
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of bimetallic nickel-based catalysts supported on activated carbon for green fuel production†
Wan Nor Adira Wan Khalitabc,
Tengku Sharifah Marliza*ac,
N. Asikin-Mijan d,
M. Safa Gamalab,
Mohd Izham Saimanab,
Mohd Lokman Ibrahimef and
Y. H. Taufiq-Yap*abg
d,
M. Safa Gamalab,
Mohd Izham Saimanab,
Mohd Lokman Ibrahimef and
Y. H. Taufiq-Yap*abg
aCatalysis Science and Technology Research Centre (PutraCat), Faculty of Science, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia. E-mail: taufiq@upm.edu.my; Fax: +60-3-89466758; Tel: +60-3-89466809
bDepartment of Chemistry, Faculty of Science, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia
cDepartment of Science and Technology, Universiti Putra Malaysia Bintulu Campus, Nyabau Road, 97008 Bintulu, Sarawak, Malaysia. E-mail: t_marliza@upm.edu.my; Fax: +60-86-855428; Tel: +60-86-855430
dDepartment of Chemical Sciences, Faculty of Science and Technology, Universiti Kebangsaan Malaysia, 43600 UKM Bangi, Selangor Darul Ehsan, Malaysia
eSchool of Chemistry and Environment, Faculty of Applied Sciences, Universiti Teknologi MARA (UiTM), 40450 Shah Alam, Selangor, Malaysia
fCentre of Nanomaterials Science, Institute of Science, Universiti Teknologi MARA (UiTM), 40450 Shah Alam, Selangor, Malaysia
gChancellery Office, Universiti Malaysia Sabah, 88400 Kota Kinabalu, Sabah, Malaysia
First published on 8th October 2020
Abstract
In this work, the catalytic deoxygenation of waste cooking oil (WCO) over acid–base bifunctional catalysts (NiLa, NiCe, NiFe, NiMn, NiZn, and NiW) supported on activated carbon (AC) was investigated. A high hydrocarbon yield above 60% with lower oxygenated species was found in the liquid product, with the product being selective toward n-(C15 + C17)-diesel fractions. The predominance of n-(C15 + C17) hydrocarbons with the concurrent production of CO and CO2, indicated that the deoxygenation pathway proceeded via decarbonylation and decarboxylation mechanisms. High deoxygenation activity with better n-(C15 + C17) selectivity over NiLa/AC exposed the great synergistic interaction between La and Ni, and the compatibility of the acid–base sites increased the removal of oxygenated species. The effect of La on the deoxygenation reaction performance was investigated and it was found that a high percentage of La species would be beneficial for the removal of C–O bonded species. The optimum deoxygenation activity of 88% hydrocarbon yield with 75% n-(C15 + C17) selectivity was obtained over 20% of La, which strongly evinced that La leads to a greater enhancement of the deoxygenation activity. The NiLa/AC reusability study showed consistent deoxygenation reactions with 80% hydrocarbon yield and 60% n-(C15 + C17) hydrocarbon selectivity within 6 runs.
1. Introduction
Environmental pollution and the depletion of fossil fuel have become twin crises in the world today. The burning of fossil fuels has contributed to an increase in carbon dioxide (CO2) levels in the atmosphere, which simultaneously increased the average temperature of the earth.1,2 Currently, fossil fuels supply 80% of the world's energy demand, and about 90% of the total supply is utilized in the transportation sector, thus, worsening the atmospheric pollution. To overcome these problems, the exploitation of alternate biofuels from non-fossil sources such as animal fats, vegetable oils and algae has become the focus.3,4 Recently, biomass-derived triglyceride-based feedstocks such as vegetable oils have gained significant attention as sustainable biodiesel feedstock due to their great abundance, easy availability and low cost.5,6 However, biodiesel has its drawbacks such as higher viscosity, rich oxygen content, high unsaturated acid content and low volatility.7,8 Higher oxygen contents cause severe engine problems like carbon deposit and lubricant thickening.9 Hence, there is a strong need to find other alternatives that have similar properties to petroleum fuel to replace transesterification technology such as the deoxygenation reaction for green diesel production.Biofuel known as green diesel consists of straight-chain hydrocarbon C12–C20 that can be derived from triglycerides via the catalytic deoxygenation process: (i) decarboxylation (–CO2) and (ii) decarbonylation (–CO, H2O) (deCOx). These methods need thermal energy with a compatible catalyst to remove the oxygenated species from the feedstock in a H2-free environment. Commonly, the obtained green diesel is virtually identical to conventional diesel; e.g., the amount of oxygen is close to zero after the elimination of oxygen in terms of CO, CO2 and H2O.10–12
The excessive production of waste cooking oil (WCO) has become a serious global concern. The European Union (EU) produces approximately 700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–1000000 tonnes of WCO annually.13 The WCO production in Asian countries, e.g., China, Indonesia, Malaysia, etc., and the Special Administrative Region (SAR), e.g., Taiwan and Hong Kong, is estimated to be 40000 tonnes per year.14,15 Improper WCO disposal could increase the levels of organic pollutants in the land and water which leads to environmental pollution.16 The proper recycling of WCO by converting to industrial products such as green diesel is considered to be an ideal solution.
000–1000000 tonnes of WCO annually.13 The WCO production in Asian countries, e.g., China, Indonesia, Malaysia, etc., and the Special Administrative Region (SAR), e.g., Taiwan and Hong Kong, is estimated to be 40000 tonnes per year.14,15 Improper WCO disposal could increase the levels of organic pollutants in the land and water which leads to environmental pollution.16 The proper recycling of WCO by converting to industrial products such as green diesel is considered to be an ideal solution.
Several catalysts like Pt/C, Pd/C, Ni/C, Ni–Co/MWCNT, Ni/MgO and ReNiMo/γ-Al2O3 have been extensively investigated for the catalytic deoxygenation of various feedstocks.17–20 According to Morgan and co-workers,17 they found that the deoxygenation of triolein, tristearin and soybean oil can be done over Pt/C, Pd/C and 20Ni/C catalysts; the 20Ni/C catalyst yielded over 80% of C8–C17 fractions. Zulkepli et al.21 confirmed that the deoxygenation of triolein over Ni promoted catalysts could result in the diesel products of (n-(C11–C20)). They suggested that the Ni promoter consists of acidic sites that increase the promotion of C–C and C–O bond cleavage and render high yields of diesel fractions. However, strong acidic sites of Ni may result in the formation of severe catalyst deactivators such as coke.22
The accumulation of active species caused by carbon deposition on a catalyst support also favours the migration of metallic particles to produce larger aggregates, resulting in poor dispersivity, thereby decreasing the deoxygenation activity.23–25 Interestingly, the incorporation of a coke inhibitor species (base or acid–base metal promoter) such as La, Fe, and Zn forming a binary system could reduce the deactivation rate.26–28 It was suggested that the synergistic interaction of the acid and basic sites originating from those metal species could promote greater catalyst stability and enhance deoxygenation activity. Instead of a base or acid–base promoter, the use of an acidic metal such as Ce, Mn, and W has been reported to be catalytically effective in promoting C–O bond cleavage activity, and some of these metals have also been shown to be good coke-suppressing agents. Coa and co-workers29 revealed that ∼5.5% of coke was formed on the CeO2/SBA-15 catalyst in the deoxygenation of WCO. Moreover, the high removal of oxygenating activity was also observed in the pyrolysis of microalgae oil (Tetraselmis sp. and Isochrysis sp.) over Ni–Ce/Al2O3 and Ni–Ce/ZrO2 catalyst and implied that the oxidative properties of CeO2 play a critical role in improving the reaction activity.30–33 Ce can also form reducible oxides that can simply gasify the carbon deposition due to its redox properties (Ce3+/Ce4+).34–36 There have been several reports on the enhancement of the deoxygenation activity over W–CaO and Mn-based catalysts and these metals also exhibited promising deoxygenation performances.37,38
Besides the metal promoters, the catalyst support also plays a major role in improving the deoxygenation of oxygenated species. Recently, some studies have focused on the deoxygenation of assorted feedstocks over a variety of carbon-supported catalysts.38–40 It appears that the use of activated carbon (AC) could inhibit coke formation and simultaneously enhance the spent catalyst life. Abdulkareem-alsultan et al.28 reported the high stability of Ag2O3–La2O3/AC with consistent deoxygenation for six runs. Similar trends also have been observed by the same researchers in the deoxygenation of WCO over CaO–La2O3/AC, high hydrocarbon yield (>72%), and n-C15 selectivity (>80%) was performed within six runs and coke formed was less than 2 wt%.41 Due to the advantages of AC and the unique properties of binary metal promoters, the present study is focused on the development of a series of binary metal oxides (NiLa, NiFe, NiZn, NiW, NiCe, and NiMn) supported on AC catalysts for the deoxygenation of WCO under a H2-free atmosphere. The outcomes of the nature of the binary metal oxides on maintaining high catalytic activity are also elucidated.
2. Experimental
2.1 Materials
Commercial activated carbon (AC) derived from charcoal was obtained from Fluka. Sulfuric acid (H2SO4) was procured from USA (J.T.Baker) (95–98 wt% purity). Nickel(II) nitrate hexahydrate (Ni(NO2)3·6H2O) with 99.0% purity and cerium(III) nitrate hexahydrate (CeN3O9·6H2O) with 99.5% purity were obtained from Acros. Lanthanum(III) nitrate hydrate (La(NO3)3·xH2O) with 99.9% purity, ammonium metatungstate hydrate ((NH4)6W12O39·xH2O) with ≥85.0% purity, zinc nitrate hexahydrate (N2O6Zn·6H2O) with 98.0% purity, and alkane and alkene standards n-(C8–C20) (with purity >98%) were obtained from Sigma Aldrich. Ferric nitrate nonahydrate (Fe(NO3)3·9H2O) with 98.0% purity was procured from Systerm, and manganese(II) acetate tetrahydrate (C4H6MnO4·4H2O) with 99.0% purity was obtained from R&M Chemical company. The GC grade n-hexane (with purity >98%) was purchased from Merck. The feedstock for this study was WCO, which was directly used without further treatment and was kindly supplied by a restaurant in Universiti Putra Malaysia, Serdang, Selangor Malaysia. Table 1 summarizes the chemical properties of the crude WCO.| Properties | Standard methods | WCO |
|---|---|---|
| a GC programs: column: zebron ZB5 ms (30 m × 0.25 mm I.D. × 0.25 μm); temperature program: starting from 50 °C and hold for three min before heating to 300 °C at 10 °C min−1. GCMS detector and injector temperature: 250 °C; flow rate: 1 mL min−1; carrier gas: helium.b Others = C11H20O, C21H42O2Si and C30H50,C26H50. | ||
| FFA content (%) | — | 13.9 |
| Acid value (mg KOH g−1) | ASTM D974 | 27.8 |
2.2 Synthesis of catalysts
Binary metal oxides supported on AC catalysts were synthesized via a wet impregnation process. Firstly, 7 g of commercial AC was activated by reflux with 95 mL of concentrated H2SO4 at 150 °C for 12 h, then it was washed with warm water until the pH of 7 was achieved. The resulting material was filtered and dried at 100 °C for 12 h. The respective aqueous Ni salt (20 wt%) and Fe salt (20 wt%) were further impregnated on AC surfaces under constant stirring for 12 h and directly dried overnight at 100 °C. The resulting solid was ground and calcined at 700 °C for 4 h under a N2 environment. This method was adapted from Aliana-Nasharuddin et al. with slight modification.42 The same procedure was applied to the impregnation of AC with a constant amount of 20 wt% of Ni, Ce, Fe, Mn, La, W and Zn salts. The catalysts were designated as NiFe/AC, NiCe/AC, NiMn/AC, NiLa/AC, NiW/AC and NiZn/AC.2.3 Characterization of catalysts
The crystallography of the prepared mixed metal oxide supported AC catalysts was determined using the Shimadzu XRD-6000 X-ray Diffractometer (XRD). The scan range was set from 2° to 80°, assisted by Cu Kα radiation (λ = 0.15406 nm, 40 kV, 30 mA).The Thermo-Finnigan Shopmatic 1990 series N2 sorption analyser was used to determine the specific surface area and pore distribution by using the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) technique. Initially, the catalysts were degassed for 12 h at 150 °C to eliminate impurities and moisture on the catalyst surface. The adsorption and desorption processes of N2 were evaluated in a vacuum chamber at −196 °C. The morphological analysis and the elemental analysis of the prepared catalyst were investigated using A LEO 1455 VP FESEM equipped with a Rayny EDX-720 energy dispersive X-ray spectrometer. The EDX analyser was launched to determine the composition of Ni, Fe, Ce, Mn, La, W, Ni, Zn, and C on the catalyst surface.
