
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptimization of double-vortex-assisted matrix solid-phase dispersion for the rapid determination of paraben preservative residues in leafy vegetables†
Chun-Ju Yanga,
Wu-Hsun Chungab and
Wang-Hsien Ding *a
*a
aDepartment of Chemistry, National Central University, Chung-Li 320, Taiwan. E-mail: wanghsiending@gmail.com; Fax: +886-3-4227664; Tel: +886-3-4227151 ext. 65905
bDepartment of Chemical Engineering, Army Academy ROC, Chung-Li 320, Taiwan
First published on 25th September 2020
Abstract
The extensive use of preservatives during the growth, transport and storage of vegetables has been a concern because of their known or suspected toxicity that jeopardizes human health. This paper reports the development of a technique that rapidly determines the presence of five paraben preservative residues in leafy vegetables using double-vortex-assisted matrix solid-phase dispersion (DVA-MSPD) and UHPLC-electrospray ionization(−)-quadrupole time-of-flight mass spectrometry detection. We simplified the original MSPD technique by eliminating the use of mortar/pestle and SPE-column procedures. The DVA-MSPD factors were screened by a multilevel categorical design, and then optimized by Box–Behnken Design plus response surface methodology. The limits of quantification were 1.2–1.8 ng g−1 (dry weight). The satisfactory average recoveries were 85–104% with RSDs less than 10%. The developed method was successfully employed for the rapid determination of selected paraben residues at trace-level in leafy vegetable samples.
1. Introduction
Parabens, a group of alkyl esters of p-hydroxybenzoic acid, are commonly employed as preservatives in foodstuff due to their broad spectrum of action against microorganisms, low cost, and high water solubility.1–3 Their widespread applications in foodstuff may be indicative of high levels of exposure to humans and as harmful to the environment. Parabens have been detected in many environmental matrices worldwide, such as in indoor air/dust, wastewater, aquatic biota, and soil/sediments, all of which have been reviewed by Piao et al.,4 and Ocaña-González et al.5 Even more worrisome, they have also been found in human urine, breast milk, and blood.6,7 Additionally, vegetables can be unexpectedly polluted by parabens through the irrigation by reclaimed wastewater. Therefore, these emerging contaminants have the potential to accumulate in roots and leaves, which can further jeopardize human health upon ingestion. Various extraction methods, such as solid–liquid extraction,2,3 QuEChERS (quick, easy, cheap, effective, rugged, and safe)1,8 and ultrasound-assisted extraction9,10 have been reported for the extraction of parabens in different vegetable samples. After extraction, various clean-up procedures, such as NH2-solid-phase extraction (SPE),2,3 multi-walled carbon nanotubes SPE,1 dispersive solid-phase extraction (d-SPE),10 and dispersive liquid–liquid microextraction (DLLME),9 have been applied. Although low concentrations of paraben residues in vegetables are assumed to not pose any acute risk, a simple and reliable method to determine their presences in vegetables would nevertheless be desirable, in order to best detect and prevent potential adverse effects caused by long-term exposure in the intake of these vegetables.Matrix solid-phase dispersion (MSPD), a popular sample pretreatment technique first reported by Barker and his group in 1989, has been widely used on a variety of semi-solid and solid biota or abiotic samples.11 The main advantage of MSPD is that extraction and clean-up can be performed in one step, so as to make the sample pretreatment procedure convenient, simple, and cost-saving. MSPD has been successfully applied to determine various organic pollutants in animal tissue, aquatic biota, and foodstuff, and these applications have been reviewed by Barker,12 Capriotti et al.,13,14 and Tu and Chen.15 Further improving on the efficiency of this method, two simplified procedures for the original MSPD have been also developed. Vortex-assisted MSPD (VA-MSPD) was first reported by Primel's group, the method that replaced the SPE-column by vortex agitation to eliminate the need for SPE-column packing and elution.16,17 The second simplified approach, known as vortex-homogenized MSPD (VH-MSPD), also utilizes vortex agitation to replace the use of a mortar and pestle for sample homogenization and blending.18,19 VH-MSPD can be used to homogenize and blend up to six samples at one time, without the time-consuming steps of cleaning the mortar and pestle between each sample preparation. In this study, we combined these two modifications, which increased sample throughput capability and simplified sample pretreatment steps, and developed a straightforward technique to rapidly determine organic contaminants in leafy vegetable samples.
The goal of this study was to develop a simple and straightforward double-vortex-assisted matrix solid-phase dispersion (DVA-MSPD) technique, for the rapid extraction of selected parabens in leafy vegetable samples. In comparison to the previous studies dealing with the analysis of parabens in vegetables, the novelty of the present work is to optimize the DVA-MSPD factors by using experimental design approaches instead of one-factor-at-a-time methodology to improve the optimization steps. These approaches can optimize factors together, minimize the number of experiments and reduce overall cost.20 In this study, Factorial Multilevel Categoric Design (FMLCD) was employed to screen and identify the important factors affecting DVA-MSPD, and then Box–Behnken Design (BBD) with response surface methodology was performed to determine the optimum points. Precision and accuracy of the developed method were evaluated, and the method's applicability and practicality for the determination trace-level of selected parabens in leafy vegetable samples were tested.
