DOI:
10.1039/D0RA06576E
(Paper)
RSC Adv., 2020,
10, 33700-33705
Development of a horseradish peroxidase-nanobody fusion protein for visual detection of ochratoxin A by dot immunoassay†
Received
29th July 2020
, Accepted 6th September 2020
First published on 11th September 2020
Abstract
Ochratoxin A (OTA) is a common cereal mycotoxin that seriously threatens food safety and public health. Herein a horseradish peroxidase-nanobody fusion protein (HRP-Nb) retaining antibody and enzyme activity was obtained after inclusion body denaturation and renaturation and enzyme reconstitution, which served both as the primary antibody and reporter enzyme and was applied to develop a membrane-based dot immunoassay (HN-DIA) for OTA visual detection. Based on the optimal experimental conditions, the HN-DIA could be finished in 10 min with a cut-off limit of 50 μg kg−1 in rice and oat samples by eye. The HN-DIA showed high selectivity for OTA and had good accuracy and reproducibility in the recovery experiments. Spiked sample analysis results of the HN-DIA and high performance liquid chromatography (HPLC) correlated well with each other. Therefore, the proposed HN-DIA has the potential for rapid screening of OTA and other small molecule pollutants in food and the environment by naked eye.
1. Introduction
Since ochratoxin A (OTA) was isolated and chemically characterized in 1965, it has been found to be widely present in all stages of the food chain.1 Toxicity research has shown that OTA is nephrotoxic, embryotoxic, teratogenic, immunotoxic, hepatotoxic, neurotoxic, genotoxic, and carcinogenic.2 Moreover, the International Agency for Research on Cancer classified OTA as a possible carcinogen to humans of group 2B in 1993.3 Therefore, OTA is one of the most common and deleterious mycotoxins. To minimize the exposure risk of OTA from food intake, many countries and international organizations have set strict maximum limits for OTA in food, which stimulates an urgent need for adequate techniques for screening of OTA.
Numerous analytical techniques have been developed for OTA detection in past decades, including instrument verification methods and antibody-based rapid screening methods, such as HPLC,4 enzyme-linked immunosorbent assay (ELISA),5 lateral flow strips,6 and biosensors.7 Among them, enzyme-based immunoassay was the most commonly used screening method due to its low cost, high sensitivity, and simple operation.8 However, the chemical labeling strategies for traditional antibody and enzyme (e.g., glutaraldehyde method and sodium periodate method) have some inherent drawbacks, including random coupling ratio and activity loss of antibody and enzyme.9
In recent years, with the progress of antibody engineering, various genetically engineered antibodies emerged as an alternative to the traditional antibody, such as antigen-binding fragment,10 single-chain variable fragment,11 and single-domain antibody (sdAb). The sdAb, also known as nanobody (Nb),12 is generally produced from the variable domain of heavy chain antibodies in Camelidaes and of the new antigen receptors of Chondrichthyes.13,14 The Nb shows many unique properties because of its special single domain structure such as high thermal stability, good water solubility, and high expression yield. Especially due to its ease of genetic manipulation, various fusion proteins of Nbs and enzymes including alkaline phosphatase,15,16 nanoluciferase17 and horseradish peroxidase18 have been generated. These fusion proteins can avoid activity loss of antibody and enzyme and heterogeneity of the conjugates caused by the traditional chemical labeling method. Therefore, they have been widely applied in the immunoassays of pesticides, toxins, viruses, and environmental pollutants as an emerging dual functional reagent. However, the horseradish peroxidase (HRP) was rarely used as a reporter for fusion expression with Nb due to its complex structure and high glycosylation level.19 To our knowledge, no HRP/Nb fusion protein has been reported for the detection of small molecules.