The acidity and basicity of the prepared catalysts were analysed using a Thermo-Finnigan TPD/R/O 1100 instrument equipped with a thermo-conductivity detector (TCD); temperature-programmed desorption – ammonia (TPD-NH3) and temperature-programmed desorption – carbon dioxide (TPD-CO2). For acidity determination, about ∼0.05 g of the catalyst was pre-treated with N2 gas for 45 min at 150 °C, and the adsorption of NH3 gas on the surface of the catalyst was continued for one hour. After that, the overloaded NH3 gas was removed with N2 gas at a flow rate of 20 mL min−1. Finally, the analysis was started during the heating of the sample from 50 °C to 950 °C with helium gas flow (30 mL min−1); the desorption of NH3 gas from the acidic sites of the catalysts was identified by TCD. The same technique was used to analyse the basicity of the catalyst with CO2 as the probe gas replacing the NH3 gas.
Thermal analysis of the catalyst was carried out using a TGA/DSC 1 Mettler Toledo thermogravimetric analyser. The sample was heated from 50 °C to 1000 °C at a heating rate of 15 °C min−1 in a N2 environment to determine the stability of the catalyst, and in an O2 environment to determine coke formation.
2.4 The catalytic deoxygenation reaction of WCO
The catalytic deoxygenation reaction of 10 g WCO was conducted in a 250 mL autoclave semi-batch reactor, containing 3 wt% catalyst, and the reactor was flushed with N2 gas to let out the O2 molecules in the system. This technique was adapted from Abdulkareem-Alsultan et al.28 The reactor was then heated to 350 °C, and the stirring was adjusted to 300 rpm and maintained for 2 h with 50 mL min−1 of N2 flow. During the reaction, the condensable products were condensed from the reactor via a cold trap with a temperature of 20 °C, and the liquid products were collected and analysed using an Agilent Technologies 7890 A Gas chromatography-flame ionization detector (GC-FID) and SHIMADZU model QP2010 Plus Gas chromatography-mass spectroscopy (GC-MS). To trap the gases released, a Tedlar gas bag was used after 1 h of reaction and subsequently analysed by using GC with thermal conductivity detector (GC-TCD).2.5 Liquid products analysis
An Agilent Technologies 7890 A GC-FID was used to quantitatively analyse the products; the alkane and alkene standards n-(C8–C20) and 1-bromohexane were used as the internal standards. The GC was equipped with an HP-5 capillary column (30 m × 0.32 mm × 0.25 μm) with helium gas as the carrier gas. Prior to analysis, a 2.0 μL aliquot of the deoxygenated liquid product was introduced into the GC injector port at 250 °C. The initial oven temperature was set at 40 °C and held for 6 min, then it was ramped up to 270 °C at the heating rate of 7 °C min−1. The total hydrocarbon yield (Y) was determined as the summation of the peak areas of alkanes and alkenes in the range of n-(C8–C20) as shown in (eqn (1)), where Aa denotes the total peak area of alkanes, Ae denotes the total peak area of alkenes, and ∑Ap denotes the total peak area for all the deoxygenized and non-deoxygenized products.43| Y = (∑Aa + ∑Ae)/(∑Ap) × 100% | (1) |
The selectivity for the hydrocarbons (HC) was determined by eqn (2), where Ah denotes the total peak area of the selected hydrocarbon, and At is the total area of hydrocarbons detected in the liquid product.42
| HC = (Ah/(∑At)) × 100% | (2) |
A SHIMADZU model QP2010 Plus GC-MS with a non-polar Zebron ZB5 ms column (30 m × 0.25 mm I.D. × 0.25 μm) with a splitless inlet was used to qualitatively investigate the WCO and deoxygenated liquid products. Briefly, the samples were diluted to 100 ppm with high-purity (>98%) GC-grade hexane. The resulting peaks observed in the GC spectrum were analysed using the National Institute of Standards and Testing Library (NIST) and the main products were matched with probabilities exceeding 95%. The gas product was analysed using an Agilent USA Model G1540 N system with a thermal conductivity detector (TCD). About 0.20 mL of the collected gas in the Tedlar bag was introduced into the gas chromatograph. Standard gases such as CO2, H2, CO, and CH4 were used to calibrate the yields of collected gases. The gas products were split using He as the carrier gas. The collected gases were calculated and compared with the percentage of the standard gas.
Furthermore, the Perkin Elmer Spectrum (PS) 100 FT-IR was used to study the functional groups of the liquid products operating in the range of 300–4000 cm−1 with a resolution of 4 cm−1.
3. Results and discussion
The XRD patterns of the AC and catalysts of the binary metal oxide-supported AC are shown in Fig. 1. The XRD of the treated AC exhibited an amorphous structure with a wide hump at 2θ = 20.28°–28.70° along with intense diffraction peaks at 2θ = 44.60°, 64.68°, and 77.55°, which are likely attributed to C.44 The presence of the amorphous carbon structure is evidence of the randomly oriented carbon sheets and this ensures that all catalysts have a high content of non-graphitic carbon in their structure. The intensity of the broad hump disappeared after it was impregnated with the active metals, suggesting the intercalation of metals implanted on the AC and the metal were successfully introduced in the support.44 Hence, it was suggested that the active metals promoted greater dispersion on the AC.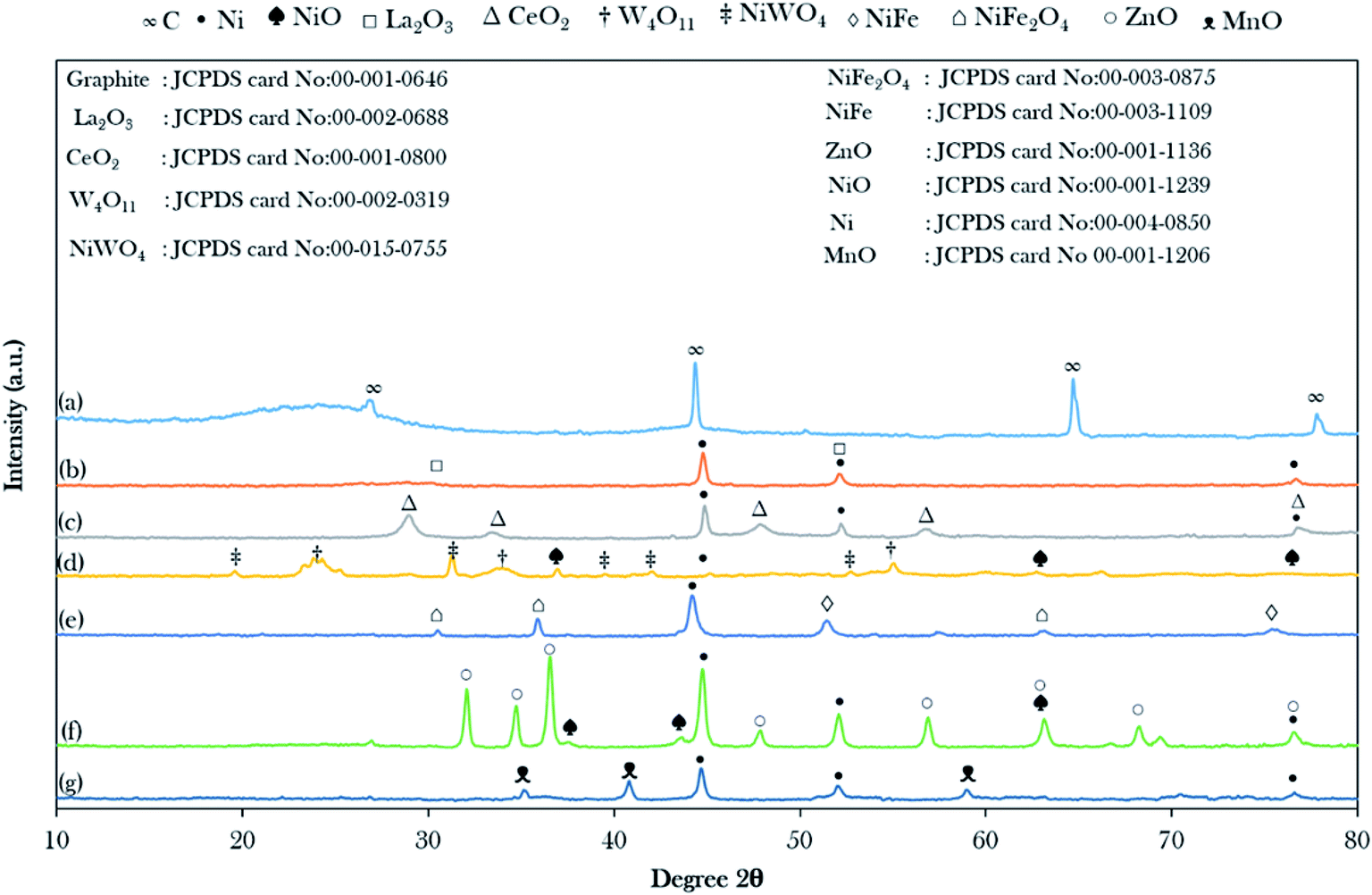 | ||
| Fig. 1 X-ray diffractograms of the binary metal oxide-supported AC: (a) activated carbon (AC), (b) NiLa/AC, (c) NiCe/AC, (d) NiW/AC, (e) NiFe/AC, (f) NiZn/AC and (g) NiMn/AC. | ||
It was observed that most of the binary metal oxide supported ACs showed the formation of the cubic metallic Ni0 at 2θ = 44.51°. In the case of NiW/AC and NiZn/AC, they showed the additional appearance of cubic NiO at 2θ = 37.44°, 43.47°, 63.20°, and 75.37°, which suggest that the lattice oxygen in W and Zn-containing catalysts possess low redox ability.45 The successful incorporation of La, W, Ce, Fe, Zn, and Mn on Ni/AC was evinced by the formation of the XRD diffraction peaks for the hexagonal structure of La2O3, cubic CeO2, tetragonal W4O11, hexagonal ZnO, and cubic MnO. Besides, NiFe/AC and NiW/AC showed the presence of the bimetallic phase for NiFe2O4, NiFe and NiWO4. However, in the case of the NiFe/AC catalyst, it was observed that the diffraction peak belonging to cubic metallic Ni0 slightly shifted from 2θ = 44.51° to 44.01° due to the addition of Fe atoms to the crystal lattice of Ni, and resulted in the formation of the NiFe alloy phase at 2θ = 51.29° and 75.34° as shown in Fig. 1e. This finding is in agreement with studies by Chen et al. and Li et al., where they reported that when Fe was added to the lattice of Ni, it caused deformation in the catalyst crystallinity and the formation of the NiFe alloy, which also resulted in the shifting of the diffraction peak of Ni0 in Ni0.9Fe0.1–Cr2O3 and Ni3Fe1/CeO2 catalysts to lower angles.46,47 It was also suggested that some of the Ni atoms were bonded to the Fe and W atoms by oxygen-bridged bonds to form Ni–Fe and Ni–W composite oxides. It is worth mentioning that the diffraction peak intensity of ZnO increased remarkably with the loading of NiO. This shows that the addition of NiO may promote the migration of ZnO crystallites to grow into larger particles, which is aligned with the report by Li et al.48
The specific surface area and pore volume of the catalysts were determined using the BET and BJH methods, and the results are tabulated in Table 2. It was noted that the AC had the highest surface area of 869 m2 g−1, and this was gradually decreased to the range of 130–485 m2 g−1 after the incorporation of active metals. This trend was in good agreement with another study that claimed that the reduction of the surface area was due to the successful impregnation of metal oxides on the surface of the AC support.49 The NiW/AC showed the lowest surface area of 130 m2 g−1, which might be due to the aggregation of W on the surface of AC that blocked the pores.50 This finding was strongly evinced by the remarkable reduction in the pore diameter and pore volume for NiW/AC, while the pore diameter and pore volume of AC were remarkably reduced from 3.24 nm to 2.78 nm and 0.70 cm3 g−1 to 0.09 cm3 g−1, respectively. Notably, indistinguishable pore diameter changes of less than 10% pore enlargement were observed for the prepared metal-loaded ACs; however, the NiCe/AC showed 62% enlargement in pore diameter. Similar findings were observed for NiCe/HZSM-5 by Balasundram and co-workers, where the pore size diameter of NiCe/HZSM-5 was remarkably enlarged when compared to the unmodified support HZSM-5.29,51 One can see that the Ce species rendered greater pore enlargement outcomes. It was reported that large pore systems may not beneficial for deoxygenation as it may induce high aromatization reactions.52
| Catalysts | N2 adsorption–desorption analysis | TPD-NH3c | TPD-CO2d | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Surface areaa (m2 g−1) | Pore size diameterb (nm) | Pore volumeb (cm3 g−1) | T (°C) | Amount of NH3 adsorbed (μmol g−1) | Total number of acidic sites (μmol g−1) | T (°C) | Amount of CO2 adsorbed (μmol g−1) | Total number of basic sites (μmol g−1) | |
| a Measured by BET analysis.b Measured by BJH analysis.c Determined by TPD-NH3 analysis.d Determined by TPD-CO2 analysis. | |||||||||
| AC | 869 | 3.24 | 0.70 | — | — | — | — | — | — |
| NiLa/AC | 336 | 3.62 | 0.30 | 635 | 2192 | 2192 | 637 | 799 | 799 |
| NiCe/AC | 380 | 8.67 | 0.10 | 568/699 | 1180/2081 | 3261 | 694 | 627 | 627 |
| NiW/AC | 130 | 2.78 | 0.09 | 958 | 18068 |
18068 |
853/952 | 1584/2093 | 3677 |
| NiFe/AC | 441 | 3.62 | 0.40 | 778 | 5981 | 5981 | 788 | 2541 | 2541 |
| NiZn/AC | 446 | 3.34 | 0.37 | 650/834 | 781/7034 | 7815 | 579/808 | 53/3123 | 3176 |
| NiMn/AC | 485 | 3.08 | 0.37 | 479/643 | 408/1226 | 1634 | 573/647/950 | 48/236/2017 | 2301 |
The surface morphology and elemental composition of all binary metal oxide-supported AC catalysts were studied by FESEM-EDX and the results are presented in Fig. 2 and Table 3, respectively. The external surface topographies of the metal-supported AC were found to vary with the dense rough surface of the AC support. The FESEM images of NiLa/AC and NiCe/AC (Fig. 2b and c) show the formation of the rough surface morphology decorated by well-developed tiny dots. In contrast, NiW/AC, NiFe/AC, and NiZn/AC displayed the formation of compact bulk structures (Fig. 2d–f). It was also worth noticing that NiW/AC also had a large cubic shape with a smooth surface belonging to W (Fig. S1†), while the NiMn/AC showed a slight agglomeration of particles.