2. Experimental
2.1. Chemicals and reagents
All chemicals and solvents used in this study were of the highest available purity and were purchased from Sigma-Aldrich (St. Louis, MO, USA), Mallinckrodt Baker (Phillipsburg, NJ, USA) and Merck (Darmstadt, Germany). Analytical standards (all greater than 98%), viz., methyl-paraben (MeP), ethyl-paraben (EtP), propyl-paraben (PrP), butyl-paraben (BuP), and benzyl-paraben (BzP) were obtained from Alfa Aesar (Lancashire, UK), and were used for the evaluation and validation of the developed method. Their structures, chemical and physical properties can be found in Table S1.† Deuterated d4-hexyl-paraben (used as an internal standard) was purchased from Toronto Research Chemicals (Ontario, Canada). The tested dispersant/sorbent used in this study were the octadecyl-bonded silica (C18) sorbent and primary secondary amine-bonded silica (PSA), which were provided by Supelco (Bellefonte, PA, USA). Oasis-HLB (hydrophilic–lipophilic balanced) reversed-phase sorbent was purchased from Waters, Inc. (Milford, MA, USA).2.2. Apparatus and instruments
The freeze-drying system CT-5000D was obtained from Panchum Scientific Corp. (Taipei, Taiwan). The vortex-blended system (Vortex-Genie 2) was purchase from Scientific Industries, Inc. (Bohemia, NY, USA). The centrifuge (CN-3600) was from Hsiangtai Machinery Co. (New Taipei, Taiwan). The Dionex UltiMate 3000 UHPLC system was from Thermo Fisher (Waltham, MA, USA), and the Bruker Compact™ LC-MS quadrupole time-of-flight mass spectrometry was purchased from Bruker Daltonik GmbH (Bremen, Germany). The Poroshell 120 EC-C18 UHPLC column was obtained from Agilent Inc. (Santa Clara, CA, USA).2.3. Sample collection and preparation
Five fresh leafy vegetables (organic Chinese cabbage, cabbage, Taiwanese lettuce, cauliflower and Bok choy) were obtained from local markets in Chung-Li City, Taiwan. Once in the laboratory, the samples were washed with deionized water. After being cut into small pieces, the samples were homogenized with a commercial blender, and the homogenate was freeze-dried using the freeze-drying system (CT-5000D) for 24 h before finally being ground into a powder form.Spiked cabbage samples were employed to evaluate and validate the developed method, which were washed by solvent and contained no target analytes. The spiked samples contained final concentrations of 10 and 250 ng g−1 (dry weight) after being spiked with the standard mixture (in methanol). After spiking, the samples were mixed well by mechanical stirring, and the methanol was left to evaporate off at room temperature in a fume hood for 2 h. Then, the samples were subjected to the DVA-MSPD procedures for method evaluation and validation.
2.4. DVA-MSPD procedure
The procedure of DVA-MSPD was accomplished under optimal extraction conditions: a powdered vegetable sample 0.2 g was dispersed and homogenized with 0.3 g of C18 (as the dispersant/clean-up co-sorbent) by vortex-blended (Vortex-Genie 2) in a centrifuge tube for 1 min (vortex-homogenization; mortar/pestle-free). Methanol (as an extracting solvent) 8 mL was added to the blended sample, and the mixture was thoroughly vortex-extracted for 5 min (vortex-extraction; SPE-column-free). The phases were then separated by centrifugation (CN-3600) at 5000 rpm for 5 min. The supernatant was collected and evaporated to dryness under a stream of nitrogen. The residue was then re-dissolved in 50 μL of 38% (v/v) of acetonitrile aqueous solution, and the target analytes were subsequently detected and quantified using UHPLC-QToF-MS.2.5. Instrumental analysis
A Dionex UltiMate 3000 UHPLC and a Bruker Compact™ LC-MS quadrupole time-of-flight mass spectrometry (QToF-MS) were applied for target analytes determination. In each case, 2.0 μL of either extract or standard solution was injected into the chromatographic system using an autosampler, and separated on a Poroshell 120 EC-C18 column (2.7 μm, 2.1 × 100 mm). The column temperature was maintained at 40 °C in a column thermostat oven. Elution was performed at a flow rate of 0.8 mL min−1 using solvent A (water) and solvent B (acetonitrile, HPLC gradient grade, ≥99.9%) as the mobile phases. The gradient profile started with 38% of B, then increased to 40% of B in 0.5 min, held for 1.6 min, and increased to 41% of B in 3 min, then increase up to 99% of B in 3.4 min, held for 0.6 min, and a reversion to the initial conditions (total 7 min: gradient elution 4.0 min and stabilization 3 min).The negative ion electrospray ionization (ESI) with full-scan mode (from m/z 50 to 400) was employed for the QToF-MS analysis, and the operating parameters were: capillary voltage of +3.5 kV; nebulizer gas pressure of 3.5 bar; dry gas flow of 10 L min−1; and dry gas temperature at 220 °C. The quantification ions of the target parabens were listed in Table 1. High resolution accurate masses were calibrated by a cluster of sodium formate ions. To increase the selectivity and sensitivity for QToF-MS detections, high resolution extracted ion chromatographic (hrEIC) traces with narrow mass window (i.e., ±5 mDa mass interval) was employed.