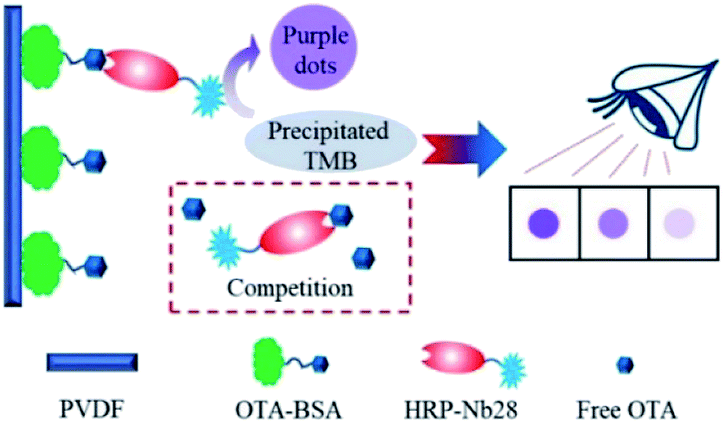
In this work, we aimed to develop an HRP/Nb fusion protein probe using the OTA-specific nanobody Nb28. The HRP-Nb28 fusion protein was efficiently expressed in the form of inclusion bodies in E. coli by optimizing the arrangement of Nb28 and HRP. In addition, we also developed an effective fusion protein inclusion body purification, renaturation and reconstruction program, and successfully prepared the HRP-Nb28 fusion protein retaining both peroxidase and antibody activity. To investigate the feasibility of HRP-Nb as a recombinant tracer in immunoassay, we developed a polyvinylidene fluoride (PVDF) membrane-based dot immunoassay using the HRP-Nb28 for visual and rapid detection of OTA in cereal (Fig. 1). The optimization of parameters including HRP-Nb28 concentration, coating antigen (OTA-BSA) concentration, ionic strength and methanol concentration in assay buffer, and competitive reaction time were detailed. Under optimal conditions, the cut-off limit of the proposed method was 50 μg kg−1 in rice and oats sample with naked eyes, showing its potential for rapid and visual analysis of OTA and the other toxic small molecules in food.
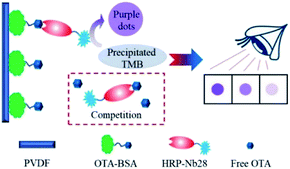 |
| | Fig. 1 Schematic diagram of the PVDF membrane-based dot immunoassay for OTA using the HRP-Nb. | |
2. Experimental
2.1. Reagent and materials
OTA and ochratoxin C (OTC) were purchased from Pribolab (Qingdao, China). Ochratoxin B (OTB) was obtained from Bioaustralis (Smithfield, NSW, Australia). Aflatoxin B1 (AFB1) and zearalenone (ZEN) were obtained from Fermentek (Jerusalem, Israel). Nonfat powdered milk, glutathione (GSH), oxidized glutathione (GSSG), and 96-well microplates were from Sangon Biotech (Shanghai, China). Precipitate TMB solution was purchased from InnoReagents (Huzhou, China). PVDF membranes were obtained from Millipore (Bedford, USA). HRP/anti-His was from CoWin Biosciences (Beijing, China). Agarose gel purification kit was purchased from TIANGEN Biotech (Beijing, China). Premix Taq enzyme was obtained from Takara (Dalian, China). Restriction endonuclease Xho I, Hind III and T4 DNA ligase were purchased from NEB (Ipswich, USA). pET25b+ vector, E. coli DH5α and E. coli Rosetta were saved in our laboratory. The recombinant pET25b+ vector with the OTA-specific Nb28 gene fragment (pET25b-Nb28) was prepared previously. The recombinant pET vector with the HRP gene fragment (pETpelHRPHis) was a gift from Dr Vitaly G. Grigorenko in Lomonosov Moscow State University.20 All inorganic chemicals and organic solvents were analytical grade.
2.2. Construction of prokaryotic expression vector
The recombinant plasmid pET25b-Nb28-HRP and pET25b-HRP-Nb28 were constructed as follows. Firstly, the gene fragments encoding Nb28 and HRP were amplified from the pET25b-Nb28 and pETpelHRPHis by PCR using paired primers (Table S1†), respectively. After agarose gel purification, the recovered gene fragments were assembled by overlap extension-polymerase chain reaction (OE-PCR) to yield full-length Nb28-HRP and HRP-Nb28 gene fragments that encode both fusion proteins (Nb28-linker-HRP and HRP-linker-Nb28) with a reference amino acid linker (GGGS)4 and restriction sites of Xho I and Hind III, respectively. The fragments were gel purified from the Xho I and Hind III restriction enzyme digested OE-PCR products, and ligated with the similarly digested prokaryotic expression vector pET-25b at a mole ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, respectively. The ligation products were transformed into the freshly prepared E. coli DH5α chemically competent cells by heat shock at 42 °C for 90 s. Then the transformed bacteria were plated on Luria-Bertani agar plates containing 50 μg mL−1 ampicillin and cultured overnight at 37 °C for ampicillin resistance screening. Positive clones of E. coli DH5α containing pET25b-Nb28-HRP or pET25b-HRP-Nb28 were randomly picked and identified by colony PCR, followed by DNA sequencing using the universal primer 5′-TAA TAC GAC TCA CTA TAG GG-3′.