 | ||
| Fig. 2 FESEM images of (a) AC, (b) NiLa/AC, (c) NiCe/AC, (d) NiW/AC, (e) NiFe/AC, (f) NiZn/AC and (g) NiMn/AC. (Magnifications ×50000). | ||
| Catalysts | Elemental surface analysis (wt%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| C | O | Ni | La | Ce | W | Fe | Zn | Mn | |
| AC | 76.7 | 23.3 | — | — | — | — | — | — | — |
| NiLa/AC | 56.6 | 19.1 | 9.0 | 15.4 | — | — | — | — | — |
| NiCe/AC | 49.6 | 18.8 | 16.7 | — | 14.8 | — | — | — | — |
| NiW/AC | 56.5 | 14.3 | 4.0 | — | — | 25.0 | — | — | — |
| NiFe/AC | 63.1 | 23.8 | 7.0 | — | — | — | 6.1 | — | — |
| NiZn/AC | 73.0 | 16.3 | 5.5 | — | — | — | — | 4.9 | — |
| NiMn/AC | 76.4 | 13.1 | 4.7 | — | — | — | — | — | 5.8 |
Theoretically, clear morphological changes in the textural morphology of AC after the addition of the active metal can be ascribed to dopant effects resulting in aggregation. The EDX analysis revealed that all the binary metal oxide-supported AC catalysts were comprised mainly of C species, which covered about 49–78 wt% from the total weight of the sample. Therefore, all of these catalysts are expected to have excellent mechanical properties during catalytic deoxygenation activity and thus increase the catalyst stability and catalytic activity.53,54 The C content was gradually reduced upon the incorporation the metals due to the addition of binary metals on the AC support.44 Overall, the elemental composition of active metal was slightly below the expected value (20 wt%) due to the major active metals being deposited within the interior of the AC support,55 which can be seen from the reduction in the pore volume in Table 2.
The density, distribution and strength of the active sites of the catalysts were investigated using TPD-NH3 and TPD-CO2 for acidic and basic properties, respectively, as shown in Fig. 3 and Table 2. It was observed that the desorption of NH3 and CO2 started at temperatures above 500 °C, indicating that all of the catalysts exhibited strongly acidic and strongly basic sites.50 The trend in the acid strength of catalysts is as follows: NiW/AC > NiZn/AC > NiFe/AC > NiCe/AC > NiLa/AC > NiMn/AC. The highest strength was demonstrated by the NiW/AC catalyst with a total acidity of 18068 μmol g−1. According to Asikin-Mijan et al., Janampelli, and Darbha, the large total acidity is due to the characteristics of the W metal.37,56 The Ni–Zn containing catalyst also has a high acidic density of 7815 μmol g−1 of active sites, which can be explained by the Ni–O–Zn domains and coordinatively unsaturated Ni2+ in the AC-supported catalyst (Fig. 1).
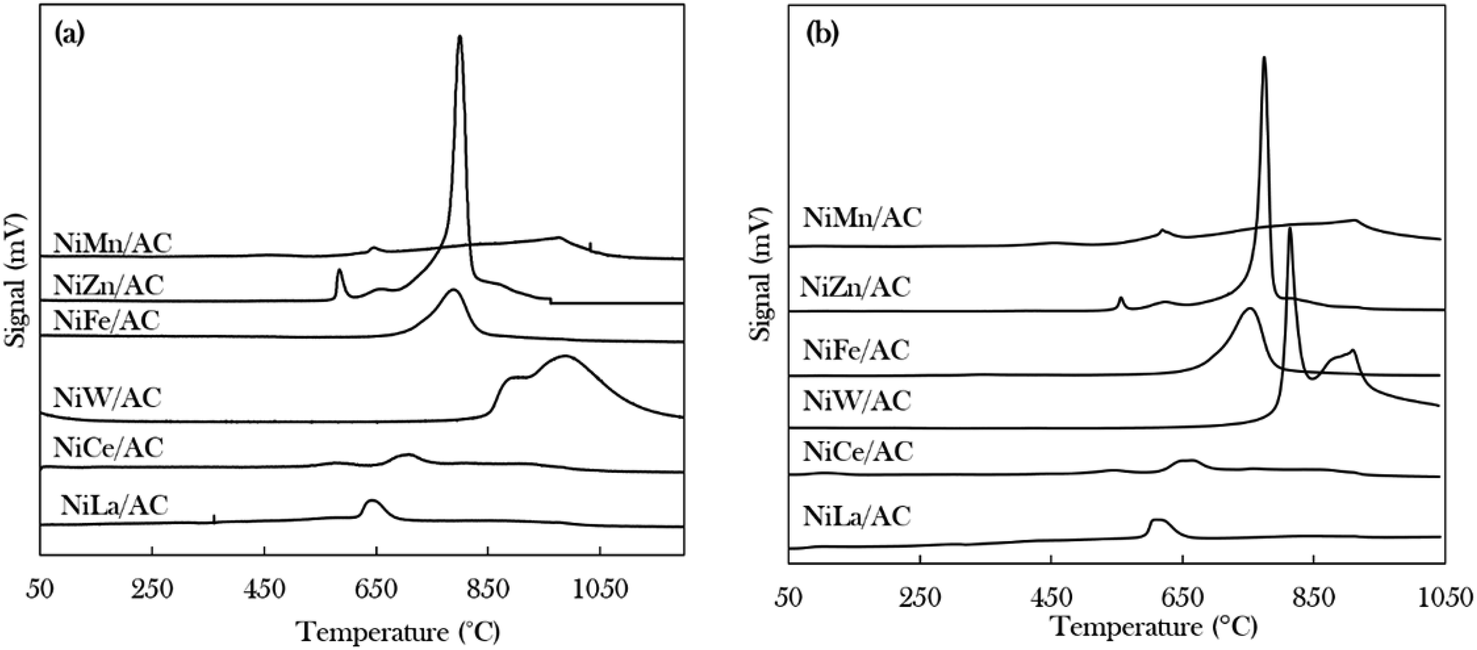 | ||
| Fig. 3 Temperature-programmed desorption patterns of (a) TPD-NH3 and (b) TPD-CO2 of binary metal oxide-supported AC catalysts. | ||
The total densities of the basic sites of prepared catalysts are of the following order: NiW/AC > NiZn/AC > NiFe/AC > NiMn/AC > NiLa/AC > NiCe/AC. The W-containing catalyst also has the highest basic density and strength, which deviates from the high acidic characteristics of W. This finding implies that the modification of Ni/AC with W can establish new acid–base properties.37 Overall, all binary metal oxide catalysts display acid–base properties; hence, it could be expected that these catalysts would have the ability to remove oxygenated species via cracking and deCOx routes.