| Analytes | RT (min) | Quantification ion (m/z) | LOD (ng g−1) | LOQ (ng g−1) |
|---|---|---|---|---|
| a RT: retention time; LOD: limits of detection; LOQ: limits of quantification. | ||||
| MeP | 0.51 | 151.0390 | 0.4 | 1.4 |
| EtP | 0.72 | 165.0546 | 0.4 | 1.2 |
| PrP | 1.20 | 179.0703 | 0.5 | 1.8 |
| BuP | 2.03 | 193.0859 | 0.4 | 1.5 |
| BzP | 2.12 | 227.0703 | 0.5 | 1.5 |
2.6. Experimental design
The Factorial Multilevel Categoric Design (FMLCD) and Box–Behnken Design (BBD) were used to perform experimental design approaches. FMLCD is a type of factorial design that is used primarily to determine if factors are important to the method, and to screen for the important factors out of many possibilities that could have affected the method. The Box–Behnken Design (BBD) was used to perform method optimization, which is one of the most commonly used response surface methodologies.21 Statistical analyses and optimization were carried out using Design-Expert 10.0.3 software from Stat-Ease, Inc. (Minneapolis, MN, USA). The same software was also used for the analysis of variance (ANOVA), and to draw three-dimensional (3D) response surface plots for BBD.3. Results and discussion
3.1. DVA-MSPD optimization
The extraction efficiency of DVA-MSPD can be affected by several factors, such as the type and amount of dispersant/clean-up co-sorbent, the type and volume of extracting solvents used, and the extraction mechanic force (such as vortex vs. ultrasonication). Based on previous studies and our experiences involving indoor dust and biota sample pretreatment using various MSPD techniques, C18, PSA and HLB sorbents have shown good extraction efficiency for polar and hydroxylated micropollutants when used as dispersant/clean-up co-sorbent.13–17 Either methanol or acetonitrile was commonly used as the extracting solvent, and either vortex or ultrasonic was often employed as an extraction mechanic force to increase the extraction efficiency.8,10,13,14,16,22To select the significant type of variables as listed above (also shown in Table 2), a FMLCD with 2 or 3 non-numeric variables was first applied for selecting the most significant factors. The experimental domain with twelve experiments in random order plus the corresponding experimental results (represented as total peak area) are illustrated in Table 2. Preliminary ANOVA results revealed that the model, the extracting solvent, the extraction mechanic force, and the interaction between the extracting solvent and extraction mechanic force were statistically significant at the 95% confidence level for FMLCD (Table S2†). As shown in Table 2 (Run #9), the highest total peak area was achieved when powdered vegetable was mixed with reversed-phase C18 as the dispersant/clean-up co-sorbent, methanol as the extracting solvent, and vortex as the extraction mechanic force. This combination produced the cleanest chromatographic profiles with the highest total peak area for all target analytes, likely due to the efficient dispersion ability of reversed-phase C18 (due to its particle size, high surface area, and porous structure) and the adsorbed interference on its surface. These results are in agreement with previous findings reported by Shao's group.23,24 In their study, reversed-phase C18 sorbent was applied as the dispersant/clean-up co-sorbent and methanol as the extracting solvent in the MSPD procedure, resulting in alkylphenol, bisphenol A and progestogens being successfully extracted and cleaned up from egg and milk samples.
| Factor | Categorical-1 | Categorical-2 | Categorical-3 |
|---|---|---|---|
| a C18: octadecyl-bonded silica sorbent; PSA: primary secondary amine bonded silica; HLB reversed-phase sorbent: hydrophilic–lipophilic balanced reversed-phase sorbent; MeOH: methanol; ACN: acetonitrile. | |||
| Sorbent | C18 | HLB | PSA |
| Extracting solvent | ACN | MeOH | |