:1, respectively. The ligation products were transformed into the freshly prepared E. coli DH5α chemically competent cells by heat shock at 42 °C for 90 s. Then the transformed bacteria were plated on Luria-Bertani agar plates containing 50 μg mL−1 ampicillin and cultured overnight at 37 °C for ampicillin resistance screening. Positive clones of E. coli DH5α containing pET25b-Nb28-HRP or pET25b-HRP-Nb28 were randomly picked and identified by colony PCR, followed by DNA sequencing using the universal primer 5′-TAA TAC GAC TCA CTA TAG GG-3′.
2.3. Expression and identification of Nb/HRP fusion proteins
The Nb28-HRP and HRP-Nb28 fusion proteins were produced by auto-induction expression according to our previously reported protocol with some modification.15 Firstly, the recombinant expression plasmids pET25b-Nb28-HRP and pET25b-HRP-Nb28 were transformed into the E. coli Rosetta chemically competent cells by heat shock, respectively. Then, the transformed cells were cultivated in 300 mL of auto-induction medium at 37 °C by shaking at 250 rpm until the OD600 reached 0.5–0.7. Afterwards, the medium was continued cultured at 25 °C with shaking at 250 rpm overnight for protein expression. Cells from induced culture medium were collected by centrifugation (4 °C, 8000g, 15 min) and suspended in 30 mL of equilibration buffer (2 mM KH2PO4, 8 mM Na2HPO4, 136 mM NaCl, 2.6 mM KCl, 1 mg mL−1 lysozyme, and 1 mM PMSF). The resuspended cells were destroyed through ultrasonication on icebath to avoid target protein denaturation. The supernatant and precipitation were separated by centrifugation at 4 °C with 10000g for 15 min. The protein fractions were analyzed with reduced 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
2.4. Purification and renaturation of inclusion body
The purification and renaturation of inclusion body were performed as follows. Firstly, the collected precipitation was successively washed with wash buffer I (10 mM Tris, pH 8.0, 1 mM EDTA, 5 mL L−1 Triton X-100, 1 M urea) and wash buffer II (10 mM Tris, pH 8.0) in accordance with m:V = 1:5 to remove impurities, where m was the content of precipitation and V was the volume of buffer. Then, the purified inclusion body was resuspended with denaturation buffer (20 mM Tris, pH 8.0, 6 M urea) in accordance with m:V = 1:10, where m was the content of the purified inclusion body and V was the volume of denaturation buffer. After the inclusion body was completely dissolved, the beta-mercaptoethanol was added with a final concentration of 20 mM and placed at 4 °C for 12 h. The supernatant was collected by centrifugation at 4 °C with 8500g for 20 min and double volume of refolding buffer (20 mM Tris, pH 8.0, 4% glycerol) was dropwise added by a peristaltic pump at 3 mL min−1 with magnetic stirring at 4 °C for 12 h. Then, the equal volume of renaturation buffer (20 mM Tris, pH 8.0, 400 mM arginine, 2 mM GSH, 4 mM GSSG, 5 mM 2,6-dimethyl-bethyl-β-cyclodextin) was dropwise added as described above. Finally, the renaturation protein was concentrated through the ultrafiltration tube.
2.5. Reconstitution of horseradish peroxidase
To recover the enzyme activity of HRP in HRP/Nb fusion proteins, the renatured inclusion body was reconstituted with hemin as described below. Briefly, the hemin stock solution (1 mM in 0.01 M KOH) was dropwise added to the renatured inclusion body solution by a peristaltic pump and placed at 4 °C for 12 h. The final concentration of the added hemin was twice that of the inclusion body. Then the mixture was dialyzed with 10 mM PBS to remove unbound hemin. The activity of HRP was detected by the color reaction of catalyzing TMB.
2.6. Procedures of the HRP-Nb28-based dot immunoassay
The dot immunoassay based on the HRP-Nb28 (HN-DIA) was performed as follows: firstly, the PVDF membrane was marked with squares (5 mm × 5 mm), followed by methanol activation and rinsing with ultrapure water. The pretreated membrane was transferred onto a pre-wetted filter paper, and 3 μL of OTA-BSA (10 μg mL−1 in PBS) was coated on the square center of the membrane at 37 °C for 2 h. After blocking with 3% (m/v) BSA in PBS at 37 °C for 1 h, the membrane was washed and dried at room temperature. Then 0.2 mL OTA standard with various concentrations (0, 10, 20, 40 and 80 ng mL−1 in 20 mM PBS containing 20% methanol) was added and mixed with an equal volume of HRP-Nb28 in a clear 1.5 mL centrifuge tube. The OTA-BSA coated membrane was immersed in the above centrifuge tube and incubated at room temperature for 5 min with gentle shaking. After rinsing with PBST, the membrane was incubated with 100 μL precipitate TMB solution for 5 min, followed by rinsing with tap water to stop the enzymatic reaction. The OTA content in sample was determined using naked eyes. The color intensity in test squares of OTA-positive and blind samples was compared with that of negative control, which presented the most intense color.