Fig. 4 displays the thermal stability profiles for AC and binary metal oxide-supported AC catalysts. The results revealed that AC rendered three stages of weight loss: 100–200 °C, 210–420 °C, and 430–850 °C. The beginning weight loss was assigned to the loss of H2O molecules at about 6.7 wt%,31,42,57 the second weight loss was associated with the decomposition of cellulose at about 11.2 wt% and the third major weight loss was about 40.5 wt%, due to the decomposition of carbonaceous compounds.58,59 In contrast to binary metal oxide-supported AC catalysts, a minor weight loss of less than 35 wt% due to carbonaceous materials was observed at decomposition temperatures above 600 °C. This finding proves that the presence of metal oxides on AC as support gives significant thermodynamic stability to the parent AC.44
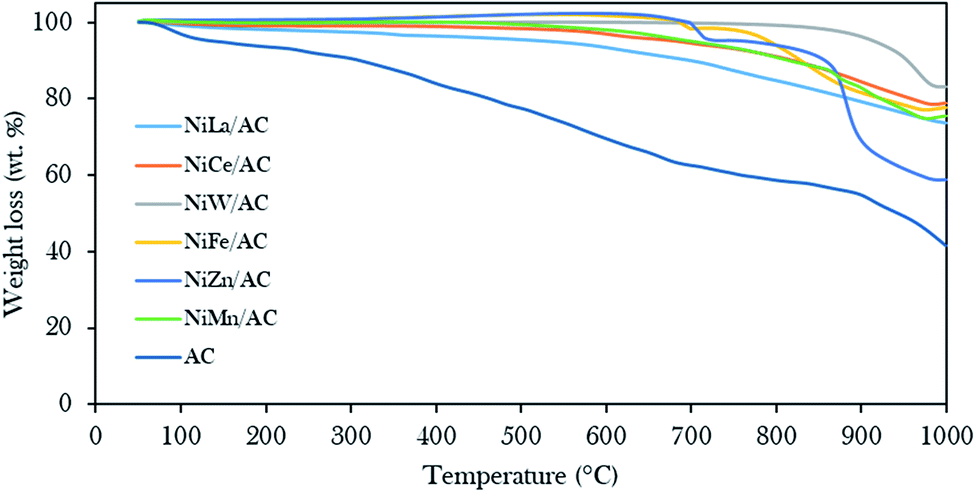 | ||
| Fig. 4 Thermal gravimetric analysis profiles of the prepared AC and binary metal oxide-supported AC catalysts. | ||
3.1 Deoxygenation activity of WCO
The deoxygenation process of WCO was conducted with a catalyst loading of 3 wt%, a reaction time of 2 h and reaction temperature of 350 °C, with stirring speed of 300 rpm in an inert N2 flow system. The hydrocarbon yield (%) and product selectivity (%) are summarized in Fig. 5. The results showed the increment in the hydrocarbon yield in the following order: blank < AC < NiW/AC < NiMn/AC < NiFe/AC < NiCe/AC < NiLa/AC < NiZn/AC. The blank test showed that the reaction was hardly taking place at 350 °C, which contributed to the hydrocarbon yield of only ∼12%. Similar behaviour was observed for the AC-catalysed deoxygenation; only 37% of the hydrocarbon yield was achieved.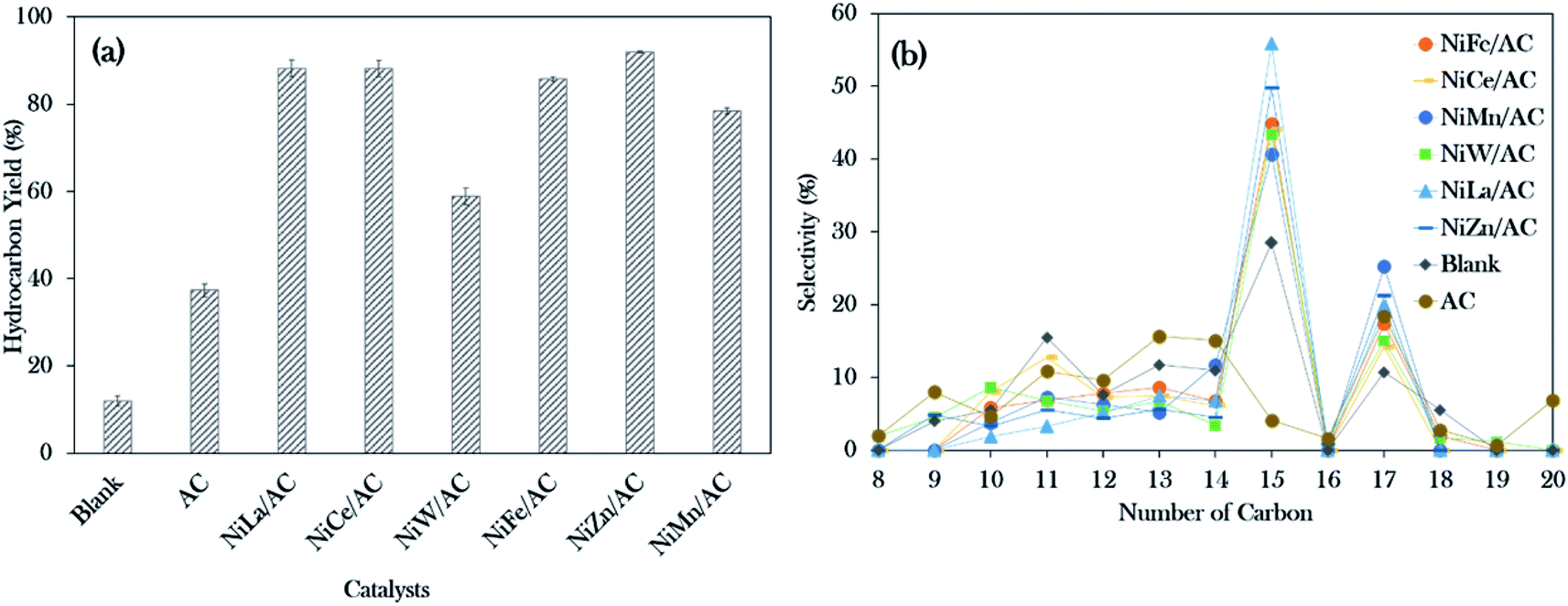 | ||
| Fig. 5 Data collected from the deoxygenation process: (a) hydrocarbon yield and (b) n-(C15 + C17) selectivity for blank and catalysed deoxygenation (operating parameters: 3 wt% of catalyst loading, 350 °C reaction temperature, 2 h reaction time under inert conditions). | ||
However, all the prepared binary mixed metal oxide-supported AC catalysts resulted in remarkably higher hydrocarbon (C8–C20) yields >75%, and only NiW/AC gave 60%. This implies that the acid–base properties were responsible for the efficient subtraction of the oxygen-bonded species reaction. According to Asikin-Mijan et al. active sites could accelerate the cracking activity and thus result in the formation of rich light gaseous compounds.37 This discovery is in line with the results of Roussel et al., who showed that the employment of NiW/silica–alumina in the hydrocracking of n-decane also resulted in the formation of linear C2 alkanes.60
Notably, NiZn/AC exhibited the highest hydrocarbon yield at 92%. This indicates that the presence of Ni–O–Zn, along with the presence of acid–base sites in NiZn/AC, was very effective in transforming the WCO to oxy-free hydrocarbon compounds.61 In the case of hydrocarbon selectivity, the blank test and AC-catalysed deoxygenation formed major light fractions of n-C8 to n-C14 with a total selectivity of about 55–66%. With the addition of active metals, the light fractions decreased to 25–37%, and the desired deCOx product n-(C15 + C17) increased remarkably to >58%. Among the binary metal oxide-supported catalysts, NiLa/AC showed the highest n-(C15 + C17) selectivity (76%), while NiCe/AC had the lowest (58%). The lowest n-(C15 + C17) selectivity by NiCe/AC strongly affirmed that the significantly large pores structures (8.67 nm as shown in Table 2) retarded the C–O bond cleavage activity. This result was in accordance with the results of Twaiq et al. who implied that large pore structured catalysts are capable of increasing the propensity for cracking activity and result in lower n-(C15 + C17) selectivity.62 Given this, it is suggested that the pore size structure is also a critical factor in determining the deCOx reaction. It is worth noting that the changes in the surface area had no effect on the n-(C15 + C17) selectivity.
Referring to the acid–base properties of the catalysts in Table 2, it was observed that the deCOx activity was limited by catalysts with higher concentrations of strong acid sites. This was revealed by the n-(C15 + C17) output, which was lowered over NiMn/AC at only 66%.
The GCMS results of the product distribution in Fig. 6a show that the total distribution of the n-(C8–C20) deoxygenated liquid catalysed by AC and binary mixed metal oxide-supported AC catalysts was in the range of 65% to 81%. The catalyst performance was of the following order arrangement: NiZn/AC > NiCe/AC > NiLa/AC = NiFe/AC > NiW/AC > NiMn/AC > AC. It can be concluded that the total hydrocarbon distribution determined by GC-FID was slightly higher as compared to the GCMS results. This is due to the presence of the hydrocarbon isomer compounds (Z) and (E), such as 5-undecene (E)- and 3-hexadecene (Z)-.41 The isomeric compounds are commonly developed from the isomerization of alkenes; the higher isomer compounds (9.88%) were obtained over NiFe/AC catalyst, due to the Fe-promoted catalyst capable of motivating the isomerization reaction.63
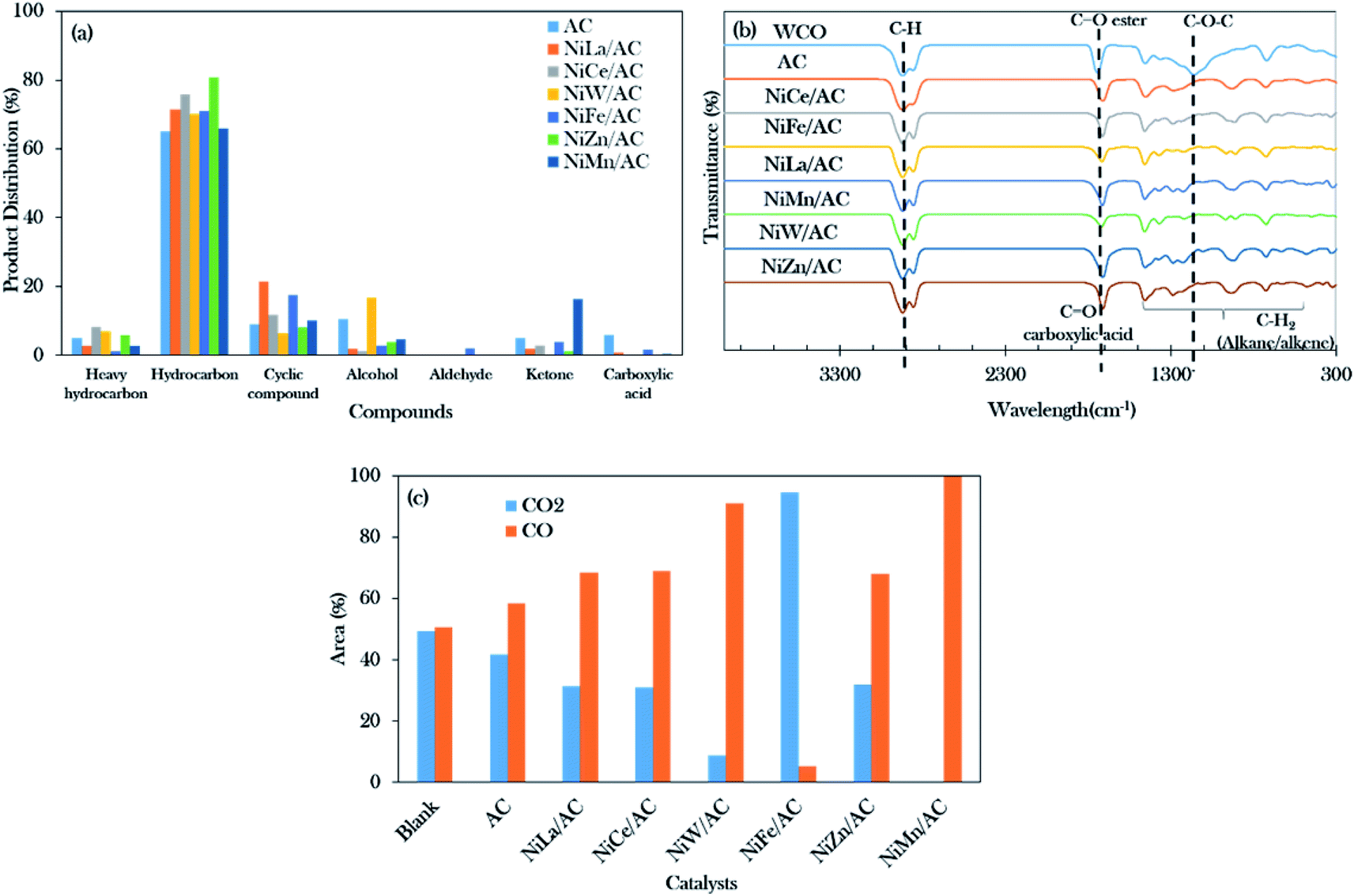 | ||
| Fig. 6 Data collected from liquid deoxygenized liquid products: (a) product distribution, (b) FTIR spectra of the WCO and deoxygenized liquid product catalysed by AC and binary mixed oxide-supported AC (c) the compositions of CO2 and CO gases over catalytic deoxygenation (operating parameters: 3 wt% of catalyst loading, 350 °C reaction temperature, 2 h reaction time under inert conditions). | ||
NiMn/AC exhibited the highest ketone product by 16%, due to the greater numbers of basic sites than acidic sites in the catalyst, which resulted in the increment of the ketonization rate.37,64,65 It was also said that the ketonization is not favourable when the catalyst co-exists with a large number of acid sites. Although the NiW/AC and NiZn/AC catalysts had the highest concentrations of basic sites (>3000 μmol g−1), the smallest amounts of ketone compounds were detected (0–1%) for both catalysts. It was further discovered that both catalysts were rich in acidic sites (>7000 μmol g−1).
Cycloalkanes such as cyclopentadecane and n-nonylcyclohexane were increased for NiLa/AC-catalysed deoxygenation, resulting in total cyclic compounds of about 21%. Li et al. reported that the La-containing catalyst favoured cyclization and aromatization reactions forming monocyclic aromatic hydrocarbons.66 It was noted that the NiLa/AC rendered side reactions; however, it was still considered to be very effective to the drive deoxygenation activity, exclusively via the deCOx reaction.
Fourier transform infrared (FTIR) spectra were used to analyse the functional groups of the deoxygenated liquid products as exhibited in Fig. 6b. It was found that the primary absorption bands of WCO were at 1120 cm−1, 1740 cm−1, and 2922 cm−1, and these were attributed to the carbonyl group (C–O–C), the ester group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), and the stretching absorption of the C–H bond, respectively.67 It also showed that the deoxygenated liquid products rendered a carboxylic acid (–COOH) group peak at 1711 cm−1, indicating that the deoxygenation of WCO was initiated by breaking the ester bond of triglycerides. The successful progression of deoxygenation activity was also in accordance with the disappearance of the carbonyl group (C–O–C) absorption band at 1120 cm−1. This resulted in correspondence with Gamal et al.44 who proposed that the occurrence of deoxygenation activity showed the reduction of intensities in the CO group and C–O absorption bands as compared to feedstock. However, Satyarthi and Srinivas68 reported that the characteristics of the C–H stretching absorption peak (2922 cm−1) could not affect the formation of n-alkanes.
O), and the stretching absorption of the C–H bond, respectively.67 It also showed that the deoxygenated liquid products rendered a carboxylic acid (–COOH) group peak at 1711 cm−1, indicating that the deoxygenation of WCO was initiated by breaking the ester bond of triglycerides. The successful progression of deoxygenation activity was also in accordance with the disappearance of the carbonyl group (C–O–C) absorption band at 1120 cm−1. This resulted in correspondence with Gamal et al.44 who proposed that the occurrence of deoxygenation activity showed the reduction of intensities in the CO group and C–O absorption bands as compared to feedstock. However, Satyarthi and Srinivas68 reported that the characteristics of the C–H stretching absorption peak (2922 cm−1) could not affect the formation of n-alkanes.