| Extraction mechanic force | Vortex | Sonication | |
| Run | Categorical-1 sorbent (A) | Categorical-2 extracting solvent (B) | Categorical-3 extraction mechanic force (C) | Total peak area (×105) |
|---|---|---|---|---|
| 1 | PSA | MeOH | Vortex | 37.49 |
| 2 | C18 | ACN | Vortex | 13.19 |
| 3 | HLB | ACN | Vortex | 10.29 |
| 4 | C18 | ACN | Sonication | 28.18 |
| 5 | C18 | MeOH | Sonication | 33.49 |
| 6 | PSA | ACN | Sonication | 25.32 |
| 7 | HLB | MeOH | Sonication | 32.66 |
| 8 | HLB | ACN | Sonication | 22.56 |
| 9 | C18 | MeOH | Vortex | 43.02 |
| 10 | HLB | MeOH | Vortex | 33.84 |
| 11 | PSA | ACN | Vortex | 13.90 |
| 12 | PSA | MeOH | Sonication | 37.49 |
In the second step, the extraction efficiency of DVA-MSPD was evaluated and optimized by BBD coupled with response surface methodology. According to the factors screened by FMLCD, three important factors were required to be optimized, which were the vortex time, the amount of C18 sorbent, and the volume of methanol. Three levels of these factors were: the vortex time (5, 10 and 15 min), the amount of C18 sorbent (0.2, 0.6 and 1 g), and the volume of methanol (5, 7.5 and 10 mL). The experimental domain with fifteen experiments in random order (containing three replicates for the central points) plus the corresponding experimental results (represented as total peak area) are illustrated in Table S3.† The significant factors of the DVA-MSPD procedure were also evaluated by ANOVA, and the results are summarized in Table S4.† The results show that the model, the amount of C18 (B), the volume of methanol (C), the interactions between the vortex time and the amount of C18 (AB), as well as the amount of C18 and the volume of methanol (BC) were statistically significant at the 95% confidence level. The F-value of the “Lack-of-Fit” was insignificant (at the 95% confidence level) which confirmed that the model fit the response variables with a near-perfect prediction. The quadratic polynomial equation to predict the extraction efficiency in terms of actual factors is as follows:
| Y = 4.14 × 106 − 2.20 × 104A − 6.34 × 105B + 2.56 × 105C − 3.57 × 105AB − 3.28 × 104AC + 2.37 × 105BC − 1.16 × 105A2 + 4.99 × 105B2 − 4.82 × 105C2 |
Accordingly, Fig. 1 shows the 3D response surface plots which indicate the interaction between two independent variables calculated by the BBD in order to examine the interactive effects of each pair of factors on extraction efficiency (as total peak area). In Fig. 1(a), the response surface obtained as a function of the amount of C18 and the vortex time with a fixed volume (8 mL) of methanol, shows that the extraction efficiency is enhanced when the amount of C18 sorbent is 0.2 g, though the vortex time has no significant effect on the extraction efficiency when increased from 5 to 15 min. As shown in Fig. 1(b), no significant effect was observed for the vortex time, but the extraction efficiency can be enhanced by increasing the extracting solvent from 5 mL to 8 mL when the amount of C18 was maintained at 0.2 g. Fig. 1(c) displays the response surface developed for the amount of C18 and the extracting solvent (methanol), with a fixed vortex time at 5 min. Fig. 1(c) shows that extraction efficiency can be enhanced significantly when the amount of C18 sorbent is 0.2 g and the volume of methanol is 8 mL, however, increasing the volume of methanol to 10 mL did not significantly affect the extraction efficiency. In conclusion, volume of the extracting solvent was the most relevant factor for extraction efficiency. The maximal spiked recovery, as calculated under the optimized conditions, ranged from 85 to 104% with an average of 98 ± 5% (as shown in Table 3: mean spiked recovery of intra-day for two spiked concentrations).
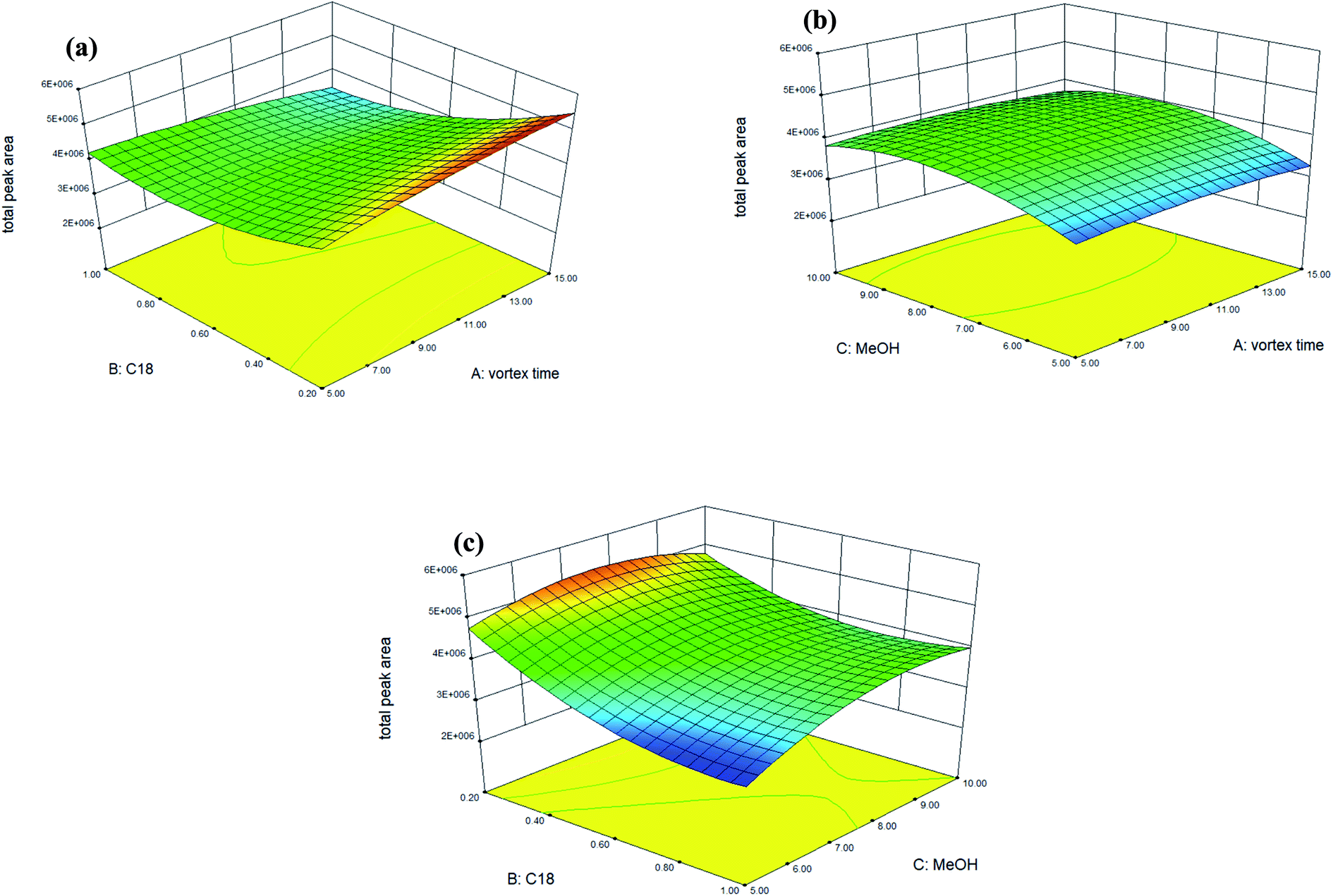 | ||