2.7. Cross-reactivity
To evaluate the selectivity of the proposed method, cross-reactivity (CR) of the HRP-Nb28 with OTA structural analogues (OTB, OTC) and some common mycotoxins (FB1, AFB1, ZEN) was determined by the HN-DIA.
2.8. Sample preparation
Cereal samples (rice and oats) for spiking and recovery study were purchased from a local supermarket in Haikou, China, which has been identified as OTA negative by LC-MS/MS analysis. The pretreatment of cereal samples for HN-DIA was performed as described previously.21 Briefly, 1 g of ground cereal sample was weighed and spiked with OTA standards of different adding levels (20, 50, 100, 200 μg kg−1). The spiked samples were mixed with 2 mL of 20 mM PBS containing 50% methanol by shaking vigorously and ultrasonication for 15 min, respectively. After centrifugation at 10000g for 5 min, the supernatant was diluted with 20 mM PBS for analysis. Meanwhile, the above supernatant was passed through a 0.45 μm filter and the filtrate was dried to concentrate under nitrogen, followed by dissolving in methanol for the HPLC with fluorescence detection (HPLC-FLD) analysis. The working conditions of HPLC-FLD for OTA were set as described previously.22 Briefly, the proportion of acetonitrile:0.4% formic acid in mobile phase was 45:55 (v/v) with a flow rate of 1 mL min−1. The injection volume was 20 mL and the column temperature was maintained at 40 °C. The excitation and emission wavelengths of fluorescence detector were set at 333 and 460 nm, respectively.
3. Results
3.1. Expression and identification of Nb/HRP fusion proteins
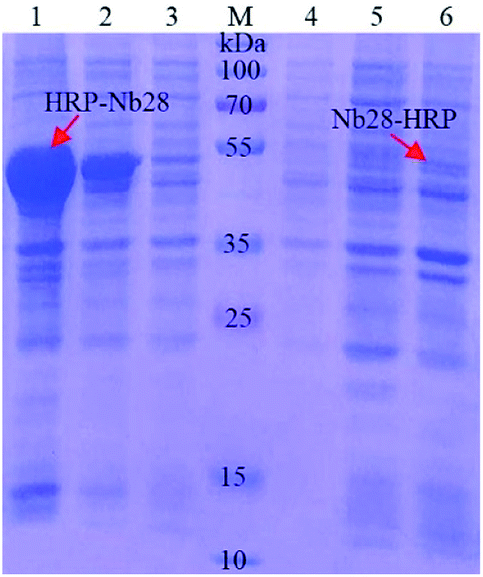
The recombinant expression vector pET25b-Nb28-HRP and pET25b-HRP-Nb28 with different arrangements of HRP and Nb28 were constructed (Fig. S1 and Table S2†) to explore the differences in expression yield. After auto-induction, the soluble fusion protein and inclusion body were characterized by SDS-PAGE. As shown in Fig. 2, a band of approximately 49 kDa was observed for both target fusion proteins HRP-Nb28 and Nb28-HRP. However, only HRP-Nb28 was expressed in a large amount as inclusion body, and the purity of HRP-Nb28 inclusion body was about 80%. These results were confirmed by the western blot assay probing with HRP/anti-His as seen in Fig. S2.† Therefore, the HRP-Nb28 was selected for further research because of its much higher yield in comparison with the Nb28-HRP.