Moreover, Fig. 6c shows the composition analyses of CO2 and CO gases collected from the reaction process. Theoretically, the deoxygenation of WCO in the absence of H2 will favour the decarboxylation and decarbonylation reactions and produce carbon gases such as CO2 and CO.44 The CO gas predominates in the majority of the binary metal oxide-supported AC catalysed reactions except for NiFe/AC. The results showed that the deoxygenation reaction of WCO over NiLa/AC, NiCe/AC, NiW/AC, NiZn/AC, and NiMn/AC followed decarbonylation pathways, while NiFe/AC favoured decarboxylation pathways.
A mechanism was proposed for the catalytic deoxygenation of WCO over Ni-based catalysts (Fig. 7). Table 1 shows that WCO contains palmitic acid, C16 (22%), and oleic acid, C18 (48%), fatty acids. The catalytic deoxygenation reaction of WCO via the decarbonylation and decarboxylation (deCOx) pathway resulted in Cn−1 hydrocarbon formation (C15 and C17) and by-products (CO2, CO and water). Fig. 7a shows the proposed reaction pathway for the deoxygenation of oleic acid. Firstly, the double bonds of oleic acid were hydrogenated to produce stearic acid as the intermediate product using in situ hydrogen that was generated via the water gas shift (WGS) reaction (pathway A).69,70 Thereafter, the deoxygenation reaction took place through the decarboxylation process by the elimination of O2 in the form of CO2, and resulted in n-heptadecane (C17H36) as the product (pathway B). The decarbonylation reaction directly produced 1-heptadecene (C17H34) through the removal of O2 in the form of CO and H2O (pathway C). Most of the liquid products produced higher yields of n-C15, hence it was proposed that the mild cracking occurred through C–C cleavage from the n-C17 fraction and produced large amounts of n-C15 (pathway D). This was in agreement with the results of Aliana-Nasharuddin et al.,42 who produced high n-C15 fractions in the catalytic deoxygenation of chicken fat oil.
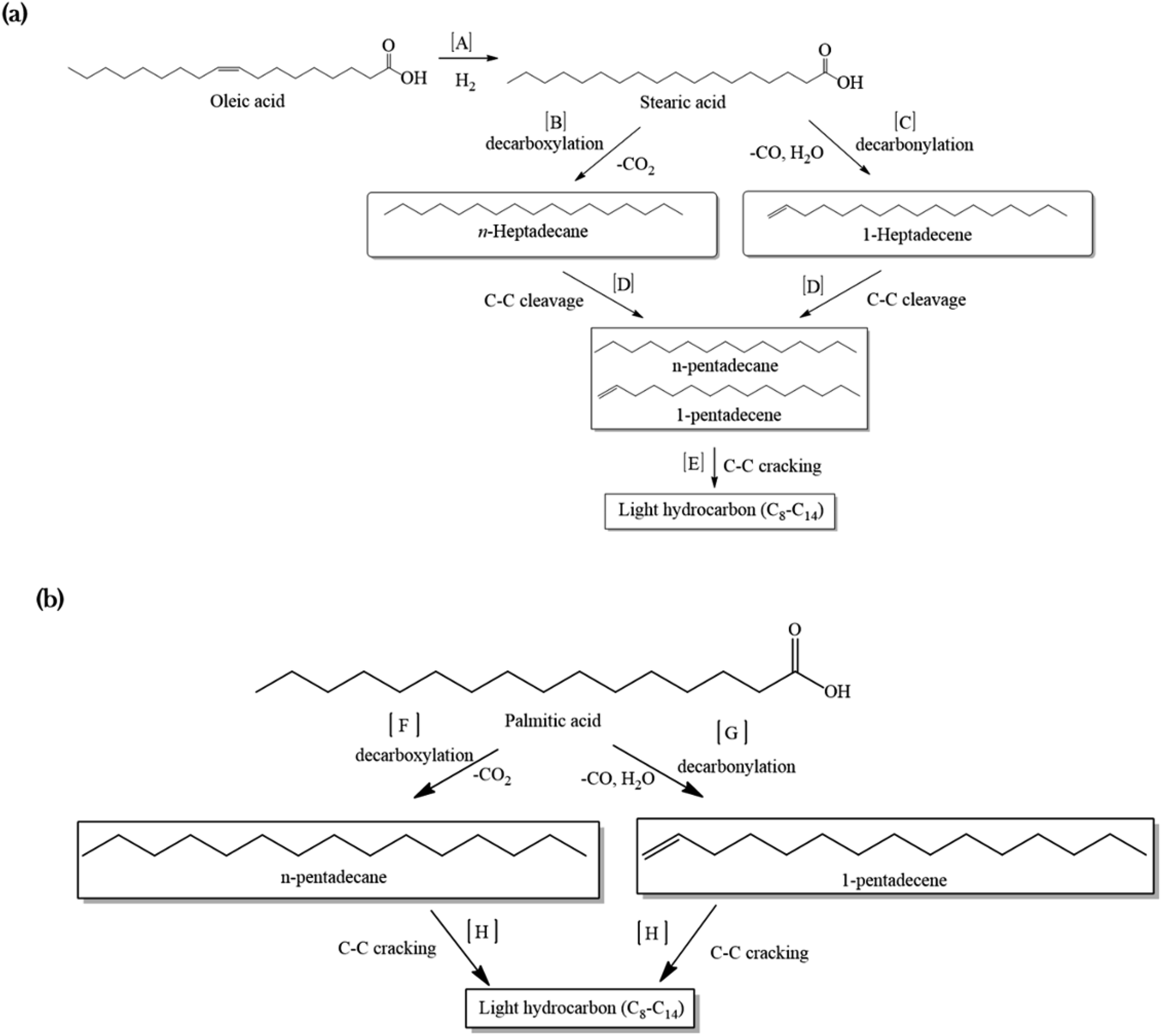 | ||
| Fig. 7 The proposed reaction mechanism for a Ni-based catalyst in the catalytic deoxygenation of WCO for (a) oleic acid and (b) palmitic acid. | ||
On the other hand, Fig. 7b shows that the deoxygenation activity of the palmitic acid resulted in n-pentadecane (C15H32) and 1-pentadecane (C15H30) by the decarboxylation and decarbonylation reaction (pathways F and G). Further C–C cracking of liquid products produced light hydrocarbons n-(C8–C14) (pathway E and H). The GC-FID results (Fig. 5) showed the formation of light hydrocarbons after the deoxygenation reaction. Hence, it can be concluded that the pathway for the deoxygenation of WCO using NiLa/AC, NiCe/AC, NiW/AC, NiZn/AC, and NiMn/AC catalyst followed the decarbonylation reaction and NiFe/AC favoured the decarboxylation reaction based on the composition of the gases Fig. 6c.
3.2 The effect of La concentration on the deoxygenation of WCO
Based on the primary catalytic screening study (Fig. 5a and b), it was found that NiLa/AC is an effective catalyst as compared to others in the catalytic deoxygenation reaction. Thus, the effect of the La concentration within the range of 5–30% was further studied to identify the concentration of La that would optimise the transformation of WCO to the diesel range under the deoxygenation activity conditions at 350 °C with 3 wt% of catalyst for 2 h reaction time and with a stirring speed of 300 rpm; the data are presented in Fig. 8a. It was found that the binary metal oxide catalyst NiLa/AC had increased the hydrocarbon yield from 75% to 89% as the concentration of La increased from 5% to 25%. However, the yield of hydrocarbon decreased when the concentration of La increased to 30%. This might be due to the excess La that saturated the surface of AC, covering the active sites of the catalyst.28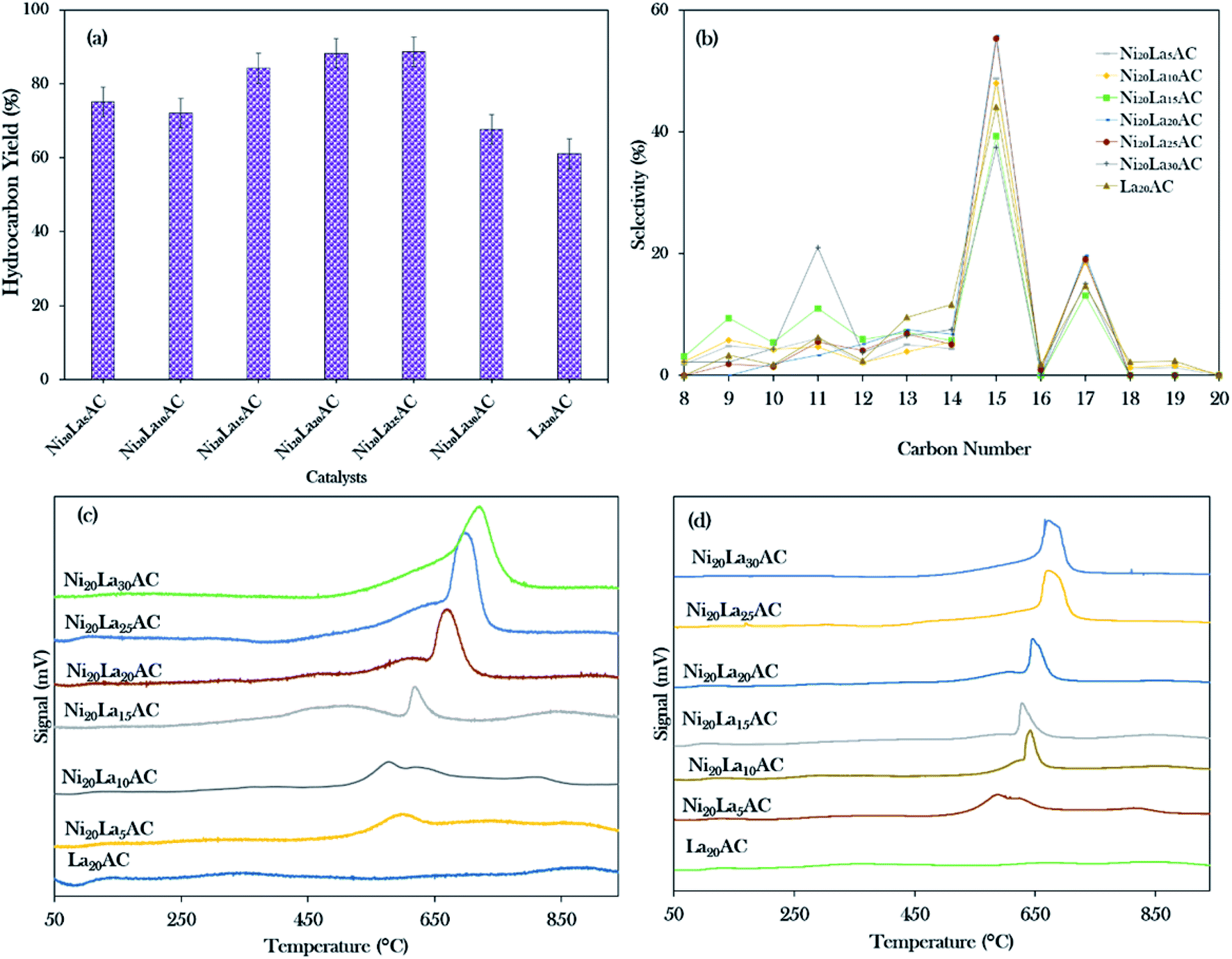 | ||
| Fig. 8 (a) Hydrocarbon yield, (b) hydrocarbon selectivity, (c) TPD-NH3, (d) TPD-CO2 for the catalytic deoxygenation of WCO over different concentrations of La (5–30 wt%). | ||
Fig. 8b shows that the majority of the deoxygenated liquid products were composed of the n-(C8 to C20) hydrocarbons. The highest total selectivity of n-(C15 + C17) followed the order Ni20La20/AC > Ni20La25/AC > Ni20La10/AC > Ni20La5/AC > La20/AC > Ni20La30/AC > Ni20La15/AC. High deCOx activity was observed for the Ni20La20AC-catalysed deoxygenation, which strongly affirmed that the moderate density of the strongly acidic sites (2192 μmol g−1) and the low density of the strongly basic sites (799 μmol g−1), as shown in Fig. 8c, d and Table 4, were beneficial for the enhancement of the deCOx activity. This finding is also in good agreement with that of Kim et al., where a large number of strongly acidic sites is not preferable to moderate and low amounts of strong acid sites, as this tends to increase the coking activity and deteriorate the catalyst stability.71 Overall, the moderate acidic strength and its density of Ni20La20/AC catalyst were attributed to the synergistic effects between the Ni and La2O3 species on the exterior surface of AC, which favoured the deCOx reaction via C–O cleavage and yielded a higher percentage of n-(C15 + C17) selectivity. Similar findings were observed by Abdulkareem-Alsultan et al.,28 who showed that the presence of the Ag2O3(10%)–La2O3(20%)/ACnano catalyst under a H2-free environment resulted in the greater hydrocarbon yield (∼89%) and selectivity for n-(C15 + C17) (∼93%). This also suggested that the rich La2O3 species induced C–O bond breaking. The higher rate of C–O bond breaking over the rich La-containing catalyst is possibly due to the formation of a large quantity of acid–base sites and also the unique characteristics of La2O3 itself.41,72