| Fig. 1 Response surface plots of the total peak area for target analytes estimated from the BBD for each pair of independent variables: (a) vortex time vs. amount of C18; (b) volume of methanol (MeOH) vs. vortex time; (c) amount of C18 vs. volume of methanol (MeOH). Experimental conditions: powdered vegetable sample 0.2 g was dispersed with 0.3 g of C18 sorbent in a centrifuge tube, and homogenized by vortex-blending for 1 min. Methanol (8 mL) was added, and the content was thoroughly vortex-extracted for 5 min. The phases were then separated by centrifugation at 5000 rpm for 5 min. | ||
| Analytes | Regression equation | Linearity range | r2 |
|---|---|---|---|
| MeP | y = 144.0 (±1.2)x + 48.9 (±29.6) | 5–500 | 0.9998 |
| EtP | y = 274.8 (±2.9)x + 544.3 (±75.0) | 5–500 | 0.9995 |
| PrP | y = 599.5 (±18.6)x + 561.8 (±47.4) | 5–500 | 0.9971 |
| BuP | y = 893.4 (±8.4)x + 50.2 (±21.3) | 5–500 | 0.9997 |
| BzP | y = 860.3 (±6.2)x + 164.7 (±57.2) | 5–500 | 0.9998 |
3.2. Validation of the proposed method
The method was validated by the evaluation of its linearity, selectivity, limits of detection (LODs), limits of quantification (LOQs), precision, and accuracy, based on the ICH Harmonised Tripartite Guideline.25To investigate the matrix effect, we first compared the calibration curves from the final extract to the standard solution (i.e., in methanol), which showed a less than 8.5% of signal suppression observed for all the target analytes. The influence of matrix effects was then further evaluated by the so-called “recovery function” method with the addition of standards to final extracts from vegetable Bok choy, as described elsewhere.19 The recovery functions were calculated by plotting the “found concentrations” versus the results for 5 concentrations that were obtained from the calibration curves. The calculated slope and intercept of the recovery functions for all target analytes were compared, respectively, with unity and zero, by means of a t-test (with confidence intervals at 95%). As listed in Table 4, the t-calculated values were all in the confidence interval of the t-tabulated value (±t(95%,df=6) = ±2.44), indicating that the results obtained by the addition of standards to the final extracts of the vegetable samples were not significantly different from the results obtained using calibration curves. Even though matrix effect did not significantly affect the calibration curves, matrix-matched calibration curves (n = 5) were applied to calculate the quantification of target analytes in order to eliminate matrix effects and to obtain satisfactory quantitative results. Each curve has a response factor covering a range from 5 to 500 ng g−1 (such as 5, 20, 100, 250, and 500 ng g−1), which is then divided by a fixed concentration of the IS (100 ng g−1). Excellent linearity was achieved within the studied concentration ranges, and the coefficients of determination (r2) were between 0.9971 and 0.9998, as shown in Table 3.
| Analytes | Intra-day | Inter-day | ||
|---|---|---|---|---|
| 10 ng g−1 | 250 ng g−1 | 10 ng g−1 | 250 ng g−1 | |
| a Average spiked recovery (accuracy, %, n = 5).b Relative standard deviation (RSD) of spiked recovery is given in parentheses (precision, %, n = 5). | ||||
| MeP | 101a(9)b | 100a(5)b | 102a(9)b | 96a(7)b |
| EtP | 85(10) | 98(4) | 102(10) | 102(5) |
| PrP | 101(9) | 98(10) | 99(8) | 90(4) |
| BuP | 97(4) | 101(5) | 98(6) | 92(7) |
| BzP | 104(8) | 96(4) | 101(5) | 90(4) |
The selectivity of the developed method was assessed by the high resolution extracted ion chromatographic (hrEIC) traces of target analytes. Accurate masses of their deprotonated molecules ([M − H]−, Table 1) were employed for both quantification and confirmation in this study. As shown in Fig. 2, no interfering peaks at or around the retention times (RTs) of the target parabens were observed, indicating that excellent selectivity can be achieved by easily identifying target analytes through their RTs.