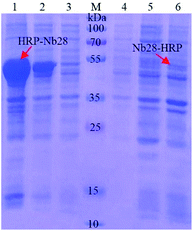 |
| | Fig. 2 SDS-PAGE analysis of the auto-induction expression of fusion proteins HRP-Nb28 (left) and Nb28-HRP (right). Lane 1 and 6: the precipitated protein of the induced E. coli cell after sonication; Lane 2 and 5: total protein of the induced E. coli cell; Lane 3 and 4: the supernatant protein of the induced E. coli cell after sonication; Lane M: prestained protein ladder. The red arrows point to the target proteins. | |
3.2. Isolation, purification and renaturation of inclusion body
The high expression and misfolding of HRP-Nb28 fusion protein in E. coli cells result in the formation of highly aggregated protein (inclusion body). To renature the HRP-Nb28 to its native and functional state, the inclusion body was sequentially isolated, purified and reconstituted. The original inclusion body obtained by centrifugation has a purity of about 80% that contains a small amount of bacteria protein as seen in Fig. 2. To improve the inclusion body purity, wash buffers I and II were used to wash the inclusion body. The EDTA, surfactant Triton X-100, and urea in wash buffer I were used to remove the lipopolysaccharide contaminants, remove the membrane proteins, and further selectively solubilize some other proteins, respectively. As shown in Fig. S3,† very few other proteins were retained in the inclusion body by SDS-PAGE analysis after the last washing with wash buffer II. The final purity of the inclusion body was improved from 80% (Lane 1, Fig. 2) to 95% (Lane 1, Fig. S3†) through two-step purification. Moreover, the HRP-Nb28 was not observed in the supernatants collected after washing the inclusion body with wash buffers I and II (Lane 2–7, Fig. S3†), indicating a very low loss of the target protein using the proposed purification strategy. After weighing, the purified inclusion body was treated with denaturation buffer, refolding buffer and renaturation buffer in turn.
3.3. Reconstitution and characterization of the HRP-Nb28 fusion protein
The structure of HRP was first solved by X-ray crystallography in 1997, which is a large alpha-helical protein and binds a heme as the redox cofactor. Therefore, the renatured inclusion body was reconstituted with heme to restore the HRP activity. After removing excess hemin by dialysis, the peroxidase activity of reconstituted inclusion body was analyzed by catalyzing TMB and compared with renatured inclusion body. As shown in Table 1, the inclusion body showed catalytic activity even at a concentration as low as 0.3125 μg mL−1 after reconstitution. Moreover, negligible interference on the color reaction caused by renatured inclusion body was observed. Thus, these results indicated the feasibility of the renaturation and reconstruction strategy in this work.
Table 1 Peroxidase activity analysis of the HRP-Nb28
| HRP-Nb28 (μg mL−1) |
HRP-Nb28 after reconstruction |
HRP-Nb28 before reconstruction |
| 10 |
3.240 |
0.074 |
| 5 |
3.143 |
0.106 |
| 2.5 |
1.545 |
0.081 |
| 1.25 |
0.750 |
0.086 |
| 0.625 |
0.360 |
0.075 |
| 0.3125 |
0.184 |
0.064 |
| 0.15625 |
0.095 |
0.070 |
| 0 |
0.069 |
0.073 |
The HRP-Nb28 can serve as a dual functional reagent with antigen binding and signal amplification capacities in immunoassay. To evaluate the antibody activity and enzyme catalytic activity of HRP-Nb28, a direct ELISA was performed to detect the OTA-BSA (1 μg mL−1 in PBS). As shown in Fig. S4,† the OD450 decreased from 0.470 to 0.189 as the concentration of HRP-Nb28 decreased from 50 to 3.125 μg mL−1, indicating that the developed HRP-Nb28 retains the antibody and enzyme activity.