| Catalysts | TPD-NH3 | TPD-CO2 | ||
|---|---|---|---|---|
| Temp. (amount of NH3 adsorbed) | Total number of acidic sites (μmol g−1) | Temp. (amount of CO2 adsorbed) | Total number of basic sites (μmol g−1) | |
| La20AC | 338 °C (469 μmol g−1) | 1177 | 364 °C (207 μmol g−1) | 558 |
| 890 °C (708 μmol g−1) | 676 °C (164 μmol g−1) | |||
| 841 °C (187 μmol g−1) | ||||
| Ni20La5AC | 635 °C (651 μmol g−1) | 1180 | 109 °C (46 μmol g−1) | 1177 |
| 870 °C (529 μmol g−1) | 641 °C (599 μmol g−1) | |||
| 874 °C (532 μmol g−1) | ||||
| Ni20La10AC | 578 °C (1403 μmol g−1) | 1403 | 635 °C (1150 μmol g−1) | 1150 |
| Ni20La15AC | 513 °C (740 μmol g−1) | 1982 | 104 °C (59 μmol g−1) | 1151 |
| 620 °C (386 μmol g−1) | 628 °C (461 μmol g−1) | |||
| 833 °C (856 μmol g−1) | 844 °C (631 μmol g−1) | |||
| Ni20La20AC | 635 °C (2192 μmol g−1) | 2192 | 637 °C (799 μmol g−1) | 799 |
| Ni20La25AC | 696 °C (3799 μmol g−1) | 3799 | 672 °C (1184 μmol g−1) | 1184 |
| Ni20La30AC | 721 °C (3606 μmol g−1) | 3606 | 666 °C (1023 μmol g−1) | 1023 |
3.3 Optimization of WCO over NiLa/AC catalyst
Based on the initial screening study, the NiLa/AC catalyst was the operative catalyst; hence, the optimization study of the NiLa/AC (Ni = 20 wt%; La = 20 wt%) catalyst was conducted by the one-variable-at-a-time (OVAT) approach. Three parameters were evaluated, namely, catalyst loading (0.5–7%), reaction time (30–180 min), and reaction temperature (300–400 °C). The effects of catalyst loading on the hydrocarbon yield and the hydrocarbon selectivity were studied at 350 °C, 2 h, and 300 rpm stirring speed in an inert atmosphere. The hydrocarbon yield and hydrocarbon selectivity n-(C15 + C17) gradually increased from 71% to 88%, and 59% to 75%, respectively, when the catalyst was added to the system from 0.5 wt% to 3 wt%. On the contrary, the hydrocarbon yield and n-(C15 + C17) selectivity decreased upon increasing the catalyst loading beyond 5 wt% as shown in Fig. 9a and b. According to Lou et al.,73 an excess of the active sites could lead to undesired secondary reactions, such as polymerization reactions; therefore, 3 wt% of catalyst loading is preferable for the deoxygenation reaction.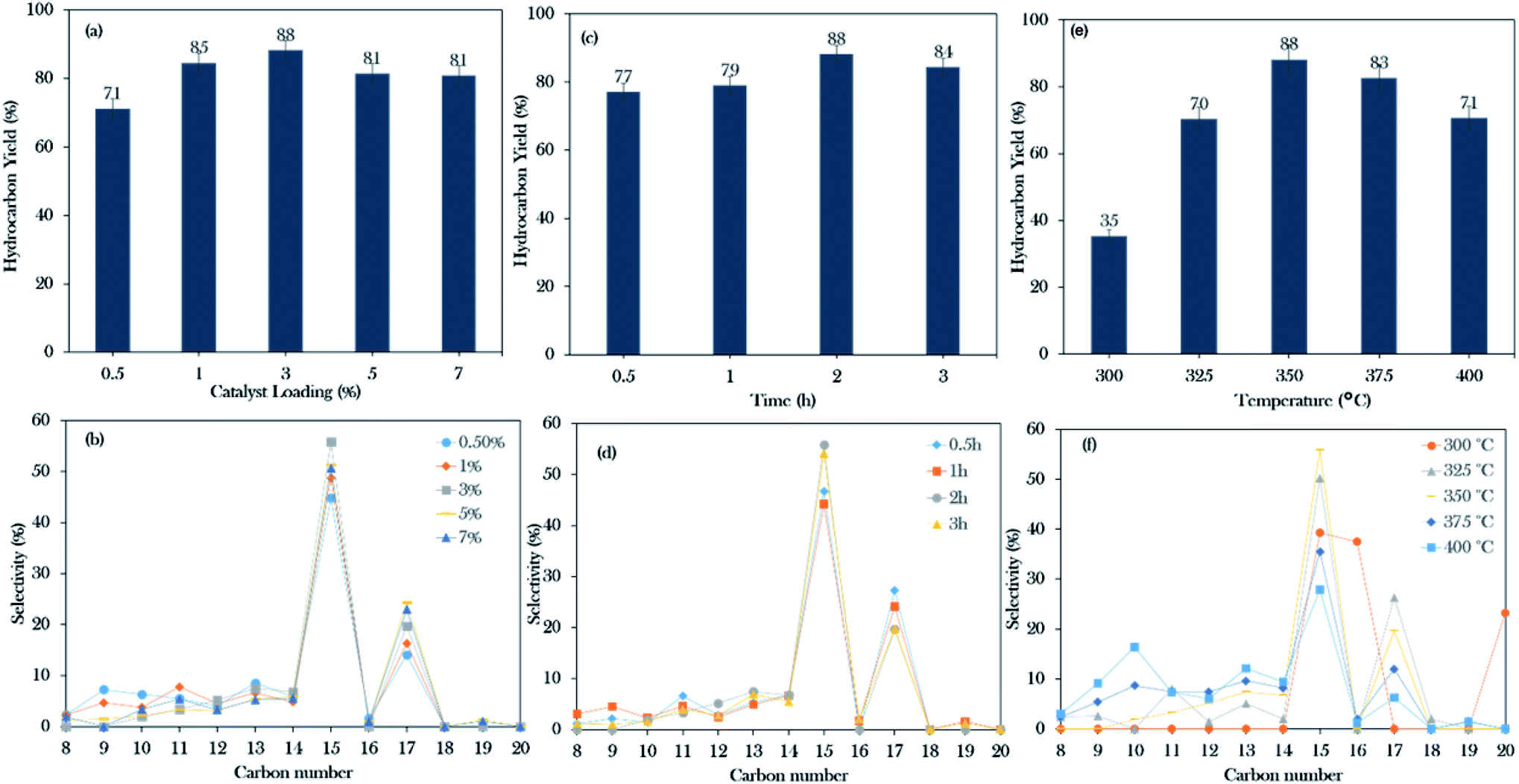 | ||
| Fig. 9 Optimization studies of WCO: (a and b) the effects of catalyst loading, parameters: 350 °C and 2 h reaction time. (c and d) The effects of reaction time, parameters: 350 °C and 3 wt% catalyst loading. (e and f) The effects of reaction temperature, parameters: 3 wt% catalyst loading and 2 h reaction time with a stirring rate of 300 rpm and under an inert atmosphere. | ||
The experiments on the effects of reaction time on deoxygenation reactions were conducted at 350 °C, 3 wt% catalyst loading, and 300 rpm stirring rate in an inert atmosphere. The collected data showed that the hydrocarbon yield gradually increased from 77% to 88% and the hydrocarbon selectivity for n-(C15 + C17) increased from 74% to 75% when the time was extended from 30 min to 2 h. Thus, it was suggested that the deoxygenation reaction still occurred for up to 2 h of reaction time. However, it was observed that beyond 3 h of reaction, it only led to a decrease in the hydrocarbon yield to 84% and the n-(C15 + C17) selectivity was reduced to 74% (Fig. 9c and d). According to Abdulkareem-Alsultan et al., this was due to the cracking of the deoxygenated liquid product and the increase in the light fraction of hydrocarbons and the consequent production of gaseous products.41 In this work, the optimum reaction duration was 2 h for an effective deoxygenation reaction.
Moreover, the influence of the temperature on the WCO was determined at 2 h reaction time, 3 wt% catalyst loading, 300 rpm stirring rate in a N2 atmosphere. Referring to Fig. 9e and f, it was observed that heating the system from 300 to 350 °C could increase the hydrocarbon yield and n-(C15 + C17) selectivity from 35–88% and 39–75%, respectively. Interestingly, the percentage of C16 and C20 fractions was significantly higher at 300 °C, due to the cracking and polymerization reactions that predominantly occurred when the deoxygenation proceeded under mild reaction condition (temperature: 270–400 °C; time: 30–240 min; catalyst loading: 0.5–9%).41,74 It should be noted that the WCO was mainly comprised of C16:0 and C18:1 (Table 1); rich C16 species may also have originated from the cracking of the C18:1 fatty acid, forming the C16 fraction. This occurrence is consistent with that of Aliana-Nasharuddin et al.,42 who suggested in their reaction mechanism scheme that the chicken fat oil (CFO) was composed mainly of C16 and C18 fatty acids. When CFO was deoxygenized via the deCOx reaction, it resulted in the formation of C15 and C17. Further cracking of C18 fatty acids also resulted in the formation of C16 fatty acids. In the case of C20, hypothetically, the C20 is formed via the polymerization reaction of short-chain hydrocarbons into a longer carbon complex.18 However, increasing the reaction temperature above 375 °C only reduced the deoxygenation activity. Bernas et al.75 revealed that the reduction of the deoxygenation activity at high temperature is due to the occurrence of the thermal cracking reaction, which drastically increased the formation of light hydrocarbons in the range of n-(C8–C13). In summary, the deoxygenation activity is very sensitive to the heat change, which could significantly affect the hydrocarbon yield and n-(C15 + C17). From this finding, 88% hydrocarbon yield and 75% hydrocarbon selectivity n-(C15 + C17) were obtained when the reactions were conducted with 3 wt% at the reaction temperature of 350 °C for a reaction time of 2 h.