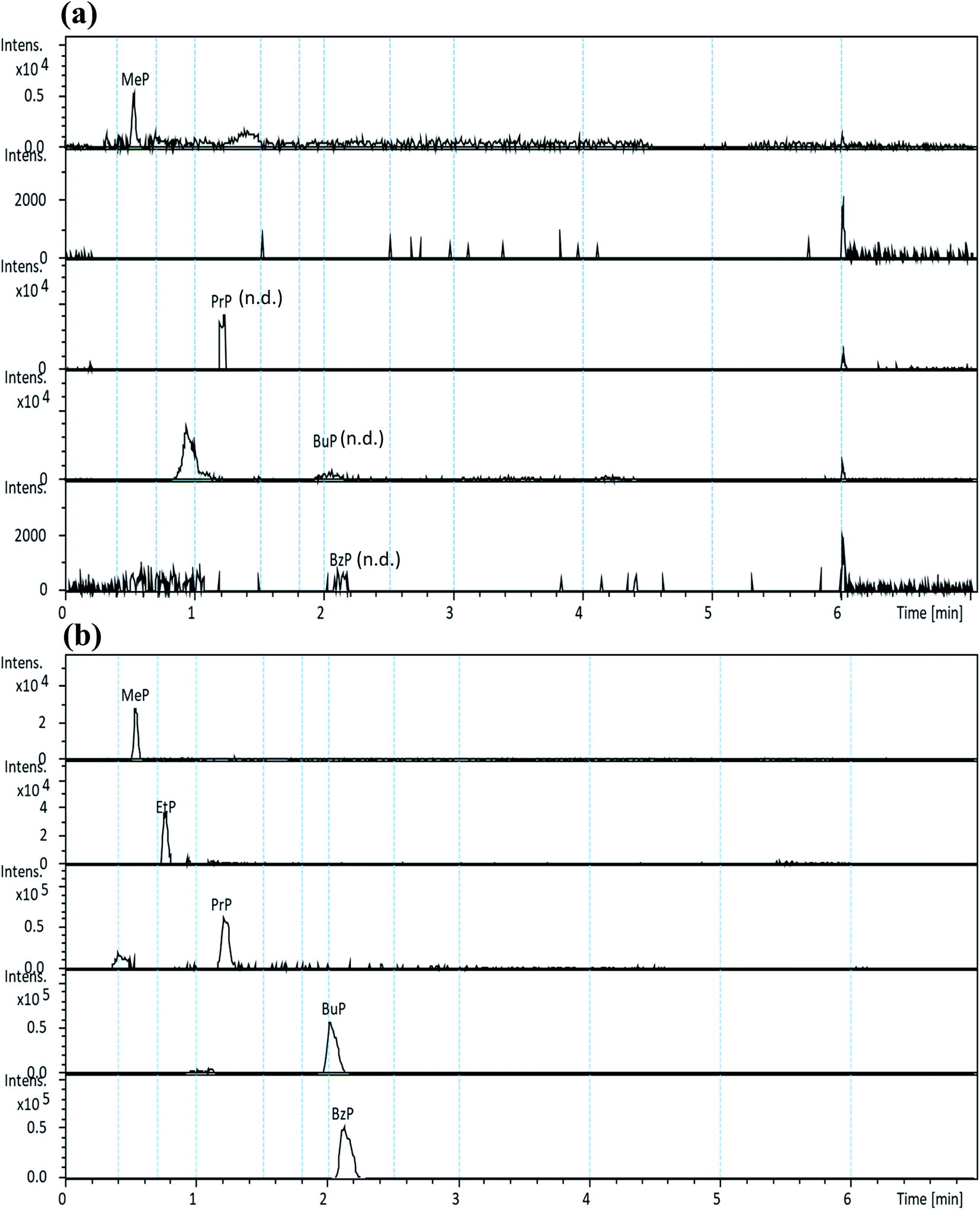 | ||
| Fig. 2 UHPLC-QToF-MS high resolution extracted ion chromatograms for (a) non-spiked, and (b) spiked samples from “cabbage” (spiked at the final concentrations of 100 ng g−1 for each analyte; n.d.: not detected). | ||
The LODs and LOQs of the method were determined for each analyte on the basis of the signal-to-noise (S/N) ratio of 3 and 10, respectively. The LODs ranged from 0.4 to 0.5 ng g−1, and values for the LOQs ranged from 1.2 to 1.8 ng g−1, as shown in Table 1.
Method precision, expressed as relative standard deviations (% RSD), was determined from intra-day and inter-day analyses. Intra-day precision (repeatability) was calculated from analyzing five consecutive spiked cabbage samples (n = 5) on the same day, and inter-day precision (within-laboratory reproducibility) was done during five successive days (n = 5). Accuracy was obtained by evaluating the percentage of average recoveries of these spiked cabbage samples. Table 4 lists the intra- and inter-day precisions and accuracies, which have values from 4 to 10% and 85 to 104%, respectively, for both low- and high-level spiked samples. Such satisfactory precisions and accuracies demonstrate that DVA-MSPD coupled with UHPLC-QToF-MS detection can achieve excellent selectivity and sensitivity, as well as high repeatability and reproducibility for the quantification of target parabens in tested vegetable samples.