3.4. HRP-Nb28-based dot immunoassay
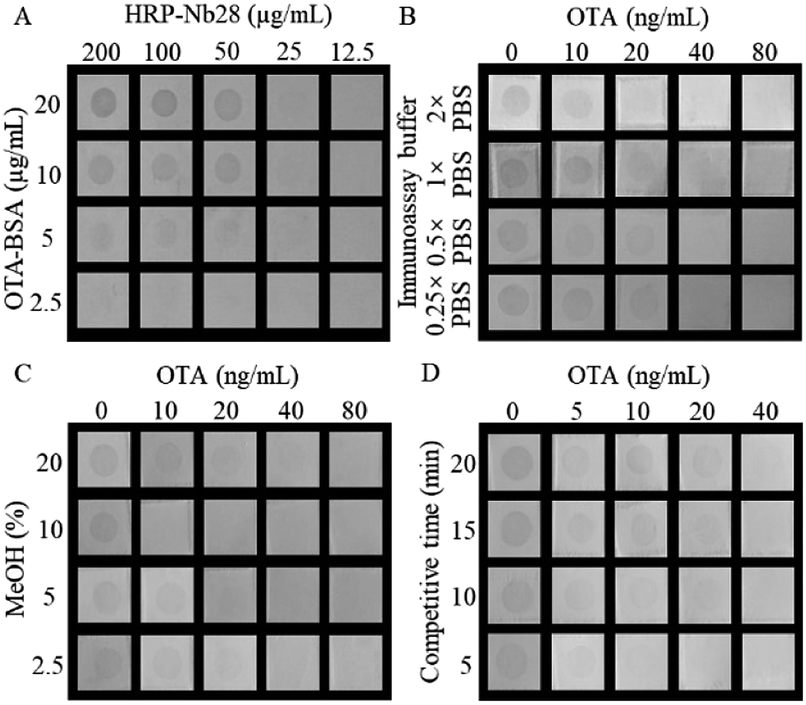
As the primary antibody and reporter molecule, the HRP-Nb28 was used to develop a dot immunoassay for visual detection of OTA. To achieve the best performance of the proposed HN-DIA, five experimental parameters including the concentrations of HRP-Nb28 and OTA-BSA, ionic strength and methanol concentration in assay buffer, and competitive reaction time were optimized. The effect evaluation was performed using the lowest cut-off limit, which was defined as the lowest concentration of OTA to achieve 100% inhibition of the antigen–antibody binding with no spot color development in the membrane square. As shown in Fig. 3A, a checkerboard titration was used to determine the optimal concentrations of OTA-BSA and HRP-Nb28. The TMB precipitate dot color intensity increased as either concentration of OTA-BSA (2.5–20 μg mL−1) or HRP-Nb28 (12.5–200 μg mL−1) increased. To obtain the lowest cut-off limit, 50 μg mL−1 of HRP-Nb28 and 10 μg mL−1 of OTA-BSA were selected. The effect of different ionic strength on the assay performance was evaluated. As shown in Fig. 3B, the dot color intensity declined with the increase of OTA concentration, and 2× PBS of ionic strength was selected for further research due to its lowest cut-off limit (20 ng mL−1). As a common solvent for lipophilic compounds, methanol could affect the antibody and enzyme activity. Therefore, assay buffers with various methanol concentrations were tested. As shown in Fig. 3C, the dot color intensity increased as the methanol concentration increased, and the lowest cut-off limit of 10 ng mL−1 OTA was observed with naked eyes when the final concentration of methanol reached 10%. Similarly, 5 min of the reaction time for OTA-BSA competitive binding to HRP-Nb28 with free OTA was selected (Fig. 3D). Based on the above optimal experimental conditions, the developed HN-DIA could be finished in 10 min with a cut-off limit of 10 ng mL−1 OTA in visual. Compared with the previously reported Nb-AP-based dot immunoassay for OTA,21 the HN-DIA is less sensitive which could be partly attributed to the activity decline of antibody and enzyme from denaturation and renaturation of the HRP-Nb28 inclusion body. Compared with the other reported membrane-based immunoassays for OTA (Table S3†), the proposed method exhibited comparable detection performance in sensitivity and speed.
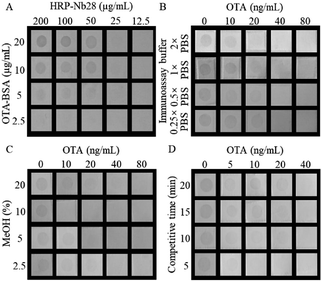 |
| | Fig. 3 Optimization of the HN-DIA for OTA. (A) Concentrations of OTA-BSA and HRP-Nb28; (B) ionic strength; (C) methanol concentration; (D) competitive reaction time. | |
3.5. Cross-reactivity
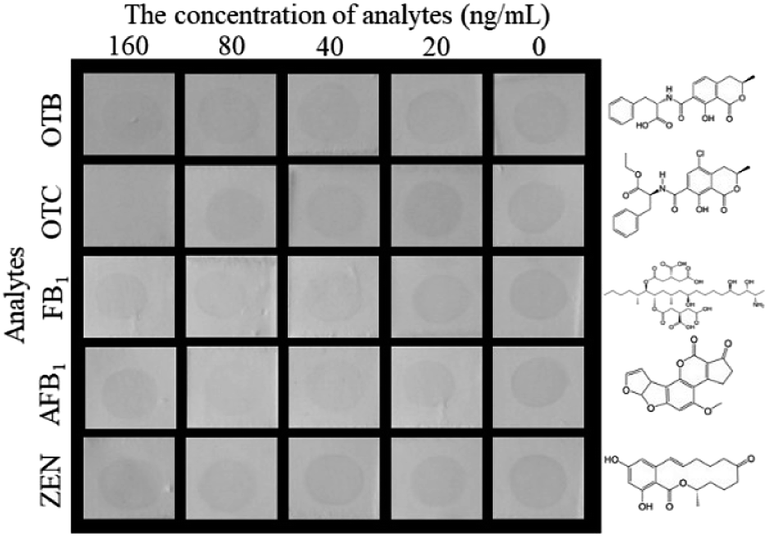
The selectivity of the proposed HN-DIA was evaluated by comparing the cross-reactivity (CR) of OTA with five common mycotoxins including OTB, OTC, FB1, AFB1 and ZEN. As shown in Fig. 4, no significant variation was observed for the dot color intensity of OTB, FB1, AFB1 and ZEN as the standard concentration reached up to 160 ng mL−1, showing the negligible CR of HRP-Nb28 with these mycotoxins. The dot disappeared when the OTC concentration increased to 160 ng mL−1, indicating that the binding of HRP-Nb28 to OTA-BSA was completely inhibited at this concentration. Thus, these results demonstrated the high selectivity of HN-DIA for OTA.
