors contributed equally. |
| § Present address: University of Nebraska Medical Center, Omaha, NE 68198, USA. |
| This journal is © The Royal Society of Chemistry 2020 |