The carryover effect of the autosampler was evaluated by analyzing a blank sample following the highest calibration standard (i.e., 500 ng g−1) injected in 6 replicates. Negligible carryover in the response (≤5% of the lowest calibration standard, i.e., 5 ng g−1) was observed at the retention time of target analytes and IS in blank samples after subsequent injections (n = 6) of the highest calibration standard.26
3.3. Method applications
The procedure was then applied to the analysis of five commonly consumed vegetables collected from local markets. The concentrations (ng g−1, dry weight) and precision of the five target analytes detected in various vegetable samples (in triplicates) are listed in Table 5. Preliminary results show that MeP was detected in all five vegetable samples (ranging from 23.6 to 68.5 ng g−1 (d.w.), and precisions varied from 0.3 to 5.5%), and PrP was only detected in organic Chinese cabbage with a concentration 9.2 ng g−1 (d.w.). Although MeP and PrP were detected in some vegetables, the content did not exceed the maximum residue of national requirements on the surfaces or peels of the fruits and vegetables (i.e., 0.012 mg g−1).27 Fig. 2 shows the typical hrEICs of UHPLC-QToF-MS for (a) non-spiked real sample (cabbage), and (b) spiked real samples from the cabbage.| Sample | MeP | EtP | PrP | BuP | BzP |
|---|---|---|---|---|---|
| a Average concentration (ng g−1, d.w., n = 3).b Relative standard deviation (RSD, n = 3) of detected concentration is given in parentheses.c Bok choy was used to evaluated the matrix effect at 95% confidence interval, and ttab value is ±t(95%,df=6) = ±2.44. | |||||
| Organic Chinese cabbage | 68.5a (2.8)b | n.d. | 9.2a (0.3)b | n.d. | n.d. |
| Cabbage | 23.6 (0.7) | n.d. | n.d. | n.d. | n.d. |
| Taiwanese lettuce | 29.3 (2.3) | n.d. | n.d. | n.d. | n.d. |
| Cauliflower | 50.9 (5.5) | n.d. | n.d. | n.d. | n.d. |
| Bok choy | 60.6 (2.4) | n.d. | n.d. | n.d. | n.d. |
| tcal-Value of matrix effectc | 1.12 | 0.93 | 1.51 | 1.19 | 0.87 |
Our results for MeP, the most commonly detected paraben preservatives in foodstuff, are similar to those reported by Liao et al. for samples collected in Albany, New York, USA,12 and Song et al. in Beijing, China.1 Liao et al. found MeP in vegetable samples from Albany with concentrations ranging from 0.041 to 69.9 ng g−1 (d.w.),3 whereas MeP was detected in vegetables with concentrations ranging from n.d. to 81.0 ng g−1 (d.w.) in Beijing, China.1 Moreover, our results for MeP have higher concentration than those in vegetable samples collected from Chongqing, China, as reported by Zhou et al.,8 and the samples collected from Seville, Spain, as reported by Aparicio et al.10 The MeP concentrations from the vegetables in Chongqing ranged from 0.9 to 5.0 ng g−1 (d.w.);8 and those in the samples from Seville ranged from 0.2 to 4.4 ng g−1 (d.w.).10 However, the results we detected for MeP are lower than the results from the vegetable samples collected in nine cities in China, as reported by Liao et al., which ranged from 0.042 to 2170 ng g−1 (d.w.) with an average of 81.1 ng g−1 (d.w.).2 Interestingly, other four paraben preservatives (i.e., EtP, PrP, BuP and BzP) with various concentrations have been reported in vegetable samples collected in both Albany, New York3 and in nine cities in China.2 This disparity can perhaps be attributed to different applications parabens in different regions or countries.
In comparison to the efficiency and simplicity of DVUA-MSPD (Table 6), our developed method requires less organic solvent, and the previously reported methods required various cleanup procedures (such as SPE, d-SPE and DLLME) after sample extraction steps. The values of LOQ for our developed method are higher than those of methods that used solid–liquid extraction and ultrasound-assisted extraction, but comparing with the extraction time, solid–liquid extraction required 60 min, and ultrasound-assisted extraction plus d-SPE cleanup required 45 min. The spiked recovery and precision for DVUA-MSPD were demonstrated to be comparable or not significantly different from those of previously reported methods.
| Extraction method | Time required | Solvent consumption | Spiked recovery | Precision | LOQ/MDL | Reference |
|---|---|---|---|---|---|---|
| a QuEChERS: quick, easy, cheap, effective, rugged, and safe; MWCNTs: multi-walled carbon nanotubes; d-SPE: dispersive solid-phase extraction; DLLME: dispersive liquid–liquid microextraction; LOQ: limits of quantification; MQL: method quantification limit; d.w.: dry weight; w.w.: wet weight. | ||||||
| DVA-MSPD | <12 min | 8 mL | 85–104% | 4–10% | LOQ (d.w.), 1.2–1.5 ng g−1 | This study |
| Solid-liquid extraction + NH2-SPE cleanup | 60 min + SPE elution | 18 mL | 82–109%, 94–112% | 8–22%, 7–22% | LOQ (d.w.), 0.01 ng g−1 | 2 and 3 |
| QuEChERS | 35 min | 25 mL | 76–117% | 0.3–14.5% | LOQ (w.w.), 1.4–2.8 ng g−1 | 8 |
| QuEChERS + MWCNTs-SPE cleanup | 25 min + SPE elution | 10 mL | 81–112% | 1–10% | LOQ (w.w.), 50 ng g−1 | 1 |
| Ultrasound-assisted extraction + d-SPE cleanup | 45 min | 3 mL | 89–126% | 1–19% | MQL (d.w.), 0.08–0.17 ng g−1 | 10 |
| Ultrasound-assisted extraction + DLLME cleanup | 20 min | 2.5 mL | 85–102% | 0.9–4.5% | MQL (d.w.), 0.1–0.5 ng g−1 | 9 |
4. Conclusions
This paper demonstrates the development of a simple and straightforward technique in which DVA-MSPD, combined with UHPLC-ESI(−)-QToF-MS, was used for the determination of selected parabens residues in various vegetable samples. The DVA-MSPD method was fully validated, as evidenced by a satisfactory linearity (r2 ≥ 0.9971), excellent selectivity (via hrEIC), low LODs (0.4–0.5 ng g−1), low LOQs (1.2–1.5 ng g−1), satisfactory accuracy (85–104%), and good precision (4–10%) for the developed method. The method only requires small volumes of solvents and small amounts of commercially available sorbents without the need for dedicated instruments. Even though we only analyzed a limited number of marketed vegetable samples, our preliminary results show that trace-levels of MeP (23.6–68.5 ng g−1) were found in the vegetables. The numerous advantages of DVA-MSPD can greatly aid future routine analysis and monitoring programs, such as surveys currently being conducted throughout Taiwan to collect in-depth information on the occurrence of parabens in vegetable samples and foodstuffs.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This study was supported by a grant from the Ministry of Science and Technology of Taiwan under contract no. MOST 108-2113-M-008-002. We wish to thank the Instrumental Center of National Central University for instrumentation support. We also thank Miss Erica Ding, who checked over the English and grammar usage within this manuscript.References
- S. Y. Song, Z. Zhang, N. Zou, R. H. Chen, L. J. Han, C. P. Pan and Y. Sapozhnikova, Food Anal. Methods, 2017, 10, 3972–3979 CrossRef.