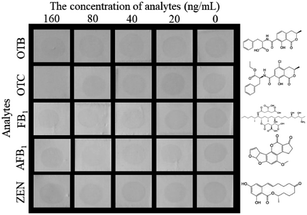 |
| | Fig. 4 The selectivity of HN-DIA for OTA detection. Serial concentrations of five common cereal mycotoxins including OTB, OTC, FB1, AFB1, and ZEN were used to replace OTA and tested by HN-DIA. The chemical structures of these mycotoxins were listed on the right. | |
3.6. Sample analysis and validation
The practicability of this proposed method was further evaluated by the spiking and recovery experiments of rice and oats. A series of OTA concentrations (20, 50, 100, 200 μg kg−1 of samples) were added to the OTA-free samples. After extraction, the supernatant was diluted 1:5 with 20 mM PBS to minimize the matrix effect (Fig. S5†) and detected by the HN-DIA. Each experiment was repeated four times on different days, and the results were judged by three persons with naked eyes. As shown in Table 2, the dot disappeared when the content of OTA was no less than 50 μg kg−1, which correlated well with the results tested by HPLC-FD. Therefore, these results demonstrated good accuracy and repeatability of the developed method.
Table 2 Analysis of the spiked rice and oat samples by HN-DIA and HPLC-FD
| Spiked OTA (μg kg−1) |
HN-DIAa (n = 4) |
HPLC-FD (μg kg−1 ± SD) |
| Rice |
Oats |
Rice |
Oats |
|
The same experiment was performed four times on different days.
Negative result with spot color development, which means that the detected OTA content is lower than the cut-off limit of HN-DIA.
Positive result with no spot color development, which means that the detected OTA content is not lower than the cut-off limit of HN-DIA.
|
| 20 |
−, −, −, −b |
−, −, −, − |
22 ± 2 |
21 ± 1 |
| 50 |
−, −, −, − |
−, −, −, − |
49 ± 4 |
47 ± 2 |
| 100 |
+, +, +, +c |
+, +, +, + |
101 ± 4 |
104 ± 6 |
| 200 |
+, +, +, + |
+, +, +, + |
220 ± 12 |
217 ± 14 |
4. Conclusion
In this study, we described the construction of an HRP-Nb fusion protein and applied it to develop a dot immunoassay for visual detection of OTA in cereal. This newly developed method has three major advantages as follows. (i) The pretreatment of samples is simple and low-cost. (ii) The HRP-Nb serves as the primary antibody and reporter molecule, which can eliminate the incubation step of secondary antibody and speed the assay time to 10 min. (iii) The detection result can be directly observed by naked eyes without using special apparatuses. However, the disadvantage of this method is that the cut-off limit is too high to meet the maximum residue limit of OTA in cereal and cereal products (5 μg kg−1) set by the European Union. The detection limit will continue to be reduced in the subsequent experiments to reach the desired level. To sum up, the developed HN-DIA has the potential for rapid and visual analysis of OTA and the other toxic small molecules in food.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Natural Science Foundation of Hainan Province (grant number 2019RC119 and 219QN149) and the National Natural Science Foundation of China (grant number 31760493 and 31901800). The APC was founded by the Scientific Research Foundation of Hainan University [grant number KYQD1631 and KYQD(ZR)1957].
References
- F. Malir, V. Ostry, A. Pfohl-Leszkowicz, J. Malir and J. Toman, Toxins, 2016, 8, 191–199 CrossRef PubMed.