3.4 The reusability and stability of the NiLa/AC catalyst
Reusability studies of a catalyst are crucial for estimating the economics of the NiLa/AC (Ni = 20 wt%; La = 20 wt%) catalyst for large scale production. The reusability of the catalyst was investigated under the favourable reaction conditions of 3 wt% catalyst loading, 2 h reaction time, 10 g of WCO, and 350 °C. The spent catalyst was reactivated by washing with hexane and it was reused for the next reaction cycle under the same reaction conditions. In this work, the deoxygenation reaction was performed for 6 consecutive runs. Surprisingly, the results revealed that the hydrocarbon yield was maintained over 80% and the hydrocarbon selectivity for n-(C15 + C17) was over 60%, as exhibited in Fig. 10a and b. It was concluded that the NiLa/AC catalyst has outstanding mechanical properties and chemical stability. This was supported by XRD patterns for the spent catalyst, where the diffraction peaks of Ni and La2O3 for the fresh and spent NiLa/AC remained after 6 consecutive runs (Fig. 11a), meaning that there were no changes in the structural phases of the active metals in each cycle.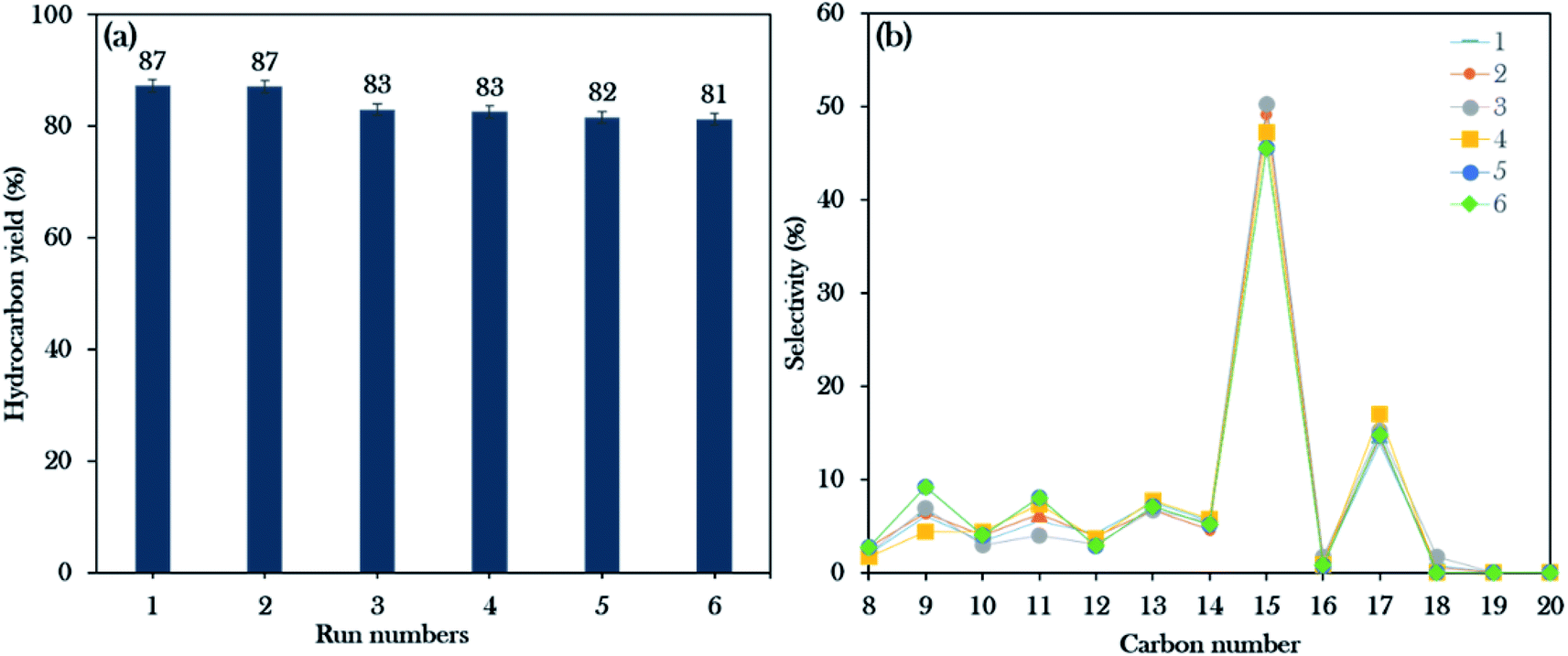 | ||
| Fig. 10 Reusability studies of the Ni20La20AC catalyst for the 6th run: (a) the hydrocarbon yield profile, (b) hydrocarbon selectivity. | ||
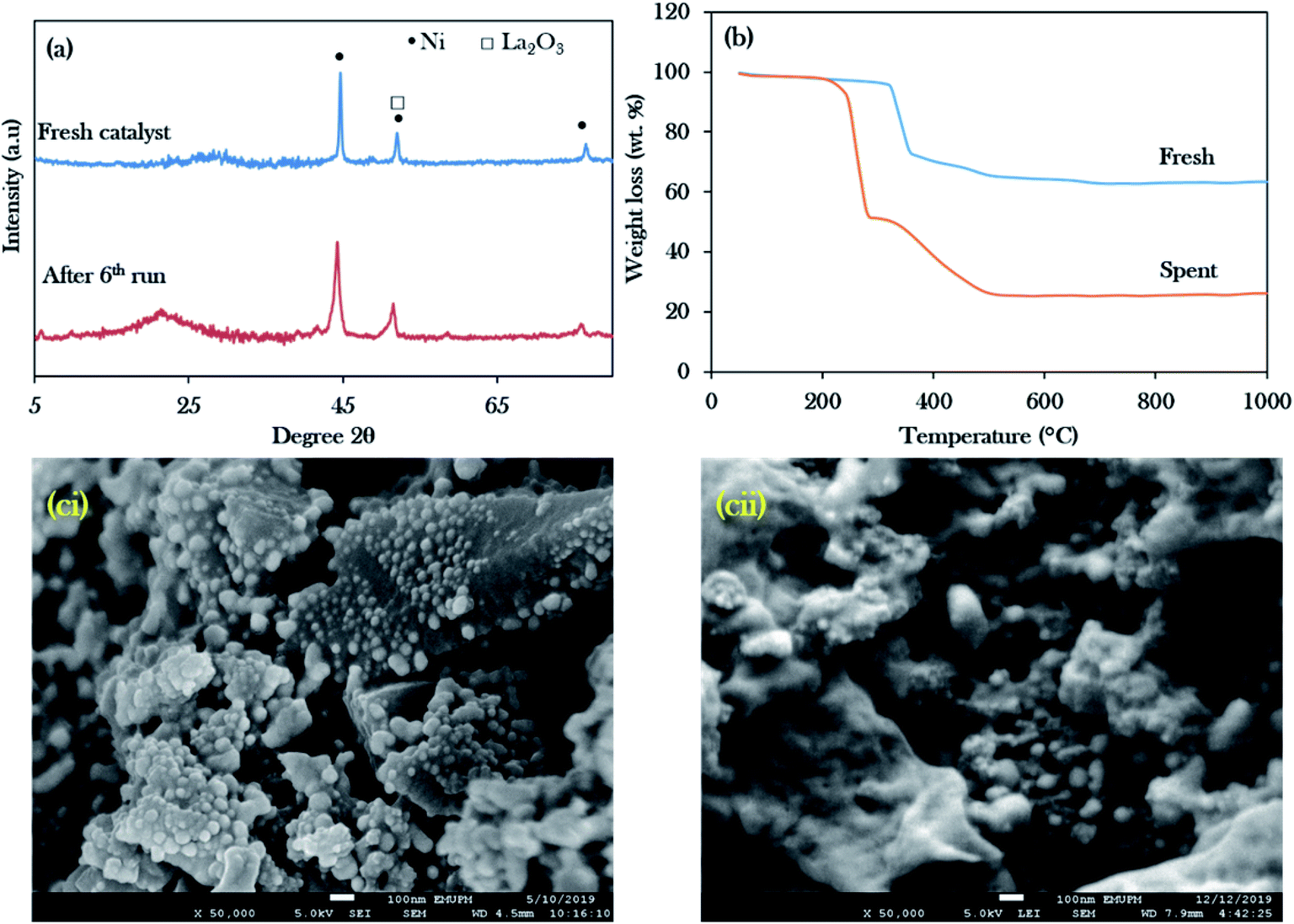 | ||
| Fig. 11 Stability studies of the Ni20La20AC catalyst for the 6th run: (a) the XRD profile, (b) TGA, (ci) FESEM image for the fresh catalyst, (cii) FESEM image for the spent catalyst. | ||
The amorphous structure of the carbon phase resulted in a weak diffraction peak at 2θ = 20–30° in the spent NiLa/AC catalyst, which intensified after deoxygenation, suggesting an increase in the formation of random aromatic carbon sheets. This can be ascribed to the increased amorphous carbon-derived coke. The amount of coke produced was evaluated by TGA analysis as shown in Fig. 11b. It was found that the main decomposition peaks for fresh NiLa/AC catalysts were between 330 °C and 506 °C; however, the spent NiLa/AC catalyst started to decompose at 225 °C to 526 °C. The early decomposition of about 47 wt% ranged from 225 °C to 325 °C, due to the oxidation of the soft coke on the surface.76 Moreover, it was noted that the spent NiLa/AC catalyst also exhibited the presence of hard coke of about 26 wt%, which decomposed between 330 °C to 750 °C; this revealed that the catalyst surface was covered by coke formation after the deoxygenation reaction.77 This finding agreed with EDX, which showed a remarkable increment in the C content in the NiLa/AC spent catalyst after 6 deoxygenation runs (Table 5); hence, it strongly implies that the spent NiLa/AC catalyst was masked by the coke.
Fig. 11ci and cii show the electron scanning images of fresh and spent NiLa/AC catalysts; the spent NiLa/AC catalyst was sintered after the reaction. It was expected that the catalysts would be exposed to high temperatures for longer times and would clamp to each other; as a result, the surface area and active sites were reduced and, consequently, deactivation occurred.78,79 Hence, the majority of the tiny dots in fresh NiLa/AC were enlarged after long-term deoxygenation reactions. This finding, evinced by Kim et al.,80 reported that sintered catalysts typically resulted in reduced catalytic activity due to the loss of active sites. Even though the NiLa/AC suffered from coke deposits and sintering, the NiLa/AC catalyst still showed consistent deoxygenation activity within 6 consecutive runs without significant reactivity loss, suggesting that the catalyst was highly stable and reusable.
4. Conclusion
The catalytic deoxygenation of WCO via deCOx pathways was studied over activated carbon derived-catalysts (NiLa/AC, NiCe/AC, NiFe/AC, NiMn/AC, NiW/AC, and NiZn/AC). Among all the prepared binary metal AC catalysts, the Ni20La20/AC catalyst showed very effective deoxygenation performance via deCOx reactions and effective exclusivity in producing n-(C15 + C17) fractions, due to the moderate density of the strongly acidic properties and the low density of the strongly basic properties. The changes in the surface the area had no effect on the formation of n-(C15 + C17) fractions. In particular, rich La (20 wt%) and Ni (20 wt%) species led to efficient deoxygenation activity. Based on the OVAT optimization study, about 88% of the hydrocarbon yield and 75% n-(C15 + C17) selectivity were observed under the favourable conditions: 3 wt% of catalyst loading, 2 h of reaction time, and 350 °C reaction temperature. It also showed excellent stability which was proven by the consistent hydrocarbon yield of over 80%, and over 60% for n-(C15 + C17) selectivity for 6 consecutive runs.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful and appreciate the financial support from Ministry of Higher Education Malaysia for Fundamental Research Grant Scheme (FRGS 2014-1), Putra Grant-IPM 9559000, Geran Putra Berimpak (GPB) UPM/800-3/3/1/GPB/2018/9658700, and Geran Putra Berimpak GBP/2019/9674500.References
- C. Kordulis, K. Bourikas, M. Gousi, E. Kordouli and A. Lycourghiotis, Appl. Catal., B, 2016, 181, 156–196 CrossRef CAS.
- K. Treusch, N. Schwaiger, K. Schlackl, R. Nagl, A. Rollett, M. Schadler, B. Hammerschlag, J. Ausserleitner, A. Huber, P. Pucher and M. Siebenhofer, React. Chem. Eng., 2018, 3, 258–266 RSC.
- E. Santillan-Jimenez and M. Crocker, J. Chem. Technol. Biotechnol., 2012, 87, 1041–1050 CrossRef CAS.
- G. Knothe, Prog. Energy Combust. Sci., 2010, 36, 364–373 CrossRef CAS.
- R. W. Gosselink, S. A. W. Hollak, S. W. Chang, J. Van Haveren, K. P. De Jong, J. H. Bitter and D. S. Van Es, ChemSusChem, 2013, 6, 1576–1594 CrossRef CAS.
- R. Shu, R. Li, B. Lin, C. Wang, Z. Cheng and Y. Chen, Biomass Bioenergy, 2020, 132, 105432 CrossRef CAS.
- P. Kumar, S. R. Yenumala, S. K. Maity and D. Shee, Appl. Catal., A, 2014, 471, 28–38 CrossRef CAS.
- M. F. Kamaruzaman, Y. H. Taufiq-Yap and D. Derawi, Biomass Bioenergy, 2020, 134, 105476 CrossRef CAS.
- X. Y. Ooi, W. Gao, H. C. Ong, H. V. Lee, J. C. Juan, W. H. Chen and K. T. Lee, Renewable Sustainable Energy Rev., 2019, 112, 834–852 CrossRef CAS.
- S. K. Kim, J. Y. Han, H. Lee, T. Yum, Y. Kim and J. Kim, Appl. Energy, 2014, 116, 199–205 CrossRef CAS.
- A. N. Kay Lup, F. Abnisa, W. M. A. Wan Daud and M. K. Aroua, J. Ind. Eng. Chem., 2017, 56, 1–34 CrossRef CAS.
- K. Jeništová, I. Hachemi, P. Mäki-Arvela, N. Kumar, M. Peurla, L. Capek, J. Wärnå and D. Y. Murzin, Chem. Eng. J., 2017, 316, 401–409 CrossRef.
- M. I. Loizides, X. I. Loizidou, D. L. Orthodoxou and D. Petsa, Recycling, 2019, 1–10 Search PubMed.
- K. Hanisah, S. Kumar and T. Ay, J. Environ. Health, 2013, 4, 76–81 Search PubMed.
- B. Peng, Q. Shu, J. Wang, G. Wang, D. Wang and M. Han, Process Saf. Environ. Prot., 2008, 86, 441–447 CrossRef CAS.
- I. Nikolopoulos, G. Kogkos, E. Kordouli, K. Bourikas, C. Kordulis and A. Lycourghiotis, Mol. Catal., 2020, 482, 110697 CrossRef CAS.
- T. Morgan, D. Grubb, E. Santillan-Jimenez and M. Crocker, Top. Catal., 2010, 53, 820–829 CrossRef CAS.
- N. Asikin-Mijan, H. V. Lee, G. Abdulkareem-Alsultan, A. Afandi and Y. H. Taufiq-Yap, J. Cleaner Prod., 2017, 167, 1048–1059 CrossRef CAS.
- H. Li, H. Ma, W. Zhao, X. Li and J. Long, Appl. Energy, 2019, 253, 113613 CrossRef CAS.
- S. Thongkumkoon, W. Kiatkittipong, U. Wetwatana, N. Laosiripojana and P. Daorattanachai, Renewable Energy, 2019, 140, 111–123 CrossRef CAS.
- S. Zulkepli, J. C. Juan, H. V. Lee, N. S. A. Rahman, P. L. Show and E. P. Ng, Energy Convers. Manage., 2018, 165, 495–508 CrossRef CAS.
- V. Balasundram, N. Ibrahim, R. M. Kasmani, M. K. A. Hamid, R. Isha, H. Hasbullah and R. R. Ali, Energy Procedia, 2017, 142, 801–808 CrossRef CAS.
- R. Zhang, Y. Wang and R. C. Brown, Energy Convers. Manage., 2007, 48, 68–77 CrossRef CAS.
- G. Wen, Y. Xu, H. Ma, Z. Xu and Z. Tian, Int. J. Hydrogen Energy, 2008, 33, 6657–6666 CrossRef CAS.
- C. Miao, O. Marin-flores, S. D. Davidson, T. Li, T. Dong, D. Gao, Y. Wang, M. Garcia-pérez and S. Chen, Fuel, 2016, 166, 302–308 CrossRef CAS.
- J. M. Gómez, M. D. Romero and V. Callejo, Catal. Today, 2013, 218–219, 143–147 CrossRef.
- N. Asikin-mijan, H. V. Lee, T. S. Marliza and Y. H. Taufiq-Yap, J. Anal. Appl. Pyrolysis, 2018, 129, 221–230 CrossRef CAS.
- G. Abdulkareem-Alsultan, N. Asikin-mijan, N. Mansir, H. V. Lee, Z. Zainal, A. Islam and Y. H. Taufiq-Yap, J. Anal. Appl. Pyrolysis, 2019, 137, 171–184 CrossRef CAS.
- X. Cao, L. Li, S. Yu, S. Liu, H. Yu, Q. Wu and A. J. Ragauskas, J. Anal. Appl. Pyrolysis, 2019, 138, 137–144 CrossRef CAS.
- S. Wang and G. Q. M. Lu, Appl. Catal., B, 1998, 19, 267–277 CrossRef CAS.
- T. Aysu, N. A. A. Rahman and A. Sanna, Energy, 2016, 103, 205–214 CrossRef CAS.
- V. Balasundram, N. Ibrahim, R. M. Kasmani, R. Isha, M. K. A. Hamid, R. R. Ali and H. Hasbullah, Appl. Energy, 2018, 220, 787–799 CrossRef CAS.
- R. Isha and P. T. Williams, J. Energy Inst., 2012, 22–28 CrossRef CAS.
- M. Akri, S. Pronier, T. Chafik, O. Achak, P. Granger, P. Simon, M. Trentesaux and C. Batiot-dupeyrat, Appl. Catal., B, 2017, 205, 519–531 CrossRef CAS.
- M. Greluk, M. Rotko and S. Turczyniak-surdacka, Appl. Catal., A, 2020, 590, 117334 CrossRef CAS.
- P. Osorio-vargas, N. A. Flores-gonzález, R. M. Navarro, J. L. G. Fierro, C. H. Campos and P. Reyes, Catal. Today, 2015, 259, 27–38 CrossRef.
- N. Asikin-Mijan, H. V. Lee, J. C. Juan, A. R. Noorsaadah, G. Abdulkareem-Alsultan, M. Arumugam and Y. H. Taufiq-Yap, J. Anal. Appl. Pyrolysis, 2016, 120, 110–120 CrossRef CAS.
- J. Fu, X. Lu and P. E. Savage, Energy Environ. Sci., 2010, 3, 311–317 RSC.
- M. Jin and M. Choi, Mol. Catal., 2019, 474, 110419 CrossRef CAS.
- K. Sun, T. C. Schulz, S. T. Thompson and H. H. Lamb, Catal. Today, 2016, 269, 93–102 CrossRef CAS.
- G. Abdulkareem-Alsultan, N. Asikin-mijan, H. V. Lee, A. S. Albazzaz and Y. H. Taufiq-Yap, Energy Convers. Manage., 2017, 151, 311–323 CrossRef.
- N. Aliana-Nasharuddin, N. Asikin-mijan, G. Abdulkareem-Alsultan, M. I. Saiman, F. A. Alharthi, A. A. Alghamdi and Y. H. Taufiq-Yap, RSC Adv., 2020, 10, 626–642 RSC.
- N. Asikin-Mijan, J. M. Ooi, G. AbdulKareem-Alsultan, H. V. Lee, M. S. Mastuli, N. Mansir, F. A. Alharthi, A. A. Alghamdi and Y. H. Taufiq-Yap, J. Cleaner Prod., 2020, 249, 119381 CrossRef CAS.
- M. S. Gamal, N. Asikin-mijan, M. Arumugam and U. Rashid, J. Anal. Appl. Pyrolysis, 2019, 144, 104690 CrossRef.
- D. Mukai, S. Tochiya, Y. Murai, M. Imori, T. Hashimoto, Y. Sugiura and Y. Sekine, Appl. Catal., A, 2013, 453, 60–70 CrossRef CAS.
- J. Chen, H. Zou, Q. Yao, M. Luo, X. Li and Z.-H. Lu, Appl. Surf. Sci., 2020, 501, 144247 CrossRef CAS.
- X. Li, B. Yan, S. Yao, S. Kattel, J. G. Chen and T. Wang, Appl. Catal., B, 2018, 231, 213–223 CrossRef CAS.
- G. Li, F. Zhang, L. Chen, C. Zhang, H. Huang and X. Li, ChemCatChem, 2015, 7, 2646–2653 CrossRef CAS.
- Y. Zheng, F. Wang, X. Yang, Y. Huang, C. Liu, Z. Zheng and J. Gu, J. Anal. Appl. Pyrolysis, 2017, 126, 169–179 CrossRef CAS.
- A. Srifa, K. Faungnawakij, V. Itthibenchapong and S. Assabumrungrat, Chem. Eng. J., 2015, 278, 249–258 CrossRef CAS.
- V. Balasundram, K. K. Zaman, N. Ibrahim, R. M. Kasmani, R. Isha, M. K. A. Hamid and H. Hasbullah, J. Anal. Appl. Pyrolysis, 2018, 134, 309–325 CrossRef CAS.
- Y. Zhang, D. Duan, H. Lei, E. Villota and R. Ruan, Appl. Energy, 2019, 251, 113337 CrossRef CAS.
- C. Tan and W. Tsai, Int. J. Hydrogen Energy, 2015, 40, 14064–14071 CrossRef CAS.
- R. Ding, Y. Wu, Y. Chen, J. Liang, J. Liu and M. Yang, Chem. Eng. Sci., 2015, 135, 517–525 CrossRef CAS.
- B. C. Ang, I. I. Yaacob and I. Nurdin, J. Nanomater., 2013, 2013, 1–6 CrossRef.
- S. Janampelli and S. Darbha, Mol. Catal., 2018, 451, 125–134 CrossRef CAS.
- G. U. O. Haiyan, M. A. Lunjie, S. Fei, Y. Gang, Z. Yanzong, D. Shihuai, Z. Jing, S. Chun and Z. Yongmei, J. Rare Earths, 2017, 35, 593–601 CrossRef.
- W. Li, K. Yang, J. Peng, L. Zhang, S. Guo and H. Xia, Ind. Crops Prod., 2008, 28, 190–198 CrossRef CAS.
- J. Jeong, H. W. Lee, S. H. Jang, S. Ryu, Y. Kim, R. Park, S.-C. Jung, J.-K. Jeon and Y.-K. Park, Catalysts, 2019, 9, 1–11 CAS.
- M. Roussel, S. Norsic, J.-L. Lemberton, M. Guisnet, T. Cseri and E. Benazzi, Appl. Catal., A, 2005, 279, 53–58 CrossRef CAS.
- M. Gousi, E. Kordouli, K. Bourikas, E. Simianakis, S. Ladas, G. D. Panagiotou, C. Kordulis and A. Lycourghiotis, Catal. Today DOI:10.1016/j.cattod.2019.02.034.
- F. A. A. Twaiq, A. R. Mohamad and S. Bhatia, Fuel Process. Technol., 2004, 85, 1283–1300 CrossRef CAS.
- E. W. Qian, N. Chen and S. Gong, J. Mol. Catal. A: Chem., 2014, 387, 76–85 CrossRef CAS.
- M. Renz, Eur. J. Org. Chem., 2005, 979–988 CrossRef CAS.
- M. Tamura, T. Tonomura, K. Shimizu and A. Satsuma, Green Chem., 2012, 14, 984–991 RSC.
- X. Li, X. Zhang, S. Shao, L. Dong, J. Zhang, C. Hu and Y. Cai, Bioresour. Technol., 2018, 259, 191–197 CrossRef CAS.
- G. Abdulkareem-Alsultan, N. Asikin-Mijan, G. Mustafa-Alsultan, H. V. Lee, K. Wilson and Y. H. Taufiq-Yap, R. Soc. Chem., 2020, 10, 4996–5009 CAS.
- J. K. Satyarthi and D. Srinivas, Energy Fuels, 2011, 25, 3318–3322 CrossRef CAS.
- H. K. G. Singh, S. Yusup, A. T. Quitain, B. Abdullah, M. Ameen, M. Sasaki, T. Kida and K. W. Cheah, Environ. Res., 2020, 186, 109616 CrossRef.
- L. Hermida, A. Z. Abdullah and A. R. Mohamed, Renewable Sustainable Energy Rev., 2015, 42, 1223–1233 CrossRef CAS.
- I. Kim, A. A. Dwiatmoko, J. Choi, D. J. Suh, J. Jae, J. Ha and J. Kim, J. Ind. Eng. Chem., 2017, 56, 74–81 CrossRef CAS.
- A. Bohre, M. I. Alam, K. Avasthi, F. Ruiz-Zepeda and B. Likozar, Appl. Catal., B, 2020, 276, 119069 CrossRef.
- Y. Lou, P. He, L. Zhao and H. Song, Fuel, 2016, 183, 396–404 CrossRef CAS.
- M. Snåre, I. Kubičková, P. Mäki-Arvela, D. Chichova, K. Eränen and D. Y. Murzin, Fuel, 2008, 87, 933–945 CrossRef.
- H. Bernas, K. Eränen, I. Simakova, A. Leino, K. Kordás, J. Myllyoja, P. Mäki-arvela, T. Salmi and D. Y. Murzin, Fuel, 2010, 89, 2033–2039 CrossRef CAS.
- N. Taufiqurrahmi, A. R. Mohamed and S. Bhatia, Chem. Eng. J., 2010, 163, 413–421 CrossRef CAS.
- S. K. Sahoo, S. S. Ray and I. D. Singh, Appl. Catal., A, 2004, 278, 83–91 CrossRef CAS.
- V. G. D. la Cruz-Flores, A. Martinez-Hernandez and M. A. Gracia-Pinilla, Appl. Catal., A, 2020, 594, 117455 CrossRef.
- J. Sehested, J. A. P. Gelten and S. Helveg, Appl. Catal., A, 2006, 309, 237–246 CrossRef CAS.
- S. Kim, E. E. Kwon, Y. T. Kim, S. Jung, H. J. Kim, G. W. Huber and J. Lee, Green Chem., 2019, 21, 3715–3743 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06302a |
| This journal is © The Royal Society of Chemistry 2020 |