- C. Y. Liao, L. X. Chen and K. Kannan, Environ. Int., 2013, 57–58, 68–74 CrossRef CAS.
- C. Y. Liao, F. Liu and K. Kannan, Environ. Sci. Technol., 2013, 47, 3918–3925 CrossRef CAS.
- C. Piao, L. Chen and Y. Wang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 569, 138–148 Search PubMed.
- J. A. Ocaña-González, M. Villar-Navarro, M. Ramos-Payán, R. Fernández-Torres and M. A. Bello-López, Anal. Chim. Acta, 2015, 858, 1–15 CrossRef.
- N. Raza, K. H. Kim, M. Abdullah, W. Raza and R. J. C. Brown, Trends Anal. Chem., 2018, 98, 161–173 CrossRef CAS.
- H. T. Zhou, H. C. Chen and W. H. Ding, J. Pharm. Biomed. Anal., 2018, 150, 469–473 CrossRef CAS.
- X. Zhou, S. Cao, X. L. Li, B. Tang, X. W. Ding, C. X. Xi, J. T. Hu and Z. Q. Chen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2015, 989, 21–26 CrossRef CAS.
- C. Abril, J. Martín, J. L. Malvar, J. L. Santos, I. Aparicio and E. Alonso, Anal. Bioanal. Chem., 2018, 410, 5155–5163 CrossRef CAS.
- I. Aparicio, J. Martín, C. Abril, J. L. Santos and E. Alonso, J. Chromatogr. A, 2018, 1533, 49–56 CrossRef CAS.
- S. A. Barker, A. R. Long and C. R. Short, J. Chromatogr. A, 1989, 475, 353–361 CrossRef CAS.
- S. A. Barker, J. Biochem. Biophys. Methods, 2007, 70, 151–162 CrossRef CAS.
- A. L. Capriotti, C. Cavaliere, P. Giansanti, R. Gubbiotti, R. Samperi and A. Laganà, J. Chromatogr. A, 2010, 1217, 2521–2532 CrossRef CAS.
- A. L. Capriotti, C. Cavaliere, P. Foglia, R. Samperi, S. Stampachiacchiere, S. Ventura and A. Laganà, Trends Anal. Chem., 2015, 71, 186–193 CrossRef CAS.
- X. J. Tu and W. B. Chen, Molecules, 2018, 23, 2767–2779 CrossRef.
- S. S. Caldas, C. M. Bolzan, E. J. de Menezes, A. L. V. Escarrone, C. M. G. Martins, A. Bianchini and E. G. Primel, Talanta, 2013, 112, 63–68 CrossRef CAS.
- G. I. Hertzog, K. L. Soares, S. S. Caldas and E. G. Primel, Anal. Bioanal. Chem., 2015, 407, 4793–4803 CrossRef CAS.
- Y. H. Chen, C. Y. Chang and W. H. Ding, J. Chromatogr. A, 2016, 1472, 129–133 CrossRef CAS.
- J. M. Chen, C. C. Yang, W. H. Chung and W. H. Ding, RSC Adv., 2016, 6, 96510–96517 RSC.
- S. T. Narenderan, S. N. Meyyanathan and V. V. S. Reddy Karri, Food Chem., 2019, 289, 384–395 CrossRef CAS.
- S. L. C. Ferreira, R. E. Bruns, H. S. Ferreira, G. D. Matos, J. M. David, G. C. Brandão, E. G. P. da Silva, L. A. Portugal, P. S. dos Reis, A. S. Souza and W. N. L. dos Santos, Anal. Chim. Acta, 2007, 597, 179–186 CrossRef CAS.
- W. H. Chung, J. S. Lin and W. H. Ding, J. Chromatogr. A, 2019, 1605, 460367 CrossRef CAS.
- B. Shao, H. Han, X. M. Tu and L. Huang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 850, 412–416 CrossRef CAS.
- Y. Yang, B. Shao, J. Zhang, Y. N. Wu and J. Ying, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 870, 241–246 CrossRef CAS.
- Harmonised tripartite guideline, ICH, Geneva, 2005, https://database.ich.org/sites/default/files/Q2_R1__Guideline.pdf Search PubMed.
- E. J. Woolf, et al., AAPS J., 2014, 16, 885–892 CrossRef CAS.
- Taiwan Food and Drug Administration, Taiwan FDA Authorization #1091300402, 2020, https://www.fda.gov.tw/tc/newsContent.aspx?cid=3%26id=26011 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05658h |
| This journal is © The Royal Society of Chemistry 2020 |