- F. Malir, V. Ostry, A. Pfohl-Leszkowicz and E. Novotna, Birth Defects Res. Part B Dev. Reproductive Toxicol., 2013, 98, 493–502 CrossRef CAS.
- A. Pfohl-Leszkowicz and R. A. Manderville, Mol. Nutr. Food Res., 2007, 51, 61–99 CrossRef CAS PubMed.
- C. Brera, S. Grossi, B. De Santis and M. Miraglia, J. Liq. Chromatogr. Relat. Technol., 2003, 26, 119–133 CrossRef CAS.
- K. Pei, Y. Xiong, B. L. Xu, K. S. Wu, X. M. Li, H. Jiang and Y. H. Xiong, Sensor. Actuator. B Chem., 2018, 262, 102–109 CrossRef CAS.
- G. Zhang, Z. Chao, Y. Huang, Y. Jiao and A. Chen, Molecules, 2018, 23, 291–297 CrossRef PubMed.
- W. Zhu, L. Li, Z. Zhou, X. Yang and K. Wang, Food Chem., 2020, 319, 126544 CrossRef CAS.
- T. H. Ha, Toxins, 2015, 7, 5276–5300 CrossRef CAS PubMed.
- P. Farka and S. Bystricky, Chem. Pap., 2010, 64, 683–695 Search PubMed.
- V. Crivianu-Gaita, A. Romaschin and M. Thompson, Biochem. Biophys. Rep., 2015, 2, 23–28 Search PubMed.
- F. Safar, A. Vahideh, T. Asghar, V. Kamal, A. K. Shiva and R. Leila, Immunopharmacol. Immunotoxicol., 2014, 36, 297–308 CrossRef PubMed.
- J. P. Salvador, L. Vilaplana and M. P. Marco, Anal. Bioanal. Chem., 2019, 411, 1703–1713 CrossRef CAS.
- E. Hejiao, H. Jessica and H. Mitchell, Antibody Therapeutics, 2020, 3, 1–9 CrossRef.
- R. J. Hoey, H. Eom and J. R. Horn, Exp. Biol. Med., 2019, 244, 1568–1576 CrossRef CAS.
- X. R. Wang, Y. Y. Wang, Y. D. Wang, Q. Chen and X. Liu, Spectrochim. Acta Mol. Biomol. Spectrosc., 2020, 226, 117617 CrossRef CAS PubMed.
- T. Q. Sun, Z. Q. Zhao, W. T. Liu, Z. H. Xu, H. W. He, B. A. Ning, Y. Q. Jiang and Z. X. Gao, Anal. Chim. Acta, 2020, 1108, 28–36 CrossRef CAS.
- W. J. Ren, Z. F. Li, Y. Xu, D. B. Wan, B. Barnych, Y. P. Li, Z. Tu, Q. H. He, J. H. Fu and B. D. Hammock, J. Agric. Food Chem., 2019, 67, 5221–5229 CrossRef CAS PubMed.
- Y. M. Sheng, K. Wang, Q. Z. Lu, P. P. Ji, B. Y. Liu, J. H. Zhu, Q. Y. Liu, Y. I. Sun, J. F. Zhang, E. M. Zhou and Q. Zhao, J. Nanobiotechnol., 2019, 17, 35 CrossRef PubMed.
- O. Spadiut and C. Herwig, Pharm. Bioproces, 2013, 1, 283–295 CrossRef.
- V. Grigorenko, T. Chubar, Y. Kapeliuch, T. Börchers, F. Spener and A. Egorova, Biocatal. Biotransform., 2009, 17, 359–379 CrossRef.
- Z. Tang, X. Wang, J. Lv, X. Hu and X. Liu, Food Contr., 2018, 92, 430–436 CrossRef CAS.
- Z. Tang, X. Liu, Y. Wang, Q. Chen, B. D. Hammock and Y. Xu, Environ. Pollut., 2019, 251, 238–245 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Table S1: the primers for constructing the recombinant plasmids pET25b-Nb28-HRP and pET25b-HRP-Nb28, Fig. S1: construction of the recombinant plasmids pET25b-Nb28-HRP and pET25b-HRP-Nb28, Fig. S2: western blot analysis of the auto-induction expression of fusion proteins HRP-Nb28, Fig. S3: SDS-PAGE analysis of the purification of HRP-Nb28 inclusion body, Fig. S4: the performance analysis of HRP-Nb28 fusion protein, Fig. S5: evaluation of the matrix effect of rice and oats samples at different dilutions on the performance of HN-DIA. See DOI: 10.1039/d0ra06576e |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab